

See Moon (doi:10.1093/awv239) for a scientific commentary on this article.
Angiotensin II type 1 receptor (AT1R) blockers, or ‘sartans’, are neuroprotective and neurorestorative. Villapol et al. show that the FDA-approved drugs candesartan and telmisartan promote morphological and functional recovery in a mouse model of traumatic brain injury. Sartans with dual AT1R-blocking and PPARγ-activating properties have therapeutic potential in patients.
Keywords: candesartan, telmisartan, inflammation, apoptosis, recovery
See Moon (doi:10.1093/awv239) for a scientific commentary on this article.
Angiotensin II type 1 receptor (AT1R) blockers, or ‘sartans’, are neuroprotective and neurorestorative. Villapol et al. show that the FDA-approved drugs candesartan and telmisartan promote morphological and functional recovery in a mouse model of traumatic brain injury. Sartans with dual AT1R-blocking and PPARγ-activating properties have therapeutic potential in patients.
Abstract
See Moon (doi:10.1093/awv239) for a scientific commentary on this article.
Traumatic brain injury frequently leads to long-term cognitive problems and physical disability yet remains without effective therapeutics. Traumatic brain injury results in neuronal injury and death, acute and prolonged inflammation and decreased blood flow. Drugs that block angiotensin II type 1 receptors (AT1R, encoded by AGTR1) (ARBs or sartans) are strongly neuroprotective, neurorestorative and anti-inflammatory. To test whether these drugs may be effective in treating traumatic brain injury, we selected two sartans, candesartan and telmisartan, of proven therapeutic efficacy in animal models of brain inflammation, neurodegenerative disorders and stroke. Using a validated mouse model of controlled cortical impact injury, we determined effective doses for candesartan and telmisartan, their therapeutic window, mechanisms of action and effect on cognition and motor performance. Both candesartan and telmisartan ameliorated controlled cortical impact-induced injury with a therapeutic window up to 6 h at doses that did not affect blood pressure. Both drugs decreased lesion volume, neuronal injury and apoptosis, astrogliosis, microglial activation, pro-inflammatory signalling, and protected cerebral blood flow, when determined 1 to 3 days post-injury. Controlled cortical impact-induced cognitive impairment was ameliorated 30 days after injury only by candesartan. The neurorestorative effects of candesartan and telmisartan were reduced by concomitant administration of the peroxisome proliferator-activated receptor gamma (PPARγ, encoded by PPARG) antagonist T0070907, showing the importance of PPARγ activation for the neurorestorative effect of these sartans. AT1R knockout mice were less vulnerable to controlled cortical impact-induced injury suggesting that the sartan’s blockade of the AT1R also contributes to their efficacy. This study strongly suggests that sartans with dual AT1R blocking and PPARγ activating properties have therapeutic potential for traumatic brain injury.
Introduction
Traumatic brain injury (TBI) is a major public health problem that can result in death or severe disability. There is a paucity of therapies at the present time, with over 30 failed clinical trials for TBI (Diaz-Arrastia et al., 2014; Wright et al., 2014). Thus, there exists an urgent demand to find novel neuroprotective and neurorestorative agents that offer significant improvement over current symptomatic treatment of TBI (Andriessen et al., 2010; McConeghy et al., 2012). After the initial lesion, complex secondary cascades worsen the initial injury. These secondary cascades, operating in minutes to days after injury produce detrimental effects including decreased cerebrovascular blood flow and nutrient access leading to cell death in the lesion core, and apoptotic, inflammatory and axonal damage in surrounding tissue (Kumar and Loane, 2012; Joseph et al., 2014; Villela et al., 2015). Drugs that reduce damage from these secondary cascades have the potential to significantly improve functional outcomes.
Our initial experiments and those of others (Timaru-Kast et al., 2012; Villapol et al., 2012) showed that administration of candesartan, an antagonist of angiotensin II type I receptors (AT1R, encoded by Atgr1), partially protected mice subjected to controlled cortical impact (CCI) injury when given before or immediately after injury. These observations strongly suggested that angiotensin II signalling through the AT1Rs may play an important role in the development and progression of TBI. It has long been established that, in addition to the classical systemic renin-angiotensin system (RAS), the brain expresses its own RAS (Saavedra, 1992; Wright and Harding, 2011). Brain angiotensin II, acting through stimulation of AT1R, regulates multiple functions, including but not restricted to cerebral circulation, hormone release, sympathetic activity, the limbic system, motor performance and sensory responses (Saavedra, 1992; Wright and Harding, 1994). Increased activation of AT1R is associated with decreased cerebral blood flow, sympathetic overdrive, and pathological responses to stress and inflammation (Saavedra et al., 2006; Ohshima et al., 2013; Villapol and Saavedra, 2015).
Consequently, decreasing AT1R activity with the AT1R antagonists, angiotensin II receptor blockers (ARBs) or sartans, is therapeutically efficacious in rodent models of cerebral haemorrhage, Parkinson’s disease, Alzheimer’s disease and brain inflammation secondary to systemic administration of bacterial endotoxin (Nishimura et al., 2000; Ito et al., 2002; Tsukuda et al., 2009; Benicky et al., 2011; Garrido-Gil et al., 2012). Sartans were also found to offer neuroprotection in animal models of stroke (Thone-Reineke et al., 2006), evidenced by an improvement in neurological outcome and brain microcirculation (Bennai et al., 1999; Ishrat et al., 2015), accompanied by a reduction in inflammation, oxidative stress, and apoptosis (Jung et al., 2007; Kozak et al., 2008). In addition, clinical trials showed that the use of sartans reduced cardiovascular mortality and incidence of stroke (Meredith et al., 2004; Lu et al., 2009; Cernes et al., 2011). These studies indicated that sartan administration ameliorated important pathogenic mechanisms common to stroke, inflammatory brain and neurodegenerative disorders and TBI, yet sartans have not been assessed for TBI in the clinic. The potential of sartans as novel therapeutic agents for TBI deserves further scrutiny.
For this study we performed CCI injury, a validated preclinical injury model, and determined the effects of administration of two different sartans, candesartan or telmisartan. Both sartans, in addition to blocking the AT1R, also activate peroxisome proliferator-activated receptor gamma (PPARγ, encoded by Pparg) (Tsukuda et al., 2009; Cernes et al., 2011). We have previously shown that the PPARγ agonist activity of candesartan contributes to the neuroprotective effect of candesartan when candesartan was administered before CCI injury (Villapol et al., 2012). We hypothesized that a sartan with greater PPARγ activity could be more beneficial to recovery than candesartan after TBI. We therefore compared the efficacy of candesartan with that of telmisartan; the sartan with the strongest PPARγ partial agonist activity (Benson et al., 2004) to determine which sartan may have the greatest beneficial activity after TBI. By interacting with two independent receptors, AT1R and PPARγ, these sartans have the potential to signal to almost every cell within the brain, and have multimodal mechanisms of action. We determined the relative contribution of AT1R blockade and PPARγ activation to the efficacy of neuroprotection by studying mice devoid of AT1R or by simultaneous administration of a PPARγ antagonist with either sartan. We also investigated whether the potential therapeutic effects of candesartan or telmisartan exhibited a clinically viable therapeutic window, were dependent on changes in blood pressure, were long lasting in duration and whether they improved cognition and motor performance.
Materials and methods
Animals, surgical procedures, drug treatments and experimental design
Animals
All animal studies were approved by the Uniformed Services University of the Health Sciences (USUHS) Institutional Animal Care and Use Committee and were conducted in accordance with the NRC guide to the Care and Use of Laboratory Animals. C57BL/6NCr male mice (NCI, Frederick, MD) weighing 20–25 g were used for most experiments. Breeding pairs of AT1a receptor (AT1aR) knockout mice (B6.129P2-Agtr1atm1Unc/J), were purchased from Jackson laboratories and bred in our animal facility. Wild-type control mice (C57BL/6J) for these experiments were also purchased from Jackson Laboratories. All mice were kept under 12:12 h light and dark cycle with access to food and water ad libitum. Nine-week-old male mice were housed in animal cages containing five mice each. Typically, surgery was done after 1 week of recovery from transportation-related stress.
Moderate controlled cortical impact
Mice were anaesthetized with isoflurane (3% induction: 2% maintance) and their heads securely fixed in a stereotactic frame. CCI injury was performed exactly as described previously (Villapol et al., 2012) above the left parietal cortex. Moderate CCI injury (coordinates; 2 mm lateral, 2 mm posterior to Bregma) at an impact depth of 1 mm, with a 2 mm diameter round impact tip (speed 3.6 m/s, dwell time 100 ms) and 12° angle to the dura mater, using an electromagnetically driven CCI injury device (Impact One stereotaxic impactor CCI, Leica) was performed. The bone flap was replaced but not sealed, the skin was sutured, and the mice were allowed to recover fully from anaesthesia before transfer to their cages. Control or naïve mice were anaesthetized, allowed to recover from anaesthesia and returned to their cages.
Drug treatment
The distinct pharmacokinetics and solubility of candesartan (Gleiter and Morike, 2002) and telmisartan (Deppe et al., 2010) led us to administer these drugs through different methods. Our prior work (Villapol et al., 2012) and that of others (Armando et al., 2002; Hamai et al., 2006; Ozacmak et al., 2007; Liu et al., 2008; Omura-Matsuoka et al., 2009; Tota et al., 2009) showed neuroprotective effects of candesartan administered by subcutaneous minipumps or by intraperitoneal injection. Candesartan (AstraZeneca, CV-11974) was suspended in physiological saline containing 0.1 N Na2CO3, pH 7.4. For the initial dose-response experiment, mice were injected with a 0.2 ml volume via a single intraperitoneal dose of candesartan (0.1, 0.5 or 1 mg/kg) after CCI injury. Mice were injected once daily until sacrifice. Control mice received intraperitoneal injections of vehicle. For longer term candesartan treatment mice were injected once, intraperitoneal at 6 h post-injury and then at 1 day post-injury implanted with minipumps (ALZET, model 1004; delivering 0.11 μl/h) with continuous infusion until sacrifice at 30 days post-injury as described (Villapol et al., 2012). Telmisartan (Sigma-Aldrich) stock was dissolved in dimethyl sulphoxide (DMSO) and diluted in distilled water. Solutions were prepared fresh daily. Telmisartan’s neuroprotective effects are usually detected after oral administration by gavage (Kasahara et al., 2010; Fukui et al., 2014; Justin et al., 2014; Yamashita et al., 2014). Thus, telmisartan was administered at 1 or 10 mg/kg once per day by oral gavage until sacrifice. Control animals received DMSO diluted in distilled water by oral gavage. The dose ranges of both drugs were selected based upon previously reported effective doses (Benicky et al., 2011; Garrido-Gil et al., 2012; Timaru-Kast et al., 2012; Villapol et al., 2012). Certain groups of mice also received the PPARγ antagonist T0070907 (2 mg/kg dissolved in saline; Sigma-Aldrich) that was administered intraperitoneally starting 1 h after CCI injury and then daily until sacrifice at 3 days post-injury.
