

Abstract
Epigenetics is a branch of science that focuses on mechanisms related to control and modification of expression of genetic material without any changes to its sequences. Such mechanisms include post-translational modifications of histones. It is widely known that carcinogenesis is related to hypoacetylation of genes that influence apoptosis, the cell cycle, cell signaling, the immunologic response, angiogenesis and occurrence of metastasis. Currently conducted research focuses on several strategies related to epigenetic therapy. One such strategy is based on the use of histone deacetylase inhibitors. This paper presents mechanisms through which these compounds work and a summary of their characteristics. It also includes a review of clinical tests related to histone deacetylase inhibitors, as well as their relationship with other chemotherapeutic methods. A better understanding of the involved mechanisms will provide a rational basis to improve the therapeutic outcome of available antitumor agents.
Keywords: epigenetics, histone deacetylase, histone deacetylase inhibitor, cancer therapy
Introduction
Epigenetics is a branch of science that focuses on mechanisms related to control and modification of expression of genetic material without any changes to its sequences. Such mechanisms include post-translational modifications of histones. Expression of genes could be related to the availability of chromatin for transcription complexes.
Activation of gene expression occurs under the influence of the processes of acetylation, phosphorylation and methylation of lysine and arginine (H3-K4, H3-K36 and H3-K79), as well as ubiquitylation of H2B-K120. Methylation of H3-K9, H3-K27 and H4-K20 and ubiquitylation of H2B-K120 inhibit expression [1].
It is widely known that carcinogenesis is related to hypoacetylation of genes that influence apoptosis, the cell cycle, cell signaling, the immunologic response and metastasis. The process of acetylation is catalyzed by histone acetyltransferase (HAT). The reverse process of deacetylation occurs with participation of histone deacetylase (HDAC) activity [2].
Histone deacetylases have been identified in mammal cells so far. They were divided into four classes, based on the homology to yeast enzymes. Class I HDACs (HDAC1-3, 8) can be mainly found in the nucleus and are related to various kinds of transcriptional repressors (Sin3, CoRest, SMRT/NCoR and NuRD). Class II HDACs (HDAC4-7, 9, 10) can be found in both the nucleus and the cytoplasm. Histone deacetylase 4 is engaged in a multiprotein transcriptional corepressor complex and is implicated in myocyte differentiation, skeletogenesis, and neuronal survival. Class III HDACs are also called sirtuins (SIRT, silent information regulators). This class of enzymes depends on the NAD cofactor. There are currently seven members of this class known to occur in humans. Sirtuins are located in the nucleus, cytoplasm and mitochondria. Histone deacetylase 11, which is sometimes called class IV, negatively regulates interleukin 10 [3].
Epigenetic therapy
Several strategies related to epigenetic therapy are currently being tested. One of these strategies is based on the use of histone deacetylase activity inhibitors (HDACi). Drugs from this group feature various mechanisms in anti-tumor therapy. One of these mechanisms is based on selective initiation of apoptosis in tumor cells through the mitochondrial pathway and by influencing the expression of proteins such as Bcl-2 (B-Cell CLL/lymphoma 2), Bcl-XL (B-cell lymphoma-extra large), XIAP (X-linked inhibitor of apoptosis) and Mcl-1 (myeloid cell leukemia sequence 1) [4], as well as through activation of the death receptors (DR4, DR5, Fas) [5].
There are also reports on the influence of HDACi on the functioning of important transcription factors, such as Stat3 (signal transducer and activator of transcription-3) and NF-κB (nuclear factor-κB) in inflammatory processes and occurrence of neoplastic lesions [6].
