

Abstract
Autophagy is an important mechanism for cellular homeostasis and survival during pathologic stress conditions in the liver, such as ischemia-reperfusion injury. In this study, we hypothesized a protective role of vitamin Din hepatic IR model. The administration of vitamin D displayed significantly preserved liver function as characterized by less histological damage and reduced serum enzymes level. We found that the protective effect was associated with ameliorated oxidative stress as manifested by the increase of antioxidant capacity and decrease of lipid peroxidation. Further, increased autophagic flux after vitamin D administration was demonstrated by the increase of protein light chain 3 (LC3) conversion both in vivo and in vitro. MEK/ERK and PTEN/PI3K/Akt/mTOR were both found critically involved in vitamin D-induced autophagy. By employing intracellular ROS and cell viability assay, we further confirmed this hypothesis with the observation that inhibition either of the MEK/ERK or PTEN/PI3K/Akt/mTOR pathway partly abolished the protective effect of vitamin D-induced autophagy, while inhibiting initiation of autophagy signaling pathway by knockdown of Beclin-1 completely reversed the protection provided by vitamin D. Collectively, the present results indicate that the protective role of vitamin D in murine hepatic IR injury is autophagy dependent, which is regulated by both MEK/ERK and PTEN/PI3K/Akt/mTOR pathway.
Keywords: Ischemia reperfusion injury, autophagy, vitamin D
Introduction
Liver ischemia reperfusion (IR) injuryis a clinically relevant condition that occurs during resection surgery, trauma, hypovolemic shock, or transplantation when liver is transiently deprived of oxygen and reoxygenated, and is a key contributing factor in liver dysfunction and failure [1]. Clinical and experimental data have established that up to 10% of early graft dysfunction and higher incidence of both acute and chronic rejection are associated with IR injury, which results in reduced long-term graft survival [2]. The pathophysiology of liver IR injury includes direct cellular damage as the result of the ischemic insult as well as delayed dysfunction and damagethat result from oxidative stress. During IR process, oxidative stress as a common event caused by excessive production of reactive oxygen species (ROS) is a critical factor implicated in cellular damage [3]. Indeed, antioxidants therapy has already been shown to be protective against IR-mediated oxidative damage in different experimental models [4].
As a cytoprotective process, autophagy can play a prosurvival role under normal physiological conditions or starvation stress. It helps to maintain cell homeostasis in nutrient-rich environments through its constitutive activity, and serves as an alternative energy source for cells under nutrient-poor conditions [5]. During the early phase of hepatic reperfusion, the mitochondria temporarily repolarize and begin generating ATP, which induces autophagy. At the same time, reperfusion of ischemic hepatocytes triggers Ca2+ and ROS accumulation in a subset of mitochondria [6]. When the capacity of autophagic clearance counter balances or surpasses reperfusion-induced injury, altered mitochondria are eliminated in a timely fashion by autophagy, and hepatocyte viability is maintained [7]. Conversely, when intra mitochondrial loading of Ca2+ and ROS exceed autophagic clearance, autophagy fails to remove all dysfunctional mitochondria and widespread onset of the mitochondrial permeability transition ensues, leading to irreversible uncoupling of oxidative phosphorylation, ATP depletion, energetic failure and ultimately hepatocyte death [7,8]. Thus, insufficient autophagy is a crucial mechanism underlying IR injury to the liver.
While 1,25-Dyhydroxyvitamin D3 (hereafter, vitamin D) is most commonly associated with the regulation of calcium homeostasis that affects bone metabolism, the broad distribution of vitamin D receptor (VDR) suggest that vitamin D may have a much broader spectrum of activity. In fact, recent studies have demonstrated that vitamin D is an important factor playing a crucial role in human homeostasis beyond calcium homeostasis [9-11]. The activation of vitamin D/VDR signaling affects various processes, including apoptosis, inflammation, immunomodulation, detoxification, and autophagy [12-14]. Therefore, vitamin D is becoming an emerging therapeutic strategy for diseases beyond bone metabolism.
However, till date, it remains unclear that how vitamin D regulates the coordination of autophagy and hepatocytes survival during IR insult.Given the fact that oxidative stress represented an important cause of cellular damage in IR injury while autophagy could lead to adaptive responses that allow cells to continue normal function in the face of oxidative stress, we think that a study targeting vitamin D in liver IR injury may provide new insights into the understanding of molecular mechanism of vitamin D/VDR signaling and provide potential therapeuticbenefits in liver IR injury. To this end, we hypothesized that vitamin D could induce autophagy through MAPK pathway and protect liver from oxidative damage caused by IR injury.
Methods
Animals and vitamin D pretreatment
Male C57BL/6 mice (10-12 week, weight 23-25 g) were obtained from Joint Ventures Sipper BK Experimental Animal (Shanghai, China). All animal experiments were performed in accordance with the guidelines of National Institute of Health for the Care and Use of Laboratory Animals, and approved by the Scientific Investigation Board of Second Military Medical University, Shanghai, China. For vitamin D pretreatment, mice were received vitamin D-sufficient diet (Solarbio, Beijing, China) at a dose of 500 IU/kg daily for 4 weeks before being subjected to IR, this dose strategy would be considered as a high daily dose when calculated to human equivalency dose [15].
Induction of liver IR
Mice were anesthetized with sodium pentobarbital (50 mg/kg, intraperitoneally). After a midline laparotomy, an atraumatic clamp (Shanghai Medical Instruments, Shanghai, China) was used to interrupt blood supply to the left lateral and median lobes of the liver (70%). After 60 minpartial hepatic ischemia, the clamp was removed to initiate hepatic reperfusion. Mice with sham surgery (no interruption of the hepatic blood flow) were used as controls. Body temperature was maintained with an adjustable heating pad at 37°C. Mice were sacrificed after the indicated periods of reperfusion, and blood and samples of the livers were taken for analysis.
Assessment of liver function
Serum AST and ALT levels were determined to assess the liver function by using a standard Modular Auto analyzer at the Central Laboratory, Changhai Hospital, Shanghai.
Histopathological examination
Hematoxylin and eosin-stained slides were prepared from routinely processed excised specimens fixed in 10% buffered formalin and processed in paraffin. Three representative sections from each liver were scored. At least 10 high-power fields (×200 and ×400) per section were examined for each sample. Histological examination was performed by two pathologists in a blinded fashion.