Experimental design and randomization
A diagram of the experimental design is shown in Fig. 1. Based on our previous experience with this TBI model, we estimated 10 animals per group were required for analysis of histological and physiological parameters after injury. A small percentage (5–10%) of the injured mice died after surgery due to complications of the injury, and there were problematic perfusions for two mice. However, none of the mice that survived were excluded post hoc. For behavioural studies, our power analysis estimated we would need at least 12 animals. We therefore included 15 mice per group for these experiments to allow for technical problems and expected casualties. Candesartan and telmisartan treatments were administered independently on different days, with each drug treatment group having its own controls. Once mice were injured, treatment was administered alternating mice between drug and vehicle, with the investigator blinded to the treatment. Control uninjured mice were similarly treated on the same day by the same investigator.
Figure 1.

Experimental design. Different groups of mice were treated daily with varying doses of candesartan or telmisartan starting at 1 h after CCI injury until sacrifice at 1 or 3 days post-injury. The therapeutic window was determined by initiating treatment at 1, 3, or 6 h after injury and assessing at 1 day post-injury. For longer-term treatment, the initial dose was administered at 6 h post-injury, then daily until sacrifice at 30 days post-injury. When required, the PPARγ inhibitor (T0070907, 2 mg/kg) was administered starting 1 h after CCI injury, and daily until sacrifice. Brains were removed to perform histological and immunocytochemical analysis. Blood pressure was measured 2 h before and 1 day post-injury, and cerebral blood flow was measured before and 1 and 3 days post-injury. Mice were tested on the rotarod 1 and 2 days before and 1 and 3 days post-injury, and with the learning and memory task, Morris water maze between 25 to 30 days post-injury.
Determination of cerebral blood flow
Changes in cerebral blood flow were measured in the pericontusional region using a laser-Doppler flowmeter (PeriFlux System 5000 LDPM, Perimed) with a flexible fibre optic extension to the LDPM probe tip 404, as previously described (Villapol et al., 2009). Cerebral blood flow was measured in anaesthetized mice just before CCI injury (baseline levels), 2 min after impact, and at 1 and 3 days post-injury. Changes in cerebral blood flow were expressed as the percentage of the baseline value recorded before CCI injury.
Determination of blood pressure
Systolic and diastolic blood pressure were monitored using the CODA™ mouse tail-cuff system that uses a volume pressure recording sensor, coupled to a PC-based data acquisition system (Kent Scientific). Conscious mice were held in a small plastic holder on a warming pad thermostatically controlled at 37°C. Ten measurements per mouse were recorded to obtain mean systolic and diastolic blood pressure of each drug treatment and control group, 2 h before (baseline) and 1 day after CCI injury as previously described (Villapol et al., 2012).
Determination of functional recovery
All behavioural studies were performed by an investigator blinded to the treatment groups.
Rotarod
Motor behaviour was assessed by ability to stay on the rod. Mice were tested 2 days before injury, and at 1 and 3 days post-injury. The rod was accelerated from 4 to 60 rpm in 2 min and the time the mice were able to stay on the rod was recorded as latency to fall in seconds.
Morris water maze
Spatial learning and memory deficits were evaluated on days 25 to 30 after CCI injury as previously described (Villapol et al., 2012). Four trials were performed every day for five consecutive days and the time taken to reach the hidden submerged platform recorded. The mice were given 60 s to locate the hidden platform, and remained on the platform for 15 s before being removed. On the final day, 1 h after the last trial, mice were put back into the tank for 1 min with the platform removed. The time spent in the quadrant where the platform had previously been located was recorded. Tracking software (ANYMaze) was used to record all movement.
Tissue preparation for histological analysis
The mice were sacrificed at 1, 3, and 30 days after CCI injury (Fig. 1). Mice were anaesthetized with ketamine/xylazine and perfused intracardially with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). Brains were removed and placed in 4% paraformaldehyde overnight, then transferred to 30% sucrose solution and stored at 4°C. Brains were cut to 30 μm thick sections using a microtome and were stored in cryoprotectant solution. Brain coronal sections through the dorsal hippocampus were selected for histological analysis (distance to Bregma −1.70 to −2.80 mm).
Cresyl-violet staining and lesion volume measurements
Cresyl-violet (0.1%, Sigma-Aldrich) was dissolved in distilled water and filtered. Every third brain section was mounted on poly-D-lysine-coated slides and stained for 20 min with cresyl-violet solution. Sections were then dehydrated for 2 min sequentially with 100, 95, 70, then 50% ethanol, cleared in xylene for another 2 min, covered with DPX mounting media (Sigma-Aldrich) and coverslipped. Lesion area was assessed on 9 to 15 brain sections spaced equidistance (every 450 µm) apart, approximately between −1.70 to −2.70 mm from Bregma. Lesion volume was obtained by multiplying the sum of the lesion areas by the distance between sections. Per cent lesion volume was calculated by dividing each lesion volume by the total ipsilateral hemisphere volume (similarly obtained by multiplying the sum of the areas of the ipsilateral hemispheres by the distance between sections).
Immunohistochemical analysis
Sections were washed three times in phosphate-buffered saline (PBS) for 5 min each, and blocked with 10% normal goat serum (NGS) in PBS with 0.1% Triton™ X-100 (PBS-T) for 1 h. The following primary antibodies were incubated at 4°C overnight in PBS-T/5% NGS: anti-GFAP, mouse monoclonal (1:2000, Millipore) or chicken polyclonal (1:400, Abcam) for astrocytes; anti-MPO, mouse monoclonal (1:100, Chemicon) for neutrophils; anti-CD68, mouse monoclonal (1:200, Chemicon) for macrophages/microglia; anti-Iba-1 rabbit polyclonal (1:750, Wako) for microglia; and anti-nitrotyrosine rabbit polyclonal (1:200, Millipore) for nitrated proteins. Sections were washed three times in PBS-T, incubated with the corresponding Alexa Fluor® 568-conjugated (red) IgG secondary antibody (1:1000, Invitrogen) for 2 h at room temperature, then rinsed with PBS and distilled water and coverslipped with ProLong® Gold antifade reagent with DAPI (Invitrogen).
Cell death assay
Sections were processed for DNA strand breaks [terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay, labelling of fragmented DNA] using the fluorescence In Situ Cell Death Detection Kit (Roche), according to the manufacturer’s instructions. TUNEL+ cells were counted in cortical layers in three to five coronal sections for each animal, with five to eight mice per group.
Densitometry analysis and cellular quantification
Images were acquired with the ×20 objective on an Olympus BX61 with attached qImaging Retiga EXi Aqua CCD camera, and iVision software (BioVision Technologies). Quantitative image analysis of the GFAP immunoreactive areas were performed on five cortical sections per animal through the level of impact site (AP: 2.0 mm) using the same densitometric analysis method as previously described (Villapol et al., 2014). Immunofluorescence intensity was calculated using the threshold method and defined as the number of pixels, divided by the total area (mm2) in the imaged field with the average background subtracted. For cellular quantification, a total of five fields per section located within the injured cortex were quantified on a minimum of three sections per mouse. The investigator was blinded to the treatment groups.
Primary microglial culture
Sprague–Dawley rats were purchased from Charles River Laboratories and housed in the USUHS animal facility. Brains were removed from postnatal Day 2 Sprague–Dawley rat pups, the cerebral hemispheres were dissected out, their meninges removed and the cortices placed in culture medium as described previously (Mitchell et al., 2014). Cells were resuspended in culture medium and were grown in plates for 24–48 h before sartan treatment. Cells were preincubated with candesartan (200 mM) or telmisartan (100 mM) for 2 h before stimulation with bacterial endotoxin (lipopolysaccharide) (10 ng/ml) for 12 h and harvesting in TRIzol® reagent (Life Technologies).
RNA isolation and quantitative PCR
RNA was isolated from microglial cultures using TRIzol® reagent according to the manufacturer’s instructions, quantified at A260 and run on a gel to verify integrity. cDNA was synthesized using SuperScript® III (Life Technologies) and SYBR® Green (Qiagen) qPCR performed in a CFX96 (Bio-Rad) with the primers: IL-1β (against Il1b) (GACCCCAAAAGATTAAGGATT; forward, AAAGAAGGTGCTTGGGTCCTC; reverse) or iNOS (against Nos2) (AAGCCCCGCTACTACTCCAT; forward, AGCTGGAAGCCACTGACACT; reverse). Target RNA expression levels were normalized to those of the housekeeping gene Gapdh. Relative changes in mRNA expression levels between control and treated samples were calculated by the delta delta threshold cycle (ΔΔCt) method using Bio-Rad CFX Manager 2.0. A minimum of four independent samples was used per group and the effects were observed in three independent experiments.
Statistical analysis
All data in this study are expressed as mean ± standard error of the mean (SEM). P < 0.05 or less was considered statistically significant. For data with a single time point or drug dosage, intergroup differences were evaluated by ordinary one-way ANOVA with Dunnett’s multiple comparison test when comparing all values to control, and Tukey’s test when comparing selected values. For analysing changes in cerebral blood flow, a two-way ANOVA with Neuman-Keuls test for multiple comparisons, for each time point, was performed. For rotarod experiments, a two-way repeated measures ANOVA was run, excluding baseline measures, followed by Fishers least significant difference test comparing the main effects of all groups. For direct comparisons between wild-type and AT1aR knockout mice, a two-tailed Student’s t-test was performed. All statistics were performed via Prism 6 software (GraphPad).
