

Abstract
The inhibitory effects of metformin have been observed in many types of cancer. However, its effect on human salivary gland carcinoma is unknown. The effect of metformin alone or in combination with pp242 (an mTOR inhibitor) on salivary adenocarcinoma cells growth were determined in vitro and in vivo. We found that metformin suppressed HSY cell growth in vitro in a time and dose dependent manner associated with a reduced expression of MYC onco-protein, and the same inhibitory effect of metformin was also confirmed in HSG cells. In association with the reduction of MYC onco-protein, metformin significantly restored p53 tumor suppressor gene expression. The distinctive effects of metformin and PP242 on MYC reduction and P53 restoration suggested that metformin inhibited cell growth through a different pathway from PP242 in salivary carcinoma cells. Furthermore, the anti-tumor efficacy of metformin was confirmed in vivo as indicated by the increases of tumor necrosis and reduced proliferation in xenograft tumors from metformin treated group. For the first time, the inhibitory effect of metformin on human salivary gland tumor cells was documented. Moreover, metformin inhibitory effects were enhanced by mTOR inhibitor suggesting that metformin and mTOR inhibitor utilize distinctive signaling pathways to suppress salivary tumor growth.
Keywords: Salivary adenocarcinoma, HSY cells, HSG cells, metformin
Introduction
Human salivary gland cancer (SGC) is one of the common malignancies found in the head and neck area [1]. SGC mostly occurs in minor and major glands [2] with some highly aggressive tumors that could invade the adjacent tissues and even spread to distant organs [1]. The vast majority of SGC are various adenocarcinomas with relatively high morbidity and poor prognosis. Patients with high-grade SG carcinoma only have a 25% 5-year survival rates [3]. The anatomic and post-operative considerations limit the efficacy of resections of SGC lesions. Furthermore, the complexity of SGC itself and resistance to radiotherapy lead to poor patient prognosis [4]. Thus more investigations on novel intervention for SGCs are needed to improve the treatment modality.
Metformin (1,1-dimethylbiguanide) is a widely used anti-type-2 diabetes medicine which reduces plasma glucose level through suppression of hepatic glucose production [5]. Epidemiological studies and accumulated evidence in tumor cell cultures and xenograft tumor models indicated the anti-tumor potential of metformin in breast, prostate, pancreas, uterus, skin, lung, liver and colon carcinogenesis [6-15]. Pre-clinical and clinical trials further support its chemopreventive potential in breast, lung and prostate cancers [16-19]. As a relatively rare tumor, research resources and interventions on SGC are limited. The broad spectrum of the anti-neoplasia effects of metformin warrants the investigation of its efficacy in SGC [20].
In malignant cells, metformin’s anti-neoplasia effects has been largely attributed to the activation of AMP-activated protein kinase (AMPK) and down-regulation of mammalian target of rapamycin (mTOR) pathways [21-23]. Our group previously demonstrated that metformin effectively targets the c-Myc oncogene to prevent the initiation and progression of prostate cancer [7]. The recent identification of the MYB-NFIB fusion in salivary gland cancers shed light on the molecular mechanism of SGC [24,25]. The clinical significance of MYB-NFIB fusion in SGC patients suggested the activation of critical MYB targets including c-MYC and BCL2, which are associated with apoptosis, cell cycle control, cell growth/angiogenesis, and cell adhesion, as key oncogenic events in the pathogenesis of SGC [24]. Indeed, the overexpression of c-Myc has also been shown in salivary gland adenoid cystic carcinoma [24,25]. Taken together, it is intriguing to explore the inhibitory effects of metformin in SGC and whether metformin targets c-Myc expression in salivary tumor to suppress its growth.
Amplification of the c-Myc oncogene is one of the predominant genetic changes in the development of human prostate cancer. Down-regulation of c-Myc may prevent or delay cancer onset. As mTOR signaling has been considered as a target in many types of tumors [26-28], new therapies mTOR inhibitors are tested in on-going clinical trials [29]. However, cancers that over-express c-Myc may become resistant to mTOR inhibition [30]. Therefore, it is postulated that metformin and mTOR inhibitors could act additively or even synergistically to inhibit cancer cell growth.
This study was designed to investigate the mechanism and molecular mediators of metformin’s anti-tumor activity in salivary gland tumor in vivo and in vitro using HSY and HSG cell lines (human salivary adenocarcinoma cell) as models.
Materials and methods
Reagents
Metformin and pp242 were purchased from Calbiochem (Darmstadt, Germany). Dimethyl sulfoxide (DMSO) was purchased from Sigma (St. Louis, MO, USA). Antibodies against p53, BCL2, caspase 3, caspase 9 and PARP, Phospho-4E-BP1, phospho-AMPK (p-AMPK), c-MYC, p-c-MYC, GAPDH and Histone H3 were purchased from Cell Signaling (Danvers, Massachusetts, USA). Non-fat dry milk was from Lab scientific (Highlands, NJ, USA). Other reagents including bovine serum albumin (BSA), polyethylene glycol 400 (PEG 400) and phenylmethanesulfonyl fluoride (PMSF) were all purchased from Fisher Scientific (Hampton, NH, USA).
Cell culture
HSY and HSG cell lines were derived from human parotid gland adenocarcinoma and submandibular gland adenocarcinoma respectively. Both cell lines were generously provided by Dr. Daniel Malamud at New York University College of Dentistry. HSY cells were maintained with Dulbecco’s Modified Eagle’s Medium (DMEM) containing 1000 mg/L glucose (Sigma, St. Louis, MO, USA), supplemented with 10% Fetal Bovine Serum (Atlanta Biologicals, GA, USA), 100 µg/mL streptomycin, 100 Units/mL penicillin (Gibco, Grand Island, NY, USA) and 100 µg/mL normocin (InvivoGen, San Diego, CA, USA) in a 37°C, 5% (v/v) CO2 and humidified incubator. While HSG cells were maintained using Minimum Essential Medium (MEM) (HyClone, Logan, UT, USA), supplemented with 2 mM Glutamine (Gibco, Grand Island, NY, USA), 1% Non Essential Amino Acids (Gibco, Grand Island, NY, USA), 10% Fetal Bovine Serum, 100 µg/mL streptomycin, and 100 units/mL penicillin. Trypsin at 0.25% in Dulbecco’s Phosphate-Buffered Saline (DPBS) was used for splitting the cells when they reached a confluence of 85%-90%.
