

Abstract
MicroRNAs (miRNAs) act as key regulators of gene expression and their dysregulation is linked to carcinogenesis and tumor progression. MiR-610 has been implicated as an anti-tumor miRNA in multiple types of cancers. However, its biological role and the underlying mechanism in colorectal cancer (CRC) have not been well explored. In this study, we report that miR-610 expression is decreased in CRC samples while ectopic expression of miR-610 suppresses cell proliferation, migration and invasion, and influences the expression of epithelial-mesenchymal transition (EMT)-associated proteins by up-regulating E-cadherin expression and down-regulating vimentin expression. Using a luciferase reporter assay, we reveal that miR-610 directly targets hepatoma-derived growth factor (HDGF) by binding to its 3’UTR. A negative correlation was also observed between miR-610 and HDGF expression in CRC tissues. Further studies show that inhibition of HDGF recapitulates the anti-tumor function of miR-610, whereas re-expression of HDGF partially abrogates the inhibitory effects of miR-610. Collectively, our findings indicate that miR-610 exerts its function by directly targeting HDGF. The miR-610/HDGF axis is a novel therapeutic target for CRC.
Keywords: miR-610, HDGF, colorectal cancer
Introduction
Colorectal cancer (CRC) is the third most common malignancy worldwide and the fourth most common cause of cancer death in the world, with more than 1 million new cases and 600,000 deaths each year [1-3]. Despite improved treatments, the death rate remained at 8.8% in 2008 [4]. A high propensity for recurrence and metastasis is the main cause of the high mortality and poor outcome [5]. The molecular mechanisms underlying the progression and development of CRC still remain poorly understood.
microRNAs (miRNAs) are short (~22 nucleotides in length), single-stranded and endogenous non-coding RNAs that negatively regulate gene expression by causing degradation of messenger RNAs (mRNAs) or by repressing their protein translation after binding to the 3’-untranslated region (3’-UTR) of mRNA [6-8]. Studies have demonstrated that dysregulation of miRNAs has been associated with the initiation and development of a wide range of human diseases, including malignancies [9-12]. MiRNAs act either as oncogenes or as tumor suppressors in various human cancers [13-15]. In CRC, miR-301a promotes, while miR-204-5p inhibits, cell proliferation, migration and invasion as well as tumor growth [16,17]. MiR-610 functions as a tumor suppressor miRNA and is deregulated in different types of cancers, including hepatocellular carcinoma (HCC), gastric cancer (GC) and lung cancer [18-20]. Recently, Li et al. reported that miR-610 was down-regulated in CRC [21]. However, the functional role and mechanism of miR-610 in CRC remain elusive.
In this study, we confirm that miR-610 is down-regulated in CRC tissues and cell lines. Over-expression of miR-610 inhibits CRC cell growth, migration, invasion and epithelial-mesenchymal transition (EMT). Furthermore, HDGF is revealed to be a direct and functional target of miR-610, which is negatively correlated with miR-610 expression in CRC tissues.
Materials and methods
Clinical tissues
Thirty-three CRC tissue samples together with their corresponding normal tissues were collected from patients who underwent surgical resection at Huashan Hospital. The tissues were frozen immediately after resection and stored in liquid nitrogen until use. Written informed consent was obtained from each patient, and all of the experiments were approved by the Medical Ethics Committee of Huashan Hospital.
Cells and culture
Human CRC cell lines (SW620, LoVo, HCT116 and SW480), and HEK293T cells were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). HEK293T cells were maintained in DMEM and other cells were cultured in RPMI1640 (GIBCO, Grand Island, NY, USA) supplemented with 10% FBS. Cells were cultured in a humidified incubator in an atmosphere of 5% CO2 at 37°C.
Lentivirus production and transduction
The lentivectors expressing miR-610 and negative control were purchased from Hanbio (Shanghai, China). These lentivectors and the packaging vectors were co-transfected into HEK293T cells with Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA, USA). Lentivirus particles were harvested 48 h after transfection. Cells were transduced with the lentivirus in the presence of polybrene (Sigma-Aldrich, St. Louis, MO, USA).
Oligonucleotide, plasmid construction and cell transfection
MiR-610 mimic, HDGF siRNA and negative controls were synthesized by RiboBio (Guangzhou, China). The hsa-miR-610 mimic includes the following: sense 5’UGAGCUAAAUGUGUGCUGGGA3’ and antisense 5’CCAGCACACAUUUAGCUCAUU3’. The miRNA mimic controls were 5’UUCUCCGAACGUGUCACGUTT 3’ and 5’ACGUGACACGUUCGGAGAATT3’.
The sequence of HDGF siRNA was: sense 5’GCCAUGUCUUCUCCCUGGA3’ and anti-sense 5’UCCAGGGAGAAGACAUGGC3’. The sequence of scrambled siRNA was: sense 5’GCAAACAUCCCAGAGGUAU3’ and anti-sense 5’AUACCUCUGGGAUGUUUGC3’. For HDGF over-expression, the full-length open reading frame of HDGF was subcloned into the pcDNA3.0 vector. Cells were transfected using Lipofectamine™ 2000 according to the manufacturer’s instructions.