Terminal dUTP nick-end labeling (TUNEL) assay
Following the protocol of manufacturer, TUNEL staining was conducted by an in situ cell death detection kit (Dead End Fluorometric TUNEL System, Promega, Madison, USA). The nuclei of all cells were counter-stained with DAPI. The number of TUNEL- and DAPI-positive nuclei was counted in six images that were chosen randomly from non-overlapping areas of each group. The data were presented as the percentage of TUNEL-positive cells.
Measurement of lipid peroxidation
Thiobarbituric acid-reactive substances (Sigma-Aldrich, St. Louis, MI, USA) wasemployed to assess the lipid peroxidation products in malondialdehyde (MDA) equivalents. In brief, liver tissues were homogenized with 0.1 mol/L sodium phosphate buffer. Two hundred microliters of liver homogenates was mixed with 5 ml reaction buffer (provided by the kit) and heated at 95°C for 60 min. Then the absorbance of the supernatant was evaluated at 532 nm via a spectrophotometric assay.
Measurement of antioxidant enzymes
Manganese-superoxide dismutase (Mn-SOD) activity and catalase activitywas determined using commercially assay kits (Beyotime, Shanghai, China). Briefly, after perfusing and rinsing with phosphate buffered saline, liver tissues were weighed and homogenized with appropriate buffers (provided by the kits). After spinning the tissue homogenates at 10,000 g for 15 min, the supernatantsamples were then determined following procedures provided by the respective manufacturers. The activities of the enzymes in the mouse liver are expressed in U/μg of the protein.
Measurement of GSH and GSSG levels
Briefly, liver tissues were weighed and homogenized with 0.1 mol/L sodium phosphate buffer. The homogenates were then centrifuged with 5% trichloroacetic acid to remove the proteins. After precipitation with 1% picric acid, the GSH level was determined in the liver homogenates using Ellman’s reagent, 5,5’-dithio-bis-(2-nitrobenzoic acid). The oxidized GSH (GSSG) level was measured by the same method in the presence of 2-vinylpyridine, and the GSH/GSSG ratio was calculated.
Real-time RT-PCR analysis
Total liver RNA was extracted using TRIzol (Invitrogen, Waltham, MA, USA) reagent according to the manufacturer’s instructions. cDNA was synthesized using oligo d(T) (Applied Biosystems, Waltham, MA, USA) and a Superscript III Reverse Transcriptase Kit (Invitrogen, Waltham, MA, USA). A StepOne Real-Time PCR System (Applied Biosystems, Waltham, MA, USA) and a SYBR RT-PCR kit (Takara, Tokyo, Japan) were used for quantitative real-time RT-PCR analysis. All reactions were conducted in a 20 µl reaction volume in triplicate. The relative expression levels for a target gene were normalized by GAPDH. Specificity was verified by melting curve analysis and agarose gel electrophoresis. Primers used RT-PCR analysis are: TNF-α (5’-AAG CCT GTA GCC CAC GTC GTA-3’; 5’-GGC ACC ACT AGT TGG TTG TCT TTG-3’); IL-2 (5’-CCA TGA TGC TCA CGT TTA AAT TTT-3’; 5’-CAT TTT CCA GGC ACT GGA GAT G-3’); IL-6 (5’-ACA ACC ACG GCC TTC CCT ACT T-3’; 5’-CAC GAT TTC CCA GAG AAC ATG TG-3’); IL-10 (5’-GCT CTTA CTG ACT GGC ATG AG-3’; 5’-CGC AGC TCT AGG AGC ATG TG-3’); and GAPDH (5’-TGA CCA CAG TCC ATG CCA TC-3’; 5’-GAC GGA CAC ATT GGG GGT AG-3’). Data were analyzed using the comparative Ct (2-ΔΔCt) method.
Myeloperoxidase (MPO) immunofluorescent analysis
Liver cryostatic sections were air-dried and fixed with acetone at -20°C. After blocking of non-specific binding with fetal calf serum for 1 hour at room temperature, the sections were incubated with primary antibody against myeloperoxidase (Beyotime Biotechnology, Shanghai, China) overnight at 4°C. After washing, liver sections were immunostained with secondary antibodies for 1 h at 37°C. Cells’ nuclei of the stained sections were marked by DAPI. The staining was imaged with a fluorescent microscope.
Transmission electron microscopy
The livers were flushed and perfused with 2.5% glutaraldehyde in 0.1 mol/L PBS and then sectioned and photographed using a transmission electron microscope (HITACHI H-800, Tokyo, Japan) at 80 kV. For autophagic vacuole quantification, 20 micrographs, primary magnification ×15,000, were taken with systematic random sampling from each sample.
Protein light chain 3 (LC3) immunofluorescent staining
Livercryostatic sections at were fixed for 15 min in 4% paraformaldehyde, followed by permeabilization with 0.25% TritonX-100 in PBS for 5 min. After blocking with fetal calf serumfor 1 h, the slides were incubated with primary antibodies (LC3B, Cell Signaling Technology, Danvers, MA) overnight at 4°C. After that, liver sections were immunostained with secondary antibodies for 1 h at 37°C. Cells’ nuclei of the stained sections were marked by DAPI. The staining was imaged with a fluorescent microscope.
Western blot analysis
Proteins from liver samples were subjected to 12%/15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane. Polyclonal rabbit anti-mouse Beclin-1, LC3B, Atg-7, SQSTM1, ERK1/2, p-ERK1/2, mTOR, p-mTOR, p70S6K, p-p70S6K, p-MEK1/2, PTEN, Akt, p-Akt, and GAPDH (Cell Signaling Technology, Danvers, MA) were used. The relative quantity of proteinswas determined using a densitometer software (ImageJ, NIH, USA).
Hepatocyte isolation and cell culture
Hepatocytes were isolated from male C57BL/6 mice by an in situ collagenase (type IV) perfusion technique and purified to >98% by repeated centrifugation at 50 g, followed by further purification over 30% Percoll. Viability at time of plating was checked using trypan blue exclusion. Highly purified hepatocytes (>98% purity and >95% viability by trypan blue exclusion) were suspended in Williams’ E medium supplemented with 10% heat-inactivated calf serum, 15 mM HEPES (pH 7.4), 16 units of insulin, 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. The cells were plated on collagen-coated cell culture dishes (3×106 cells/6-cm dish) or plates (2.5×105 cells/well in 6-well plates) and cultured overnight at 37°C under normoxic conditions (21% O2).
