

Abstract
Pulmonary arterial hypertension (PAH) is a cardiovascular disorder characterized by elevated pulmonary artery pressure as a result of arterial wall thickening. Patients are 3–4 times more likely to be women than men. This gender discrepancy demonstrates a need for an animal model with similar sex differences. 4,4′-Methylenedianiline (DAPM) is an aromatic amine used industrially in the synthesis of polyurethanes. Chronic, intermittent treatment of male and female rats with DAPM resulted in medial hyperplasia of pulmonary arterioles, exclusively in females, coupled to increases in pulmonary arterial pressures. Significant increases in plasma levels of endothelin-1 (ET-1) and serotonin, but decreases in nitrite , were observed in females treated with DAPM. A decrease was observed in the serum ratio of the estrogen metabolites 2-hydroxyestradiol (2-OHE1)/16α-hydroxyestrogen (16α-OHE1). In females, ET-1, , and 2-OHE1/16α-OHE1 were significantly correlated with peak pressure gradient, an indirect measure of pulmonary arterial pressure. Expression of the serotonin transport protein (SERT) was significantly higher in the arteries of DAPM-treated females. In vitro, DAPM induced human pulmonary vascular smooth muscle cell proliferation and serotonin uptake, both of which were inhibited by treatment with the estrogen receptor antagonist ICI 182,780 or the selective serotonin reuptake inhibitor fluoxetine. DAPM also induced the release of serotonin from human pulmonary endothelial cells in culture, which is blocked by ICI 182,780. Taken together, this suggests that DAPM-mediated dysregulation of serotonin transport is estrogen-receptor dependent. Thus, DAPM-induced PAH pathology may be a new tool to clarify the sex selectivity of PAH disease pathogenesis.
Keywords: pulmonary arterial hypertension; 4,4′-methylenedianiline; serotonin; serotonin transporter; estrogen receptor
Pulmonary hypertension (PH) is a cardiovascular condition defined by increased pressure in the pulmonary arteries at or above 25 mmHg. It can occur under a variety of conditions, often as a secondary complication to an existing disease state. Pulmonary arterial hypertension (PAH) is a markedly severe form of PH. Although PAH can arise for a variety of reasons, the end result is right heart failure and death. It is estimated that 50 000–100 000 people in the United States have PAH and 10–15 per 1 million are diagnosed annually (McGoon and Miller, 2012). Additionally, women are more likely to develop PAH than men, at a ratio of almost 4 to 1 (Frost et al., 2011).
Although it is clear that some patients find substantial improvement in their health and quality of life through the use of current treatments, there is no cure. Thus, the identification of the underlying mechanisms of PAH pathogenesis is critical for the development of new curative strategies.
In an effort to uncover the causes of PAH, several rat models have been established. However, as is typically the case with animal models, all have limitations. Traditionally, chronic exposure to hypoxia (Rabinovitch et al., 1979) or treatment with the plant alkaloid monocrotaline (MCT; Chesney et al., 1974; Hayashi and Lalich, 1967; Rosenberg and Rabinovitch, 1988) has been used to induce PH in rats. However, PH due to chronic hypoxia in animals may not be truly representative of human PAH, due to the fact that hypoxia-induced PH is classified by the WHO as clinically distinct from PAH (Group 3 vs Group 1; Simonneau et al., 2013). More recently, the Sugen model has been developed. This model pairs chronic hypoxia with the vascular endothelial growth factor receptor antagonist Sugen5416 (Tarasevicine-Stewart et al., 2001; Sakao and Koichiro, 2010).
Although certain aspects may make one model preferable to another, all lack any demonstrated sex specificity in the development of PAH. Male rats treated with MCT are just as likely to develop PAH as females (Kay et al., 1967; Meyrick et al., 1980; Rosenberg and Rabinovitch 1988), and the Sugen model was characterized in male rats (Tarasevicine-Stewart et al., 2001; Toba et al., 2014). This is potentially problematic due to the fact that the majority of the human PAH patient population is female. Thus, there is a need for a model that addresses this discrepancy and reveals the underlying connections between PAH pathogenesis and the female sex.
In general, the links between serotonin (5-HT) and PAH pathogenesis have been well established, but yet not completely understood. There is mounting evidence that 5-HT transport could be a link between sex and PAH. For example, mice deficient in the expression of the 5-HT transport protein SERT resist developing PAH (Eddahibi et al., 2000), whereas female mice over-expressing SERT developed PAH, but males did not (White et al., 2011).
4,4′-Methylenedianiline (4,4′-diaminodiphenylmethane (DAPM)) is an aromatic diamine commonly used in the synthesis of polyurethanes and epoxy resins. In high doses (100–250 mg/kg) DAPM treatment in rodents results in hepatobiliary toxicity (Bailie et al., 1993; Kanz et al., 1992). Previously, we have found that oral doses of DAPM (25 mg/kg), which fall at the lowest end of the threshold for hepatic toxicity (25–75 mg/kg; Bailie et al., 1993), can induce vascular medial hyperplasia in the vascular beds in the lungs (Dugas et al., 2004). In this study, we sought to further characterize the effects of low doses of DAPM on the pulmonary vascular in female rats that we first reported in 2004 and to clarify why females may have increased susceptibility to DAPM. It is our belief that effects we observed in female rats suggest that DAPM could be utilized as a novel, chemical-induced, female-specific, model for PAH.
We had several objectives for the work reported here. Our first objective was to analyze and describe the morphological components of DAPM-induced PAH pathophysiology in DAPM-treated male versus female rats. Because the lesion development appeared to involve mainly vascular smooth muscle cell proliferation within the medial layer of the vessel wall, we further hypothesized that the female predominance of the observed effects was due to an alteration in 5-HT transport in vascular smooth muscle. Thus, our second objective was to test whether 5-HT transport contributes to increased female susceptibility to the effects of DAPM on the pulmonary vasculature. Finally, biochemical studies with separate isolates of normal human pulmonary artery cells from both female and males were used to investigate whether DAPM-induced effects on 5-HT release and uptake were mediated in part by interaction with the estrogen receptor (ER).
MATERIALS AND METHODS
Animals
Male and female Sprague-Dawley rats (8–12 weeks of age) were purchased from Harlan (Indianapolis, Indiana). Animals were maintained in an AALAC-accredited animal facility, housed in a 12-h light-dark cycle with ad libitum access to food and water. Animal care and use was in accordance with NIH guidelines and the LSUHSC Institutional Animal Care and Use Committee approved all procedures in advance. Rats were treated with vehicle (35% ethanol) or 25 mg/kg 4,4′-methylenedianiline (DAPM; Sigma-Aldrich, St Louis, Missouri) by oral gavage once weekly for 8, 10, 12, 16, 17, or 22 weeks, over the course of 6 different studies. At the initiation of experiments, all groups contained 6–8 animals; however, some animals were lost during the course of the studies. Thus, at the time of animal sacrifice, each group contained 4–6 animals.