Results
Determination of dose response and therapeutic window
To determine the lowest, most effective dose of each sartan we treated mice 1 hour after CCI injury and sacrificed at 1 day post-injury to assess lesion volume. We determined responses to doses spanning the clinical therapeutic range for candesartan (0.1 to 1 mg/kg) and for telmisartan (1 and 10 mg/kg) (Stangier et al., 2000; Cabaleiro et al., 2013). Lower doses of candesartan (0.1 and 0.5 mg/kg) produced larger reductions in lesion volume than the highest dose (1 mg/kg) (Fig. 2A). Both low and high doses of telmisartan were equally effective in reducing the lesion volume (Fig. 2B). We therefore proceeded using the lowest effective dose of each drug; 0.1 mg/kg candesartan and 1 mg/kg telmisartan. To determine the maximum therapeutic window after injury when these drugs retained efficacy, candesartan or telmisartan were administered either at 1, 3 or 6 h after CCI injury, and animals were sacrificed at 1 day post-injury. At all time points, sartan treatment resulted in significant reduction in lesion volume as compared to injured mice treated with vehicle (Fig. 2C and D). Therefore the therapeutic window for administration of either telmisartan or candesartan after injury in mice is at least 6 h. To determine if there could be a longer therapeutic window, we initiated treatment 24 h after injury and treated once daily until sacrifice at 3 days post-injury. There was no significant reduction in lesion volume with this treatment paradigm (Supplementary Fig. 1A). To investigate whether any sartan drug altered blood pressure, we measured systolic and diastolic blood pressures after treatment with vehicle, candesartan (0.1 and 1 mg/kg) or telmisartan (1 and 10 mg/kg) before and 1 day after CCI injury. Neither dose of candesartan nor telmisartan significantly altered blood pressure when compared to vehicle treated injured mice (Supplementary Table 1).
Figure 2.
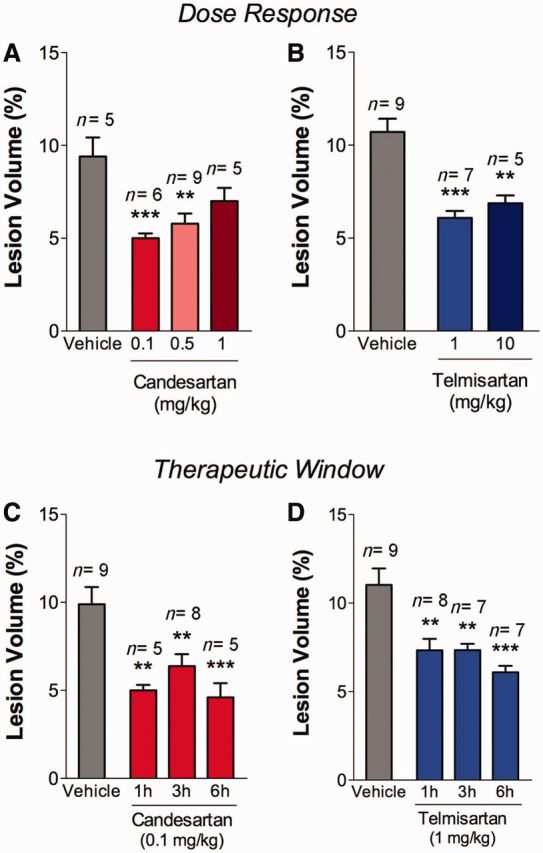
Sartan treatment reduces cortical lesion volume when administered up to 6 h after brain injury. (A and B) Sartan was administered 1 h after injury and assessed at 1 day post-injury. Lower doses of candesartan (0.1 or 0.5 mg/kg) reduced the lesion volume compared to vehicle alone; this neurorestorative effect was not observed at a higher dose (1 mg/kg). (B) Higher (10 mg/kg) and lower (1 mg/kg) doses of telmisartan reduced the lesion volume compared to vehicle administration. (C and D) Candesartan (0.1 mg/kg) or telmisartan (1 mg/kg) reduced the lesion volume when administered at 1, 3 or 6 h after injury as determined at 1 day post-injury. Mean ± SEM, n = 5–9, *P < 0.05, **P < 0.01, ***P < 0.001, sartan versus vehicle-treated mice.
Effect of candesartan and telmisartan in CCI-induced reduction of cerebral blood flow
As cerebrovascular reactivity is often impaired after TBI, and sartans have neurovascular protective properties (Guan et al., 2011) we wanted to determine whether administration of either sartan would alter the reduced cerebrovascular flow that occurs after TBI. Indeed, CCI injury significantly reduced cerebral blood flow in the perilesional cortex, by ∼40% during the initial minutes after cortical impact. Cerebral blood flow was still low at 3 days post-injury (Fig. 3). Administration of candesartan significantly ameliorated the reduction in cerebral blood flow, raising the cerebral blood flow back to baseline rates at 1 and 3 days post-injury (Fig. 3). The beneficial effect of telmisartan on cerebral blood flow was seen only at 3 days post-injury.
Figure 3.

Candesartan and telmisartan treatment improve recovery of cerebral blood flow. Cerebral blood flow was decreased in the injured cerebral cortex 2 min after CCI injury. Candesartan (0.1 mg/kg) administered 6 h after injury significantly ameliorated the CCI-induced decrease in cerebral blood flow at 1 and 3 days post-injury and telmisartan (1 mg/kg) at 3 days post-injury compared with vehicle-treated mice. Data were expressed as a per cent of baseline values. Mean ± SEM, n = 8-10/group, **P < 0.001, ***P < 0.0001, candesartan versus vehicle-treated mice; #P < 0.05, telmisartan versus vehicle-treated mice.
Effects of candesartan and telmisartan on CCI-induced inflammation and cell injury
In mice that have been injured, there is significant inflammation and increased glial reactivity in the perilesional area (Villapol et al., 2014). This reaction includes infiltration of macrophages (CD68+) and neutrophils (MPO+) together with activation of microglia (also CD68+) and reactive astrocytes (GFAP+) (Fig. 4A, E and I). Injury-induced signalling cascades lead to large numbers of apoptotic cells (visualized by TUNEL staining). Reactive oxygen species combine with nitric oxide to form peroxynitrite that nitrosylates tyrosine residues in proteins (detected with nitrotyrosine antiserum) (Fig. 4M). Naive control mice did not show expression of these markers (data not shown). Both candesartan and telmisartan administered 6 h after CCI injury significantly reduced the number of CD68 + and MPO + cells in the perilesional cortex (Fig. 4B–D and F–H). Additionally, candesartan and telmisartan significantly reduced the number of TUNEL and nitrotyrosine + cells, both in the perilesional area and in the lesion core (Fig. 4N, O, Q and R). Only candesartan, but not telmisartan, induced a decrease in the amount of GFAP immunoreactivity (Fig. 4J and K). Thus, telmisartan and candesartan ameliorate many markers of inflammation and neuronal injury at acute times after CCI injury. However, when the therapeutic window of administration was extended to 24 h after injury, sartan treatment did not yield a reduction in the number of apoptotic cells, microglial density, or astrogliosis analysed at 3 days post-injury (Supplementary Fig. 1B–D).
Figure 4.
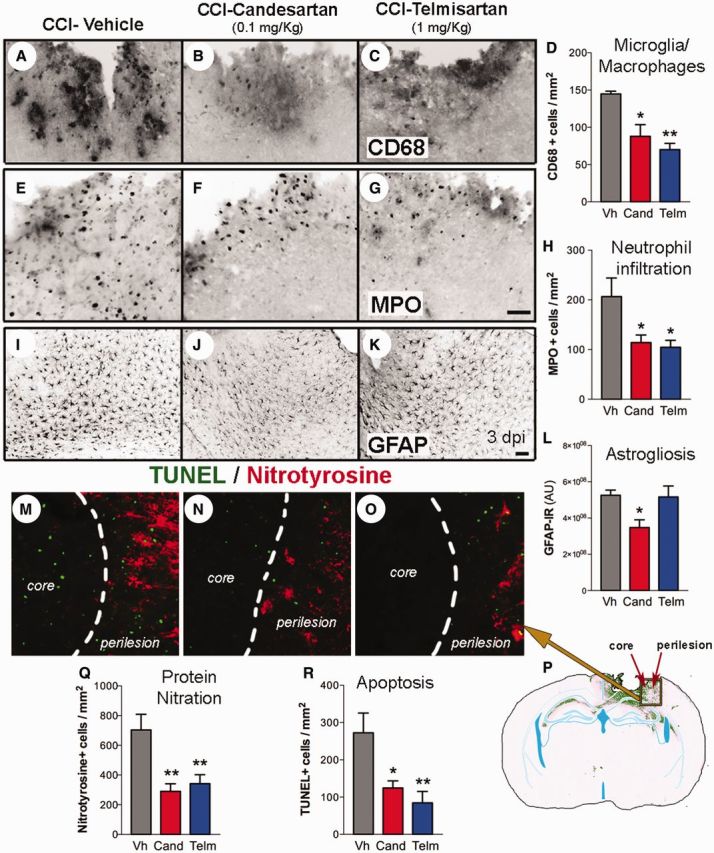
Candesartan or telmisartan reduced CCI-induced inflammation and apoptosis. Mice were administered either candesartan (Cand, 0.1 mg/kg) (B, F, J and N), telmisartan (Telm, 1 mg/kg) (C, G, K and O) or vehicle (Vh) (A, E, I and M) at 6 h post-injury and sacrificed at 3 days post-injury. Candesartan and telmisartan reduced the number of activated microglia and infiltrating macrophages (CD68 +) (A–D) and the number of neutrophils (myeloperoxidase, MPO+) (E–H) compared with vehicle-treated mice in the perilesional cortical region. Only candesartan reduced the GFAP+ staining (for reactive astrocytes) (I–L). (M–R) Candesartan and telmisartan also reduced the number of cells stained for nitrotyrosinalated proteins (red), a marker of oxidative stress, and apoptotic cells (TUNEL+, green) in the lesion core and perilesional cortex. (M–O) Images are magnifications of the core and perilesional cortical areas taken from the area indicated by the square that is marked on a representative coronal brain section (P). Quantitative analysis of nitrotyrosine + (Q) and TUNEL + cells (R) in the ipsilateral cortex. Scale bars: A–K = 50 μm; M–O = 20 μm. Quantitative data (D, H, L, Q, R) are represented using mean ± SEM, n = 5–8. *P < 0.05, **P < 0.01 candesartan or telmisartan versus vehicle-treated mice.