RNA extraction and real-time PCR (qRT-PCR)
Total RNA was extracted using TRIZOL reagent (Invitrogen). MiRNAs were isolated using the miRNeasy Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocol. cDNA was synthesized from total RNA using the Prime-Script® RT Reagent Kit (Takara, Dalian, China), and from miRNA using One Step PrimeScript miRNA cDNA Synthesis Kit (Takara) [22]. QRT-PCR was performed with the SYBR green Premix Ex Taq II (Takara). The 2-comparative (2-ΔΔCt) method was used to calculate the relative target mRNA or miRNA expression levels. U6 and β-actin were used as endogenous controls.
Cell viability assay
MTT assay was performed to measure cell viability. Cells were seeded at a density of 2 × 103 cells/well into 96-well plates and cultured for 1-5 days. At the indicated time-points, 20 μL of MTT (Sigma-Aldrich) solution (0.5 mg/mL) was added to each well and incubated for another 4 h at 37°C. Then the medium was carefully removed and 100 μL of dimethyl sulfoxide (DMSO, Sigma-Aldrich) was added to each well to dissolve the formazan crystals. The absorbance was measured at a wavelength of 490 nm.
Colony formation assay
Transfected cells were cultured in 6-well plates at 1,000 cells per well and grown for 14 days. After fixation with methanol, colonies were stained with 0.1% crystal violet for 15 min and washed. The number of colonies was counted under a light microscope.
Migration and invasion assays
Migration and invasion assays were performed using a transwell system (8 μm pore size; Millipore, Billerica, MA, USA). A suspension of 2.5 × 105 cells in serum-free medium was seeded into the upper chamber with or without Matrigel® (Invitrogen). Medium containing 20% FBS was used as a chemoattractant in the lower chamber. After incubation for 24 h, cells on the upper surface of the filter were removed using cotton buds, and cells adhering to the lower surface of the inserts were fixed in methanol for 15 minutes, and then stained with 0.1% crystal violet for 30 minutes. The number of cells was counted in five random fields per well under a light microscope.
Western blotting
Cells were lysed in cell lysis buffer (Cell Signaling Technology, Danvers, MA, USA). The total protein concentration was measured with the BCA method (Pierce, Rockford, IL, USA). Equal amounts of protein were electrophoresed in 10% SDS-PAGE and transferred onto PVDF membranes (Millipore). After blocking with 5% non-fat dry milk in TBST, the membranes were incubated overnight at 4°C with the following antibodies: anti-E-cadherin antibody (1:1,000; Cell Signaling Technology), anti-Vimentin antibody (1:1,000; Abcam, Cambridge, MA, USA), anti-HDGF antibody (1:2,000; Abcam) and anti-GAPDH antibody (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA, USA), and then with horseradish peroxidase-conjugated secondary antibodies. Protein bands were detected with enhanced chemiluminescence (ECL; Thermo Scientific, Rockford, IL, USA).
Animal model
HCT116 cells (2 × 106) transduced with lentivirus expressing miR-610 or negative control vector were subcutaneously injected into BALB/c nu/nu female mice (4-5 weeks old). Tumor size was monitored every three days and the tumor growth curve was recorded accordingly. Tumor volume (V) was calculated by the formula: V (mm3) = 1/2 (length × width2). After 21 days, mice were killed and xenograft tumors were collected. All animal experiments were approved by the Animal Care and Use Committee of Anhui Medical University, China.
Immunohistochemistry
Tissues were embedded in paraffin. Consecutive sections (4 μm thick) were deparaffinized with xylene and rehydrated through a graded series of ethanol. Antigen retrieval was performed by heating in sodium citrate buffer pH 6.0. The sections were blocked with 3% hydrogen peroxide (H2O2) for 20 minutes and then in 10% goat serum for 5 minutes. The sections were incubated with anti-E-cadherin antibody (1:400; Cell Signaling Technology), anti-vimentin antibody (1:200, Abcam) and anti-Ki67 antibody (1:400; Cell Signaling Technology) overnight at 4°C, and then incubated with HRP-conjugated secondary antibodies for 30 minutes. Color was developed using 3,3-diaminobenzidine to examine the enzymatic reaction, and hematoxylin was used as a counterstain.
Dual-luciferase assay
The 3’UTR of HDGF containing the wild-type or mutant putative binding site was cloned into the psiCHECK2 vector (Promega, Madison, WI, USA). miR-610 or control mimic at a concentration of 50 nM together with 0.5 μg of wild-type or mutant HDGF 3’UTR vector were co-transfected into HEK293T cells using Lipofectamine™ 2000. At 48 h after transfection, cells were lysed, and firefly and Renilla luciferase activities were measured using a dual-luciferase reporter assay system (Promega). The firefly activity was used for normalization.