To simulate tissue ischemia, anoxia conditions were achieved by placing the hepatocytes into an airtight modular incubator chamber (Billups-Rothenburg, Del Mar, CA) gassed with 5% CO2 and 95% N2. After 4 hours, anoxic hepatocytes were returned for reoxygenation in the normoxic incubator. Hepatocytes incubated under normoxic conditions served as controls. To dissolve reagents, DMSO (final concentration <0.1%) was present in the buffer of all groups. Some hepatocytes were pretreated before anoxia reoxygenation (AR) with Vitamin D (50 nM) alone or in combination with Bafilomycin A1 (100 nM) or wortmannin (10 μM) or Rapamycin (100 nM) or U0126 (5 μM) or LY294002 (100 μM), respectively.
siRNA transfection and cell viability assay
Before transfection, the medium was changed to fresh medium containing FBS but without antibiotics. Beclin-1 or scramble siRNA (Cell Signaling technology, Danvers, MA, USA) were added to the culture medium for 48-h incubation using siRNA transfection reagent (Sigma-Aldrich, St Louis, MO, USA)according to the manufacturer’s instructions.
Twenty-four hours after AR, the cell viability was measured with CCK-8 (Dojindo Molecular Technologies, Kumamoto, Japan) according to the instruction of the manufacturer. The countings were repeated three times.
Measurements of intracellular ROS
The level of cytosolic ROS was quantified with an oxygen radical-sensitive probe, DCFH-DA staining (Beyotime, Shanghai, China) as previously described [16]. Briefly, hepatocytes were treated with AR in the presence of vitamin D, U0126, LY294002, or Beclin-1 siRNA. Then the cells were washed with PBS and incubated with 20 μM DCFH-DA for 30 min. Relative fluorescent intensities were quantified using a flow cytometer.
Statistical analysis
We used the computer software GraphPad Prism 5 (La Jolla, CA) for data analysis. The data that were obtained from two groups were analyzed using an unpaired Student’s t test or a Mann-Whitney test (two tailed). ANOVA was used to compare the four groups, followed by Bonferroni’s multiple comparisons test to assess the statistical significance between treated and untreated groups in all experiments. All of the data are expressed as mean ± SD. In every case, p<0.05 was considered to be statistically significant.
Results
Vitamin D pretreatment protects livers against IR injury
To assess the impact of vitamin Don hepatic IRI, we first induced warm hepatic IR using a well-established two-lobe IR model, and different groups were subjected to 60 minutes of partial hepatic ischemia. At different time points post reperfusion, serum ALT and AST concentrations were collected and showed in Figure 1A. The serum levels of ALT and AST were significantly higher in the IR group than those in the IR+Vit D group, which peaked at 6 hours post reperfusion (3606.17±445.07 versus 2760.67±392.04 [P<0.01] for sALT, and 3761.67±542.95 versus 2766.67±527.25 [P<0.01] for sAST). The serum transaminases levels remained elevated at 24 hours post reperfusion and recovered to normal level at 48 hours. We also examined the histopathological changes in liver tissues. The sham-operation mice with or without vitamin D treatment showed no noticeable effects on liver histology, and exhibited normal morphology. Large necrotic areas as well as hemorrhagic change were evident in IR group livers, whereas the hepatic architecture of IR+Vit D group livers was better preserved with small and nonconfluent necrotic areas and attenuated hemorrhage (Figure 1B). Hepatocellular damage was graded according to Suzuki’s criteria (score, 5.875±0.30 in IR group compared to 4.25±0.31 in IR+Vit D group, P<0.05) (Figure 1C).Quantitative analysis of TUNEL positive hepatocytes showed that the number of apoptotic hepatocytes in mice insulted by IR was higher than that in control mice. Treatment with vitamin D significantly reduced the number of apoptotic cells in the liver (Figure 1D).
Figure 1.

Protective effect of vitamin D in liver IR injury. A. Hepatic function was assessed by measuring serum ALT and AST concentrations. B. Six hours after reperfusion, sham and IR livers pretreated with vehicle and vitamin D were fixed in 10% buffered formalin and processed in paraffin. 4-μm sections were cut and stained with Hematoxylin and eosin. C. The histological degree of hepatic IR injury was assessed according to the Suzuki’s criteria. D. Liver paraffin sections were analyzed for apoptosis using DAPI and TUNEL assay, apoptotic index indicates that the vitamin D treated group had significantly less TUNEL-positive cells when compared to the control group. Data represent mean ± S.D. (*p<0.05, **p<0.01).
Vitamin D pretreatment decreased IR-induced oxidative stress and inflammatory milieuin the livers
Oxidative stress is an important phenomenon in the mechanisms of liver injury owing to its ability to modulate inflammatory responses [17]. To dissect the underlying mechanisms by which vitamin D protects liver against IR-induced injury, the status of oxidative stress in liver tissues was determined. It was interestingly noted that the level of lipid peroxidation product MDA was significantly elevated in IR mice compared to those in sham-operated control mice. The elevation of MDA was reduced by vitamin D treatment, demonstrating that vitamin D treatment inhibits IR-induced oxidative stress in the livers. To demonstrate whether the reduced oxidative stress relevant tovitamin D treatment is associated with enhanced antioxidant capacity, we examined hepatic Mn SOD, catalase activity, and GSH/GSSG ratio. As expected, livers originated from mice withvitamin D treatment showed significantly higher activities of Mn SOD, catalase, and GSH/GSSG ratio after IR induction as compared with those of control mice, suggesting that vitamin D treatment enhanced the antioxidant capability of hepatocytes (Figure 2A). Given the fact that cytokines such as TNF-α, IL-6, IL-2 and IL-10 play critical roles in IR-induced hepatic injury, we therefore further compared their expressions between vitamin D treated mice and control mice. Interestingly, mRNAs for TNF-α, IL-6 and IL-2 were significantly higher in control mice as compared with that of vitamin D treated mice. In sharp contrast, IL-10 was significantly higher in vitamin D treated mice six hours after IR induction (Figure 2B). By indirect immunhistochemical labelling, we also detected an early decrease in the number of MPO+ cells when treated with vitamin D six hours after IR (Figure 2C).