Using pneumothorax under anesthesia (50 mg/kg pentobarbital, i.p.), all animals were euthanized 24 h after the final treatment (ie, week 13). Once euthanasia was ensured, the lungs were gravity inflated through the trachea using 10% buffered formalin. Lungs and liver were collected and fixed in 10% buffered formalin.
Morphometric analysis
Lung tissue specimens were fixed overnight in 4% buffered formalin and were paraffin embedded. Serial histological sections were cut so that the pulmonary arteries could be viewed in cross-section. Hematoxylin-stained lung sections were photographed using a high resolution digital camera interfaced to a Motic B1 series DMB1-223 light microscope. Vessels in the range of 100–600 µm were selected for analysis. The surface areas (mm2) of each vessel (wall plus lumen and lumen only) were determined under 20× magnification using ImageJ software. The wall area was calculated as the difference between the 2 measures. The ratio of the wall-to-lumen area of each vessel was then determined. Three to five sections per animal (6–12 vessels per section) were analyzed. An increase in wall-to-lumen area ratio was considered indicative of PAH pathology.
Ultrasound
Pulmonary arterial pressures were determined weekly by using ultrasound (VisualSonics, Vevo 770), 48-h post-treatment, in male and female rats. In preparation for ultrasound analysis, rats were anesthetized with 5% isoflurane administered in 1 l/min oxygen, were shaved over their heart and ultrasound gel was applied. To ensure the rats remained stabilized while under anesthesia, a heart rate of 300 beats/min was maintained throughout the procedure. Using the Pulse-Wave Doppler mode, the heart was visualized on ultrasound with a 710B probe and the pulmonary arterial measurements (peak pulmonary valve gradient, peak pulmonary valve velocity, mean pulmonary valve velocity, and mean pulmonary valve gradient) were calculated via the Velocity Time Interval function. For comparison, a baseline measure was taken in each prior to initial DAPM treatment (week 0).
Right heart catheterization and Fulton index
Right ventricular systolic pressure (RVSP) was determined via right heart catheterization. Female rats were treated with vehicle (35% ethanol) or DAPM (25 mg/kg) once weekly for 10 weeks. Twenty-four hours after the final dose, the rats were anesthetized with isofluorane, intubated, and mechanically ventilated (2% isofluorane in O2, 3 l/min). The chest cavity was opened to expose the heart, with care taken to not disturb the lungs. A Millar solid state mouse pressure catheter was then introduced into the right ventricle (RV) through a small hole (made with a 23-gauge needle) in the right ventricular wall. After a 2-min stabilization period, right ventricular pressures were measured as an index of pulmonary arterial pressure. Euthanasia was then be confirmed by pneumothorax and the whole heart was removed. The atria were carefully dissected away, and the RV was separated from the left ventricle and septum (LV + S). The tissues were weighed and utilized for analysis of RV hypertrophy. The Fulton Index for each heart was determined [RV/(LV + S)]. The RV/total body weight for each animal was also calculated.
Western blot analysis for SERT protein levels in pulmonary arteries
Segments of pulmonary artery were taken at the conclusion of the ultrasound study and were frozen at −80°C. Pulmonary arteries from vehicle- and DAPM-treated females were homogenized in RIPA buffer containing protease inhibitors and the protein concentration in each sample was determined using the BCA protein assay (Thermo Scientific, Rockford, Illinois). Equivalent amounts of protein were loaded into lanes of a Novex 4–20% Tris-glycine gradient gel (Invitrogen, Carlsbad, California). The gel was electrophoresed for 2 h at 125 V and then transferred onto a PVDF membrane for 1 h at 100 V. The membrane was blocked for 1 h at 4°C. After washing twice, the membrane was incubated overnight with 1:100 SERT polyclonal antibody (rabbit anti-rat; Millipore, Temecula, California) or 1:500 GAPDH polyclonal antibody (rabbit anti-rat; Santa Cruz Biotechnology, Dallas, Texas) in phosphate-buffered saline (PBS)/Tween buffer. After 2 more washes, the membrane was incubated for 1 h with a 1:20 000 dilution of secondary antibody (anti-rabbit, GE Healthcare, Piscataway, New Jersey). The blot was then washed twice more and developed using the ECL plus detection system (GE Healthcare). The blots were developed on X-ray film, the film was imaged and densitometric analysis was performed using BioRad Quantity One software. To confirm that equivalent amounts of protein were loaded into each well, the bands were normalized to total GAPDH protein.
Measurement of plasma analytes
After euthanasia was ensured, blood was drawn from the vena cava into 10-ml EDTA tubes that were then centrifuged for the collection of plasma.
Plasma endothelin-1 (ET-1), estradiol and serotonin (5-HT) levels were determined by enzyme-linked immunosorbent assay using kits obtained from SABiosciences (Valencia, California), Cayman Chemical Company (Ann Arbor, Michigan), and Enzo Life Sciences (Farmingdale, New York), respectively.
Plasma brain natriuretic peptide (BNP) levels were measured by enzyme immunoassay using a RayBio Human/Mouse/Rat Brain Natriuretic Peptide EIA Kit (RayBiotech, Norcross, Georgia).
Total nitrite was determined by measuring both nitrite and nitrosothiols (as a storage form of NO) using a Sievers 280i Nitric Oxide Analyzer. NO metabolite stabilizing solution (21 ml of KFeCN solution, 2.5 ml of NEM solution and 1.5 ml of NP40) was added to plasma aliquots at a 1:5 ratio (volume:volume) immediately after centrifugation and prior to analysis. Nitrate levels were not determined.
Measurements of plasma 2-hydroxyestrone and 16α-hydroxyestrone
Plasma levels of the estradiol metabolites 2-hydroxyestrone (2-OHE1) and 16α-hydroxyestrone (16α-OHE1) were determined by enzyme immunoassay with an Estramet Urinary Estrogen Metabolite kit (Immuna Care Corporation, Blue Bell, Pennsylvania). The kit was adapted to measure plasma levels as described by Jernström et al. (2003).
Cell culture
HPAVSMC and human pulmonary artery endothelial cells (HPAEC) isolated from healthy, pre-menopausal women and healthy men were purchased from Cell Applications, Inc. (San Diego, California) and Invitrogen (Carlsbad, California), respectively. Cells were grown to confluence in DMEM containing 10% FBS (HPAVSMC) and MCDB131 containing 10% FBS (HPAEC).
Note that because an estrogenic effect of DAPM was expected, estrogenic components, such as phenol red, were removed from the cell culture medium during experiments. Also, all FBS used was from the same lot, so that the serotonin concentration is conserved between experiments.