Histone deacetylase inhibitor also influence regulation of the cell cycle through blocking it at the G1/S point. This is a result of the fact that drugs from this group interact with the genes p21 as well as CDK2 (cyclin-dependent kinase 2) and CDK4 (cyclin-dependent kinase 4) [7]. It is assumed that HDACi can also have an anti-angiogenic and inhibitory effect on the occurrence of metastasis through modulation of expression of the coding genes VEGF (vascular endothelial growth factor), bFGF (basic fibroblast growth factor), HIF-1α (hypoxia-inducible factor-1α), angiopoietins, eNOS (endothelial nitric-oxide synthase), MMPs (matrix metalloproteinases) and TIMPs (tissue inhibitors of metalloproteinases) [3, 7–9].
Histone deacetylase inhibitor cause accumulation of reactive oxygen species (ROS) and caspase activation in transformed but not normal cells [10].
Moreover, HDACi can contribute to inhibition of expression of various proteins which participate in repair of DNA, such as RAD50, RAD51 and Mre11 (meiotic recombination 11) [11].
Moreover, there is growing evidence that HDACi may increase immunogenicity of neoplastic cells through an influence on the expression of MHC (major histocompatibility complex), CD40, CD80 and CD86 molecules and intercellular adhesion molecule 1 (ICAM-1) [7, 12].
Systematics of histone deacetylase inhibitor compounds
There are several ways to systematize HDACi compounds. The first division is based on their chemical constitution. HDACi include hydroxamic acid derivatives, short-chain aliphatic (fatty) acids, benzoic acid amides, cyclic tetrapeptides, and epoxides. The second division is based on their activity in relation to various classes of HDAC [13].
Histone deacetylase inhibitor in cancer therapy
Examples below present various applications of HDACi in cancer therapy attempts.
Vorinostat (suberoylanilide hydroxamic acid, SAHA, trade name Zolinza, MSK-390) is a derivative of hydroxamic acid. It was the first HDACi compound certified by the FDA (Food and Drug Administration) authorities to be used in refractory cutaneous T-cell lymphoma (CTCL) therapy. It is a class I HDAC inhibitor (HDAC1, HDAC2, HDAC3) and a class II inhibitor (HDAC6) [14]. One of the first clinical studies where vorinostat was used in cancer therapy was a phase I study conducted by Kelly et al. [15] in a group of 73 patients with hematologic malignancies. The maximum tolerated dose was 400 mg once a day and 200 mg twice a day for continuous daily dosing and 300 mg twice a day for 3 consecutive days per week dosing. The compound demonstrated good bioavailability of 43%. Twenty-two patients (30%) remained under observation for 4 to 37+ months. There was 1 complete response, 3 partial responses, and 2 unconfirmed partial responses [15]. A study conducted by Olsen et al. [16] on 74 patients suffering from CTCL demonstrated unfavorable side-effects of vorinostat, such as diarrhea, fatigue, nausea, and anorexia. The ORR (objective response rate) was 29.7% overall, and 29.5% in stage IIB or higher patients [16].
Romidepsin (depsipeptide, trade name Istodax, FK228, FR901228) is a cyclic tetrapeptide isolated for the first time from Chromobacterium violaceum bacterial strains. This compound mainly inhibits activity in the HDAC class I. Pre-clinical trials proved it to have a strong cardiotoxic effect. The first clinical phase study conducted by Sandor et al. [17] on 37 patients did not confirm that effect, though it showed cardiac arrhythmia of the patients. In that trial, romidepsin was administered on day 1 and day 5 of a 21-day cycle as a 4-h i.v. infusion through a central venous catheter that had healed for at least 7 days after placement. One patient achieved a partial response. Similarly to vorinostat, romidepsin was authorized by the FDA to be used in CTCL treatment [17].
Belinostat (PXD101) is a member of the hydroxamic acid derivative family, affecting all HDAC classes [18]. A clinical phase I trial conducted by Steele et al. [19] on patients with advanced stages of solid tumors showed that the maximum tolerated dose was 1000 mg/m2/day (a total of 46 patients were given belinostat in one of six doses ranging from 150 to 1200 mg/m2/day). Stabilization of the disease was achieved in 39% of cases [19].