Statistical analysis
Statistical analysis was performed using SPSS 17.0 software. Data are expressed as mean ± standard deviation. Correlation between HDGF and miR-610 expression was analyzed by Spearman’s correlation. Student’s t-test was performed to compare two groups, and P values < 0.05 were considered significant.
Results
MiR-610 is down-regulated in CRC tissues and inhibits CRC cell proliferation and clonogenic ability
To investigate the expression pattern of miR-610 in CRC, we first examined miR-610 expression in 33 pairs of CRC tissues and their matched adjacent noncancerous tissues. QRT-PCR results demonstrated that miR-610 expression levels were significantly lower in CRC tissues than in the corresponding adjacent noncancerous tissues (Figure 1A). We next investigated miR-610 expression in four CRC cell lines (SW480, LoVo, HCT116 and SW620) by qRT-PCR and found that miR-610 was expressed in all four CRC cell lines (Figure 1B).
Figure 1.
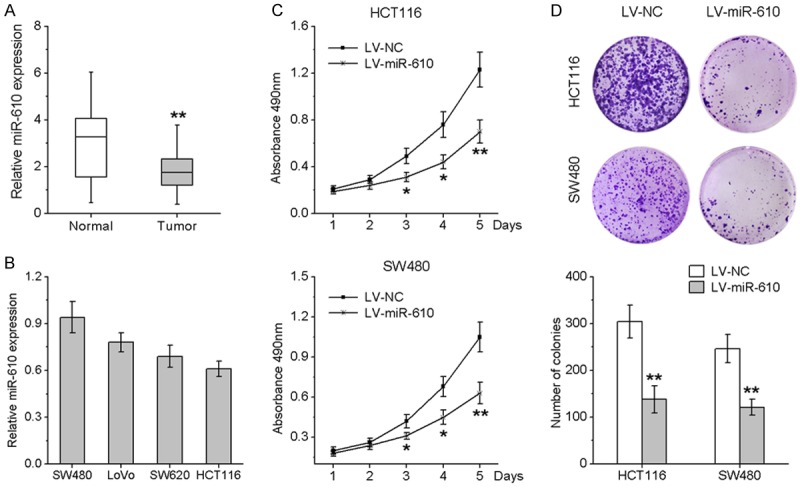
miR-610 is down-regulated in CRC tissues and inhibits CRC cell proliferation and clonogenic ability. A. miR-610 expression in CRC tissues compared with their adjacent normal tissues. B. Endogenous expression of miR-610 in 4 CRC cell lines by qRT-PCR. C. HCT116 and SW480 cells were stably infected with lentivirus expressing miR-610 or negative control. Cell viability was validated by MTT assay. D. Effects of ectopic expression of miR-610 on the clonogenic ability of transfected cells. *P < 0.05, **P < 0.01.
To investigate the biological function of miR-610 in CRC cells, we used the lentivirus system to establish stable HCT116 and SW480 cell lines over-expressing miR-610. Ectopic expression of miR-610 resulted in a significant decrease in cell viability, as detected by MTT assays (Figure 1C). To further verify the inhibitory effects of miR-610 on CRC cell proliferation, we applied the clonogenic assay. As illustrated in Figure 1D, cells transduced with lentivirus encoding miR-610 exhibited significantly reduced clonogenic ability. Collectively, these findings demonstrate that miR-610 functions as a tumor suppressor miRNA and inhibits CRC cell growth in vitro.
MiR-610 suppresses the migration and invasion of CRC cells
We evaluated the effects of miR-610 on CRC cell migration and invasion, a key determinant of cancer progression and metastasis. As indicated in Figure 2A and 2B, the cells treated with lentivirus coding for miR-610 displayed a significant reduction in migratory activity compared with the negative control group. Consistent with these data, the invasive capacities of HCT116 and SW480 cell lines were remarkably repressed by infection with lentivirus encoding miR-610 compared with negative control cells (Figure 2A and 2B). To further explore whether the inhibitory effect of miR-610 on migration and invasion was mediated though mesenchymal-to-epithelial transition (MET), western blotting was performed to investigate the expression of several MET marker proteins in CRC cells. Ectopic expression of miR-610 increased epithelial marker (E-cadherin) expression and decreased mesenchymal marker (vimentin) expression (Figure 2C and 2D). Taken together, these results suggest that miR-610 can curb CRC cell migration and invasion mediated via MET.
Figure 2.