Figure 2.

Vitamin D pretreatment decreased IR-induced oxidative stress and inflammatory milieu in the livers. Livers were harvested six hours after reperfusion. A. The Mn SOD activity, catalase activity, and GSH/GSSG ratio were decreased, while MDA was increased by IR injury. Vitamin D, by itself, did not affect these variables but ameliorated the effects of IR injury. B. The TNF-α, IL-6 and IL-2 were significantly higher in control mice as compared with that of vitamin D treated mice. In sharp contrast, IL-10 was significantly higher in vitamin D treated mice after IR induction. C. Double staining of liver sections with monoclonal antibodies against MPO and DAPI, followed by fluorescence immunodetection in sections of mice livers. Data represent mean ± S.D. (*p<0.05, **p<0.01).
Vitamin Dinduces hepatic autophagyboth in normal situation and IR insult in vivo
Because IR-induced oxidative stresswas alleviated by vitamin D treatment, we next asked if vitamin D could enhance the capacity of autophagic clearance, which would counterbalance the ROS accumulation caused by dysfunctional mitochondria. To test the autophagic response following vitamin D treatment, we first examined the accumulation of autophagosomes in hepatocytes by transmission electron microscopy by evaluating the amount of autophagic vacuoles per 100 μm cytoplasmic area (Figure 3A). Compared with the basal level of 3.17±0.65 in the sham control, much more autophagic vacuoles (9.67±1.36, p<0.01) were seen in vitamin D-treated mice liver tissues. In contrast, the number of autophagic vacuoles experienced an increase following hepatic IR insult, to 7.5±0.89 (p<0.05), which were increased to 21.5±2.9 (p<0.01) by vitamin D preconditioning. Next, we determined LC3B expression using fluorescence immunostaining technique (LC3B-positive cells shown as green fluorescence normalized to the DAPI stained nucleus number). As shown in Figure 3B, vitamin D pretreatment significantly increased the LC3B-positive cells both in normal situation and following IR insult. To confirm the above results, we further evaluated the expression of autophagy markers LC3I/II, Beclin-1, and Atg-7. Immunoblotting results revealed overtly elevated levels of LC3II, Beclin-1, and Atg-7 following IR insult, and this elevation was much more significant withintervention with vitamin D during IR insult (Figure 3C). Thus, autophagy is induced by vitamin D both in normal situation and IR insult.
Figure 3.

Vitamin D induces hepatic autophagy both in normal situation and IR insult in vivo. Livers were harvested six hours after reperfusion. A. The result by transmission electron microscopy showed that vitamin D induced hepatic autophagy both in normal and IR situation. Arrowheads indicate autophagic vesicles. B. LC3 puncta formation was assayed by immunofluorescence. Administration of vitamin D significantly induced hepatic autophagy both in normal and IR situation. C. Western blot analysis of key molecules involved in autophagy revealed that vitamin D induced LC3-I to LC3-II conversion and subsequent autophagy. Data represent mean ± S.D. (*p<0.05, **p<0.01).
Vitamin D increases hepatocellular autophagic flux in vitro
The dynamic process of autophagy includes initiation, elongation, maturation and degradation, which is also called autophagic flux. An increase of LC3 lipidation may result from increased formation of autophagosomes or decreased degradation of autophagosomes. Tofurther confirm whether vitamin D enhanced autophagic fluxby increasing formation of autophagosomes or decreasing degradation of autophagosomes in anoxia reoxygenation treated hepatocytes, we treated hepatocytes with vitamin D in the presence or absence of bafilomycin A1 (an inhibitor of autophagosomes and lysosome fusion) or wortmannin (an inhibitor of autophagosomes initiation) or rapamycin (an autophagy activator). As shown in Figure 4A, bafilomycin A1 increased LC3 lipidation and SQSTM1 levels (a autophagy-related protein that degraded in lysosomes after autophagosomes fuse with lysosomes), and more importantly, vitamin D plus bafilomycin A1 further upregulated SQSTM1 levels. Wortmannin attenuated vitamin D-induced LC3 II upregulation and SQSTM1 levels, whereas rapamycin further increased LC3 lipidation but potentiated vitamin D-induced downregulation of SQSTM1 levels. Collectively, these data suggest that vitamin D increased autophagic flux rather than blocking the fusion of autophagosomes with lysosomes (Figure 4B, 4C).
Figure 4.
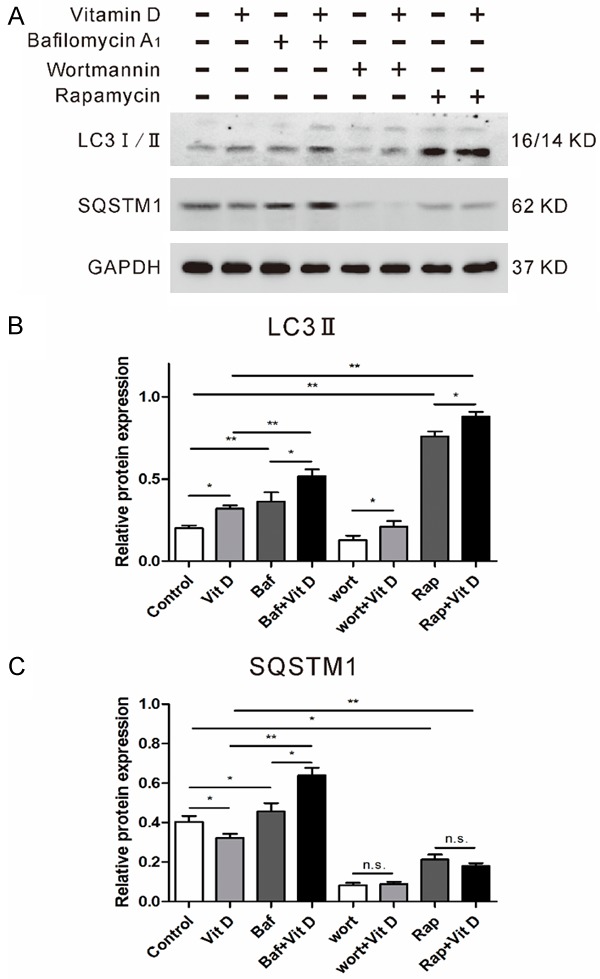
Vitamin D increases hepatocellular autophagic flux in vitro. (A) Hepatocytes were treated with vitamin D in the presence/absence of bafilomycin A1, wortmannin or rapamycin. The protein samples were collected eight hours after the reoxygenation. Relative LC3-II (B) and SQSTM1 (C) level were determined by densitometry and normalized to GAPDH levels. Data represent mean ± S.D. (*p<0.05, **p<0.01).