Mocetinostat (MGCD0103) is a benzoic acid amide, a class I and IV HDAC inhibitor [20]. One of the first phase I clinical trials was conducted by Garcia-Manero et al. [21] on 29 patients diagnosed with leukemia and myelodysplastic syndromes (MDS). The trial proved that the maximum tolerated doze was 60 mg/m2. Higher doses resulted in such side-effects as fatigue, nausea, vomiting and diarrhea [21].
Table 1 shows characteristics of HDACi and a review of neoplasms treated in the clinical trials where the mentioned compounds were used [based on [22, 23]].
Table 1.
Characteristics of selected histone deacetylase inhibitor
| HDAC inhibitors | Other common identifiers | Class | Structure | Cancer type (e.g.) |
|---|---|---|---|---|
| Vorinostat | Suberoylanilide hydroxamic acid, SAHA, Zolinza® (Merck Corporate, USA), a, MK0683 | hydroxamic acids | 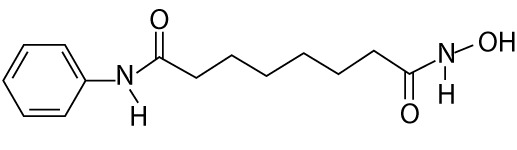 |
leukemia (e.g. blastic phase chronic myelogenous leukemia; recurrent adult acute lymphoblastic leukemia; recurrent adult acute myeloid leukemia), lymphoma (e.g. follicular lymphoma marginal zone lymphoma mantle cell lymphoma, Hodgkin's lymphoma), prostate cancer, small intestine cancer, relapsed non-small cell lung cancer, breast adenocarcinoma, colorectal carcinoma, advanced thyroid carcinoma |
| Panobinostat | LBH589 | hydroxamic acids |  |
prostate cancer, Hodgkin's lymphoma, melanoma, neuroendocrine tumors, thyroid carcinoma, myeloma, colon cancer, hepatocellular carcinoma, head and neck cancer, lymphoblastic leukemia |
| Trichostatin A | – | hydroxamic acids | 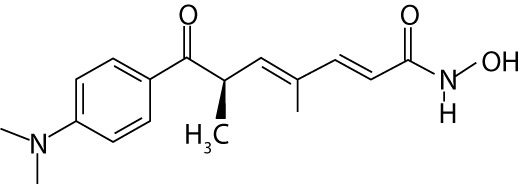 |
– |
| Belinostat | PXD101 | hydroxamic acids |  |
cutaneous T-cell lymphoma; peripheral T-cell lymphoma; non-Hodgkin's lymphoma, non-small-cell lung carcinoma, thymoma, soft tissue sarcomas |