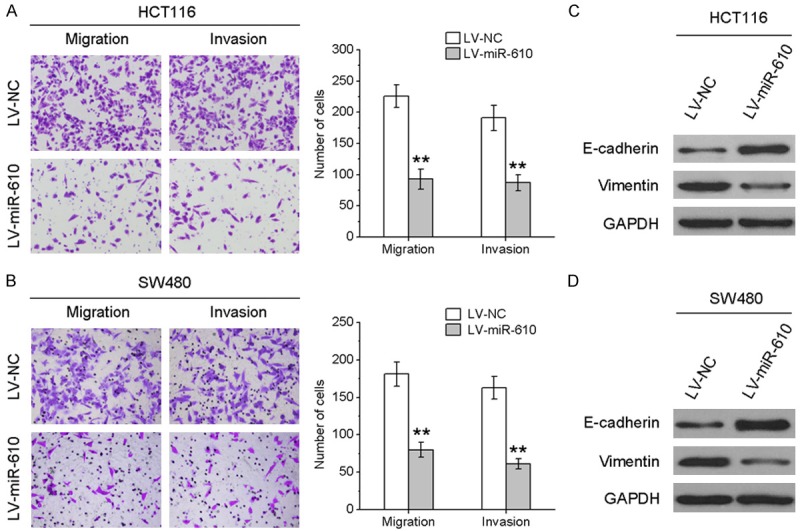
miR-610 suppresses the migration and invasion of CRC cells. Transwell migration and invasion assays were performed on HCT116 (A) and SW480 (B) cells infected with lentivirus encoding miR-610 or negative control. E-cadherin and vimentin protein expression levels in HCT116 (C) and SW480 (D) cells with and without miR-610 over-expression were analyzed by western blotting. *P < 0.05, **P < 0.01.
MiR-610 inhibits the growth of CRC cell xenografts in nude mice
Since the above experiments demonstrated that miR-610 functions as a tumor suppressor miRNA in CRC cells, we tested whether miR-610 over-expression could inhibit tumor growth in nude mice. HCT116 cells infected with lentivirus encoding miR-610 or negative control were used to establish CRC xenograft models in nude mice. Up to 21 days after cell transplantation, the tumors formed by miR-610-over-expressing HCT116 cells grew more slowly than the control tumors, and the average tumor weight in the control group was significantly heavier than that in the miR-610-over-expressing group (Figure 3A). Furthermore, the growth curves of tumors over time showed a significant difference between the two groups (Figure 3B). In addition, we observed lower expression of E-cadherin and vimentin, and fewer Ki-67 positive cells in tumor tissues derived from miR-610-over-expressing cells, as determined by IHC analysis (Figure 3C). Taken together, these results indicate that miR-610 inhibits CRC cell tumorigenic ability.
Figure 3.
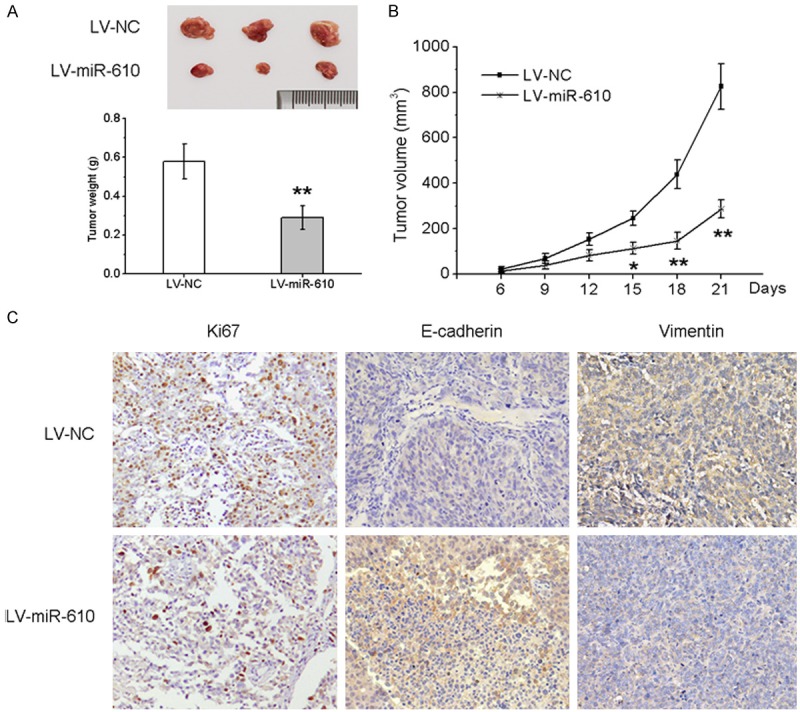
miR-610 inhibits the growth of CRC cell xenografts in nude mice. Lentivirus expressing miR-610 or negative control stably infected HCT116 cells were subcutaneously injected into nude mice. A. Representative photographs of tumors and the mean tumor weights of each group. B. Growth curves for tumor volumes at the indicated times. C. Immunohistochemical (IHC) staining of Ki67, E-cadherin and vimentin in tumor sections. *P < 0.05, **P < 0.01.