Vitamin D activates mTORC1 independent of MEK/ERKin hepatocytes following AR insult
The MEK/ERK/mTOR pathway is an important signaling pathway that regulates autophagic activity. Generally, upregulation of MEK/ERK results in inhibition of mTORC1 pathway and subsequently autophagic activation [18]. To better understand the cell signaling mechanisms, we determined the effect of vitamin D on the MEK/ERK and the mTOR pathway. As shown in Figure 5A, vitamin D elevated the phosphorylation of ERK1/2 while inhibited the phosphorylation of mTOR and p70S6K, a substrate of mTORC1, in hepatocytes following AR insult. However, vitamin D intake failed toaffect these pathways in sham-operated group (Figure 5C, 5D), indicating that vitamin D effectthis pathway under stress condition rather than in normal physiological conditions. Pretreatment of hepatocytes with the MEK1/2 inhibitor U0126 increased phosphorylation of MEK1/2 (Figure 5H), but inhibited the phosphorylation of the downstream substrates ERK1/2 (Figure 5I). However, U0126 did not have any noticeable effect on the vitamin D-induced reduction of mTORC1 as measured by the level of mTOR and p70S6K phosphorylation (Figure 5F, 5G), indicating that the MAPK/ERK does not play a critical role in the vitamin D-induced mTORC1 reduction.
Figure 5.

Vitamin D activates mTORC1 independent of MEK/ERK. (A) MEK/ERK and mTORC1 are positioned upstream of autophagic signaling after AR insult. Relativep-ERK (B), p-mTOR (C), and p-p70S6K (D) levels were determined by densitometry and normalized to each total protein levels. (E) Employing the MEK1/2 inhibitor, U0126, did not have any noticeable effect on the vitamin D-induced reduction of p-mTOR and p-p70S6K. Relativep-mTOR (F), p-p70S6K (G), p-MEK (H), and p-ERK (I) levels were determined by densitometry and normalized to each total protein levels. Data represent mean ± S.D. (*p<0.05, **p<0.01).
Vitamin D-induced mTORC1 activation is dependent on PTEN-mediated Akt downregulation
Phosphatase and tensin homologue on chromosome 10 gene (PTEN) is a multiphosphatase tumor suppressor which blocks the downstream activity of PI3K/AKT/mTOR signaling by degrading PIP-3 [19]. We investigated the role of PTEN in AR-insulted hepatocytes (Figure 6A). As shown in Figure 6B, vitamin D downregulated the expression of PTEN after AR-insult and subsequently upregulated the phosphorylation of Akt. Unlike decreased vitamin D-induced PTEN expression promoted activation of Akt which leaded to the down-regulation of mTORC1 activity as manifested by decreased phosphorylation of p70S6K, in the present of LY294002 (a PI3K/Akt pathway inhibitor), the ability of vitamin D to activate Akt and inhibitmTORC1 was abolished (Figure 6C, 6D).
Figure 6.

PTEN and ERK are essential for the protective role of vitamin D. (A) vitamin D downregulated the expression of PTEN and upregulated the phosphorylation of Akt after AR-insult. However, with the addition of LY294002, the expression of p-p70S6K could not be obviously reduced by vitamin D. Relative PTEN (B), p-Akt (C), and p-p70S6K (D) levels were determined by densitometry and normalized to GAPDH or each total protein levels. (E) Western blot showing decrease in the expression of Beclin-1 when hepatocytes were transfected with Beclin-1 targeting siRNA. (F) Results of the CCK-8 assay showing a significant protective effect of vitamin D on hepatocytes while U0126, LY294002, and Beclin-1 siRNA treatment caused an aggravation of cell viability in spite of vitamin D presence. (G) DCFH-DA fluorescence measurements of cellular ROS levels showed significantly lower ROS level in vitamin D group compared with that in control group. However, this reduction was reversed by U0126, LY294002, and Beclin-1 siRNA treatment, respectively.
PTEN and ERK are essential for the protective role of vitamin D
As PTEN/PI3K/Akt/mTORC1 and MEK/ERK signaling are both play a critical role in autophagy [20,21], the above results prompted us to further testthe impact of both pathways on cell viability and ROS generation. We chose to use Beclin-1 in examining whether knockdown of autophagy has effect on hepatocytes. A significant inhibition of Beclin-1 at protein levels (>90%) was observed following siRNA treatment (Figure 6E). Notably, cell viability assessed by CCK-8 showed no significant differences among vitamin D, U0126, LY294002, and Beclin-1 siRNA treatment on hepatocytes before AR insult. After AR insult, vitamin D showed a significant protective effect on hepatocytes while U0126, LY294002, and Beclin-1 siRNA treatment caused an aggravation of cell viability in spite of vitamin D presence (Figure 6F). We then determined intracellular ROS generation and it was demonstrated that in the absence of AR-insult, vitamin D nor U0126 nor LY294002 nor Beclin-1 siRNA could alter the intracellular ROS level. In contrast, the ROS level was significantly higher in the AR-insulted hepatocytes (Figure 6G). As expected, hepatocytes with vitamin D treatment showed significantly lower ROS level compared with control hepatocytes. However, this reduction was reversed by U0126, LY294002, and Beclin-1 siRNA treatment, respectively (26%, 37.67%, and 59.17%). These results suggest that both PTEN/PI3K/Akt/mTORC1 and MEK/ERK signaling contribute to the clearance ability of vitamin D-induced autophagy.
Discussion
The hormonally active form of vitamin D, 1,25-Dyhydroxyvitamin D3, influences the expression of various genes, whose products not only are involved in the bone and calcium metabolism but are able to interact with a wide range of nonclassic organs and target tissues, including the heart and liver [22,23]. Experimental and preliminary clinical evidence has demonstrated that vitamin D is critically involved in cell proliferation, differentiation and immunomodulation. As reported in literature, vitamin D is able to reduce the damage following H2O2-mediated stress in a dose- and time-dependent manner, through the decrease of anion superoxide generation and apoptotic cells [24]. The present research explains the relationship between vitamin D and antioxidation is a consequence of its activity in the autophagic signaling process. Activation of autophagy by vitamin D partially counteract the negative effects on hepatocytes triggered by ischemia-reperfusion.