| Oxamflatin | hydroxamic acids | 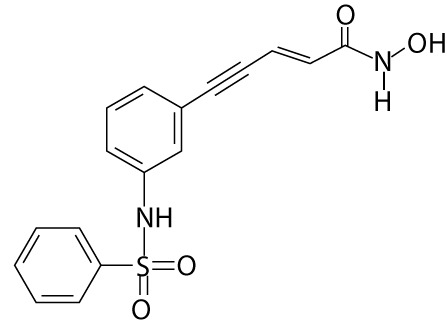 |
– | |
| Dacinostat | LAQ824, NVP-LAQ824 | hydroxamic acids | 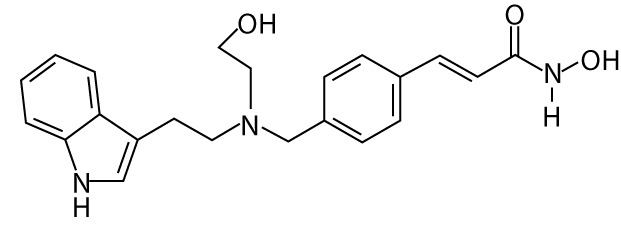 |
– |
| Scriptaid | GCK1026 LAQ824 | hydroxamic acids | 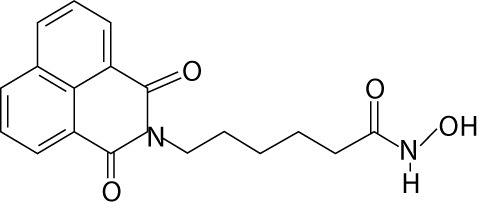 |
– |
| Pyroxamide | – | 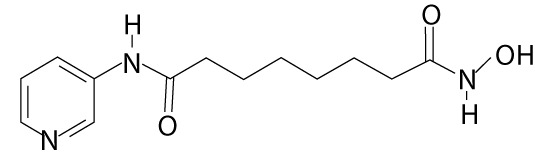 |
leukemia (e.g. chronic myeloid leukemia); lymphoma (e.g. chronic lymphocytic), small intestine cancer | |
| Tubacin | hydroxamic acids | 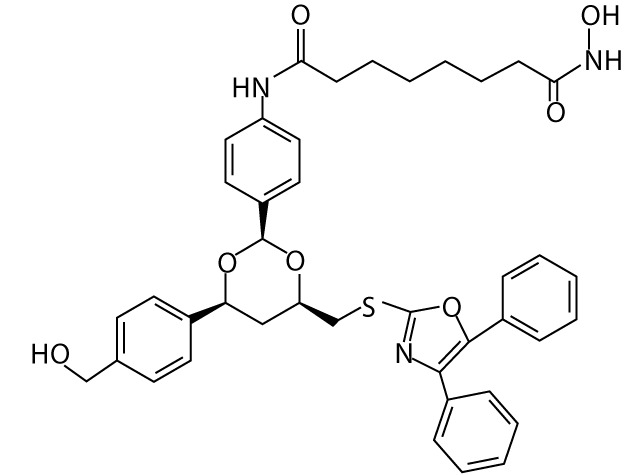 |
– | |
| Entinostat | MS-27-275, MS-275, SNDX-275 | benzamides |  |
recurrent non-small cell lung cancer, lymphoma (e.g. Hodgkin's lymphoma, adult acute monocytic leukemia) melanoma, leukemia, breast cancer, recurrent colon cancer, recurrent rectal cancer |
| Mocetinostat | MGCD0103 | benzamides | 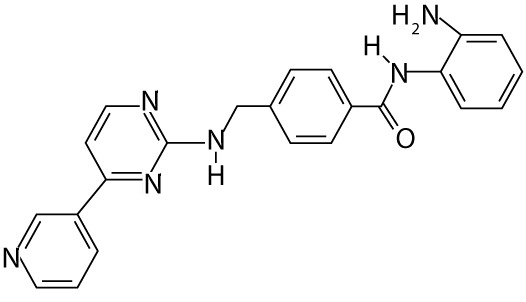 |
myelodysplastic syndrome |
| Tacedinaline | CI-994 | 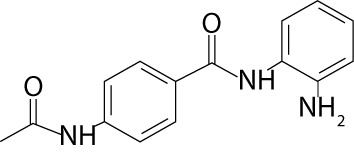 |
multiple myeloma, non-small cell lung cancer, pancreatic cancer | |
| Valproic acid | Depakene (Banner Pharmacaps, USA; AbbVie, USA), Depakote (AbbVie, USA), Depakote ER (AbbVie, USA), Depakote Sprinkle (AbbVie, USA) | small molecular weight carboxylates | 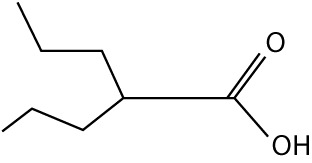 |
lung cancer, recurrent non-small cell lung cancer, head and neck cancer, colorectal cancer, neuroblastoma, oral cavity cancer, oropharyngeal cancer, pancreatic cancer, pediatric brain tumor, glioma, anaplastic astrocytoma, medulloblastoma |
| Sodium butyrate | – | small molecular weight carboxylates | 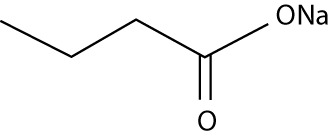 |
– |
| Romidepsin | Depsipeptide, Istodax® (Celgene Corporation, USA), FK228, FR901228 | cyclic tetrapeptides | 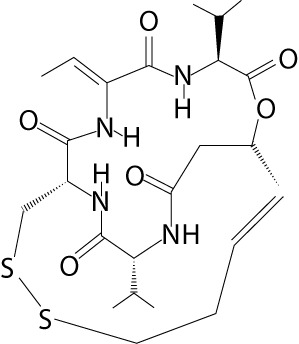 |