Cell viability assay
HSY cells and HSG cells were seeded at 0.75 × 106/ml into 12 well plates. Metformin was added to the medium at a range of concentrations from 0 to 8 mM for 24 hours, 48 hours up to 72 hours. Cell density determined by crystal violet assay as previously described was used as an indicator for the overall cell growth [31]. The same assays were performed to assess the cell number in the cultures that received the combined treatment with PP242 (0.5 µM [32]) and metformin, PP242, metformin as indicated. Images were taken prior to the crystal violet assay using EVOS cell imaging system (Life technologies, Carlsbad, CA, USA).
Flow cytometry
For cell cycle: HSY cells were treated with PBS or 4 mM metformin for 48 hours. After ribonuclease A treatment, Propidium iodide at 50 µg/mL [33] was used to stain the cells, FACSCalibur instrument (BD Biosciences, San Jose, CA, USA) and Modfit software were used to analyze the cell cycle.
For apoptosis: HSY cells were treated with PBS or 4 mM metformin for 48 hours. Cells were washed and fixed. FITC Annexin V and propidium iodide (BD Pharmingen, Franklin Lakes, NJ, USA) were used to evaluate apoptosis. Apoptotic cells were analyzed by FloJo (FLOJO LLC. Ashland, OR, USA).
Protein extraction and Western blot
Cells from both cell lines were cultured and treated as indicated. Whole proteins were extracted by using RIPA buffer (Pierce, Rockford, IL USA) supplemented with protease inhibitor cocktail tablets (Sigma) and 1 mM PMSF. Protein concentration was determined by BCA protein assay (Pierce Biotechnology, Rockford, IL, USA). Even amounts of protein were loaded to 10% tris-glycin gel (Novex, CA, USA), followed by standard tris-glycin SDS-polyacrylamide gel electrophoresis and transferred to the PVDF membranes. Non-fat milk or BSA at 5% were used to block the membranes [34]. β-actin and GAPDH were used as internal loading control for total protein, while Histone H3 was used as a control for nuclear protein. Bio-Rad ChemiDoc™ XRS+ System with Image Lab™ Software (Hercules, California, USA) was used to detect the signals.
In vivo nude mice xenograft experiments
Six-week-old male nude mice were purchased from the Jackson Laboratory and maintained in NYU animal facility under the protocol appro-ved by Division of Laboratory Animal Resources (DLAR). HSY cells at 4 ×106 in 100 µL Matrigel (BD Biosciences, Franklin Lakes, NJ, USA) were subcutaneously injected into the flanks of each nude mouse [32]. Mice were randomly assigned into four treatment groups (N=6) and received daily intraperitoneal injections of PBS, metformin (350 mg/Kg), PP242 (5 mg/Kg in 30% PEG 400) or a combination of metformin and PP242 accordingly [35]. Tumor size was measured once a week and volume was calculated based on the formula: 0.5 × width2 × length [32]. After sacrificing the experimental animals, tumor tissues were collected and prepared for histochemistry. Hematoxylin and eosin (H&E) and immunohistochemistry (IHC) Ki-67 staining were performed by Histology Core in NYU Langone Medical Center. All slides were then scanned by Leica SCN400F at a 400 × magnification.
Statistical analysis
Two-tailed student’s t-test was used to compare the difference between two experiment groups. Analysis of variance (ANOVA) was used when study subjects were more than 2 groups, followed by the Bonferroni t-test. Paired-comparison was applied in in-vivo tumor size analysis. A value of P < 0.05 was deemed to statistically significant.
Results
Metformin inhibits salivary adenocarcinoma cell growth in vitro
To explore the anti-tumor effect of metformin on HSY and HSG cells, we carried out the cell proliferation assay by using crystal violet staining to determine the number of both cells that were subjected to the treatment of metformin at different concentrations for 24, 48 or 72 hours. The result showed that metformin significantly reduced the proliferation of HSY cells at 48 h in a dose-dependent manner (Figure 1A-C) and similarly, high doses of metformin also reduced tumor burden more effectively than lower doses (Figure S1). Meanwhile, photomicrographs of the cells in each group also showed the inhibitory effect of metformin on tumor cell growth as indicated by the flatness of the cells (Figure 1D). Similar trend was observed HSG cells, in which cell number was reduced by approximately 30% after in 48 hours treatment with 4 mM metformin compare to PBS group (Figure 1E-H). To determine if the inhibition of salivary gland carcinoma cell growth by metformin was mediated through cell cycle arrest, flow cytometry analysis was performed to determine the metformin impact on cell cycle distribution after a 48 h treatment. Notably, metformin treatment increased the proportion of HSY cells in the S and G2 phases while decreased proportion of cells in G1 phase (Figure 2A, 2B). Consistently, levels of cyclin-dependent kinase (CDK) complex composed of CDK1 and cyclin B, which regulates the check-point at S/G2 phase in mammalian cell cycle, were reduced. As indicated by the Western blots for CDK1, as well as its activator cyclin B, both proteins were down-regulated by metformin treatment in HSY cells (Figure 2C). Similar trend was also observed in HSG cells after 48 hours metformin treatment (Figure S2). Taken together, our results indicate that metformin can suppress the proliferation of salivary gland tumor cells through cell cycle arrest at S/G2.
Figure 1.