Assessment of cell proliferation
HPAVSMC were seeded into 96-well plates. When the HPAVSMC reached ∼80% confluence, cells were washed with PBS. To synchronize cell cycle, cells were growth arrested by incubation in phenol red-free DMEM containing 0.1% FBS for 72 h. The medium was then removed and the cells stimulated with DMEM containing 5 nM 5-HT and treated with 0.5, 1, 2.5, 5, or 10 μM DAPM, or vehicle (0.05% ethanol). In some experiments, cells were co-treated with 5 nM ICI 182 780, a nonselective ER antagonist, or 10 μM fluoxetine, a selective serotonin reuptake inhibitor (SSRI). Cells were then incubated at 37°C and were assayed for proliferation at 24, 48, or 72 h. Proliferation was determined by counting cells using a hemocytometer, and by measuring DNA synthesis using the BrDU (5-bromo-2′-deoxyuridine) Labeling and Detection Kit I (Roche Molecular Biochemicals, Indianapolis, Indiana).
Measurement of 5HT uptake
HPVSMC were seeded in 6-well plates and allowed to reach ∼80% confluence. Then, the medium was removed, the cells were rinsed with PBS and were growth arrested by incubation in phenol-red free DMEM containing 0.1% FBS for 72 h. The medium was then replaced with phenol red-free DMEM containing 5 nM 5-HT and the cells were incubated with 10 μM DAPM ± 5 nM ICI 182 780 (Tocris Biosciences, Minneapolis, Minnesota) or 10 μM fluoxetine (Sigma-Aldrich, St. Louis, Missouri) for 24 h. [3H]-5-HT uptake was determined as described by Dodson et al. (2004). Radioactivity was measured by liquid scintillation counting and 5-HT uptake was normalized for protein content.
Measurement of 5-HT release
HPAEC were seeded in 6-well plates and allowed to reach ∼80% confluence. The medium was then removed and cells rinsed with PBS. Cells were then incubated with new medium containing 10 μM DAPM, 10 μM DAPM + 5 nM ICI 182 780, 5 nM ICI 182 780 or vehicle for 24 h. Spent medium was collected and assayed for 5-HT using a human 5-HT ELISA kit (Abnova, Taipei City, Taiwan).
Statistical analysis
Values given in figures and tables represent means ± SEM. Statistical analysis was performed using Prism 6 for Mac OS X (Graphpad Software, Inc.). Statistical significance was determined using 2-way analysis of variance (ANOVA), 1-way ANOVA or Student’s t test, where appropriate. Significance between individual data points was determined using Newman-Keuls multiple comparisons test. In all cases, p < .05 was accepted as statistical significance.
RESULTS
Morphometric Analysis of Pulmonary Arteries
The pulmonary arteries of vehicle-treated male and female rats appear normal and exhibited a thin-walled medial morphology (Fig. 1a and c, respectively). The pulmonary arteries of DAPM-treated females exhibited markedly thickened and proliferated medial layers (Fig. 1d); however, the pulmonary arteries of DAPM-treated males had morphologies similar to vehicle-treated animals (Fig. 1b). No neointima was observed.
FIG. 1.

Representative cross-sections of pulmonary arteries from male and female rats treated with 25 mg/kg DAPM or ethanol vehicle. Pulmonary arteries from DAPM-treated males (b) did not differ in appearance from pulmonary arteries of vehicle-treated males (a), after 12 weeks of treatment. The pulmonary arteries of females treated with DAPM have thicker walls, and narrower lumens (d), compared with those from females treated with ethanol vehicle (c), after 12 weeks of treatment. ×10 magnification.
When the wall-to-lumen area ratios were calculated, the ratios for DAPM-treated females increased over time, culminating in a higher wall-to-lumen ratio for DAPM- compared with vehicle-treated females after 17 weeks (3.6-fold increase over vehicle; p < .05; Fig. 2). The ratios at 17 weeks were also significantly higher than those calculated for 8, 10, and 12 weeks of DAPM treatment (4.3-, 3.2-, and 2-fold, respectively; Fig. 2b). No such changes were observed between male treatment groups (Fig. 2a). Initially, the female 17 week study was intended to continue through 22 weeks, but was shortened due to animal mortality. As a result, subsequent studies were shortened to ensure all animals would remain viable and did not exhibit moribund behavior for the entirety of the study.
FIG. 2.

Wall area to lumen area ratios calculated for pulmonary arteries of rats treated with DAPM (25 mg/kg), or ethanol, for 8, 12, 16, or 22 weeks (male rats; a) and 8, 10, 12, or 17 weeks (female rats; b). Data are expressed as mean ± SEM, n = 3–6 animals. *Indicates significant difference from 17 weeks vehicle treatment, p < .05. φ Indicates significant difference from 8 weeks DAPM treatment, p < .05.
Hemodynamic Measurements
Pulmonary arterial pressures were determined weekly by ultrasound over the course of a 12-week study. At 9 weeks, the peak pulmonary valve gradient measure of DAPM-treated females was increased over that of vehicle-treated females and remained elevated through the end of the study (p < .05; Fig. 3b). No change was observed between male treatment groups (Fig. 3a).
FIG. 3.

Pulmonary peak pressure gradient—determined weekly by ultrasound—for DAPM (25 mg/kg)- or ethanol-treated male (a) and female (b) rats. Data expressed as mean ± SEM, n = 4–6 animals. *Indicates significant difference from vehicle-treated animals, p < .05.
Once it was determined that 9 weeks treatment was sufficient for observing a significantly increased pulmonary arterial pressures in DAPM-treated female rats, a subsequent study was conducted whereby right ventricular pressures were determined directly via right heart catheterization in female rats after once weekly treatment with DAPM or vehicle for 10 weeks. Because these were meant only to confirm our Doppler measurements of increased pulmonary arterial pressures (assessed as peak pressure gradients), only female animals were utilized. When compared with vehicles, a significant increase in RVSP (1.25-fold over vehicle treatment; Table 1) was observed in DAPM-treated rats.
TABLE 1.
Measures of right ventricular pressures and right ventricular hypertrophy in female rats treated with DAPM (25 mg/kg) or ethanol vehicle, once a week for 10 weeks.
| RVSP (mmHg) | RV Weight (g) | Fulton Index RV/(LV + S) | RV/Body Weight | |
|---|---|---|---|---|
| Vehicle | 19.1 ± 0.64 | 0.113 ± 0.011 | 0.159 ± 0.014 | 0.426 ± 0.037 |
| DAPM | 23.6 ± 1.55* | 0.156 ± 0.009* | 0.213 ± 0.015* | 0.583 ± 0.037* |
Data expressed as means ± SEM, n = 5–6 animals per group.
*Indicates significant difference from vehicle-treated animals, p < .05.
RVSP, right ventricular systolic pressure; RV, right ventricle; LV, left ventricle; S, septum.
Right Ventricular Hypertrophy
We calculated the Fulton Index (RV/[LV+S]) and found that the ratio of the weight of the RV to the left ventricle plus septum (LV+S) was increased in female rats treated with DAPM compared with those treated with vehicle after 10 weeks (1.34-fold over vehicle; p < .05; Table 1). Furthermore, BNP levels were significantly elevated in the DAPM treatment group (Fig. 4).
FIG. 4.