lymphoma (e.g. cutaneous B-cell non-Hodgkin, cutaneous T-cell lymphoma, peripheral T-cell lymphoma), non-small cell lung cancer, pancreatic cancer, leukemia (e.g. B-cell chronic lymphocytic leukemia), gastrinoma, glucagonoma, insulinoma |
| Trapoxin A | – | cyclic tetrapeptides | 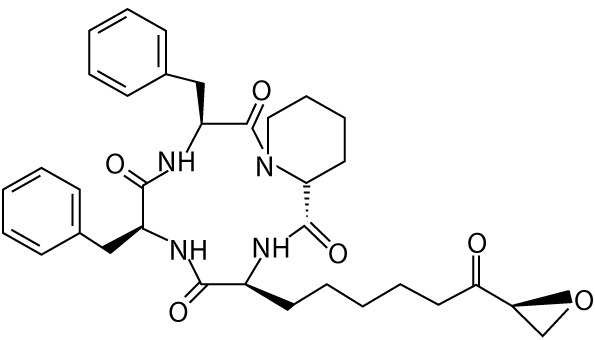 |
– |
| Apicidin | – | cyclic tetrapeptides | 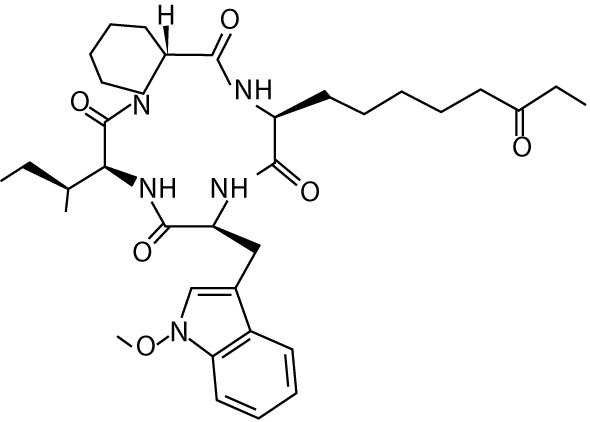 |
– |
Despite a number of promising results of those trials, further solutions are sought in order to increase efficiency of clinical applications of HDACi. One of the ways to do so is combining HDACi with other chemotherapeutic methods.
The first example of such combination is application of azacitidine with various types of HDACi (e.g. entinostat, mocetinostat, panobinostat, valproic acid, vorinostat) [23]. Azacitidine is an analog of pyrimidine nucleotide and a DNA methyltransferase inhibitor (DNMTi) [24]. The process of methylation is based on covalent modification of cytosines through binding a methyl group from S-adenosylmethionine (SAM) to the fifth carbon atom of that compound in a catalytic reaction of DNA methyltransferase (DNMT). Distortion in the process of DNA methylation can lead to oncogenesis. This process may be related to hypermethylation of CpG islands of the suppressor genes or to a decrease in the level of methylation (hypomethylation) within the promoters of genes involved in proliferation and differentiation of cells [1]. The combination of HDACi and DNMTi is currently the focus of several clinical trials related to the treatment of such neoplasms as leukemia, non-small cell lung cancer, and multiple myeloma [23].
Another strategy is combination of a HDACi compound such as vorinostat with β-phenylethyl isothiocyanate, a natural compound capable of depleting glutathione, which significantly enhanced the cytotoxicity of vorinostat in leukemia cell lines and primary leukemia cells [25].