Metformin inhibited human salivary gland cell growth in vitro. A-C: HSY cell viability assay. HSY cells were plated and cultured in DMEM medium and received metformin at different concentrations 0 mM, 2 mM, 4 mM, or 8 mM for three different time points. Crystal violet assays were done, the OD values were read via plate reader. (ANOVA were used to determine the significant difference. **P < 0.01, ***P < 0.001). D: Images (20 ×) were captured on 48 hours treated group prior to cell viability assay. E-H: HSG cell viability assay. HSG cells were cultured in MEM medium and treated by metformin as previous described in HSY cells. (ANOVA were used to determine the significant difference. ***P < 0.001).
Figure 2.
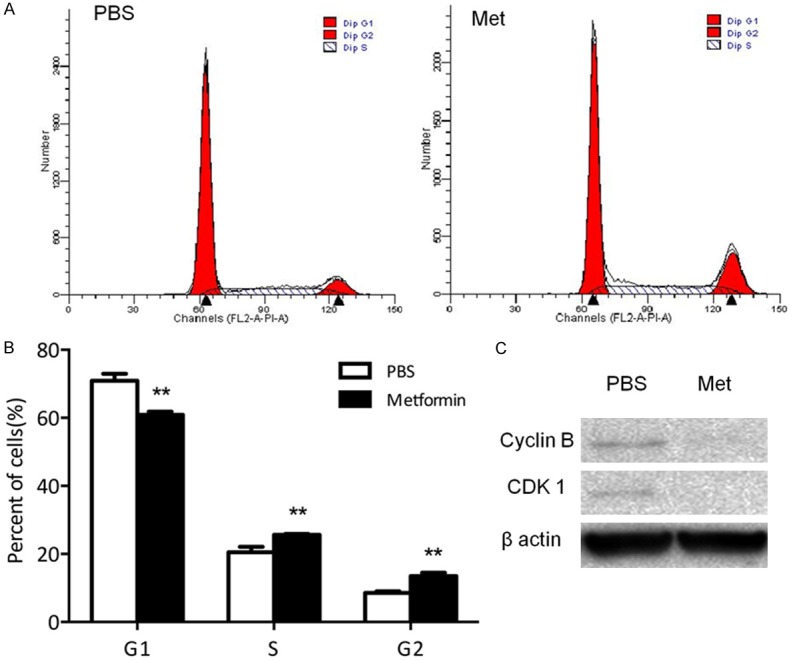
Metformin arrested human salivary gland carcinoma cell cycle in S/G2 phases. A, B: HSY cells were treated with PBS or 4 mM metformin for 48 hours. PI was used to stain the cells. **P < 0.01. C: HSY cells were treated for 48 h and the protein was harvested, then the cell cycle markers were tested by Western blot.
MTOR inhibitor PP242 enhanced metformin inhibitory effects
PP242, a novel mTOR inhibitor that inhibits both mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) [36], recently has been tested widely as a therapeutic modality for cancer patients. As metformin’s anti-tumor effect has been attributed to its ability to inhibit mTOR signaling, we compared the effects of metformin and PP242 in both HSY and HSG cells. Cell viability assays indicated that metformin played a more profound inhibitory role in cell proliferation than PP242 did (Figure 3A, Figure S3A). When combined with PP242, metformin’s inhibitory effect was enhanced (Figure 3A, Figure S3A). Previous reports showed activation of mTOR pathway in a subset of salivary gland tumors [37,38]. As expected, the levels of the mTOR pathway mediator (4E-BP/eIF4E) were reduced by PP242 and metformin. The stimulation of p-AMPK was also verified in metformin treated HSY and HSG cells. Photomicrographs of the cell morphology showed the same trend of cell-flattening (Figure 3B). Further tests revealed that metformin, but not PP242, can induce the expression of p53 as well as reduce the expression of c-Myc (Figure 3C, Figure S4). The same reduction effect of metformin on c-MYC expression was also confirmed in HSG cells (Figure S3B). The induction of p53 by metformin also supported our previous observation that HSY cells were arrested at G2 phase with metformin treatment. This result implied that metformin exerted its inhibitory effects through another pathway in addition to induction of mTOR signaling. By reducing c-Myc expression, metformin could circumvent drug-resistance to mTOR inhibitor associated with c-Myc overexpression in malignant cells.
Figure 3.
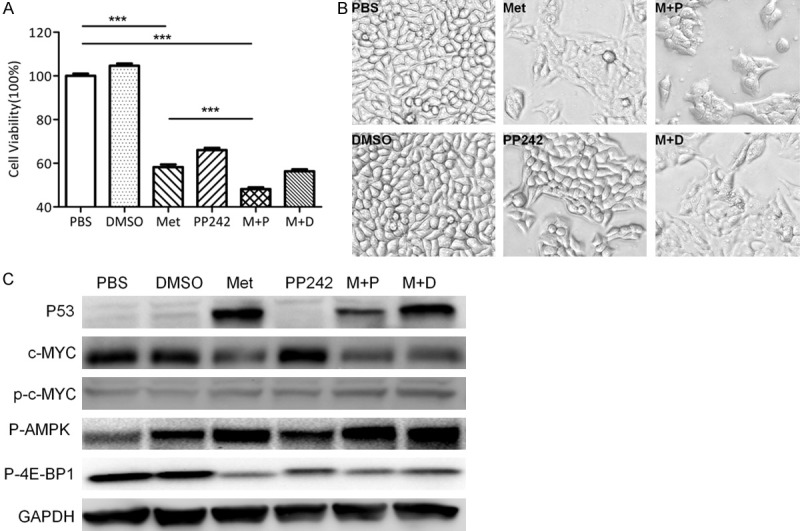
Metformin but not PP242 reduced c-MYC expression in human salivary gland cells. A: HSY cells were evenly plated in 24 well plates and treated in 6 groups for 48 h: PBS, DMSO: 0.5 µl/mL, Metformin: 4 mM, PP242: 0.5 µM, Metformin+PP242: 4 mM+ 0.5 µM, Metformin+DMSO: 4 mM+ 0.5 µL/mL. B: Images (200 ×) were captured prior to CV assay. C: Total protein was extracted from HSY cells, Phospho-4E-BP1, p-AMPK, p53 and MYC levels were tested by Western blot.