It is now well recognized that a protective stimulus can be applied at the onset of reperfusion to attenuate reperfusion injury. Although controversial, excessive ROS is considered to independently mediate tissue damage by strong cellular oxidizing potential leading to mitochondrial injury [25]. In the present study it has been clearly demonstrated that vitamin D administration antecedent to IR induction provides protection to liver. After 70% hepatic ischemia, vitamin D pretreated mice displayed significantly preserved liver function as characterized by less histological damage and reduced serum enzymes level. We further demonstrated that the protective effect was associated with ameliorated oxidative stress as manifested by the increase of antioxidant capacity (higher SOD and catalase activities) and decrease of lipid peroxidation (less generation of MDA and larger GSH/GSSH ratio). By employing electron microscopy and immunofluorescent staining, our studies revealed that vitamin D administration significantly induced autophagy, which removes dysfunctional mitochondria and counter balance intra mitochondrial ROS generation [26].
Autophagy is a cellular self-defense response involving the lysosomal degradation of cytoplasmic organelles or cytosolic components [27]. Although evidence showed a significant increasein autophagic response after vitamin D treatment, increasing LC3 lipidation or autophagosomes may result from either an enhancement of autophagosomal formation or inhibition of autophagosomal degradation, or may be caused by autophagy-independent mechanisms [28]. Therefore, both generation and degradation of autophagosomes must be taken into consideration to determine autophagic flux. In this study, we showed that vitamin D decreased the level of SQSTM1 after IR insult, but has the opposite effect in the presence of bafilomycin A1, suggesting that vitamin D increases the formation/maturation of autophagosomes rather than blocks the fusion of the autophagosomes and lysosomes. Furthermore, wortmannin attenuated vitamin D-promoted LC3 lipidation and SQSTM1 level while rapamycin upregulated LC3 lipidation and yet downregulated SQSTM1 level. These results provide additional evidence that vitamin D promotes autophagic initiation instead of inhibiting degradation.
The mammalian target of rapamycin (mTOR) is anevolutionarily conserved Ser/Thr protein kinase that exists in two distinct complexes, mTORC1 and mTORC2. mTORC1 functions as a convergence point for many upstream stimuli and pathways, including MEK/ERK and PI3K/AKT, to regulate autophagy as well as other cellular activities [29]. In contrast to previous reports of ERK-dependent mTORC1 activation [30,31], we showed that ERK, the well-known upstream mTORC1 activator, did not play a critical role in VDR signaling to the mTOR pathway.Rather, the vitamin D-induced mTORC1 activation is dependent on the activation of PI3K/Akt pathway by downregulation of PTEN. Our data presented in the current report are in agreement with previous studies in PTEN as the key negative regulator of the prosurvival PI3K/Akt signaling pathways [32,33]. It is reported that PTEN regulates LPS-induced TLR4 signaling and protects from endotoxic shock through a PI3K/Akt-dependent signaling. Activation of PI3K/Akt negatively regulates NF-κB and the expression of inflammatory genes in macrophages, whereas deletion of PTEN in macrophages results in diminished inflammation in response to TLR4 signaling [34]. Moreover, in IR injury models, a short-term PTEN downregulation is observed and identified as a protective physiological response to IR injury [35].
Previous studies have shown that autophagy plays a pivotal role in vitamin D-mediated innate immunity [36-38]. In our studies, we found that pretreatment with vitamin D remarkably enhanced autophagic flux and attenuated reperfusion injury in vivo and in vitro. Figure 7 depictsthat MEK/ERK and PTEN/PI3K/Akt/mTOR both critically involved in vitamin D-induced autophagy. By employing intracellular ROS and cell viability assay, we further confirmed this hypothesis with the observation that inhibitioneither of the MEK/ERK or PTEN/PI3K/Akt/mTOR pathway partly abolished the protective effect of vitamin D-induced autophagy, while inhibiting initiation of autophagy signaling pathway by knockdown of Beclin-1 completely reversed the protection provided by vitamin D.
Figure 7.
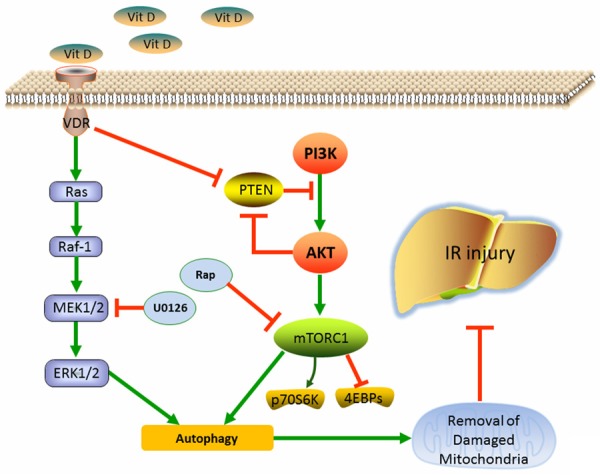
A proposed model of vitamin D-induced autophagy.
In summary, our data resulted from a hepatic IR model provide experimental evidence for the effect of antecedent vitamin D exposure on liver and hepatocytes protection against IR-induced injury, with enhanced autophagic flux observed in vivo and in vitro. Moreover, our work suggests that the protective effect is autophagy dependent, which is regulated by both MEK/ERK and PTEN/PI3K/Akt/mTOR pathway.
Acknowledgements
This study was supported by the National Nature Science Foundation of China (No. 81270550).
Disclosure of conflict of interest
None.