MiR-610 down-regulates HDGF expression by directly targeting its 3’UTR
Using bioinformatics algorithms, HDGF was predicted to be a potential target of miR-610. To identify whether HDGF was a direct target of miR-610, we performed a dual-luciferase reporter assay. A human HDGF 3’UTR fragment with the putative miR-610 binding site or the mutant site was cloned into the psiCHECK2 vector (Figure 4A), and then the wild-type or mutant HDGF 3’UTR vector was co-transfected into HEK293T cells with miR-610 mimic or controls. As indicated in Figure 4B, miR-610 remarkably reduced the luciferase activity of wild-type HDGF 3’UTR vector, whereas the luciferase activity of mutant HDGF 3’UTR vector abolished the suppressive effect. We also confirmed that miR-610 dramatically reduced HDGF mRNA and protein levels (Figure 4C and 4D). To further investigate the potential relationship between HDGF and miR-610 in CRC tissues, HDGF expression was validated. As shown in Figure 4E, HDGF mRNA expression was increased in CRC tissues and inversely correlated with the endogenous levels of miR-610. Taken together, our data showed that miR-610 negatively modulated HDGF expression by directly binding to its 3’UTR.
Figure 4.
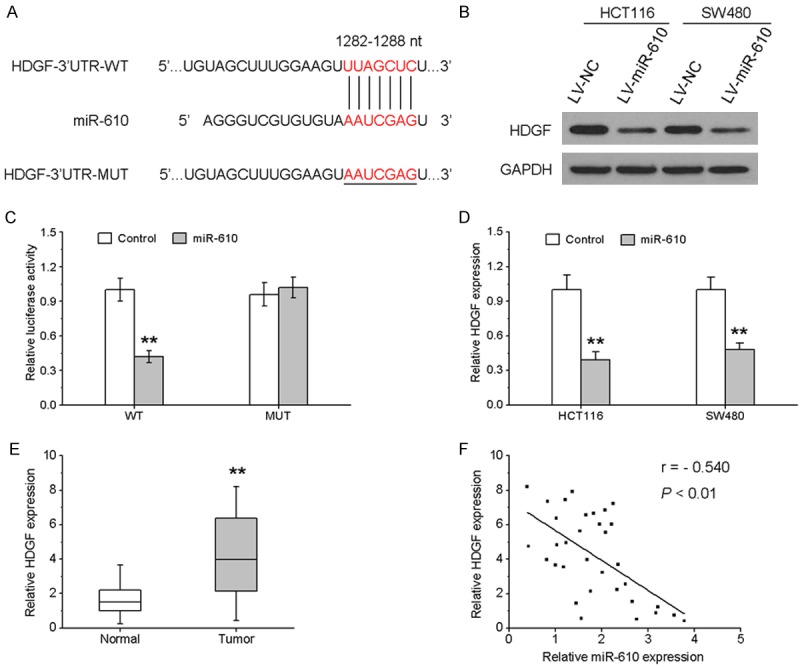
miR-610 down-regulates HDGF expression by directly targeting its 3’UTR. A. Schematic representation of the construction of wild-type or mutant HDGF 3’UTR vector. B. The luciferase reporter assay was performed in HEK293T cells co-transfected with wild-type or mutant HDGF 3’UTR vector with miR-610 mimic or controls. C and D. HDGF protein and mRNA expression in HCT116 and SW480 cells treated with lentivirus-expressing miR-610 or negative control were measured by western blotting and qRT-PCR, respectively. E. The mRNA expression of HDGF in 33 paired CRC samples and adjacent noncancerous tissues. F. Scatter plots showing the negative correlation between miR-610 and HDGF mRNA expression in CRC tissues. *P < 0.05, **P < 0.01.
Inhibition of HDGF caused similar effects to miR-610 overexpression
We further investigated whether HDGF knockdown could have a suppressive effect similar to miR-610 overexpression. MTT and Transwell assays showed that HDGF knockdown could significantly inhibit cell proliferation (Figure 5A), migration (Figure 5B) and invasion (Figure 5C) abilities of CRC cells. Western blotting showed that downregulation of HDGF enhanced E-cadherin and suppressed vimentin expression (Figure 5D).
Figure 5.