Metformin induces cell apoptosis in human salivary gland tumor cells
Because the induction of apoptosis could also contribute to the reduction in cell numbers, next we determined metformin effect on HSY cell apoptosis using PI and Annexin-V double staining and followed by flow cytometry analysis. Our results showed that metformin treatment significantly induced both early stage and late stage apoptosis in HSY cells (Figure 4A, 4B). As apoptosis cascade is mainly triggered by the cleavage of protein members in the caspase family, we further evaluated the protein levels of the total and cleaved apoptotic effectors by Western blots. Our results showed that metformin treatment led to the cleavages of Caspase 3, caspase 9 as well as PARP. Further Western blot showed that in addition to the effect on mediators of apoptosis, metformin also reduced the levels of pro-survival protein Bcl-2 in both HSY and HSG cells (Figure 4C, 4D, Figure S5). These data support the role of metformin in stimulating salivary gland tumor cell apoptosis.
Figure 4.
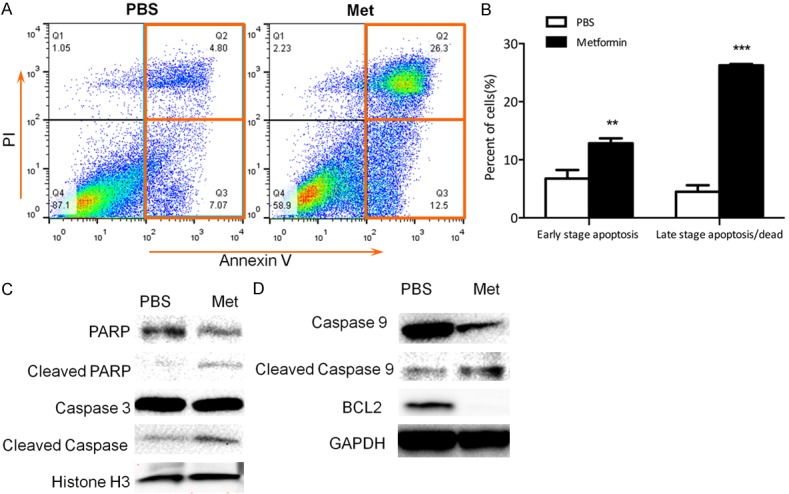
Metformin stimulated apoptosis in human salivary gland cells. (A, B) HSY cells were treated with 4 mM metformin for 48 hours. Annexin V and PI were used to stain the cells. **P < 0.01, ***P < 0.001. (C, D) Western blot using both nuclear and total protein with relevant apoptosis markers (C: nuclear protein).
Metformin inhibits tumor proliferation in nude mice xenograft in vivo
To further investigate the anti-tumor effect of metformin in vivo, we established tumor xenograft model with HSY cells in nude mice through subcutaneous injection. Tumor volumes were measured weekly to establish the tumor growth curves over the treated period. As shown in Figure 5A, metformin started to restrain HSY tumor growth as early as 3-weeks after treatment. When mice were sacrificed after 6-weeks of treatment, all the tumors were harvested for further analysis. Metformin administration significantly reduced the weight of tumors in mice compared to that of the control group (Figure 5B, 5C). PP242 mono-therapy only moderately reduced tumor burdens which could partially due to its inadequate solubility in solutions without DMSO. Despite the fact that the combining PP242 treatment with metformin in vitro indicated an enhanced inhibitory effect in HSY cell growth, the enhancement of reduction in tumor growth was not statistically significant in vivo. Interestingly, when the tumor sections were examined by H&E staining, profound inhibitory efficacy of metformin was demonstrated by the massive necrosis inside the tumor tissues (Figure 6A), which indicated the induction of tumor cell death associated with metformin administration. Furthermore, immunohistochemistry for cell proliferation marker Ki67 revealed that metformin induced a marked reduction of HSY cell proliferation (Figure 6B, 6C). Taken together, these in vivo data confirmed the potential therapeutic effect of metformin for salivary gland adenocarcinomas.
Figure 5.
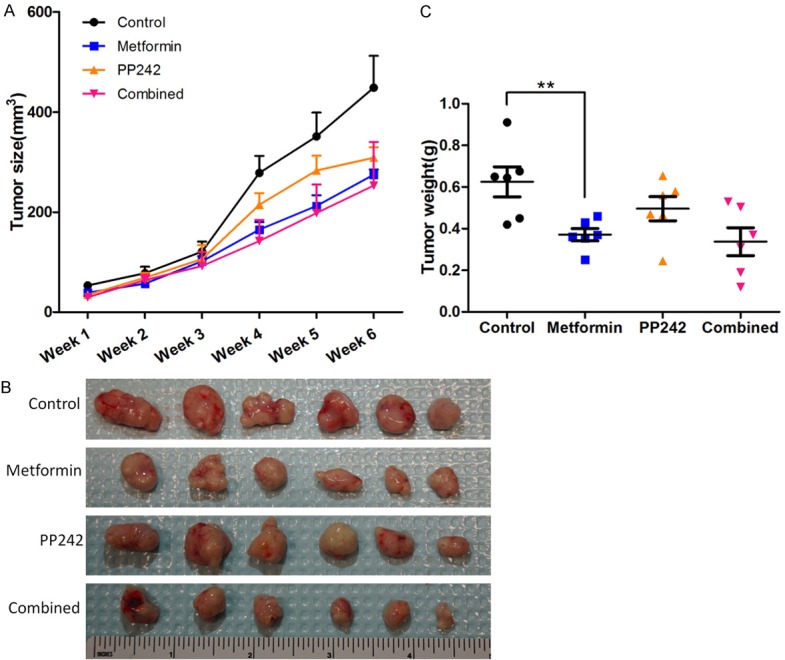
Metformin and PP242 reduced tumor burden in xenograft model. A: Mice were treated in 4 different groups. (Control: PBS, metformin: 350 mg/Kg, PP242: 5 mg/Kg in 30% PEG 400, combined: metformin 350 mg/Kg + PP242: 5 mg/Kg). Tumor size was measured every week and the volume was calculated by 0.5 × lengh × width2. B: Gross tumors removed from animals after 6 weeks treatment. C: Tumor weight valued instantly after dissection from euthanized animals. **P < 0.01.