Participation of PPARγ in candesartan and telmisartan neurorestoration
Activation of PPARγ significantly reduces inflammation (Bordet et al., 2006). As both sartans showed powerful anti-inflammatory activity we wanted to determine the relative contribution of their PPARγ partial agonist activity to their efficacy after CCI injury. We therefore administered a selective PPARγ antagonist (T0070907; 2 mg/kg) to mice 1 h after injury followed by either vehicle, telmisartan or candesartan at 6 h post-injury. T0070907 treatment increased the lesion volume of vehicle treated mice at 3 days post-injury by 2% (Fig. 5). However, this difference was not significant and was not reproducible (data not shown). When either candesartan or telmisartan was administered together with T0070907, the resultant lesion volume of the injured brain was significantly larger than when candesartan or telmisartan were administered alone. Thus, antagonizing PPARγ activity reduced the neurorestorative effect of the sartans, suggesting that their efficacy in treating mice with TBI is partially dependent on PPARγ agonist activity.
Figure 5.
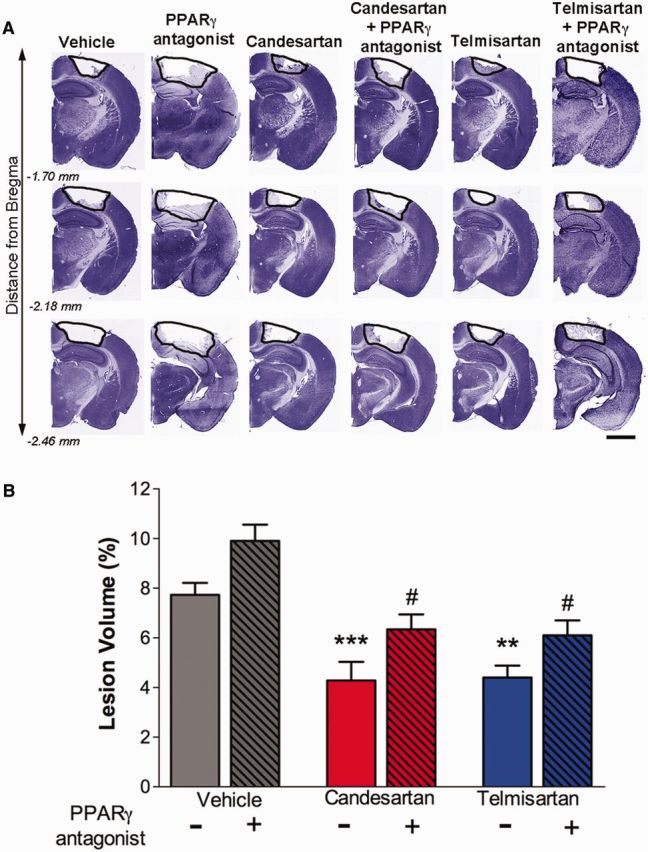
Sartans ability to reduce lesion size is partially dependent on PPARγ activation. Mice were injured by CCI injury, treated 1 h post-injury with either PPARγ antagonist (2 mg/kg, T0070907) or vehicle, and administered vehicle, telmisartan (1 mg/kg) or candesartan (0.1 mg/kg) at 6 h post-injury. Mice were sacrificed at 3 days post-injury and brain sections stained with cresyl-violet. (A) Representative brains sections from different treatment groups of mice indicating the lesion area (black line), comprised of the cavity and oedematous regions. Scale bar = 200 μm. (B) Quantitative determination of the lesion volume showing that either sartan reduces the lesion volume, but co-treatment with T0070907 reverses this effect. Data are mean ± SEM, n = 8–15. ***P < 0.001, **P < 0.01 candesartan or telmisartan versus vehicle; #P < 0.05 groups versus PPARγ antagonist.
To investigate the anti-inflammatory activity of the sartans more specifically we examined activation of microglia after CCI injury. Injury induced a dramatic alteration in microglial morphology, in the perilesional cortex (Fig. 6B–G). Large numbers of hypertrophic microglia with a bushy amoeboid appearance were detected in vehicle-treated mice at 3 days post-injury (Fig. 6B). The number of Iba-1+ microglia was significantly reduced by candesartan or telmisartan administration (Fig. 6C, D, F and G) with the microglial morphology less amoeboid in these brains (Fig. 6c, d, f and g). Administration of T0070907 after CCI injury significantly increased the number of Iba-1+ activated microglia in vehicle-treated mice (Fig. 6B and E). When T0070907 was administered before candesartan or telmisartan, the number of Iba-1+ cells was greater than either sartan alone and not significantly different than vehicle-treated mice after injury (Fig. 6F–H). Thus, the PPARγ agonist activity of both candesartan and telmisartan contributed to the ability of these drugs to dampen microglial activation after injury.
Figure 6.

Candesartan and telmisartan reduce microglial activation after CCI injury. Mice were injured by CCI, treated 1 h post-injury with either PPARγ antagonist (2 mg/kg, T0070907) or vehicle, and administered vehicle, telmisartan (1 mg/kg) or candesartan (0.1 mg/kg) at 6 h post-injury, then daily until sacrifice at 3 days post-injury. (A) Representative images from brain sections from perilesional cortex at −1.70 mm from Bregma immunostained with Iba-1 antibody. Five fields per brain section were counted. Scale bar = 500 μm. (B–G) CCI-induced increases in microglial (Iba-1+) density and hypertrophic morphology in vehicle-treated mice (B) were reduced by candesartan (C) or telmisartan (D) treatments. The PPARγ antagonist enhanced CCI-induced microglia activation (E) and partially decreased the candesartan (F) and telmisartan protective effects (G). Small inset images (b–g) are magnified to reveal details of the hypertrophic/bushy activated microglial cells. Scale bars for B–G = 50 μm; b–g = 20 μm. (H) Quantitative analysis of the density of Iba-1+ cells for each group. Data are means ± SEM, n = 5–12. **P < 0.01, ***P < 0.001 candesartan, or telmisartan versus vehicle; #P < 0.05, ##P < 0.01, ###P < 0.001, PPARγ antagonist treatment versus same treatment group without antagonist. (I and J) Candesartan and telmisartan reduce lipopolysaccharide-induced inflammatory gene expression in primary microglia. Primary microglia were treated with candesartan or telmisartan for 2 h before addition of lipopolysaccharide (LPS, 10 ng/ml) for 12 h before harvesting. QPCR analysis shows (I) iNOS (Nos2) and (J) IL-1β (Il1b) mRNA expression in different conditions. Data are mean ± SEM, n = 4–8. ****P < 0.0001, ***P < 0.001, *P < 0.05 treatments versus lipopolysaccharide alone.
To determine whether sartans reduced microglial activation directly or indirectly, we examined the effect of these drugs on microglial activation in primary culture. We treated primary cortical microglia with candesartan or telmisartan and 2 h later activated them with lipopolysaccharide (10 ng/ml) for 12 h. Lipopolysaccharide treatment of microglia induced very high levels of Nos2 (also known as iNOS) and Il1b mRNA (Fig. 6I and J). Candesartan pretreatment significantly reduced, and telmisartan pretreatment completely abolished lipopolysaccharide-induced Nos2 mRNA expression (Fig. 6I and J). On the other hand, while telmisartan significantly reduced lipopolysaccharide-induced Il1b expression, candesartan was without effect (Fig. 6I and J). Thus, sartans have a direct anti-inflammatory action on microglia.
Effect of candesartan and telmisartan on functional recovery
To determine whether sartan treatment altered the functional recovery from injury, we assessed mice for motor and cognitive function at different time points. Motor performance, determined as the latency to fall from a rotating rod, was determined at baseline, 1 and 3 days post-injury. Control naïve mice, but not CCI-injured mice improved their motor performance over the course of 3 days (Fig. 7A and B). Treatment of injured mice with 1 mg/kg telmisartan, but not 10 mg/kg, improved motor performance at 1 and 3 days post-injury, although not significantly so (P = 0.13 telmisartan versus vehicle-treated injured mice). This effect was significantly diminished by PPARγ blockade (P = 0.03) (Fig. 7B). As there were significant, random differences between the groups before injury, we omitted baseline values in the analysis of post-injury drug treatments on motor performance. There was no observed difference between 1 and 3 day post-injury values. Candesartan treatment did not have a significant effect on motor function at these time points (Fig 7A). Thus, telmisartan, but not candesartan, trended towards improved motor function after CCI injury, an effect that was dependent on its ability to activate PPARγ.
Figure 7.