Plasma BNP (pg/ml) levels in female rats treated weekly with DAPM (25 mg/kg), versus vehicle-treated female rats, for 10 weeks. *p < .05, n = 5–6 animals.
Plasma Analytes
In females, plasma levels of ET-1 were elevated in DAPM- compared with vehicle-treated rats (p < .05). No difference was observed among male treatment groups. When compared with the vehicle-treated females, plasma nitrite levels (indicative of nitric oxide levels) were significantly reduced. Additionally, the vehicle-treated females had higher nitrite levels than their male counterparts (p < .05; Table 2).
TABLE 2.
Plasma ET-1, serotonin and nitrite in male and female rats treated once a week for 12 weeks with DAPM (25 mg/kg) or ethanol vehicle
| ET-1 (pg/ml) |
Serotonin (ng/ml) |
Nitrite (nM) |
||||
|---|---|---|---|---|---|---|
| Male | Female | Male | Female | Male | Female | |
| Vehicle | 6.53 ± 0.57 | 9.16 ± 1.27 | 43.1 ± 6.33 | 54.2 ± 3.71 | 117 ± 7.64 | 283 ± 58.7** |
| DAPM | 7.85 ± 1.37 | 14.5 ± 0.39* | 60 ± 7.87 | 99.8 ± 19.8* | 157 ± 30.5 | 140 ± 14.8* |
Data expressed as means ± SEM, n = 4–6 animals per group.
*Indicates significant difference from vehicle-treated animals of the same sex, p < .05. **Indicates significant difference from males of same treatment group, p < .05.
DAPM-treated females were found to have higher plasma 5-HT levels than those treated with vehicle (p < .05; Table 2). No difference in treatment groups was observed for the males.
DAPM treatment did not produce a significant change in plasma estrogen levels in females. The ratio of the estrogen metabolites 2-hydroxyestrone (2-OHE1) and 16α-hydroxyestrone (16α-OHE1) was found to be trending downward in the DAPM-treated animals, but the decrease was not significant (p = .07; Table 3). However, although there was no change in levels of 2-OHE, the measured level of 16α-OHE1 was increased by 22% (p < .05).
TABLE 3.
Plasma estradiol and the ratio of the estrogen metabolites 2-OHE1 (2-hydroxyestrone) and 16α-OHE1 (16α-hydroxyestrone) in female rats treated with DAPM (25 mg/kg) or ethanol vehicle once a week for 12 weeks
| 17β-Estradiol (pg/ml) | 2-OHE1 (pg/ml) | 16α-OHE1(pg/ml) | 2-OHE1/16α-OHE1 | |
|---|---|---|---|---|
| Vehicle | 44.2 ± 4.70 | 233.9 ± 8.58 | 339.4 ± 7.78 | 0.692 ± 0.035 |
| DAPM | 40.4 ± 3.28 | 251.6 ± 3.83 | 413.5 ± 1.04* | 0.609 ± 0.008 |
Data expressed as means ± SEM, n = 4–6 animals per group.
*Indicates significant difference from vehicle-treated animals, p < .05.
SERT Expression in the Pulmonary Arteries
SERT protein expression was increased by 67% in the pulmonary arteries of DAPM-treated females compared with those treated with vehicle (p < .05; Fig. 5).
FIG. 5.

Representative Western blots illustrating SERT protein expression in the pulmonary arteries of male and female rats treated with DAPM (25 mg/kg) or ethanol, for 12 weeks. Each lane represents a sample from a single animal. GAPDH was used as a loading control, after the membrane was stripped. Relative density of SERT bands was quantified and normalized to corresponding GAPDH bands. Data expressed as means ± SEM, n = 4–6 animals/group. *Indicates significant difference between DAPM-treated groups.
HPAEC 5-HT Release
In HPAEC isolated from healthy women, DAPM treatment increased the release of 5-HT into the culture medium (p < .05). Treatment with the nonselective ER antagonist ICI 182 780 blocked 5-HT release (Fig. 6a). DAPM treatment did not stimulate 5-HT release in healthy male HPAEC isolates (Fig. 6b).
FIG. 6.
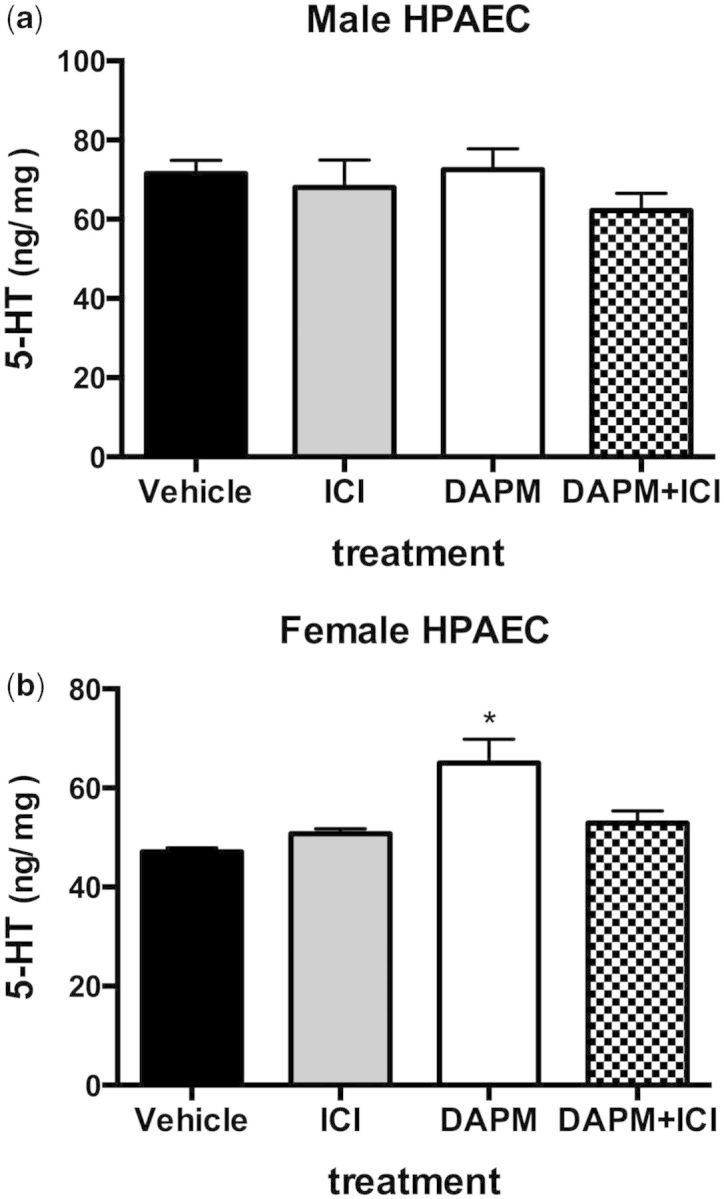
5-HT release from HPAEC females (a) or males (b) isolates. Cells were treated with ethanol vehicle, 5 nM ICI 182 780, 10 µM DAPM, or DAPM+ICI, for 24 h. 5-HT in culture media was determined by ELISA. Data are expressed as mean ± SEM, n = 3 experiments. *Indicates significant difference from ethanol vehicle treatment, p < .05.