Figure 6.

Metformin reduced human salivary gland cell proliferation in vivo. A. H&E staining. Lower magnification (4 ×) and higher magnification (40 ×) images were captured in PBS and metformin treated group. The yellow arrow indicates the boundary between tumor cells and necrotic cells. B. IHC for Ki67 on tumor sections from both groups. C. Values of Ki67-positive ratio based on 40 × magnification images.
Discussion
Metformin, with an established treatment efficacy and a good safety profile, is an inexpensive and most commonly prescribed therapy for patients with type-2 diabetes. Our previous study showed that metformin suppressed prostate cancer through down-regulation of c-Myc expression [7]. C-Myc oncogene is frequently deregulated in many human cancers and it is estimated that 450,000 Americans are diagnosed with a c-Myc-dependent cancer each year. The regulatory effects of metformin action on c-Myc provide a new insight into the broad efficacy and mechanism of metformin as an anti-neoplastic agent.
Salivary gland cancers are associated with high morbidity accompanied with a low 5-year survival rate and yet have no standard effective treatments. The lack of comprehensive understanding of the pathways controlling SGC growth hinders the identification of effective therapy in this field. Recently, several studies showed that salivary gland tumors frequently have recurring chromosome translocations resulting in pathogenetically relevant fusion oncogenes [25,37,38]. The downstream gene products of these fusions could be important targets for the development of new therapeutic strategies. Interestingly, the fusion gene products stimulate c-Myc expression [25] or directly work with c-Myc [38] to promote cancer growth and spread by activating basic transformation pathways.
While metformin’s anti-neoplastic effects have been demonstrated in many other types of cancers, the current study is the first one showing metformin’s anti-neoplastic properties in SGC. As over-expression of c-Myc was observed in SGC patient samples as well as in HSY cells, it is not a surprise that metformin exerted inhibitory effect in HSY cells accompanied with strikingly reduced levels of c-Myc. One of the obstacles that restrict the effect of mTOR inhibitors, which have been used widely for treatment of many types of cancers [26-28,39], could be the resistance related to c-Myc oncogene over-expression in tumor cells [30,40]. In this regard, we anticipate that metformin may enhance the efficacy of PP242 by reducing c-Myc expression. Unlike another mTOR inhibitor rapamycin which mainly targets mTOR complex I, PP242 targets both mTOR complex 1 and complex 2 and demonstrates a better inhibitory efficacy [41]. Indeed, in vitro the combination of metformin and PP242 yielded the most suppression in both HSY and HSG cell growth as compared to the mono-treatment groups. However, the advantage of the combined treatment was not significant in vivo. PP242 mono-therapy only moderately reduced tumor burdens which could be partially due to its inadequate solubility in solutions without DMSO. Therefore, despite that the combining PP242 treatment with metformin in vitro indicated an enhanced inhibitory effect in HSY cell growth, the enhancement of reduction in tumor growth was not statistically significant in vivo. Considering the solubility of PP242 could limit its efficacy because of the dosage used in current experiment, an enhanced inhibitory efficacy of metformin may be achievable with P242 at a higher dose or with another mTOR inhibitor. Interestingly, metformin down-regulated c-Myc expression and restored the expression of genome stability guardian p53 [42,43]; PP242 did not affect c-Myc and p53 expression levels. Thus, PP242 and metformin may regulate different cellular molecules to exert their inhibitory effects.
The inhibitory effects of metformin on HSY and HSG cells growth are contributed by its ability to induce cell cycle arrest as well as apoptosis which is consistent with previous studies. However, instead of arresting cells in G1 phase, as we and others have observed in prostate cancer [7,44] and breast cancer cells [45-47], metformin treatment caused HSY cell and HSG cell cycle arrest in G2 phase. S/G2 phase arrest may indicate an interruption in the process of mitochondrial energy generation and similar observation was reported in endometrial cancer cells, in which metformin inhibited cell growth via cell cycle arrest in G2/M phase [48]. Therefore, in different cell context, metformin may inhibit cell proliferation via an unknown mechanism. It is known that the activation of cyclin B-CDK1 complex is required for cells progress from G2 to mitotic phase [49]. Consistently, we observed decreased expression of cyclin B and CDK1 in metformin-treated HSY cells which supported the flow cytometry analysis of G2 cell cycle arrest before proceeding to mitosis.
The apoptosis assay revealed that metformin also induced HSY cell apoptosis. Metformin stimulated the pro-apoptotic caspase cascade initiated by caspase-9 activation and cleavage, followed by the cleavage of caspase 3 and PARP. Meanwhile, the pro-survival marker BCL2 was decreased in metformin-treated HSY cells. Collectively, activation of apoptosis pathway and the suppression of pro-survival protein led to the cell suicide process, and eventually restricted the expansion of HSY cells.
In the HSY xenograft tumor experiment, metformin treatment reduced tumor burden and increased the size of necrotic areas inside the tumors. In contrast, tumors from PBS-treated mice maintained tumor structure with a limited central necrotic area in the core of tumors. Staining of proliferation marker Ki-67 confirmed the inhibition of proliferation by metformin since the tumor cells in metformin group had a significantly smaller population of Ki-67 positive cells as compared to the PBS group. No metastasis was observed in metformin-treated group, while one out of 6 mice in PBS group demonstrated liver metastases indicated that metformin treatment reduced HSY tumor invasion in vivo (Figure S6). This finding supports further exploration at the capability of metformin to suppress tumor metastasis, which has been demonstrated in several cancers [50-52].