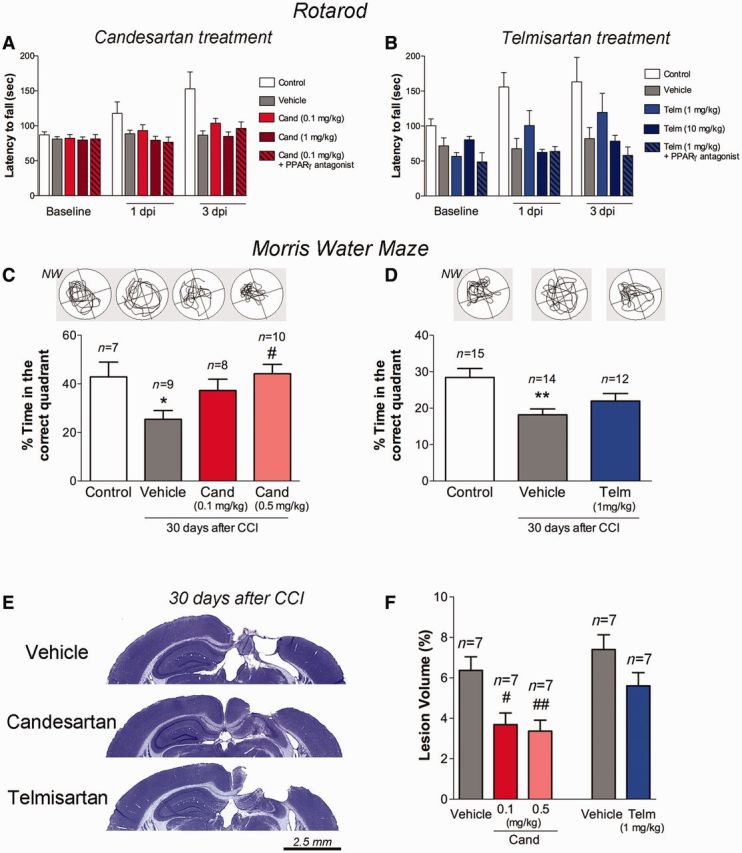
Candesartan and telmisartan protect motor performance, reduce cortical damage and protect cognition. Recovery of mice treated with either candesartan (Cand) (A and C) or telmisartan (Telm) (B and D) was assessed by (A and B) rotarod or (C and D) Morris water maze task. (B) Telmisartan improvement of motor function was not statistically different from injured mice treated with vehicle, (P = 0.13). The addition of a PPARγ antagonist significantly decreased the 1 mg/kg telmisartan effect (P = 0.03). Overall differences among groups were statistically significant (P = 0.003) but did not alter from 1 day post-injury to 3 days post-injury (P = 0.99 Group × Time interaction). Control group (n = 10), injured vehicle (n = 9), Cand (n = 8) and Telm (n = 8). (C) Candesartan 0.5 mg/kg led to a greater ability to learn and recall the location of a hidden platform in the Morris water maze, compared to vehicle-treated mice at 30 days post-injury (P = 0.019). Mice treated with 0.1 mg/kg candesartan did not behave significantly different than those treated with vehicle (P = 0.26). (D) Telmisartan-treated mice behaved similarly to vehicle-treated mice in this task after injury (P = 0.456), but no longer significantly different than control uninjured mice (P = 0.092). (E) Representative cresyl-violet images (scale bar = 2.5 mm) and (F) their quantification demonstrate the reduction in the cortical cavity at 30 days post-injury, in brains from candesartan but not telmisartan treated mice. Data are mean ± SEM, **P < 0.01, *P < 0.05 injured mice versus control; ##P < 0.01, #P < 0.05 candesartan/telmisartan versus vehicle.
To determine whether sartans altered cognitive function at a later time point, we assessed performance of the mice in the Morris water maze from 25 to 30 days post-injury. In the probe trial at 30 days post-injury, injured mice spent significantly less time in the correct quadrant than naïve mice (Fig. 7C and D). Injured mice treated with 0.5 mg/kg of candesartan significantly recovered cognitive function when compared to vehicle-treated mice (P = 0.0189), similar to control uninjured mice. Mice treated with only 0.1 mg/kg candesartan were not significantly different from either vehicle-treated injured mice (P = 0.26) or control uninjured mice (P = 0.84). Thus, 0.1 mg/kg candesartan treatment had a beneficial effect, but not as strong as 0.5 mg/kg candesartan treatment (Fig 7C). Injured mice treated with telmisartan (1 mg/kg) also showed partial recovery; their time spent in the correct quadrant during the probe trial was not significantly different from either vehicle-treated injured mice (P = 0.456) or control uninjured mice (P = 0.092) (Fig. 7D). Examination of the brains taken from these mice sacrificed at 30 days post-injury, showed that candesartan had a more beneficial effect at this longer time point, significantly reducing the lesion volume when administered either at 0.1 mg/kg or 0.5 mg/kg (Fig 7E and F). Telmisartan (1 mg/kg) did not significantly reduce lesion volume at 30 days post-injury. Thus, candesartan, but not telmisartan treatment, reduced lesion volume and improved cognitive function at 30 days post-injury.
Effects of AT1aR deletion on blood pressure, cerebral blood flow, inflammation, apoptosis and motor performance
To investigate the contribution of angiotensin II signalling through the AT1R to the secondary cascades after TBI, we compared the response of mice lacking the AT1R with wild-type mice after injury. In mice there are two AT1 receptors; AT1a (encoded by Agtr1a) and AT1b (encoded by Agtr1b) (Sasamura et al., 1992). In the brain, angiotensin II effects are transmitted mostly through AT1aR activation, making the AT1aR knockout an appropriate model to study the role of brain AT1R (Johren and Saavedra, 1996). CCI injury to AT1aR knockout mice resulted in a smaller lesion volume at 3 days post-injury, in comparison to that in wild-type mice (Fig. 8C and D). Additionally, AT1aR knockout mice had significantly fewer TUNEL + cells (Fig. 8E), and a dramatic drop in GFAP staining for reactive astrocytes (Fig. 8F) in comparison to wild-type mice at 3 days post-injury. However, we did not find any significant differences in the number of CD68, Iba-1 or MPO-immunoreactive cells between AT1aR knockout and wild-type mice (Supplementary Fig. 2). The differences between AT1aR knockout and wild-type mice were not significantly changed after PPARγ blockade with T0070907 (data not shown). As previously reported (Sumners et al., 2013), AT1aR knockout mice have lower systolic and diastolic blood pressures compared to wild-type mice, and this difference was maintained at 1 day post-injury (Fig. 8A). There was no difference in the injury-induced reduction of cerebral blood flow in the pericontusional cortex between AT1aR knockout mice and wild-type mice (Fig. 8B). AT1aR knockout mice demonstrated better performance on the rotarod before injury but did not improve their performance with time, unlike the uninjured mice, so at later time points there was no significant difference between AT1aR knockout injured mice and wild-type uninjured mice (Fig. 8G). Thus, the absence of AT1a receptors is beneficial for some important aspects of the recovery from injury, suggesting that angiotensin II signalling through the AT1aR has detrimental consequences for the brain after trauma.
Figure 8.
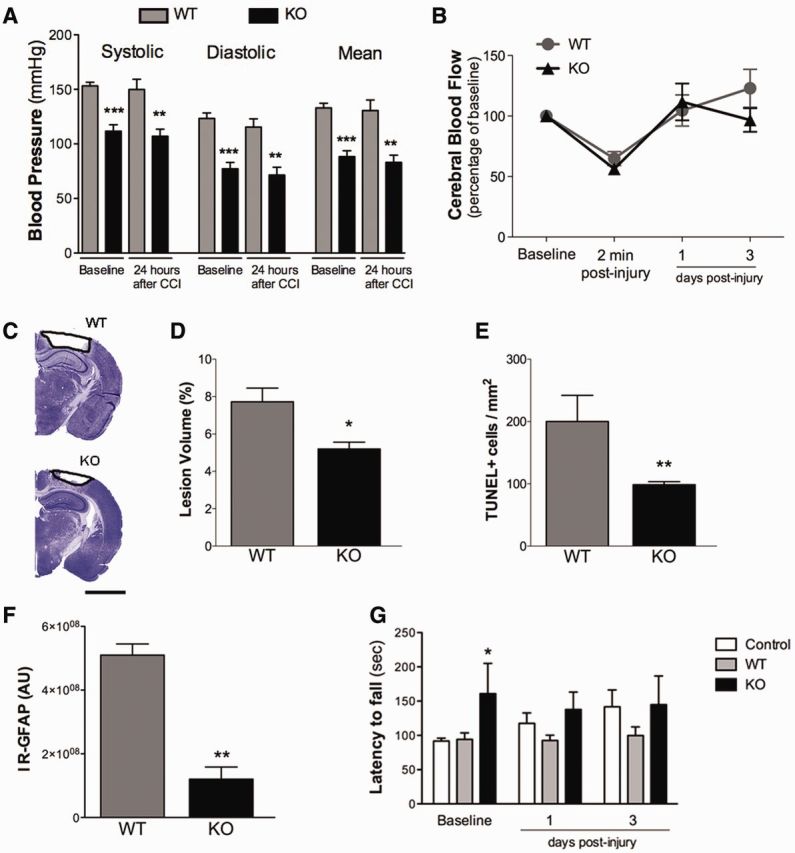
Genetic deletion of AT1R results in decreased vulnerability to brain injury. (A) Systolic, diastolic and mean blood pressures in AT1aR knock-out (KO) mice before and 24 h after CCI injury were reduced compared with those of wild-type (WT) mice. (B) AT1R deletion did not alter the cerebral blood flow after CCI injury compared to wild-type mice. (C) Representative Cresyl-violet brain sections show lesion volume, quantified in (D) is reduced in AT1aR knockout mice compared with wild-type mice. (E) AT1aR knockout mice had fewer apoptotic cells (TUNEL+) at 3 days post-injury. (F) AT1aR knockout mice exhibited a strong reduction in astrogliosis, measured by GFAP immunoreactivity (IR-GFAP) at 3 days post-injury. (G) AT1aR knockout mice exhibited better motor performance before injury (baseline levels) compared with wild-type mice. Data are expressed as mean ± SEM, n = 8–10 (for rotarod task), n = 4–6 (histology), ***P < 0.001, **P < 0.01, *P < 0.05 AT1aR knockout versus wild-type mice.
Discussion
We found that two sartans, candesartan and telmisartan, improve functional and morphological recovery when administered up to 6 h following TBI in the mouse. The beneficial effects of these drugs include acute and long-term reduction of lesion volume, enhancement of cognitive and motor function, protection of cerebral blood flow, and reduction in inflammation and the amount of activated microglia and astrocytes. For both candesartan and telmisartan, effects occur at non-hypotensive doses, a benefit as a decline in blood pressure immediately after TBI may worsen the outcome (Andriessen et al., 2010). Although both drugs have somewhat similar effects initially, candesartan’s actions seem more beneficial at more chronic time points, suggesting that candesartan has better long-term benefit. Mechanistically, our data indicate that sartan treatment affects two different signalling pathways to produce an improvement in recovery from TBI, AT1R blockade and PPARγ activation. Thus, these preclinical data show that sartan treatment is a promising therapeutic for the treatment of TBI.