References
- 1.Yang J, Wang X, Song S, Liu F, Fu Z, Wang Q. Near-Term Anti-CD25 Monoclonal Antibody Administration Protects Murine Liver from Ischemia-Reperfusion Injury Due to Reduced Numbers of CD4+ T Cells. PLoS One. 2014;9:e106892. doi: 10.1371/journal.pone.0106892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fondevila C, Busuttil RW, Kupiec-Weglinski JW. Hepatic ischemia/reperfusion injury--a fresh look. Exp Mol Pathol. 2003;74:86–93. doi: 10.1016/s0014-4800(03)00008-x. [DOI] [PubMed] [Google Scholar]
- 3.Yu HC, Qin HY, He F, Wang L, Fu W, Liu D, Guo FC, Liang L, Dou KF, Han H. Canonical notch pathway protects hepatocytes from ischemia/reperfusion injury in mice by repressing reactive oxygen species production through JAK2/STAT3 signaling. Hepatology. 2011;54:979–988. doi: 10.1002/hep.24469. [DOI] [PubMed] [Google Scholar]
- 4.Tewari A, Mahendru V, Sinha A, Bilotta F. Antioxidants: The new frontier for translational research in cerebroprotection. J Anaesthesiol Clin Pharmacol. 2014;30:160–171. doi: 10.4103/0970-9185.130001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol. 2005;6:439–448. doi: 10.1038/nrm1660. [DOI] [PubMed] [Google Scholar]
- 6.Kim JS, Wang JH, Lemasters JJ. Mitochondrial permeability transition in rat hepatocytes after anoxia/reoxygenation: role of Ca2+-dependent mitochondrial formation of reactive oxygen species. Am J Physiol Gastrointest Liver Physiol. 2012;302:G723–731. doi: 10.1152/ajpgi.00082.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Czaja MJ, Ding WX, Donohue TM Jr, Friedman SL, Kim JS, Komatsu M, Lemasters JJ, Lemoine A, Lin JD, Ou JH, Perlmutter DH, Randall G, Ray RB, Tsung A, Yin XM. Functions of autophagy in normal and diseased liver. Autophagy. 2013;9:1131–1158. doi: 10.4161/auto.25063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang JH, Ahn IS, Fischer TD, Byeon JI, Dunn WA Jr, Behrns KE, Leeuwenburgh C, Kim JS. Autophagy suppresses age-dependent ischemia and reperfusion injury in livers of mice. Gastroenterology. 2011;141:2188–2199. e2186. doi: 10.1053/j.gastro.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walentowicz-Sadlecka M, Sadlecki P, Walentowicz P, Grabiec M. [The role of vitamin D in the carcinogenesis of breast and ovarian cancer] . Ginekol Pol. 2013;84:305–308. doi: 10.17772/gp/1581. [DOI] [PubMed] [Google Scholar]
- 10.Korf H, Decallonne B, Mathieu C. Vitamin D for infections. Curr Opin Endocrinol Diabetes Obes. 2014;21:431–436. doi: 10.1097/MED.0000000000000108. [DOI] [PubMed] [Google Scholar]
- 11.Yuk JM, Shin DM, Lee HM, Yang CS, Jin HS, Kim KK, Lee ZW, Lee SH, Kim JM, Jo EK. Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe. 2009;6:231–243. doi: 10.1016/j.chom.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 12.Klotz B, Mentrup B, Regensburger M, Zeck S, Schneidereit J, Schupp N, Linden C, Merz C, Ebert R, Jakob F. 1,25-dihydroxyvitamin D3 treatment delays cellular aging in human mesenchymal stem cells while maintaining their multipotent capacity. PLoS One. 2012;7:e29959. doi: 10.1371/journal.pone.0029959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mirzakhani H, Al-Garawi A, Weiss ST, Litonjua AA. Vitamin D and the development of allergic disease: how important is it? Clin Exp Allergy. 2015;45:114–25. doi: 10.1111/cea.12430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alfawaz HA, Bhat RS, Al-Ayadhi L, El-Ansary AK. Protective and restorative potency of Vitamin D on persistent biochemical autistic features induced in propionic acid-intoxicated rat pups. BMC Complement Altern Med. 2014;14:416. doi: 10.1186/1472-6882-14-416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tao Q, Wang B, Zheng Y, Jiang X, Pan Z, Ren J. Vitamin D Prevents the Intestinal Fibrosis Via Induction of Vitamin D Receptor and Inhibition of Transforming Growth Factor-Beta1/Smad3 Pathway. Dig Dis Sci. 2015;60:868–75. doi: 10.1007/s10620-014-3398-6. [DOI] [PubMed] [Google Scholar]
- 16.Yang YH, Li B, Zheng XF, Chen JW, Chen K, Jiang SD, Jiang LS. Oxidative damage to osteoblasts can be alleviated by early autophagy through the endoplasmic reticulum stress pathway--implications for the treatment of osteoporosis. Free Radic Biol Med. 2014;77:10–20. doi: 10.1016/j.freeradbiomed.2014.08.028. [DOI] [PubMed] [Google Scholar]
- 17.Elias-Miro M, Jimenez-Castro MB, Rodes J, Peralta C. Current knowledge on oxidative stress in hepatic ischemia/reperfusion. Free Radic Res. 2013;47:555–568. doi: 10.3109/10715762.2013.811721. [DOI] [PubMed] [Google Scholar]
- 18.Wang PR, Wang JS, Zhang C, Song XF, Tian N, Kong LY. Huang-Lian-Jie-Du-Decotion induced protective autophagy against the injury of cerebral ischemia/reperfusion via MAPKmTOR signaling pathway. J Ethnopharmacol. 2013;149:270–280. doi: 10.1016/j.jep.2013.06.035. [DOI] [PubMed] [Google Scholar]
- 19.Piguet AC, Dufour JF. PI(3)K/PTEN/AKT pathway. J Hepatol. 2011;54:1317–1319. doi: 10.1016/j.jhep.2010.12.013. [DOI] [PubMed] [Google Scholar]
- 20.Tong Y, Huang H, Pan H. Inhibition of MEK/ERK activation attenuates autophagy and potentiates pemetrexed-induced activity against HepG2 hepatocellular carcinoma cells. Biochem Biophys Res Commun. 2015;456:86–91. doi: 10.1016/j.bbrc.2014.11.038. [DOI] [PubMed] [Google Scholar]