In conclusion, our study provides evidence to support a pivotal role of metformin in suppressing SGC growth in vitro and in vivo, which suggests the need to consider the application of metformin as neo-adjuvant therapy in SGC cancer patients. Future validation of our findings with more SGC tumor cell lines and the enhanced efficacy of metformin and an mTOR inhibitor with better solubility in vivo will be the next step. If proven effective, metformin will eventually become a promising therapeutic strategy for SGC patients.
Acknowledgements
This study has been supported by New York University Start-up funding and National Cancer Institute (CA172894, CA180277) to X. Li.
Disclosure of conflict of interest
None.
Abbreviations
- AMPK
AMP-activated protein kinase
- BSA
bovine serum albumin
- DMEM
Dulbecco’s Modified Eagle’s Medium
- DMSO
dimethyl sulfoxide
- DPBS
Dulbecco’s Phosphate-Buffered Saline
- MEM
Minimum Essential Medium
- mTOR
mammalian target of rapamycin
- PMSF
phenylmethanesulfonyl fluoride
- PEG 400
polyethylene glycol 400
- PI
propidium iodide
- SGC
salivary gland cancer
Supporting Information
References
- 1.Eveson JW, Cawson RA. Salivary gland tumours. A review of 2410 cases with particular reference to histological types, site, age and sex distribution. J Pathol. 1985;146:51–58. doi: 10.1002/path.1711460106. [DOI] [PubMed] [Google Scholar]
- 2.Evans HL, Luna MA. Polymorphous low-grade adenocarcinoma: a study of 40 cases with long-term follow up and an evaluation of the importance of papillary areas. Am J Surg Pathol. 2000;24:1319–1328. doi: 10.1097/00000478-200010000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Chen MM, Roman SA, Sosa JA, Judson BL. Predictors of survival in carcinoma ex pleomorphic adenoma. Head Neck. 2014;36:1324–1328. doi: 10.1002/hed.23453. [DOI] [PubMed] [Google Scholar]
- 4.Lima SS, Soares AF, de Amorim RF, Freitas Rde A. [Epidemiologic profile of salivary gland neoplasms: analysis of 245 cases] . Braz J Otorhinolaryngol. 2005;71:335–340. doi: 10.1016/S1808-8694(15)31332-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cusi K, Consoli A, DeFronzo RA. Metabolic effects of metformin on glucose and lactate metabolism in noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1996;81:4059–4067. doi: 10.1210/jcem.81.11.8923861. [DOI] [PubMed] [Google Scholar]
- 6.Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007;67:10804–10812. doi: 10.1158/0008-5472.CAN-07-2310. [DOI] [PubMed] [Google Scholar]
- 7.Akinyeke T, Matsumura S, Wang X, Wu Y, Schalfer ED, Saxena A, Yan W, Logan SK, Li X. Metformin targets c-MYC oncogene to prevent prostate cancer. Carcinogenesis. 2013;34:2823–2832. doi: 10.1093/carcin/bgt307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deriabina ON, Plotnikova NA, Anisimov VN. [Melatonin and metformin inhibit skin carcinogenesis induced by benz(a)pyrene in mice] . Vopr Onkol. 2010;56:583–587. [PubMed] [Google Scholar]
- 9.Checkley LA, Rho O, Angel JM, Cho J, Blando J, Beltran L, Hursting SD, DiGiovanni J. Metformin inhibits skin tumor promotion in overweight and obese mice. Cancer Prev Res (Phila) 2014;7:54–64. doi: 10.1158/1940-6207.CAPR-13-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Memmott RM, Mercado JR, Maier CR, Kawabata S, Fox SD, Dennis PA. Metformin prevents tobacco carcinogen--induced lung tumorigenesis. Cancer Prev Res (Phila) 2010;3:1066–1076. doi: 10.1158/1940-6207.CAPR-10-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quinn BJ, Dallos M, Kitagawa H, Kunnumakkara AB, Memmott RM, Hollander MC, Gills JJ, Dennis PA. Inhibition of lung tumorigenesis by metformin is associated with decreased plasma IGF-I and diminished receptor tyrosine kinase signaling. Cancer Prev Res (Phila) 2013;6:801–810. doi: 10.1158/1940-6207.CAPR-13-0058-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhalla K, Hwang BJ, Dewi RE, Twaddel W, Goloubeva OG, Wong KK, Saxena NK, Biswal S, Girnun GD. Metformin prevents liver tumorigenesis by inhibiting pathways driving hepatic lipogenesis. Cancer Prev Res (Phila) 2012;5:544–552. doi: 10.1158/1940-6207.CAPR-11-0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tajima K, Nakamura A, Shirakawa J, Togashi Y, Orime K, Sato K, Inoue H, Kaji M, Sakamoto E, Ito Y, Aoki K, Nagashima Y, Atsumi T, Terauchi Y. Metformin prevents liver tumorigenesis induced by high-fat diet in C57Bl/6 mice. Am J Physiol Endocrinol Metab. 2013;305:E987–998. doi: 10.1152/ajpendo.00133.2013. [DOI] [PubMed] [Google Scholar]
- 14.Hosono K, Endo H, Takahashi H, Sugiyama M, Uchiyama T, Suzuki K, Nozaki Y, Yoneda K, Fujita K, Yoneda M, Inamori M, Tomatsu A, Chihara T, Shimpo K, Nakagama H, Nakajima A. Metformin suppresses azoxymethane-induced colorectal aberrant crypt foci by activating AMP-activated protein kinase. Mol Carcinog. 2010;49:662–671. doi: 10.1002/mc.20637. [DOI] [PubMed] [Google Scholar]
- 15.Shimomoto T, Luo Y, Ohmori H, Chihara Y, Fujii K, Sasahira T, Denda A, Kuniyasu H. Advanced glycation end products (AGE) induce the receptor for AGE in the colonic mucosa of azoxymethane-injected Fischer 344 rats fed with a high-linoleic acid and high-glucose diet. J Gastroenterol. 2012;47:1073–1083. doi: 10.1007/s00535-012-0572-5. [DOI] [PubMed] [Google Scholar]