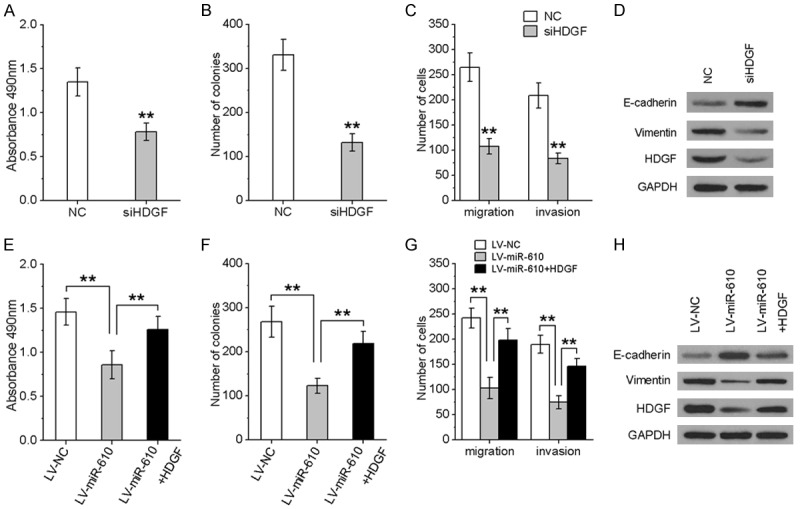
miR-610 inhibited CRC cell proliferation, migration and invasion by targeting HDGF. HCT116 cells were transfected by HDGF siRNA or HDGF expression vector to inhibit or restore HDGF expression. MTT (A, E), colony formation (B, F), Transwell migration and invasion assays (C, G) were used to analyze CRC cell viability, migration and invasion. (D, H) Western blot of HDGF, E-cadherin and vimentin in the indicated cells. All experiments were repeated three times. *P < 0.05, **P < 0.01.
Inverse modulation of HDGF expression by miR-610 is vital for the tumor-suppressive effects of miR-610
Given that miR-610 was able to repress HDGF expression, we wondered whether HDGF could affect miR-610-mediated inhibitory effects on CRC progression. Hence, we evaluated the effect of miR-610 on CRC cell proliferation, migration and invasion in the presence of HDGF expression. As shown in Figure 5E-H, over-expression of HDGF rescued the suppression of miR-610-induced CRC cell proliferation, migration and invasion. Furthermore, reintroduction of HDGF could abrogate miR-610-induced E-cadherin up-regulation and vimentin down-regulation (Figure 5H). Collectively, these results indicate that miR-610 suppresses the proliferation, migration and invasion of CRC cells and EMT phenotypes by negatively modulating HDGF expression.
Discussion
In this study, down-regulation of miR-610 is observed in CRC clinical specimens and cell lines. Ectopic miR-610 expression represses CRC cell proliferation, migration and invasion in vitro, as well as tumor growth in vivo. We also found that ectopic expression of miR-610 reverses the EMT by enhancing E-cadherin expression and inducing a loss of vimentin. Further study showed that HDGF is a direct target gene of miR-610, and its expression inversely correlated with miR-610 expression in CRC clinical specimens. Over-expression of HDGF rescued the inhibitory effects of miR-610 in CRC cells. Collectively, our data suggest an essential role of miR-610 in CRC progression.
MiR-610 has been reported to be abnormally expressed in many types of cancers and involved in the initiation and progression of malignancies. Zeng et al. reported that miR-610 was under-expressed in HCC tissues and cell lines, which correlated with HCC progression and patient survival, while reduced miR-610 levels suppressed HCC cell proliferation and cycle progression in vitro and inhibited HCC cell tumorigenicity in vivo by activating wnt/β-catenin signaling [18]. Wang et al. found that miR-610 was down-regulated in GC, and inhibited the migration and invasion of GC cells by repressing vasodilator-stimulated expression phosphoprotein (VASP) [19]. Jin et al. showed that miR-610 played a suppressor role in lung cancer by influencing cell proliferation, migration and invasion [20]. Consistent with these reports, miR-610 expression is found to be significantly down-regulated in CRC tissues and cell lines. Ectopic miR-610 expression effectively represses CRC cell proliferation, migration, invasion and EMT, and inhibits tumorigenicity in nude mice.
To further understand the underlying mechanisms participating in the miR-610-mediated inhibitory effects on cell proliferation, migration and invasion in CRC cells, we identified HDGF as a direct candidate target of miR-610 using public prediction algorithms. HDGF, a heparin-binding growth factor, was originally purified from culture medium conditioned by HuH7 hepatoma cells [23]. It is over-expressed in the early development of various organs, including cardiovascular, kidneys and liver, while its expression is rapidly reduced after birth [24]. Growing evidence demonstrates that HDGF is highly expressed in different types of malignancies and is linked to tumor progression and poor prognosis. HDGF is over-expressed in glioma samples, and silencing of HDGF significantly decreases the proliferation, migration, invasion and tumorigenesis of glioma cells, enhances E-cadherin expression and reduces vimentin expression [25]. Knock-down of HDGF expression inhibits anchorage-independent growth and invasion of non-small cell lung cancer (NSCLC) cells and represses blood formation in NSCLC tumors [26]. In CRC, HDGF expression gradually increases during carcinogenesis, while knock-down of HDGF can down-regulate survivin, activate the mitochondrial pathway, and induce apoptosis in LoVo cells [27]. In contrast, HDGF over-expression suppresses nordihydroguaiaretic acid (NDGA)-induced apoptosis by the mitochondrial pathway, and mitigates the inhibitory effect of NDGA on tumor growth [28]. In this study, we show that miR-610 represses HDGF expression through targeting its 3’UTR, and demonstrate that high HDGF expression is associated with low miR-610 levels in CRC. Knockdown of HDGF mimics the effects of miR-610 over-expression, while ectopic expression of HDGF reverses the antitumor effect of miR-610. These results suggest that HDGF is a direct and functional target of miR-610.