As candesartan and telmisartan act through a combination of PPARγ agonist activity and AT1R blockade (Saavedra, 2012; Villapol and Saavedra, 2015), we attempted to determine the relative contribution of each pathway to the efficacy of both drugs. The AT1R is mainly located in neurons, vascular endothelial and smooth muscle cells and astrocytes in the mature brain (Saavedra, 2005). Microglia in culture express AT1R, while activation of microglia in vivo may induce AT1R expression (Li et al., 2009; Wu et al., 2013; Rodriguez-Perez et al., 2015). The reduced vulnerability of AT1R knockout mice to CCI injury in comparison to wild-type mice suggests that the brain’s endogenous RAS is activated by TBI and that AT1R signalling is detrimental to recovery from TBI. AT1R signalling has been well studied in peripheral vascular cells, and it is likely that similar mechanisms produce harmful effects of angiotensin II signalling through the AT1R in the brain. AT1R signalling generates significant reactive oxygen species, through activation of NADPH oxidase, the major source of non-mitochondrial reactive oxygen species (Rodriguez-Perez et al., 2015). AT1R activation also activates NF-κB dependent transcription, and hence induces transcription of several pro-inflammatory cytokines, as well as stimulation of several different kinases that themselves participate in the propagation of inflammatory responses and apoptotic pathways (Villapol and Saavedra, 2015). AT1R signalling therefore could enhance many of the harmful pathways that are activated after TBI, worsening outcomes. Sartans, through inhibition of AT1R are therefore directly neuroprotective as well as indirectly through reducing the amount of inflammation and reactive oxygen species that exists after injury.
The cerebrovasculature is significantly impacted by injury. Following CCI injury, the blood–brain barrier is dramatically opened, but even in closed head injury there is significant leakiness of the blood–brain barrier (Steckelings and Unger, 2012; Umschweif et al., 2014). AT1R signalling can alter the properties of vascular endothelial cells to increase blood–brain barrier permeability (Fleegal-DeMotta et al., 2009; Zhang et al., 2010). Sartan treatment after TBI may therefore assist in the closure of the blood–brain barrier through blocking AT1R signalling (Pelisch et al., 2013). AT1R activity in vascular smooth muscle cells promotes vasoconstriction, so blockade of these receptors should promote increased cerebral blood flow. Indeed, we did find sartan treatment improved cerebral blood flow after injury (Fig. 3). Given the proportional relationship between mean arterial pressure and cerebral blood flow, it was perhaps surprising that we found improved cerebral blood flow at sartan doses that did not decrease either systolic or diastolic pressure. Sartans may therefore be acting to reduce intracranial pressure and/or decrease cerebrovascular resistance. It has been previously demonstrated that sartan treatment reverses arterial stiffness and restores cerebral blood flow autoregulation in spontaneous hypertensive rats (Nishimura et al., 2000; Zhou et al., 2006). Further, the blockage of cerebrovascular AT1R significantly helped to maintain cerebral blood flow levels following ischaemia (Ito et al., 2002).
However, there was no difference in cerebral blood flow between wild-type and AT1R knockout mice. This may be attributed to compensatory changes during development of the AT1R knockout mice. In various models of vascular distress sartan treatment can provide neurovascular protection and be proangiogenic through the increased expression of BDNF and VEGF (Kozak et al., 2009; Guan et al., 2011; Alhusban et al., 2013; Soliman et al., 2014). Administration of sartans also protects the endothelium through inducing eNOS (encoded by Nos3) expression and reducing the expression of iNOS (Nos2) in vitro and in vivo (Bennai et al., 1999). Thus, sartan treatment, by blockade of the AT1R, may protect the cerebrovasculature through numerous mechanisms after TBI.
PPARγ activation is an important component of the efficacy of both candesartan and telmisartan because neuroprotection by both compounds is decreased to a substantial degree, by PPARγ blockade (Figs 5 and 6). Activation of PPARγ is strongly neuroprotective in numerous models of neurological disease and stroke (Kapadia et al., 2008; Gillespie et al., 2011). Exogenous PPARγ agonists mediate their PPARγ-dependent effects mainly through reduction of microglial and astrocyte activation with a subsequent reduction in inflammatory cytokine and chemokine expression (Kapadia et al., 2008). Although it has been assumed that PPARγ activation by sartans is independent of AT1R blockade, there are reports of an inverse balance between AT1R activity and PPARγ activation (Tham et al., 2002). However, we did not find any effect of PPARγ antagonists on the reduced vulnerability of AT1R knockout mice to TBI (data not shown), suggesting that these two pathways function separately to improve recovery from TBI. We have previously reported that CCI injury increases PPARγ mRNA expression in the brain (Villapol et al., 2012). Our results here show that inhibiting PPARγ activity is partially detrimental for brain recovery (although not significantly so).
There were differential effects of the sartans on functional recovery. Telmisartan, but not candesartan, partially improved motor function as assessed by performance on the rotarod (Fig. 7). This effect was removed by co-administration of the PPARγ antagonist. However, telmisartan’s benefit to functional recovery was restricted to this early time point. Given that candesartan administration improved cognitive function up to 1 month after injury, it was surprising that candesartan administered after injury did not have more of an effect on motor function. We have previously shown that candesartan administration before injury improved rotarod performance (Villapol et al., 2012). At 30 days post-injury mice treated with candesartan showed improved function in the Morris water maze, a test of learning and spatial memory. This effect correlated with a significant reduction in lesion volume that led to conservation of the majority of the hippocampus, an area of the brain associated with spatial working memory. Although this protection of brain parenchyma could explain the improvement in cognitive function, there is also evidence that interfering with the brain’s RAS is beneficial to learning and memory. Thus, sartan treatment improves cognitive function in various mouse models of disease (Tota et al., 2009; Tsukuda et al., 2009; Wang et al., 2014) and in patients receiving sartans for chronic hypertension (Fogari et al., 2003; Hajjar et al., 2013). Additionally, a large retrospective study revealed that patients receiving chronic sartan treatment for hypertension had reduced incidence of Alzheimer’s disease (Li et al., 2010), although this has not been replicated in all populations (Hsu et al., 2013). Autopsy studies have also shown that patients on sartans had less Alzheimer’s disease-related pathology (Hajjar et al., 2012). Although the reasons behind these effects are no doubt complex, some of the same mechanisms may function after TBI to promote improved cognitive performance with sartan treatment.
Individual sartans have different pharmacological profiles (Schupp et al., 2004; Saavedra, 2012; Michel et al., 2013). Telmisartan, in addition to its AT1R blocking properties, is a very effective partial PPARγ activator (Benson et al., 2004; Schupp et al., 2004; Mogi et al., 2008). Candesartan has little PPARγ activating effects in cell culture systems, (Benson et al., 2004; Schupp et al., 2004) but is an effective PPARγ activator when administered in vivo (Zorad et al., 2006; Villapol et al., 2012). Candesartan has a higher binding potency to the AT1R, and a slower dissociation than telmisartan, suggesting that it might be a better AT1R antagonist, even if a worse PPARγ agonist (Cernes et al., 2011). In our experiments telmisartan had slightly better short-term benefits, but by 1 month after injury, effects were significantly less than those of candesartan. Thus, it is possible that at more acute time points, the stronger PPARγ agonist has greater benefit, but as the injury progresses, the importance of AT1R blockade is more prominent. However, these drugs have different routes of administration due to their different solubility (Gleiter and Morike, 2002; Deppe et al., 2010). Telmisartan was administered once daily by oral gavage, whereas candesartan, after the initial injection, for treatment longer than 3 days, was administered by continuous subcutaneous minipump. So although the half-life of telmisartan is longer than for candesartan, it is possible that the continuous infusion of candesartan provided some benefit.
Both sartans showed efficacy when administered up to 6 h post-injury. This time window allows for realistic clinical application. The recent failed PROTECT trial of progesterone, administered the first drug dose within 4 h of injury (Wright et al., 2014). Although sartan administration at 24 h post-injury did not significantly improve histological outcome measures at 3 days post-injury (Supplementary Fig. 1), it is possible that some time between 6 and 24 h post-injury may still be effective for a first dose of sartan. We will address this question in future studies. Candesartan and telmisartan do cross the blood–brain barrier, although this has only been shown directly in humans for telmisartan (Nishimura et al., 2000; Noda et al., 2012). Although to date sartans have not been adequately tested in neurodegenerative or traumatic brain disorders, there is clear clinical evidence that sartan administration is therapeutically effective in brain ischaemia and to prevent stroke (Zanchetti and Elmfeldt, 2006; Hajjar et al., 2013). Our preclinical observations indicate that two well characterized and safe sartans, candesartan and telmisartan, have pleiotropic neurorestorative effects, providing strong support for initiation of a clinical trial to determine their efficacy in patients suffering from TBI.
Acknowledgements
We thank Dr Tim O'Neil for his assistance with the blood pressure measurements and Dr Cara Olsen for statistical advice. The opinions and assertions contained herein are the private opinions of the authors and are not to be construed as reflecting the views of the Uniformed Services University of the Health Sciences or the US Department of Defense. The authors declare no competing financial interests.
Glossary
Abbreviations
- AT1R
angiotensin II type 1 receptor
- CCI
controlled cortical impact
- PPARγ
proliferator-activated receptor gamma
- TBI
traumatic brain injury
Funding
This work was supported by a Department of Defense, Center for Neuroscience and Regenerative Medicine (CNRM) grant to A.J.S. S.V. was supported by a CNRM postdoctoral fellowship. J.M.S. was supported by the Intramural Research Program at the National Institute of Mental Health, NIH (MH 002762-16).
Supplementary material
Supplementary material is available at Brain online.