- 16.Thompson AM. Molecular pathways: preclinical models and clinical trials with metformin in breast cancer. Clin Cancer Res. 2014;20:2508–2515. doi: 10.1158/1078-0432.CCR-13-0354. [DOI] [PubMed] [Google Scholar]
- 17.DeCensi A, Puntoni M, Gandini S, Guerrieri-Gonzaga A, Johansson HA, Cazzaniga M, Pruneri G, Serrano D, Schwab M, Hofmann U, Mora S, Aristarco V, Macis D, Bassi F, Luini A, Lazzeroni M, Bonanni B, Pollak MN. Differential effects of metformin on breast cancer proliferation according to markers of insulin resistance and tumor subtype in a randomized presurgical trial. Breast Cancer Res Treat. 2014;148:81–90. doi: 10.1007/s10549-014-3141-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leiter A, Sablinski T, Diefenbach M, Foster M, Greenberg A, Holland J, Oh WK, Galsky MD. Use of crowdsourcing for cancer clinical trial development. J Natl Cancer Inst. 2014;106 doi: 10.1093/jnci/dju258. [DOI] [PubMed] [Google Scholar]
- 19.Fasano M, Della Corte CM, Capuano A, Sasso FC, Papaccio F, Berrino L, Ciardiello F, Morgillo F. A Multicenter, open-label phase ii study of metformin with erlotinib in second-line therapy of stage iv non-small-cell lung cancer patients: treatment rationale and protocol dynamics of the metal trial. Clin Lung Cancer. 2015;16:57–9. doi: 10.1016/j.cllc.2014.06.010. [DOI] [PubMed] [Google Scholar]
- 20.Rego DF, Pavan LM, Elias ST, De Luca Canto G, Guerra EN. Effects of metformin on head and neck cancer: A systematic review. Oral Oncol. 2015;51:416–22. doi: 10.1016/j.oraloncology.2015.01.007. [DOI] [PubMed] [Google Scholar]
- 21.Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond) 2012;122:253–270. doi: 10.1042/CS20110386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miranda VC, Barroso-Sousa R, Glasberg J, Riechelmann RP. Exploring the role of metformin in anticancer treatments: a systematic review. Drugs Today (Barc) 2014;50:623–640. doi: 10.1358/dot.2014.50.9.2229920. [DOI] [PubMed] [Google Scholar]
- 23.Martin M, Marais R. Metformin: a diabetes drug for cancer, or a cancer drug for diabetics? J. Clin. Oncol. 2012;30:2698–2700. doi: 10.1200/JCO.2012.42.1677. [DOI] [PubMed] [Google Scholar]
- 24.Stenman G. Fusion oncogenes in salivary gland tumors: molecular and clinical consequences. Head Neck Pathol. 2013;7(Suppl 1):S12–19. doi: 10.1007/s12105-013-0462-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Persson M, Andren Y, Mark J, Horlings HM, Persson F, Stenman G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc Natl Acad Sci U S A. 2009;106:18740–18744. doi: 10.1073/pnas.0909114106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X, Lai P, Zhang Z, Huang M, Wang L, Yin M, Jin D, Zhou R, Bai X. Targeted inhibition of mTORC2 prevents osteosarcoma cell migration and promotes apoptosis. Oncol Rep. 2014;32:382–388. doi: 10.3892/or.2014.3182. [DOI] [PubMed] [Google Scholar]
- 27.Arena F. Clinical implications of recent studies using mTOR inhibitors to treat advanced hormone receptor-positive breast cancer. Cancer Manag Res. 2014;6:389–395. doi: 10.2147/CMAR.S56802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mao JH, Kim IJ, Wu D, Climent J, Kang HC, DelRosario R, Balmain A. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science. 2008;321:1499–1502. doi: 10.1126/science.1162981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Atreya CE, Ducker GS, Feldman ME, Bergsland EK, Warren RS, Shokat KM. Combination of ATP-competitive mammalian target of rapamycin inhibitors with standard chemotherapy for colorectal cancer. Invest New Drugs. 2012;30:2219–2225. doi: 10.1007/s10637-012-9793-y. [DOI] [PubMed] [Google Scholar]
- 30.Muellner MK, Uras IZ, Gapp BV, Kerzendorfer C, Smida M, Lechtermann H, Craig-Mueller N, Colinge J, Duernberger G, Nijman SM. A chemical-genetic screen reveals a mechanism of resistance to PI3K inhibitors in cancer. Nat Chem Biol. 2011;7:787–793. doi: 10.1038/nchembio.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li X, Koh AJ, Wang Z, Soki FN, Park SI, Pienta KJ, McCauley LK. Inhibitory effects of megakaryocytic cells in prostate cancer skeletal metastasis. J Bone Miner Res. 2011;26:125–134. doi: 10.1002/jbmr.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burd R, Wachsberger P. Application of radiotherapy and chemotherapy protocols to pre-clinical tumor models. Curr Protoc Pharmacol. 2007 doi: 10.1002/0471141755.ph1407s38. Chapter 14: Unit 14.7. [DOI] [PubMed] [Google Scholar]
- 33.Riccardi C, Nicoletti I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat Protoc. 2006;1:1458–1461. doi: 10.1038/nprot.2006.238. [DOI] [PubMed] [Google Scholar]