In conclusion, this study demonstrates that the expression of miR-610 is low in CRC tissues and cell clines, and that miR-610 inhibits cell growth, migration and invasion of CRC cells by targeting HDGF. These findings may provide new insights into the molecular mechanisms of CRC progression and a novel, potential therapeutic strategy for CRC.
Acknowledgements
This study was supported by a grant from the Shanghai Science and Technology Department of Medicine guiding biomedical projects (No. 134119a1400).
Disclosure of conflict of interest
None.
References
- 1.Haggar FA, Boushey RP. Colorectal cancer epidemiology: incidence, mortality, survival, and risk factors. Clin Colon Rectal Surg. 2009;22:191–197. doi: 10.1055/s-0029-1242458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wolpin BM, Mayer RJ. Systemic treatment of colorectal cancer. Gastroenterology. 2008;134:1296–1310. doi: 10.1053/j.gastro.2008.02.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 4.Sung JJ, Lau JY, Goh KL, Leung WK. Increasing incidence of colorectal cancer in Asia: implications for screening. Lancet Oncol. 2005;6:871–876. doi: 10.1016/S1470-2045(05)70422-8. [DOI] [PubMed] [Google Scholar]
- 5.Xu L, Zhang Y, Wang H, Zhang G, Ding Y, Zhao L. Tumor suppressor miR-1 restrains epithelial-mesenchymal transition and metastasis of colorectal carcinoma via the MAPK and PI3K/AKT pathway. J Transl Med. 2014;12:244. doi: 10.1186/s12967-014-0244-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu B, Qu J, Xu F, Guo Y, Wang Y, Yu H, Qian B. MiR-195 suppresses non-small cell lung cancer by targeting CHEK1. Oncotarget. 2015;6:9445–9456. doi: 10.18632/oncotarget.3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lujambio A, Lowe SW. The microcosmos of cancer. Nature. 2012;482:347–355. doi: 10.1038/nature10888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ricci EP, Limousin T, Soto-Rifo R, Rubilar PS, Decimo D, Ohlmann T. miRNA repression of translation in vitro takes place during 43S ribosomal scanning. Nucleic Acids Res. 2013;41:586–598. doi: 10.1093/nar/gks1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Y, Lv Z, He G, Wang J, Zhang X, Lu G, Ren X, Wang F, Zhu X, Ding Y, Liao W, Liang L. The SOX17/miR-371-5p/SOX2 axis inhibits EMT, stem cell properties and metastasis in colorectal cancer. Oncotarget. 2015;6:9099–9112. doi: 10.18632/oncotarget.3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yan J, Gumireddy K, Li A, Huang Q. Regulation of mesenchymal phenotype by MicroRNAs in cancer. Curr Cancer Drug Targets. 2013;13:930–934. doi: 10.2174/15680096113136660098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiang W, He J, Huang C, Chen L, Tao D, Wu X, Wang M, Luo G, Xiao X, Zeng F, Jiang G. miR-106b-5p targets tumor suppressor gene SETD2 to inactive its function in clear cell renal cell carcinoma. Oncotarget. 2015;6:4066–4079. doi: 10.18632/oncotarget.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salah Z, Arafeh R, Maximov V, Galasso M, Khawaled S, Abou-Sharieha S, Volinia S, Jones KB, Croce CM, Aqeilan RI. miR-27a and miR-27a* contribute to metastatic properties of osteosarcoma cells. Oncotarget. 2015;6:4920–4935. doi: 10.18632/oncotarget.3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang Y, Liu C, Yang J, Liu G, Feng F, Tang J, Hu L, Li L, Jiang F, Chen C, Wang R, Yang Y, Jiang X, Wu M, Chen L, Wang H. MiR-20a triggers metastasis of gallbladder carcinoma. J Hepatol. 2013;59:518–527. doi: 10.1016/j.jhep.2013.04.034. [DOI] [PubMed] [Google Scholar]