HPAVSMC 5-HT Uptake
HPAVSMC isolated from women were stimulated to take up [3H]-5-HT when exposed to DAPM (Fig. 7). This effect was abolished upon treatment with the SSRI fluoxetine (p < .05; Fig. 7a). The DAPM-induced [3H]-5-HT uptake was also inhibited by the presence of ICI 182 780 (p < .05; Fig. 7b).
FIG. 7.

Effect of fluoxetine (Fluox; a) or ICI (b) on the DAPM-induced uptake of [3H]-5-HT by HPAVSMC female isolates. Cells were treated with ethanol vehicle, 5 nM Fluox (or 5 nM ICI), 10 µM DAPM, or DAPM+fluoxetine (or DAPM+ICI), for 24 h. Then, [3H]-5-HT added to the cells. Cells were rinsed then lysed and [3H]-5-HT in lysates was determined by scintillation counter. Radioactivity was expressed at desiccations per minute and normalized to total protein (mg/ml) of each lysate sample. Data are expressed as means ± SEM, n = 3 experiments. *Indicates significant difference from ethanol vehicle treatment, p < .05.
HPAVSMC Proliferation
HPAVSMC proliferation was assessed by increase in cell number 24 h after treatment with 6 increasing doses of DAPM: 0, 0.5, 1, 2.5, 5, and 10 µM for HPAVSMC isolates from women (Fig. 8b); 0, 2.5, 5, 10, 25, and 50 µM for HPAVSMC isolates from men (Fig. 8a). Female HPAVSMC proliferation was increased at the 2.5 and 5 µM doses of DAPM (p < .05). It was observed that doses above 10 µM were toxic to the cells, which, in response to treatment, began to lift off the plate. Male HPAVSMC did not proliferate in response to DAPM treatment, even when the dose was increased 10-fold over that which was administered to female cell isolates (Fig. 8a).
FIG. 8.

DAPM-induced HPAVSMC proliferation, as indicated by increase in cell number. Male isolates (a) and female isolates (b). Cells were incubated with a range of DAPM doses for 24 h. Data are expressed as means ± SEM, n = 3–4 experiments. *Indicates significant difference from ethanol vehicle treatment, p < 0.05.
The effects of fluoxetine and ICI 182 870 on DAPM-induced HPAVSMC proliferation were determined by BrDU incorporation (Fig. 9). DAPM treatment alone showed a significant increase in BrDU incorporation compared with cotreatment with ICI 182 780 or fluoxetine at all 5 concentrations of DAPM: 0.5, 1, 2.5, 5, and 10 µM.
FIG. 9.

Effects of Fluox or ICI on DAPM-induce HPAVSMC proliferation (female isolates only). Cell proliferation in DAPM, DAPM + ICI and DAPM + Fluox treatment groups was determined by BrDU incorporation and presented as percentages of BrDU incorporation measured in vehicle treated cells. Data are expressed as means ± SEM, n = 3 experiments. *Indicates significant difference from DAPM + Fluox treatment group, p < .05. φ Indicates significant difference from DAPM + ICI treatment group, p < .05.
DISCUSSION
For every man diagnosed with PAH, there are∼4 women (Badesch et al., 2010). Several studies with experimental models of PAH have reported sex related effects in their results, but no model has completely elucidated the female predisposition for developing PAH. Furthermore, although over 30 agents have been found to be effective in treating disease progression in experimental PAH, these achievements have not translated into clinical efficacy (Stenmark et al., 2009). In order to better understand PAH disease pathogenesis per se, and to elucidate why women are particularly more susceptible, new animal models are needed.
We have found that the synthetic chemical compound DAPM induces a PAH pathophysiology almost exclusively in female rats. Furthermore, given the experimental evidence for a possible relationship between PAH and 5-HT and sex, we hypothesized that these DAPM-induced effects were related to 5-HT transport.
The narrowing and occlusion of precapillary pulmonary vessels, primarily due to aberrant vascular smooth muscle cell proliferation, characterize PAH pathology. We observed that DAPM treatment induced PAH-like pulmonary artery vascular smooth muscle cell hyperplasia, and narrowing of pulmonary vessels, in female rats (Figs. 1 and 2). Moreover, these changes coincided with increased pulmonary arterial pressures (Fig. 3).
PAH is fatal due to the stress and strain that elevated pulmonary arterial pressure imparts to the RV of the heart. When the pressure in the pulmonary circulation is increased, a compensatory cardiac hypertrophy is triggered, whereby the cardiomyocytes increase in size in order to meet the increasing demands on the muscle, resulting in an enlarged RV. We found this to be the case in DAPM-treated females (Table 1). Interestingly, we found that changes in RVSP and the Fulton Index occurred at the same time (ie, 10 weeks) as our observations of significant elevations in pulmonary artery peak pressure gradients, but prior to significant increases were observed for the calculated wall-to-lumen ratios. Rather than speculate that these findings reflect temporal events in the disease pathogenesis, we posit that compared with morphometric analyses for vascular wall thickening, RVSP and the Fulton Index may simply be more sensitive indicators of DAPM-induced effects on the pulmonary vasculature.
With regard to changes in the right heart, cardiomyocytes also respond to stress by releasing pro-BNP, which is converted into its active form BNP in the plasma. Thus, increases in plasma BNP levels are indicative of cardiac dysfunction (Omland et al., 1996; Tulevski et al., 2001). PAH patients have significantly elevated plasma BNP and pro-BNP (Gan et al., 2006; Leutche et al., 2004; Nagaya et al., 1998). These increases are significantly correlated to other clinically relevant hemodynamic parameters associated with PAH (Leutche et al., 2005), including the severity and overall survival of PAH patients (Andreassen et al., 2006; Nagaya et al., 2000). Our DAPM-treated female rats exhibited increased levels of plasma BNP (Fig. 4), thus confirming that DAPM induces a cardiac dysfunction reminiscent of that observed in humans.
Plasma biomarkers can also be used to characterize vascular dysfunction. In the case of PAH, endothelial cell stress is thought to contribute to the dysfunctional hyperplastic state of the vascular smooth muscle cells within pulmonary arteries (Xu et al., 2011). A common indicator of endothelial dysfunction is ET-1 synthesis. ET-1 can induce vasoconstriction and down-regulate the expression of NO synthase, resulting in reduced NO production. Elevated ET-1 in PAH patients is significantly correlated to disease severity (Rubens et al., 2001). Additionally, ET-1 receptor antagonism is an effective treatment in a portion of the PAH patient population (Davie et al., 2009). Again, only DAPM-treated female rats displayed changes in PAH-related clinical plasma biomarkers (Table 2).