- 21.Dong F, Mo Z, Eid W, Courtney KC, Zha X. Akt inhibition promotes ABCA1-mediated cholesterol efflux to ApoA-I through suppressing mTORC1. PLoS One. 2014;9:e113789. doi: 10.1371/journal.pone.0113789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andersen LB, Przybyl L, Haase N, von Versen-Hoynck F, Qadri F, Jorgensen JS, Sorensen GL, Fruekilde P, Poglitsch M, Szijarto I, Gollasch M, Peters J, Muller DN, Christesen HT, Dechend R. Vitamin d depletion aggravates hypertension and target-organ damage. J Am Heart Assoc. 2015;4 doi: 10.1161/JAHA.114.001417. pii: e001417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garcia-Alvarez M, Pineda-Tenor D, Jimenez-Sousa MA, Fernandez-Rodriguez A, Guzman-Fulgencio M, Resino S. Relationship of vitamin D status with advanced liver fibrosis and response to hepatitis C virus therapy: a metaanalysis. Hepatology. 2014;60:1541–1550. doi: 10.1002/hep.27281. [DOI] [PubMed] [Google Scholar]
- 24.Polidoro L, Properzi G, Marampon F, Gravina GL, Festuccia C, Di Cesare E, Scarsella L, Ciccarelli C, Zani BM, Ferri C. Vitamin D protects human endothelial cells from H(2) O(2) oxidant injury through the Mek/Erk-Sirt1 axis activation. J Cardiovasc Transl Res. 2013;6:221–231. doi: 10.1007/s12265-012-9436-x. [DOI] [PubMed] [Google Scholar]
- 25.Jaeschke H. Reactive oxygen and mechanisms of inflammatory liver injury: Present concepts. J Gastroenterol Hepatol. 2011;26(Suppl 1):173–179. doi: 10.1111/j.1440-1746.2010.06592.x. [DOI] [PubMed] [Google Scholar]
- 26.Vucicevic L, Misirkic-Marjanovic M, Paunovic V, Kravic-Stevovic T, Martinovic T, Ciric D, Maric N, Petricevic S, Harhaji-Trajkovic L, Bumbasirevic V, Trajkovic V. Autophagy inhibition uncovers the neurotoxic action of the antipsychotic drug olanzapine. Autophagy. 2014;10:2362–2378. doi: 10.4161/15548627.2014.984270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giron MD, Vilchez JD, Shreeram S, Salto R, Manzano M, Cabrera E, Campos N, Edens NK, Rueda R, Lopez-Pedrosa JM. beta-Hydroxy-beta-Methylbutyrate (HMB) Normalizes Dexamethasone-Induced Autophagy-Lysosomal Pathway in Skeletal Muscle. PLoS One. 2015;10:e0117520. doi: 10.1371/journal.pone.0117520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen G, Ke Z, Xu M, Liao M, Wang X, Qi Y, Zhang T, Frank JA, Bower KA, Shi X, Luo J. Autophagy is a protective response to ethanol neurotoxicity. Autophagy. 2012;8:1577–1589. doi: 10.4161/auto.21376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–131. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kong B, Wu W, Cheng T, Schlitter AM, Qian C, Bruns P, Jian Z, Jager C, Regel I, Raulefs S, Behler N, Irmler M, Beckers J, Friess H, Erkan M, Siveke JT, Tannapfel A, Hahn SA, Theis FJ, Esposito I, Kleeff J, Michalski CW. A subset of metastatic pancreatic ductal adenocarcinomas depends quantitatively on oncogenic Kras/Mek/Erk-induced hyperactive mTOR signalling. Gut. 2015 doi: 10.1136/gutjnl-2014-307616. gutjnl-2014-307616. [DOI] [PubMed] [Google Scholar]
- 31.Sun L, Li T, Wei Q, Zhang Y, Jia X, Wan Z, Han L. Upregulation of BNIP3 mediated by ERK/HIF-1alpha pathway induces autophagy and contributes to anoikis resistance of hepatocellular carcinoma cells. Future Oncol. 2014;10:1387–1398. doi: 10.2217/fon.14.70. [DOI] [PubMed] [Google Scholar]
- 32.Kamo N, Ke B, Busuttil RW, Kupiec-Weglinski JW. PTEN-mediated Akt/beta-catenin/Foxo1 signaling regulates innate immune responses in mouse liver ischemia/reperfusion injury. Hepatology. 2013;57:289–298. doi: 10.1002/hep.25958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peyrou M, Bourgoin L, Poher AL, Altirriba J, Maeder C, Caillon A, Fournier M, Montet X, Rohner-Jeanrenaud F, Foti M. Hepatic PTEN deficiency improves muscle insulin sensitivity and decreases adiposity in mice. J Hepatol. 2015;62:421–429. doi: 10.1016/j.jhep.2014.09.012. [DOI] [PubMed] [Google Scholar]
- 34.Sahin E, Haubenwallner S, Kuttke M, Kollmann I, Halfmann A, Dohnal AM, Chen L, Cheng P, Hoesel B, Einwallner E, Brunner J, Kral JB, Schrottmaier WC, Thell K, Saferding V, Bluml S, Schabbauer G. Macrophage PTEN regulates expression and secretion of arginase I modulating innate and adaptive immune responses. J Immunol. 2014;193:1717–1727. doi: 10.4049/jimmunol.1302167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zu L, Zheng X, Wang B, Parajuli N, Steenbergen C, Becker LC, Cai ZP. Ischemic preconditioning attenuates mitochondrial localization of PTEN induced by. Am J Physiol Heart Circ Physiol. 2011;300:H2177–86. doi: 10.1152/ajpheart.01138.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yao T, Ying X, Zhao Y, Yuan A, He Q, Tong H, Ding S, Liu J, Peng X, Gao E, Pu J, He B. Vitamin d receptor activation protects against myocardial reperfusion injury through inhibition of apoptosis and modulation of autophagy. Antioxid Redox Signal. 2015;22:633–650. doi: 10.1089/ars.2014.5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharma K, Goehe RW, Di X, Hicks MA 2nd, Torti SV, Torti FM, Harada H, Gewirtz DA. A novel cytostatic form of autophagy in sensitization of non-small cell lung cancer cells to radiation by vitamin D and the vitamin D analog, EB 1089. Autophagy. 2014;10:2346–2361. doi: 10.4161/15548627.2014.993283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu S, Zhang YG, Lu R, Xia Y, Zhou D, Petrof EO, Claud EC, Chen D, Chang EB, Carmeliet G, Sun J. Intestinal epithelial vitamin D receptor deletion leads to defective autophagy in colitis. Gut. 2015;64:1082–94. doi: 10.1136/gutjnl-2014-307436. [DOI] [PMC free article] [PubMed] [Google Scholar]