- 14.Ma F, Song H, Guo B, Zhang Y, Zheng Y, Lin C, Wu Y, Guan G, Sha R, Zhou Q, Wang D, Zhou X, Li J, Qiu X. MiR-361-5p inhibits colorectal and gastric cancer growth and metastasis by targeting staphylococcal nuclease domain containing-1. Oncotarget. 2015;6:17404–17416. doi: 10.18632/oncotarget.3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Minna E, Romeo P, De Cecco L, Dugo M, Cassinelli G, Pilotti S, Degl’Innocenti D, Lanzi C, Casalini P, Pierotti MA, Greco A, Borrello MG. miR-199a-3p displays tumor suppressor functions in papillary thyroid carcinoma. Oncotarget. 2014;5:2513–2528. doi: 10.18632/oncotarget.1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fang Y, Sun B, Xiang J, Chen Z. MiR-301a promotes colorectal cancer cell growth and invasion by directly targeting SOCS6. Cell Physiol Biochem. 2015;35:227–236. doi: 10.1159/000369690. [DOI] [PubMed] [Google Scholar]
- 17.Yin Y, Zhang B, Wang W, Fei B, Quan C, Zhang J, Song M, Bian Z, Wang Q, Ni S, Hu Y, Mao Y, Zhou L, Wang Y, Yu J, Du X, Hua D, Huang Z. miR-204-5p inhibits proliferation and invasion and enhances chemotherapeutic sensitivity of colorectal cancer cells by downregulating RAB22A. Clin Cancer Res. 2014;20:6187–6199. doi: 10.1158/1078-0432.CCR-14-1030. [DOI] [PubMed] [Google Scholar]
- 18.Zeng XC, Liu FQ, Yan R, Yi HM, Zhang T, Wang GY, Li Y, Jiang N. Downregulation of miR-610 promotes proliferation and tumorigenicity and activates Wnt/beta-catenin signaling in human hepatocellular carcinoma. Mol Cancer. 2014;13:261. doi: 10.1186/1476-4598-13-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang J, Zhang J, Wu J, Luo D, Su K, Shi W, Liu J, Tian Y, Wei L. MicroRNA-610 inhibits the migration and invasion of gastric cancer cells by suppressing the expression of vasodilator-stimulated phosphoprotein. Eur J Cancer. 2012;48:1904–1913. doi: 10.1016/j.ejca.2011.11.026. [DOI] [PubMed] [Google Scholar]
- 20.Jin J, Li C, You J, Zhang B. [miR-610 suppresses lung cancer cell proliferation and invasion by targeting GJA3] . Zhonghua Zhong Liu Za Zhi. 2014;36:405–411. [PubMed] [Google Scholar]
- 21.Li E, Ji P, Ouyang N, Zhang Y, Wang XY, Rubin DC, Davidson NO, Bergamaschi R, Shroyer KR, Burke S, Zhu W, Williams JL. Differential expression of miRNAs in colon cancer between African and Caucasian Americans: implications for cancer racial health disparities. Int J Oncol. 2014;45:587–594. doi: 10.3892/ijo.2014.2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dong R, Liu X, Zhang Q, Jiang Z, Li Y, Wei Y, Yang Q, Liu J, Wei JJ, Shao C, Liu Z, Kong B. miR-145 inhibits tumor growth and metastasis by targeting metadherin in high-grade serous ovarian carcinoma. Oncotarget. 2014;5:10816–10829. doi: 10.18632/oncotarget.2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakamura H, Izumoto Y, Kambe H, Kuroda T, Mori T, Kawamura K, Yamamoto H, Kishimoto T. Molecular cloning of complementary DNA for a novel human hepatoma-derived growth factor. Its homology with high mobility group-1 protein. J Biol Chem. 1994;269:25143–25149. [PubMed] [Google Scholar]
- 24.Enomoto H, Yoshida K, Kishima Y, Okuda Y, Nakamura H. Participation of hepatoma-derived growth factor in the regulation of fetal hepatocyte proliferation. J Gastroenterol. 2002;37(Suppl 14):158–161. doi: 10.1007/BF03326437. [DOI] [PubMed] [Google Scholar]
- 25.Song Y, Hu Z, Long H, Peng Y, Zhang X, Que T, Zheng S, Li Z, Wang G, Yi L, Liu Z, Fang W, Qi S. A complex mechanism for HDGF-mediated cell growth, migration, invasion, and TMZ chemosensitivity in glioma. J Neurooncol. 2014;119:285–295. doi: 10.1007/s11060-014-1512-4. [DOI] [PubMed] [Google Scholar]
- 26.Zhang J, Ren H, Yuan P, Lang W, Zhang L, Mao L. Down-regulation of hepatoma-derived growth factor inhibits anchorage-independent growth and invasion of non-small cell lung cancer cells. Cancer Res. 2006;66:18–23. doi: 10.1158/0008-5472.CAN-04-3905. [DOI] [PubMed] [Google Scholar]
- 27.Liao F, Dong W, Fan L. Apoptosis of human colorectal carcinoma cells is induced by blocking hepatoma-derived growth factor. Med Oncol. 2010;27:1219–1226. doi: 10.1007/s12032-009-9362-1. [DOI] [PubMed] [Google Scholar]
- 28.Liao F, Liu M, Lv L, Dong W. Hepatomaderived growth factor promotes the resistance to anti-tumor effects of nordihydroguaiaretic acid in colorectal cancer cells. Eur J Pharmacol. 2010;645:55–62. doi: 10.1016/j.ejphar.2010.07.033. [DOI] [PubMed] [Google Scholar]