It is paradoxical that women develop PAH more often than men, because the primary female hormone, estrogen, has demonstrated vascular-protective properties. Pre-menopausal women are less likely to develop cardiovascular disease than men; however, incidence increases in post-menopausal women (Stampfer et al., 1991). Estrogen is thought to confer protection, in part, due to its inhibitory effects on vascular smooth muscle cell proliferation (Farhat et al., 1996; Geraldes et al., 2002; Lavigne et al., 1999). Estrogen is also protective in animal models of PH. Moreover, estradiol administration prevented smooth muscle cell hyperplasia in both chronic hypoxia-induced and MCT-induced PAH in rats (Farhat et al., 1993, 1995). These findings in animals are at odds with the preponderance of females in the human PAH patient population. Moreover, PAH is observed in both women of childbearing age and in those on estrogen replacement (Miller et al., 1987 ; Morse et al., 1999). Thus, the estrogen paradox is a significant challenge to our understanding of the development of PAH.
In our female rats, DAPM treatment did not significantly alter circulating levels of estradiol (Table 3). However, we also measured levels of the estrogen metabolites 2-OHE1 and 16α-OHE1, which are major estrogen metabolites (albeit not the only ones). These two are of particular interest, because 2-OHE1 has weak and/or antiestrogenic effects (Scheinder et al., 1984) whereas 16α-OHE1 has been shown to be mitogenic (Telang et al., 1992). Epidemiological studies have revealed that women with a high 2-OHE1/16α-OHE1 ratio have reduced risk of developing breast cancer (Fowke et al., 2003; Meilhan et al., 1998; Muti et al., 2000). Therefore, it is possible that a change in circulating estrogen metabolites, having actions different from those mediated by the parent compound, could play a role in PAH pathogenesis. Although we did not observe significant differences in the ratio of the 2 metabolites, DAPM-treated females did exhibit significantly higher plasma levels of the proproliferative metabolite 16α-OHE1 (Table 3). It is possible that changes in the estrogen metabolite profile are responsible for some the effects of DAPM observed in these females. Further investigation into the relationship between DAPM and estrogen is on-going in our laboratory.
DAPM treatment also resulted in a significant increase in circulating levels of 5-HT in female rats (Table 2). Additionally, these females exhibited increased expression of SERT in their pulmonary arteries (Fig. 5). Considering the established links between 5-HT and PAH, we hypothesized that DAPM-related effects could be mediated by 5-HT. PAH patients have elevated plasma 5-HT levels (Hervé et al., 1995) and human pulmonary artery smooth muscle cells (HPAVSMC)isolated from PAH patients are more sensitive to the proproliferative effects of 5-HT than are normal HPAVSC (Eddahibi et al., 2001). Also, PAH patients have increased expression of the SERT mRNA in their lungs, and SERT appears to be particularly localized in the media of the pulmonary arteries, suggesting there is an increase in 5-HT uptake in the pulmonary vasculature (Eddahibi et al., 2001). Additionally, the use of certain drugs that interfere with 5-HT transport in the pulmonary vasculature, such as the appetite suppressants fenfluramine and aminorex (Rothman et al., 1999), have been correlated to increased incidence of PAH (Abenhaim et al., 1996; Kay et al., 1971).
We found evidence that DAPM may be inducing PAH pathology in female rats by disrupting the balance of available 5-HT and 5-HT uptake in the pulmonary circulation. Therefore, we sought to test whether DAPM would induce similar effects in human pulmonary artery cells in culture. DAPM-induced proliferation was observed in the female isolates of HPAVSMC (Fig. 8b). This is likely due, in part, to DAPM-induced uptake of 5-HT in female HPAVSMC (Fig. 7). Interestingly, male HPAVSMC did not proliferate in response to DAPM (Fig. 8a). This data are in agreement with our observations of medial hyperplasia in DAPM-treated female but not male rats. Even at doses 10-fold higher, the number of male cells did not increase. We had previously observed similar sex selectivity in aortic smooth muscle cells isolated from rats (Hebert et al., 2011). In that case, both male and female cells proliferated in response to DAPM; however, the female response was significantly higher than that of the male isolates. Additionally, higher doses of DAPM were required to elicit responses in aortic cells. We also found that DAPM-induced 5-HT release from HPAEC was also specific to female cell isolates (Fig. 6). The present and previous observations suggest that cells of the pulmonary vessels are especially susceptible to the proproliferative effects of DAPM, and that the female vasculature in general is uniquely sensitive DAPM. Moreover, data presented here suggest that the sex-specific effects of DAPM may be related to increased 5-HT levels and 5-HT transport.
Suppression of DAPM-induced responses in both female HPAEC and HPAVSMC with the ER inhibitor ICI 182 780 indicates that the effects of DAPM are at least partially mediated via one or more of the ERs. The next step will be to determine which ER is primarily responsible for mediating the effects of DAPM, and if DAPM treatment alters the ratio of ER expression in endothelial and/or smooth muscle cells.
To summarize, we have observed PAH pathology in female rats treated with DAPM. There is evidence to suggest that DAPM induces this pathology via 5-HT-related mechanisms. This is borne out by our in vitro data demonstrating DAPM-induced 5-HT release and uptake in female HPAVSMC. DAPM treatment also promoted female HPAVSMC proliferation. We propose that DAPM-induced PAH pathology in rats is a novel, relevant chemical model of PAH and has potential as a valuable tool in furthering our understanding of the sex-selective nature of PAH disease pathogenesis, hopefully leading to the development of more effective therapies.
FUNDING
This work was supported by the American Heart Association (14GRNT20490300).