There are also reports of combining HDACi with proteasome inhibitors, for example bortezomib. Other ongoing clinical trials include a combination of panobinostat and bortezomib in peripheral T cell lymphoma, multiple myeloma, and pancreatic cancer [23, 26].
Another tested combination is application of HDACi with DNA-damaging drugs. An example of such a method is karenitecin (KTN, a compound from the topoisomerase I inhibitor group). Phase I and II clinical trials conducted by Daud et al. [27] on patients with diagnosed stage IV melanoma aged 29–89 used various combinations of valproic acid (in 30–90 mg/kg/day doses) with karenitecin (in 0.8 and 1 mg/m2/day doses). They proved that the maximum tolerated dose of valproic acid was 75 mg/kg/day combined with a 1 mg/m2/day dose of KTN. Stabilization of disease progression was noted in 39% (13 out of 33) of cases [27].
Summary
Histone deacetylase inhibitors are compounds that are more and more frequently used in cancer therapy. The future of HDACi application may involve combined therapy strategies. It is also necessary to improve our understanding of the effects caused by this group of drugs. The majority of currently available studies focus on the changes in gene function caused by inhibition of histone acetylation. For a more comprehensive understanding, side effects of application of these compounds should also be noted. They are the result of toxicity of HDAC and DNMT inhibitors and the lack of a selective effect on particular genes. It is expected that a combination of HDAC and DNMT inhibitors with more conventional chemo- and radiotherapy will bring success to cancer therapy.
The authors declare no conflict of interest.
References
- 1.Kwinta Ł. Epigenetyka czerniaka. Contemp Oncol (Pozn) 2008;2:45–50. [Google Scholar]
- 2.Sigalotti L, Covre A, Fratta E, et al. Epigenetics of human cutaneous melanoma: setting the stage for new therapeutic strategies. J Transl Med. 2010;8:56. doi: 10.1186/1479-5876-8-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen S, Sang N. Histone deacetylase inhibitors: the epigenetic therapeutics that repress hypoxia-inducible factors. J Biomed Biotechnol. 2011;2011:197946. doi: 10.1155/2011/197946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gillespie S, Borrow J, Zhang XD, Hersey P. Bim plays a crucial role in synergistic induction of apoptosis by the histone deacetylase inhibitor SBHA and TRAIL in melanoma cells. Apoptosis. 2006;11:2251–65. doi: 10.1007/s10495-006-0283-6. [DOI] [PubMed] [Google Scholar]
- 5.Insinga A, Monestiroli S, Ronzoni S, et al. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat Med. 2005;11:71–6. doi: 10.1038/nm1160. [DOI] [PubMed] [Google Scholar]
- 6.Yuan ZL, Guan YJ, Chatterjee D, Chin YE. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science. 2005;307:269–73. doi: 10.1126/science.1105166. [DOI] [PubMed] [Google Scholar]
- 7.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–84. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 8.Michaelis M, Michaelis UR, Fleming I, et al. Valproic acid inhibits angiogenesis in vitro and in vivo. Mol Pharmacol. 2004;65:520–7. doi: 10.1124/mol.65.3.520. [DOI] [PubMed] [Google Scholar]
- 9.Deroanne CF, Bonjean K, Servotte S, et al. Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene. 2002;21:427–36. doi: 10.1038/sj.onc.1205108. [DOI] [PubMed] [Google Scholar]