- 34.Dusik V, Senthilan PR, Mentzel B, Hartlieb H, Wulbeck C, Yoshii T, Raabe T, Helfrich-Forster C. The MAP kinase p38 is part of Drosophila melanogaster’s circadian clock. PLoS Genet. 2014;10:e1004565. doi: 10.1371/journal.pgen.1004565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anisimov VN. Do metformin a real anticarcinogen? A critical reappraisal of experimental data. Ann Transl Med. 2014;2:60. doi: 10.3978/j.issn.2305-5839.2014.06.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoang B, Benavides A, Shi Y, Yang Y, Frost P, Gera J, Lichtenstein A. The PP242 mammalian target of rapamycin (mTOR) inhibitor activates extracellular signal-regulated kinase (ERK) in multiple myeloma cells via a target of rapamycin complex 1 (TORC1)/eukaryotic translation initiation factor 4E (eIF-4E)/RAF pathway and activation is a mechanism of resistance. J Biol Chem. 2012;287:21796–21805. doi: 10.1074/jbc.M111.304626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Z, Chen J, Gu Y, Hu C, Li JL, Lin S, Shen H, Cao C, Gao R, Li J, Ha PK, Kaye FJ, Griffin JD, Wu L. Aberrantly activated AREG-EGFR signaling is required for the growth and survival of CRTC1-MAML2 fusion-positive mucoepidermoid carcinoma cells. Oncogene. 2014;33:3869–3877. doi: 10.1038/onc.2013.348. [DOI] [PubMed] [Google Scholar]
- 38.Amelio AL, Fallahi M, Schaub FX, Zhang M, Lawani MB, Alperstein AS, Southern MR, Young BM, Wu L, Zajac-Kaye M, Kaye FJ, Cleveland JL, Conkright MD. CRTC1/MAML2 gain-of-function interactions with MYC create a gene signature predictive of cancers with CREB-MYC involvement. Proc Natl Acad Sci U S A. 2014;111:E3260–3268. doi: 10.1073/pnas.1319176111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blaser B, Waselle L, Dormond-Meuwly A, Dufour M, Roulin D, Demartines N, Dormond O. Antitumor activities of ATP-competitive inhibitors of mTOR in colon cancer cells. BMC Cancer. 2012;12:86. doi: 10.1186/1471-2407-12-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shepherd C, Banerjee L, Cheung CW, Mansour MR, Jenkinson S, Gale RE, Khwaja A. PI3K/mTOR inhibition upregulates NOTCH-MYC signalling leading to an impaired cytotoxic response. Leukemia. 2013;27:650–660. doi: 10.1038/leu.2012.285. [DOI] [PubMed] [Google Scholar]
- 41.Ono A, Oike R, Okuhashi Y, Takahashi Y, Itoh M, Nara N, Tohda S. Comparative effects of PP242 and rapamycin on mTOR signalling and NOTCH signalling in leukemia cells. Anticancer Res. 2013;33:809–813. [PubMed] [Google Scholar]
- 42.Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene. 1999;18:3004–3016. doi: 10.1038/sj.onc.1202746. [DOI] [PubMed] [Google Scholar]
- 43.Karanika S, Karantanos T, Li L, Corn PG, Thompson TC. DNA damage response and prostate cancer: defects, regulation and therapeutic implications. Oncogene. 2014;34:2815–22. doi: 10.1038/onc.2014.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Colquhoun AJ, Venier NA, Vandersluis AD, Besla R, Sugar LM, Kiss A, Fleshner NE, Pollak M, Klotz LH, Venkateswaran V. Metformin enhances the antiproliferative and apoptotic effect of bicalutamide in prostate cancer. Prostate Cancer Prostatic Dis. 2012;15:346–352. doi: 10.1038/pcan.2012.16. [DOI] [PubMed] [Google Scholar]
- 45.Queiroz EA, Puukila S, Eichler R, Sampaio SC, Forsyth HL, Lees SJ, Barbosa AM, Dekker RF, Fortes ZB, Khaper N. Metformin induces apoptosis and cell cycle arrest mediated by oxidative stress, AMPK and FOXO3a in MCF-7 breast cancer cells. PLoS One. 2014;9:e98207. doi: 10.1371/journal.pone.0098207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hadad SM, Hardie DG, Appleyard V, Thompson AM. Effects of metformin on breast cancer cell proliferation, the AMPK pathway and the cell cycle. Clin Transl Oncol. 2014;16:746–752. doi: 10.1007/s12094-013-1144-8. [DOI] [PubMed] [Google Scholar]
- 47.Zhuang Y, Miskimins WK. Cell cycle arrest in Metformin treated breast cancer cells involves activation of AMPK, downregulation of cyclin D1, and requires p27Kip1 or p21Cip1. J Mol Signal. 2008;3:18. doi: 10.1186/1750-2187-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takahashi A, Kimura F, Yamanaka A, Takebayashi A, Kita N, Takahashi K, Murakami T. Metformin impairs growth of endometrial cancer cells via cell cycle arrest and concomitant autophagy and apoptosis. Cancer Cell Int. 2014;14:53. doi: 10.1186/1475-2867-14-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choi HJ, Fukui M, Zhu BT. Role of cyclin B1/Cdc2 up-regulation in the development of mitotic prometaphase arrest in human breast cancer cells treated with nocodazole. PLoS One. 2011;6:e24312. doi: 10.1371/journal.pone.0024312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu B, Li S, Sheng L, Zhu J, Gu L, Shen H, La D, Hambly BD, Bao S, Di W. Metformin inhibits the development and metastasis of ovarian cancer. Oncol Rep. 2012;28:903–908. doi: 10.3892/or.2012.1890. [DOI] [PubMed] [Google Scholar]
- 51.Rattan R, Graham RP, Maguire JL, Giri S, Shridhar V. Metformin suppresses ovarian cancer growth and metastasis with enhancement of cisplatin cytotoxicity in vivo. Neoplasia. 2011;13:483–491. doi: 10.1593/neo.11148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vazquez-Martin A, Oliveras-Ferraros C, Cufi S, Del Barco S, Martin-Castillo B, Lopez-Bonet E, Menendez JA. The anti-diabetic drug metformin suppresses the metastasis-associated protein CD24 in MDA-MB-468 triple-negative breast cancer cells. Oncol Rep. 2011;25:135–140. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.