REFERENCES
- Abenhaim L., Moride Y., Brenot F., Rich S., Benichou J., Kurz X., Higenbottam T., Oakley C., Wouters E., Aubier M., et al. (1996). Appetite-suppressant drugs and the risk of primary pulmonary hypertension. International Primary Pulmonary Hypertension Study Group. N. Engl. J. Med. 335, 609–616. [DOI] [PubMed] [Google Scholar]
- Andreassen A. K., Wergeland R., Simonsen S., Geiran O., Guevara C., Ueland T. (2006). N-terminal pro-B-type natriuretic peptide as an indicator of disease severity in a heterogeneous group of patients with chronic precapillary pulmonary hypertension. Am. J. Cardiol. 98, 525–529. [DOI] [PubMed] [Google Scholar]
- Badesch D. B., Raskob G. E., Elliot G., Krichman A. M., Farber H. W., Frost A. E., Barst R. J., Benza R. L., Liou T. G., Turner M., et al. (2010). Pulmonary arterial hypertension: Baseline characteristics from the REVEAL Registry. Chest 137, 376–387. [DOI] [PubMed] [Google Scholar]
- Bailie M. B., Mullaney T. P., Roth R. A. (1993). Characterization of Acute 4,4′-Methylene Dianiline Hepatotoxicity in Rats. Environ Health Perspect 124, 25–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesney C. F., Allen J. R., Hsu I. C. (1974). Right ventricular hypertrophy in monocrotaline pyrrole treated rats Exp Mol Pathol 20, 257–268. [DOI] [PubMed] [Google Scholar]
- Davie N. J., Schermuly R. T., Weissmann N., Grimminger F., Ghofrani H. A. (2009). The science of endothelin-1 and endothelin receptor antagonists in the management of pulmonary arterial hypertension: current understanding and future studies. Euro. J. Clin. Invest. 39(Suppl. 2), 38–49. [DOI] [PubMed] [Google Scholar]
- Dodson A. M., Anderson G. M., Rhoden K. J. (2004). Serotonin uptake and metabolism by cultured guinea pig airway smooth muscle cells. Pulm. Pharmacol. Ther. 17, 19–25. [DOI] [PubMed] [Google Scholar]
- Dugas T. R., Kanz M. F., Hebert V. Y., Hennard K. L., Hanlin L., Santa Cruz V., Conklin D., Boor P. J. (2004). Vascular medial hyperplasia following chronic, intermittent exposure to 4,4’-methylenedianiline. Cardiovasc. Toxicol. 4, 85–96. [DOI] [PubMed] [Google Scholar]
- Eddahibi S., Hanoun N., Lanfumey L., Lesch K. P., Rafferstin B., Hamon M., Adnot S. (2000). Attenuated hypoxic pulmonary hypertension in mice lacking the 5-hydroxytryptamine transporter gene. J. Clin. Invest. 105, 1555–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddahibi S., Humbert M., Fadel E., Raffestin B., Darmin M., Capron F., Simonneau G., Dartevelle P., Hamon M., Adnot S. (2001). Serontonin transporter overexpression is responsible for pulmonary artery smooth muscle cell hyperplasia in primary pulmonary hypertension. J. Clin. Invest. 108, 1141–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhat M. Y., Chen M. F., Bhatti T., Iqbal A., Cathapermal S., Ramwell P. W. (1993). Protection by oestradiol against the development of cardiovascular changes associated with monocrotaline pulmonary hypertension in rats. Br. J. Pharmacol. 110, 719–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhat M. Y., Lavigne M. C., Ramwell P. W. (1996). The vascular protective effects of estrogen. FASEB J. 10, 615–624. [PubMed] [Google Scholar]
- Farhat M. Y., Roman C. D., Shaker M., Lavigne M., Massaro D., Ranwell P. W. (1995). Protection by oestradiol 17β against the development of hyposic pulmonary hypertension in the rat. Endothelium 3, 201–207. [Google Scholar]
- Fowke J. H., Qi D., Bradlow H. L., Shu X. O., Gao Y. T., Cheng J. R., Jin F., Zheng W. (2003). Urinary estrogen metabolites and breast cancer: differential pattern of risk found with pre- versus post-treatment collection. Steroids, 68, 65–72. [DOI] [PubMed] [Google Scholar]
- Frost A. E., Badesch D. B., Barst R. J., Benza R. L., Elliot G., Farber H. W., Krichman A., Liou T. G., Raskob G. E., Wason P., Feldkircher K., Turner M., McGoon M. D. (2011). The changing pictures of patients with pulmonary arterial hypertension in the United States. Chest 139, 128–137. [DOI] [PubMed] [Google Scholar]
- Gan C. T., McCann G. P., Marcus J. T., van Wolferen S. A., Twisk J. W., Boonstra A., Postmus P. E., Vonk-Noordegraaf A. (2006). NT-pro-BNP reflects right ventricular structure and function in pulmonary hypertension. Eur. Respir. J. 28, 1190–1194. [DOI] [PubMed] [Google Scholar]
- Geraldes P., Sirois M. G., Bernatchez P. N., Tanguay J. F. (2002). Estrogen regulation of endothelial and smooth muscle cell migration and proliferation: Role p38 and p42/44 mitogen-activated protein kinase. Atherioscler. Thromb. Vasc. 22, 1585–1590. [DOI] [PubMed] [Google Scholar]
- Hayashi Y., Lalich J. J. (1967). Renal and pulmonary alterations induced in rats by a single injection of monocrotaline. Proc Soc Exp Biol Med 124, 392–396. [DOI] [PubMed] [Google Scholar]
- Hebert V. Y., Jones B. C., Mifflin R. C., Dugas T. R. (2011). Role of COX-2 in the bioactivation of methylenedianiline and in its proliferative effects in vascular smooth muscle cells. Cardiovasc. Toxicol. 11, 316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hervé P., Launay J. M., Scrobohaci M. L., Brenot F., SImonneau G., Petitpretz P., Poubeau P., Cerrina J., Duroux P., Drouet L. (1995). Increased plasma serotonin in primary pulmonary hypertension. Am. J. Med. 99, 249–254 [DOI] [PubMed] [Google Scholar]
- Jernström H., Klug T. L., Sepkovic D. W., Bradlow H. L., Narod S. A. (2003). Predictors of the plasma ratio of 2-hydroxyestrone to 16α-hydroxyestrone among pre-menopausal, nulliparous women from four ethnic groups. Carcinogenesis 24, 991–1005. [DOI] [PubMed] [Google Scholar]
- Kanz M. F., Gunasena G. H., Kaphalia L., Hammond D. K., Syed Y. A. (1998). A Minimally Toxic Dose of Methylene Dianiline Injures Biliary Epithelial Cells in Rats. Toxicology and Applied Pharmacology 150, 414–426. [DOI] [PubMed] [Google Scholar]
- Kay J. M., Harris P., Heath D. (1967). Pulmonary hypertension produced in rats by ingestion of Crotalaria spectabilis seeds. Thorax 22, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay J. M., Smith P., Heath D. (1971). Aminorex and the pulmonary circulation. Thorax 26, 262–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavigne M. C., Ramwell P. W., Clarke R. (1999). Inhibition of estrogen receptor function promotes porcine coronary artery smooth muscle cell proliferation. Steroids 64, 472–480. [DOI] [PubMed] [Google Scholar]
- Leutche H. H., Holzapfel M., Baumgartner R. A., Ding I., Neurohr C., Vogeser M., Kolbe T., Schwaiblmair M., Behr J. (2004). Clinical significance of brain natriuretic peptide in primary pulmonary hypertension. J. Am. Coll. Cardiol. 43, 764–770. [DOI] [PubMed] [Google Scholar]