- 10.Ungerstedt JS, Sowa Y, Xu WS, et al. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2005;102:673–8. doi: 10.1073/pnas.0408732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee JH, Choy ML, Ngo L, Foster SS, Marks PA. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc Natl Acad Sci U S A. 2010;107:14639–44. doi: 10.1073/pnas.1008522107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Magner WJ, Kazim AL, Stewart C, et al. Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. J Immunol. 2000;165:7017–24. doi: 10.4049/jimmunol.165.12.7017. [DOI] [PubMed] [Google Scholar]
- 13.Federico M, Bagella L. Histone deacetylase inhibitors in the treatment of hematological malignancies and solid tumors. J Biomed Biotechnol. 2011;2011:475641. doi: 10.1155/2011/475641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iwamoto M, Friedman EJ, Sandhu P, Agrawal NG, Rubin EH, Wagner JA. Clinical pharmacology profile of vorinostat, a histone deacetylase inhibitor. Cancer Chemother Pharmacol. 2013;72:493–508. doi: 10.1007/s00280-013-2220-z. [DOI] [PubMed] [Google Scholar]
- 15.Kelly WK, O'Connor OA, Krug LM, et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J Clin Oncol. 2005;23:3923–31. doi: 10.1200/JCO.2005.14.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olsen EA, Kim YH, Kuzel TM, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25:3109–15. doi: 10.1200/JCO.2006.10.2434. [DOI] [PubMed] [Google Scholar]
- 17.Sandor V, Bakke S, Robey RW, et al. Phase I trial of the histone deacetylase inhibitor, depsipeptide (FR901228, NSC 630176), in patients with refractory neoplasms. Clin Cancer Res. 2002;8:718–28. [PubMed] [Google Scholar]
- 18.Plumb JA, Finn PW, Williams RJ, Bandara MJ, Romero MR, Watkins CJ, La Thangue NB, Brown R. Pharmacodynamic response and inhibition of growth of human tumor xenografts by the novel histone deacetylase inhibitor PXD101. Mol Cancer Ther. 2003;2:721–8. [PubMed] [Google Scholar]
- 19.Steele NL, Plumb JA, Vidal L, et al. A phase 1 pharmacokinetic and pharmacodynamic study of the histone deacetylase inhibitor belinostat in patients with advanced solid tumors. Clin Cancer Res. 2008;14:804–10. doi: 10.1158/1078-0432.CCR-07-1786. [DOI] [PubMed] [Google Scholar]
- 20.Prince HM, Bishton MJ, Harrison SJ. Clinical studies of histone deacetylase inhibitors. Clin Cancer Res. 2009;15:3958–69. doi: 10.1158/1078-0432.CCR-08-2785. [DOI] [PubMed] [Google Scholar]
- 21.Garcia-Manero G, Assouline S, Cortes J, et al. Phase 1 study of the oral isotype specific histone deacetylase inhibitor MGCD0103 in leukemia. Blood. 2008;112:981–9. doi: 10.1182/blood-2007-10-115873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ai T, Cui H, Chen L. Multi-targeted histone deacetylase inhibitors in cancer therapy. Curr Med Chem. 2012;19:475–87. doi: 10.2174/092986712798918842. [DOI] [PubMed] [Google Scholar]
- 23. http://clinicaltrials.gov/
- 24.Ren J, Singh BN, Huang Q, et al. DNA hypermethylation as a chemotherapy target. Cell Signal. 2011;23:1082–93. doi: 10.1016/j.cellsig.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 25.Hu Y, Lu W, Chen G, et al. Overcoming resistance to histone deacetylase inhibitors in human leukemia with the redox modulating compound β-phenylethyl isothiocyanate. Blood. 2010;116:2732–41. doi: 10.1182/blood-2009-11-256354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pei XY, Dai Y, Grant S. Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clin Cancer Res. 2004;10:3839–52. doi: 10.1158/1078-0432.CCR-03-0561. [DOI] [PubMed] [Google Scholar]
- 27.Daud AI, Dawson J, DeConti RC, et al. Potentiation of a topoisomerase I inhibitor, karenitecin, by the histone deacetylase inhibitor valproic acid in melanoma: translational and phase I/II clinical trial. Clin Cancer Res. 2009;15:2479–87. doi: 10.1158/1078-0432.CCR-08-1931. [DOI] [PubMed] [Google Scholar]