References
- Alhusban A, Kozak A, Ergul A, Fagan SC. AT1 receptor antagonism is proangiogenic in the brain: BDNF a novel mediator. J Pharmacol Exp Ther 2013; 344: 348–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andriessen TM, Jacobs B, Vos PE. Clinical characteristics and pathophysiological mechanisms of focal and diffuse traumatic brain injury. J Cell Mol Med 2010; 14: 2381–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armando I, Terron JA, Falcon-Neri A, Takeshi I, Hauser W, Inagami T, et al. Increased angiotensin II AT(1) receptor expression in paraventricular nucleus and hypothalamic-pituitary-adrenal axis stimulation in AT(2) receptor gene disrupted mice. Neuroendocrinology 2002; 76: 137–47. [DOI] [PubMed] [Google Scholar]
- Benicky J, Sanchez-Lemus E, Honda M, Pang T, Orecna M, Wang J, et al. Angiotensin II AT(1) receptor blockade ameliorates brain inflammation. Neuropsychopharmacology 2011; 36: 857–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennai F, Morsing P, Paliege A, Ketteler M, Mayer B, Tapp R, et al. Normalizing the expression of nitric oxide synthase by low-dose AT1 receptor antagonism parallels improved vascular morphology in hypertensive rats. J Am Soc Nephrol 1999; 10 (Suppl 11): S104–15. [PubMed] [Google Scholar]
- Benson SC, Pershadsingh HA, Ho CI, Chittiboyina A, Desai P, Pravenec M, et al. Identification of telmisartan as a unique angiotensin II receptor antagonist with selective PPARgamma-modulating activity. Hypertension 2004; 43: 993–1002. [DOI] [PubMed] [Google Scholar]
- Bordet R, Ouk T, Petrault O, Gele P, Gautier S, Laprais M, et al. PPAR: a new pharmacological target for neuroprotection in stroke and neurodegenerative diseases. Biochem Soc Trans 2006; 34(Pt 6): 1341–6. [DOI] [PubMed] [Google Scholar]
- Cabaleiro T, Roman M, Ochoa D, Talegon M, Prieto-Perez R, Wojnicz A, et al. Evaluation of the relationship between sex, polymorphisms in CYP2C8 and CYP2C9, and pharmacokinetics of angiotensin receptor blockers. Drug Metab Dispos 2013; 41: 224–9. [DOI] [PubMed] [Google Scholar]
- Cernes R, Mashavi M, Zimlichman R. Differential clinical profile of candesartan compared to other angiotensin receptor blockers. Vasc Health Risk Manag 2011; 7: 749–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deppe S, Boger RH, Weiss J, Benndorf RA. Telmisartan: a review of its pharmacodynamic and pharmacokinetic properties. Expert Opin Drug Metab Toxicol 2010; 6: 863–71. [DOI] [PubMed] [Google Scholar]
- Diaz-Arrastia R, Kochanek PM, Bergold P, Kenney K, Marx CE, Grimes CJ, et al. Pharmacotherapy of traumatic brain injury: state of the science and the road forward: report of the Department of Defense Neurotrauma Pharmacology Workgroup. J Neurotrauma 2014; 31: 135–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleegal-DeMotta MA, Doghu S, Banks WA. Angiotensin II modulates BBB permeability via activation of the AT(1) receptor in brain endothelial cells. J Cereb Blood Flow Metab 2009; 29: 640–7. [DOI] [PubMed] [Google Scholar]
- Fogari R, Mugellini A, Zoppi A, Derosa G, Pasotti C, Fogari E, et al. Influence of losartan and atenolol on memory function in very elderly hypertensive patients. J Hum Hyperten 2003; 17: 781–5. [DOI] [PubMed] [Google Scholar]
- Fukui Y, Yamashita T, Kurata T, Sato K, Lukic V, Hishikawa N, et al. Protective effect of telmisartan against progressive oxidative brain damage and synuclein phosphorylation in stroke-resistant spontaneously hypertensive rats. J Stroke Cerebrovasc Dis 2014; 23: 1545–53. [DOI] [PubMed] [Google Scholar]
- Garrido-Gil P, Joglar B, Rodriguez-Perez AI, Guerra MJ, Labandeira-Garcia JL. Involvement of PPAR-gamma in the neuroprotective and anti-inflammatory effects of angiotensin type 1 receptor inhibition: effects of the receptor antagonist telmisartan and receptor deletion in a mouse MPTP model of Parkinson's disease. J Neuroinflammation 2012; 9: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie W, Tyagi N, Tyagi SC. Role of PPARgamma, a nuclear hormone receptor in neuroprotection. Indian J Biochem Biophys 2011; 48: 73–81. [PubMed] [Google Scholar]
- Gleiter CH, Morike KE. Clinical pharmacokinetics of candesartan. Clin Pharmacokinet 2002; 41: 7–17. [DOI] [PubMed] [Google Scholar]
- Guan W, Somanath PR, Kozak A, Goc A, El-Remessy AB, Ergul A, et al. Vascular protection by angiotensin receptor antagonism involves differential VEGF expression in both hemispheres after experimental stroke. PloS One 2011; 6: e24551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajjar I, Brown L, Mack WJ, Chui H. Impact of Angiotensin receptor blockers on Alzheimer disease neuropathology in a large brain autopsy series. Arch Neurol 2012; 69: 1632–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajjar I, Hart M, Chen YL, Mack W, Novak V, Chiu H, et al. Antihypertensive therapy and cerebral hemodynamics in executive mild cognitive impairment: results of a pilot randomized clinical trial. J Am Geriatr Soc 2013; 61: 194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamai M, Iwai M, Ide A, Tomochika H, Tomono Y, Mogi M, et al. Comparison of inhibitory action of candesartan and enalapril on brain ischemia through inhibition of oxidative stress. Neuropharmacology 2006; 51: 822–8. [DOI] [PubMed] [Google Scholar]
- Hsu CY, Huang CC, Chan WL, Huang PH, Chiang CH, Chen TJ, et al. Angiotensin-receptor blockers and risk of Alzheimer's disease in hypertension population–a nationwide cohort study. Circ J 2013; 77: 405–10. [DOI] [PubMed] [Google Scholar]
- Ishrat T, Pillai B, Soliman S, Fouda AY, Kozak A, Johnson MH, et al. Low–dose candesartan enhances molecular mediators of neuroplasticity and subsequent functional recovery after ischemic stroke in rats. Mol Neurobiol 2015; 51: 1542–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Yamakawa H, Bregonzio C, Terron JA, Falcon-Neri A, Saavedra JM. Protection against ischemia and improvement of cerebral blood flow in genetically hypertensive rats by chronic pretreatment with an angiotensin II AT1 antagonist. Stroke 2002; 33: 2297–303. [DOI] [PubMed] [Google Scholar]
- Johren O, Saavedra JM. Expression of AT1A and AT1B angiotensin II receptor messenger RNA in forebrain of 2-wk-old rats. Am J Physiol 1996; 271(1 Pt 1): E104–12. [DOI] [PubMed] [Google Scholar]
- Joseph JP, Mecca AP, Regenhardt RW, Bennion DM, Rodriguez V, Desland F, et al. The angiotensin type 2 receptor agonist Compound 21 elicits cerebroprotection in endothelin-1 induced ischemic stroke. Neuropharmacology 2014; 81: 134–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung KH, Chu K, Lee ST, Kim SJ, Song EC, Kim EH, et al. Blockade of AT1 receptor reduces apoptosis, inflammation, and oxidative stress in normotensive rats with intracerebral hemorrhage. J Pharmacol Exp Ther 2007; 322: 1051–8. [DOI] [PubMed] [Google Scholar]
- Justin A, Sathishkumar M, Sudheer A, Shanthakumari S, Ramanathan M. Non-hypotensive dose of telmisartan and nimodipine produced synergistic neuroprotective effect in cerebral ischemic model by attenuating brain cytokine levels. Pharmacol Biochem Behav 2014; 122: 61–73. [DOI] [PubMed] [Google Scholar]
- Kapadia R, Yi JH, Vemuganti R. Mechanisms of anti-inflammatory and neuroprotective actions of PPAR-gamma agonists. Front Biosci 2008; 13: 1813–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara Y, Taguchi A, Uno H, Nakano A, Nakagomi T, Hirose H, et al. Telmisartan suppresses cerebral injury in a murine model of transient focal ischemia. Brain Res 2010; 1340: 70–80. [DOI] [PubMed] [Google Scholar]
- Kozak A, Ergul A, El-Remessy AB, Johnson MH, Machado LS, Elewa HF, et al. Candesartan augments ischemia-induced proangiogenic state and results in sustained improvement after stroke. Stroke 2009; 40: 1870–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak W, Kozak A, Johnson MH, Elewa HF, Fagan SC. Vascular protection with candesartan after experimental acute stroke in hypertensive rats: a dose-response study. J Pharmacol Exp Ther 2008; 326: 773–82. [DOI] [PubMed] [Google Scholar]
- Kumar A, Loane DJ. Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav Immun 2012; 26: 1191–201. [DOI] [PubMed] [Google Scholar]
- Li JJ, Lu J, Kaur C, Sivakumar V, Wu CY, Ling EA. Expression of angiotensin II and its receptors in the normal and hypoxic amoeboid microglial cells and murine BV-2 cells. Neuroscience 2009; 158: 1488–99. [DOI] [PubMed] [Google Scholar]
- Li NC, Lee A, Whitmer RA, Kivipelto M, Lawler E, Kazis LE, et al. Use of angiotensin receptor blockers and risk of dementia in a predominantly male population: prospective cohort analysis. BMJ 2010; 340: b5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Kitazato KT, Uno M, Yagi K, Kanematsu Y, Tamura T, et al. Protective mechanisms of the angiotensin II type 1 receptor blocker candesartan against cerebral ischemia: in-vivo and in-vitro studies. J Hypertens 2008; 26: 1435–45. [DOI] [PubMed] [Google Scholar]
- Lu GC, Cheng JW, Zhu KM, Ma XJ, Shen FM, Su DF. A systematic review of angiotensin receptor blockers in preventing stroke. Stroke 2009; 40: 3876–8. [DOI] [PubMed] [Google Scholar]
- McConeghy KW, Hatton J, Hughes L, Cook AM. A review of neuroprotection pharmacology and therapies in patients with acute traumatic brain injury. CNS Drugs 2012; 26: 613–36. [DOI] [PubMed] [Google Scholar]
- Meredith PA, Murray LS, McMurray JJ. A putative placebo comparison of the SCOPE and LIFE trials. J Renin Angiotensin Aldosterone Syst 2004; 5: 59–63. [DOI] [PubMed] [Google Scholar]