- Leutche H. H., Holzapfel M., Baumgartner R. A., Neurohr C., Vogeser M., Behr J. (2005). Characterization of brain natriuretic peptide in long-term follow-up of pulmonary arterial hypertension. Chest 128, 2368–2374. [DOI] [PubMed] [Google Scholar]
- McGoon M. D., Miller D. P. (2012). REVEAL: A contemporary US pulmonary arterial hypertension registry. Eur. Respir. Rev. 21, 8–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meilhan E. N., De Stavola B., Allen D. S., Fentiman I., Bradlow H. L., Sepkovic D. W., Kuller L. H. (1998). Do urinary oestrogen metabolites predict breast cancer? Guernsey III cohort follow-up. Br. J. Cancer. 78, 1250–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyrick B., Gamble W., Reid L. (1980). Development of Crotalaria pulmonary hypertension: hemodynamic and structural study. Am J Physiol 39(Heart Circ Physiol 8), H692–H702. [DOI] [PubMed] [Google Scholar]
- Miller M. H. (1987). Pulmonary hypertension, systemic lupus erythematosis, and the contraceptive pill: another report. Ann. Rheum. Dis. 46, 159–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse J. H., Horn E. M., Barst R. J. (1999). Hormone replacement therapy: A possible risk factor in carriers of familial primary pulmonary hypertension. Chest 116, 847. [DOI] [PubMed] [Google Scholar]
- Muti P., Bradlow H. L., Micheli A., Krogh V., Freudenheim J. L., Schüneman H. J., Stanulla M., Yang J., Sepkovic D. W., Trevisan M., et al. (2000). Estrogen metabolism and risk of beast cancer: A prospective study of the 2:16α-hydroxyestrone ratio in premenopausal and postmenopausal women. Epidemiology 11, 635–640. [DOI] [PubMed] [Google Scholar]
- Nagaya N., Nishikimi T., Okano Y., Uemastu M., Satoh T., Kyotani S., Kuribayashi S., Hamada S., Kakishita M., Nakanishi N., et al. (1998). Plasma brain natriuretic peptide levels increase in proportion to the extent of right ventricular dysfunction in pulmonary hypertension. J. Am. Coll. Cardiol. 31, 202–208. [DOI] [PubMed] [Google Scholar]
- Nagaya N., Nishikimi T., Uemastsu M., Satoh T., Kyotani S., Sakamaki F., Kakishita M., Fukushima K., Okano Y., Nakanishi N., et al. (2000). Plasma brain natriuretic peptide as a prognostic indicators in patients with primary pulmonary hypertension. Circulation 102, 865–870. [DOI] [PubMed] [Google Scholar]
- Omland T., Aakvaag A., Bonarjee V. V., Caidahl K., Lie R. T., Nilsen D. W., Sundsfjord J. A., Dickstein K. (1996). Plasma brain natriuretic peptide as an indicator of left ventricular systolic function and long-term survival after acute myocardial infarction. Comparison with plasma atrial natriuretic peptide and N-terminal proatrial natriuretic peptide. Circulation 93, 1963–1969. [DOI] [PubMed] [Google Scholar]
- Rabinovitch M., Gamble W., Nadas A. S., Miettinen O. S., Reid L. (1979). Rat pulmonary circulation after chronic hypoxia: hemodynamic and structural features. Am J Physiol 236, H818–H827. [DOI] [PubMed] [Google Scholar]
- Rosenberg H. C., Rabinovitch M. (1988). Endothelial injury and vascular reactivity in monocrotaline pulmonary hypertension. Am J Physiol 255(Heart Circ Physiol 24), H1484–H1491. [DOI] [PubMed] [Google Scholar]
- Rothman R. B., Ayestas M. A., Dersch C. M., Baumann M. H. (1999). Aminorex, fenfluramine, and chlorphentermine are serotonin transporter substrates. Implications for primary pulmonary hypertension. Circulation 100, 869–875 [DOI] [PubMed] [Google Scholar]
- Rubens C., Ewert R., Halank M., Wensel R., Orzechowski H., Schultheiss H., Hoeffken G. (2001). Big endothelin-1 and endothelin-1 plasma levels are correlated with severity of primary pulmonary hypertension. Chest 120, 1562–1569. [DOI] [PubMed] [Google Scholar]
- Scheinder J., Huh M. M., Bradlow H. L., Fishman J. (1984). Antiestrogen action of 2-hydroxyestrone on MCF-7 human breast cancer cells. J. Biol. Chem. 259, 4840–4845. [PubMed] [Google Scholar]
- Sakao S., Tatsumi K. (2011). The Effects of Antioangiogenic Compound SU4516 in a Rat Model of Pulmonary Arterial Hypertension. Respiration 81, 253–261. [DOI] [PubMed] [Google Scholar]
- Simonneau G., Gatzoulis M. A., Adatia I., Celermajer D., Denton C., Ghofrani A., Sanchez M. A. G., Kumar R. K., Landzberg M., Machado R. F., et al. (2013). Updated Clinical Classification of Pulmonary Hypertension. J Am Coll Cardiol 62, D34–D41. [DOI] [PubMed] [Google Scholar]
- Stampfer M. J., Colditz G. A., Willett W. C., Mansen J. E., Rosner B., Speizer F. E., Hennekens C. H. (1991). Postmenopausal estrogen therapy and cardiovascular disease. Ten-year follow-up from the nurse’s health study. N. Engl. J. Med. 325, 756–762. [DOI] [PubMed] [Google Scholar]
- Stenmark K. R., Meyrick B., Galie N., Mooi W. J., McMurtty I. F. (2009). Animal models of pulmonary arterial hypertension: the hope of etiological discovery and pharmacological cure. Am. J. Physiol. Lung. Cell. Mol. Physiol. 297, L1013–L1032. [DOI] [PubMed] [Google Scholar]
- Taraseviciene-Stewart L., Kasahara Y., Alger L., Hirth P., McMahon G., Waltenberger J., Voelkel N. F., Tuder R. M. (2001). Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell proliferation and severe pulmonary hypertension. FASEB J 15, 427–438. [DOI] [PubMed] [Google Scholar]
- Telang N. T., Suto A., Wong G. Y., Osborne M. P., Bradlow H. L. (1992). Induction by estrogen metabolite 16 alpha-hydroxyestrone of genotoxic damage and aberrant proliferation in mouse mammary epithelial cells. J. Natl. Cancer Inst. 84, 634–638. [DOI] [PubMed] [Google Scholar]
- Toba M., Alzoubi A., O'Neill K. D., Gairhe S., Matsumoto Y., Oshima K., Abe K., Oka M., McMurtry I. F. (2014). Temporal hemodynamic and histological progression in Sugen5416/hypoxia/normoxia-exposed pulmomnary arterial hypertensive rats. Am J Physiol Heart Circ Physiol 306, H243–H250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulevski I. I., Groenink M., van der Wall E. E., van Veldhuisen D. J., Boomsma F., Stoker J., Hirsh A., Lemkes J. S., Mulder B. J. M. (2001). Increased brain and atrial natriuretic peptides in patients with chronic right ventricular pressure overload: Correlation between plasma neurohormones and right ventricular dysfunction. Heart 86, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White K., Dempsie Y., Nilsen M., Wright A. F., Loughlin L., MacLean M. R. (2011). The serotonin transporter, gender, and 17β oestradiol in the development of pulmonary arterial hypertension. Cardiovasc. Res. 90, 373–382. [DOI] [PubMed] [Google Scholar]
- Xu W., Erzurum S. C., (2011). Endothelial cell energy metabolism, proliferation and apoptosis in pulmonary hypertension. Compr. Physiol. 1, 357–372. [DOI] [PMC free article] [PubMed] [Google Scholar]