- Michel MC, Foster C, Brunner HR, Liu L. A systematic comparison of the properties of clinically used angiotensin II type 1 receptor antagonists. Pharmacol Rev 2013; 65: 809–48. [DOI] [PubMed] [Google Scholar]
- Mitchell K, Shah JP, Tsytsikova LV, Campbell AM, Affram K, Symes AJ. LPS antagonism of TGF-beta signaling results in prolonged survival and activation of rat primary microglia. J Neurochem 2014; 129: 155–68. [DOI] [PubMed] [Google Scholar]
- Mogi M, Li JM, Tsukuda K, Iwanami J, Min LJ, Sakata A, et al. Telmisartan prevented cognitive decline partly due to PPAR-gamma activation. Biochem Biophys Res Commun 2008; 375: 446–9. [DOI] [PubMed] [Google Scholar]
- Nishimura Y, Ito T, Hoe K, Saavedra JM. Chronic peripheral administration of the angiotensin II AT(1) receptor antagonist candesartan blocks brain AT(1) receptors. Brain Res 2000; 871: 29–38. [DOI] [PubMed] [Google Scholar]
- Noda A, Fushiki H, Murakami Y, Sasaki H, Miyoshi S, Kakuta H, et al. Brain penetration of telmisartan, a unique centrally acting angiotensin II type 1 receptor blocker, studied by PET in conscious rhesus macaques. Nucl Med Biol 2012; 39: 1232–5. [DOI] [PubMed] [Google Scholar]
- Ohshima K, Mogi M, Horiuchi M. Therapeutic approach for neuronal disease by regulating renin-angiotensin system. Curr Hypertens Rev 2013; 9: 99–107. [DOI] [PubMed] [Google Scholar]
- Omura-Matsuoka E, Yagita Y, Sasaki T, Terasaki Y, Oyama N, Sugiyama Y, et al. Postischemic administration of angiotensin II type 1 receptor blocker reduces cerebral infarction size in hypertensive rats. Hypertens Res 2009; 32: 548–53. [DOI] [PubMed] [Google Scholar]
- Ozacmak VH, Sayan H, Cetin A, Akyildiz-Igdem A. AT1 receptor blocker candesartan-induced attenuation of brain injury of rats subjected to chronic cerebral hypoperfusion. Neurochem Res 2007; 32: 1314–21. [DOI] [PubMed] [Google Scholar]
- Pelisch N, Hosomi N, Mori H, Masaki T, Nishiyama A. RAS inhibition attenuates cognitive impairment by reducing blood- brain barrier permeability in hypertensive subjects. Curr Hypertens Rev 2013; 9: 93–8. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Perez AI, Borrajo A, Rodriguez-Pallares J, Guerra MJ, Labandeira-Garcia JL. Interaction between NADPH-oxidase and Rho-kinase in angiotensin II-induced microglial activation. Glia 2015; 63: 466–82. [DOI] [PubMed] [Google Scholar]
- Saavedra JM. Brain and pituitary angiotensin. Endocr Rev 1992; 13: 329–80. [DOI] [PubMed] [Google Scholar]
- Saavedra JM. Brain angiotensin II: new developments, unanswered questions and therapeutic opportunities. Cell Mol Neurobiol 2005; 25: 485–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saavedra JM. Angiotensin II AT(1) receptor blockers as treatments for inflammatory brain disorders. Clin Sci (Lond) 2012; 123: 567–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saavedra JM, Benicky J, Zhou J. Angiotensin II: multitasking in the brain. J Hypertens Suppl 2006; 24: S131–7. [DOI] [PubMed] [Google Scholar]
- Sasamura H, Hein L, Krieger JE, Pratt RE, Kobilka BK, Dzau VJ. Cloning, characterization, and expression of two angiotensin receptor (AT-1) isoforms from the mouse genome. Biochem Biophys Res Commun 1992; 185: 253–9. [DOI] [PubMed] [Google Scholar]
- Schupp M, Janke J, Clasen R, Unger T, Kintscher U. Angiotensin type 1 receptor blockers induce peroxisome proliferator-activated receptor-gamma activity. Circulation 2004; 109: 2054–7. [DOI] [PubMed] [Google Scholar]
- Soliman S, Ishrat T, Pillai A, Somanath PR, Ergul A, El-Remessy AB, et al. Candesartan induces a prolonged proangiogenic effect and augments endothelium-mediated neuroprotection after oxygen and glucose deprivation: role of vascular endothelial growth factors A and B. J Pharmacol Exp Ther 2014; 349: 444–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stangier J, Su CA, Schondorfer G, Roth W. Pharmacokinetics and safety of intravenous and oral telmisartan 20 mg and 120 mg in subjects with hepatic impairment compared with healthy volunteers. J Clin Pharmacol 2000; 40(12 Pt 1): 1355–64. [PubMed] [Google Scholar]
- Steckelings UM, Unger T. Angiotensin II type 2 receptor agonists–where should they be applied? Expert Opin Investig Drugs 2012; 21: 763–6. [DOI] [PubMed] [Google Scholar]
- Sumners C, Horiuchi M, Widdop RE, McCarthy C, Unger T, Steckelings UM. Protective arms of the renin-angiotensin-system in neurological disease. Clin Exp Pharmacol Physiol 2013; 40: 580–8. [DOI] [PubMed] [Google Scholar]
- Tham DM, Martin-McNulty B, Wang YX, Wilson DW, Vergona R, Sullivan ME, et al. Angiotensin II is associated with activation of NF-kappaB-mediated genes and downregulation of PPARs. Physiol Genomics 2002; 11: 21–30. [DOI] [PubMed] [Google Scholar]
- Thone-Reineke C, Steckelings UM, Unger T. Angiotensin receptor blockers and cerebral protection in stroke. J Hypertens Suppl 2006; 24: S115–21. [DOI] [PubMed] [Google Scholar]
- Timaru-Kast R, Wyschkon S, Luh C, Schaible EV, Lehmann F, Merk P, et al. Delayed inhibition of angiotensin II receptor type 1 reduces secondary brain damage and improves functional recovery after experimental brain trauma*. Crit Care Med 2012; 40: 935–44. [DOI] [PubMed] [Google Scholar]
- Tota S, Kamat PK, Awasthi H, Singh N, Raghubir R, Nath C, et al. Candesartan improves memory decline in mice: involvement of AT1 receptors in memory deficit induced by intracerebral streptozotocin. Behav Brain Res 2009; 199: 235–40. [DOI] [PubMed] [Google Scholar]
- Tsukuda K, Mogi M, Iwanami J, Min LJ, Sakata A, Jing F, et al. Cognitive deficit in amyloid-beta-injected mice was improved by pretreatment with a low dose of telmisartan partly because of peroxisome proliferator-activated receptor-gamma activation. Hypertension 2009; 54: 782–7. [DOI] [PubMed] [Google Scholar]
- Umschweif G, Liraz-Zaltsman S, Shabashov D, Alexandrovich A, Trembovler V, Horowitz M, et al. Angiotensin receptor type 2 activation induces neuroprotection and neurogenesis after traumatic brain injury. Neurotherapeutics 2014; 11: 665–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villapol S, Bonnin P, Fau S, Baud O, Renolleau S, Charriaut-Marlangue C. Unilateral blood flow decrease induces bilateral and symmetric responses in the immature brain. Am J Pathol 2009; 175: 2111–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villapol S, Byrnes KR, Symes AJ. Temporal dynamics of cerebral blood flow, cortical damage, apoptosis, astrocyte-vasculature interaction and astrogliosis in the pericontusional region after traumatic brain injury. Front Neurol 2014; 5: 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villapol S, Saavedra JM. Neuroprotective effects of angiotensin receptor blockers. Am J Hypertens 2015; 28; 289–99. [DOI] [PubMed] [Google Scholar]
- Villapol S, Yaszemski AK, Logan TT, Sanchez-Lemus E, Saavedra JM, Symes AJ. Candesartan, an angiotensin II AT(1)-receptor blocker and PPAR-gamma agonist, reduces lesion volume and improves motor and memory function after traumatic brain injury in mice. Neuropsychopharmacology 2012; 37: 2817–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villela D, Leonhardt J, Patel N, Joseph J, Kirsch S, Hallberg A, et al. Angiotensin type 2 receptor (AT2R) and receptor Mas: a complex liaison. Clin Sci (Lond) 2015; 128: 227–34. [DOI] [PubMed] [Google Scholar]
- Wang J, Pang T, Hafko R, Benicky J, Sanchez-Lemus E, Saavedra JM. Telmisartan ameliorates glutamate-induced neurotoxicity: roles of AT(1) receptor blockade and PPARgamma activation. Neuropharmacology 2014; 79: 249–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright DW, Yeatts SD, Silbergleit R, Palesch YY, Hertzberg VS, Frankel M, et al. Very early administration of progesterone for acute traumatic brain injury. N Engl J Med 2014; 371: 2457–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright JW, Harding JW. Brain angiotensin receptor subtypes in the control of physiological and behavioral responses. Neurosci Biobehav Rev 1994; 18: 21–53. [DOI] [PubMed] [Google Scholar]
- Wright JW, Harding JW. Brain renin-angiotensin–a new look at an old system. Prog Neurobiol 2011; 95: 49–67. [DOI] [PubMed] [Google Scholar]
- Wu CY, Zha H, Xia QQ, Yuan Y, Liang XY, Li JH, et al. Expression of angiotensin II and its receptors in activated microglia in experimentally induced cerebral ischemia in the adult rats. Mol Cell Biochem 2013; 382: 47–58. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Zhai Y, Kurata T, Hishikawa N, Morimoto N, Ohta Y, et al. Strong improvement of apolipoprotein E/low-density lipoprotein receptor signals by telmisartan in poststroke spontaneously hypertensive stroke resistant. J Stroke Cerebrovasc Dis 2014; 23: 2240–9. [DOI] [PubMed] [Google Scholar]
- Zanchetti A, Elmfeldt D. Findings and implications of the Study on COgnition and Prognosis in the Elderly (SCOPE) - a review. Blood Press 2006; 15: 71–9. [DOI] [PubMed] [Google Scholar]
- Zhang M, Mao Y, Ramirez SH, Tuma RF, Chabrashvili T. Angiotensin II induced cerebral microvascular inflammation and increased blood-brain barrier permeability via oxidative stress. Neuroscience 2010; 171: 852–8. [DOI] [PubMed] [Google Scholar]
- Zhou J, Pavel J, Macova M, Yu ZX, Imboden H, Ge L, et al. AT1 receptor blockade regulates the local angiotensin II system in cerebral microvessels from spontaneously hypertensive rats. Stroke 2006; 37: 1271–6. [DOI] [PubMed] [Google Scholar]
- Zorad S, Dou JT, Benicky J, Hutanu D, Tybitanclova K, Zhou J, et al. Long-term angiotensin II AT1 receptor inhibition produces adipose tissue hypotrophy accompanied by increased expression of adiponectin and PPARgamma. Eur J Pharmacol 2006; 552: 112–22. [DOI] [PMC free article] [PubMed] [Google Scholar]