

Abstract
Background and Aims: The heart in diabetic state is sensitive to myocardial ischemia reperfusion (mI/R) injury. In the present study, we investigated the potential mechanisms of modulating mI/R injury in diabetic state. Methods: Diabetic db/db mice and control non-diabetic mice were administrated with mI/R injury or sham operation. Mouse atrial-derived cardiac cell line HL-1 subjected to hypoxia-reoxygenation (H/R) was used as in vitro model of I/R injury to the heart. Results: Compared with normal mice, mI/R elevated the levels of myocardial infarct size, apoptosis and TXNIP expression (in mRNA and protein) in diabetic mice. Myocardial miR-135a expression level was reduced in diabetic mice regardless of mI/R treatment or not. MiR-135a overexpression protected myocardial cells from mI/R injury in diabetic mice. In vitro, high glucose incubation contributed to a significant down-regulation of miR-135a and up-regulation of TXNIP in cells with or without H/R treatment. Luciferase reporter assay showed that TXNIP was a target gene of miR-135a. MiR-135a overexpression protected HL-1 cells from H/R injury in high glucose condition, while this effect was reversed by up-regulated TXNIP. Conclusion: miR-135a protects against mI/R injury by decreasing TXNIP expression in diabetic state.
Keywords: Myocardial ischemia/reperfusion injury, diabetes, miR-135a, TXNIP, myocardial infarction
Introduction
Ischemic heart disease is one of the leading cause of death worldwide. Although blood reperfusion is required for restoring myocardial oxygen supply and improving function of ischemic myocardium, it will contribute to a more severe myocardial injury after reperfusion than pure ischemia, which termed as myocardial ischemia/reperfusion (mI/R) injury [1]. Diabetes is a high risk factor of congestive heart failure and ischemic heart disease [2,3]. The mortality in myocardial infarction patients with diabetesis three to four times as high as in nondiabetic patients [4-6]. In addition, the incidence of re-infarction in myocardial infarction patients with diabetes is also increased significantly. Large body of evidences have demonstrated that diabetic heart is more sensitive to mI/R injury [7,8]. Therefore, the potential mechanisms of mI/R injury under the diabetic status are significant to enable us to develop the new therapeutic strategy for preventing ischemic heart disease.
MicroRNAs (miRNAs), a type of small non-coding RNA molecule containing about 22 nucleotides, are implicated in negative regulation of gene expression in various physiological processes through binding 3’untranslated regions (3’UTR) of target genes [9-11]. Emerging studies have indicated the role of miRNAs in various cardiovascular disease, including ischemic heart disease [11,12]. Some miRNAs such as miR-34a, miR-155, miR-153, miR-143 and miR-135a have been reported to be a regulator of cardiac repair or myocardial injury [13-17]. It has shown that miRNAs contribute to mI/R injury through modulating several key signaling pathways in cell survival, cell cycle and cell apoptosis [13,18]. However, thestudies of mI/R injury in diabetic model is limited. Moreover, Type 1 diabetic model is commonly used in the existing researches. It is necessary to investigate the mechanisms of I/R injury in the setting of Type 2 diabetes mellitus (T2DM), which encompasses roughly 90% of diabetic patients.
Thioredoxin-interacting protein (TXNIP), also known as thioredoxin binding protein-2 or vitamin D3-up-regulated protein-1, is a regulator of cellular redox state and associated with cell apoptosis [19]. The experiments have indicated that high glucose up-regulates TXNIP expression levels, and TXNIP down-regulation functions as alleviating mI/R injury in rats exposed to high glucose via reducing intracellular oxidative stress [20,21]. Therefore, the concept of TXNIP in protecting myocardial cells is widely established. However, the relationship between miRNAs and TXNIP in mI/R injury in diabetic status remains to belargely elucidated.
In the present study, we firstly investigated the expression levels of some miRNAs in mI/R injury in diabetic mice,and the results demonstrated that only miR-135a was significantly up-regulated. TXNIP was considered as a potential target molecule of miR-135a, and the mechanism underlying myocardial ischemia-reperfusion injury in diabetic status was illustrated.
Materials and methods
Animals
Adult male diabetic C57BL/KsJdb/db mice and corresponding back-ground nondiabeticdb/m mice at 12 weeks of age, purchased from Nanjing Peng Sheng Biological Technology Development Co., Ltd, were used to establish mI/R injury model. The animal-related study was approved by Institutional AnimalCare and Use Committee of the Anhui Provincial Hospital affiliated to Anhui Medical University. The animal procedures were performed with the Guide for the Care and Use of Laboratory Animals.
Myocardial ischemia/reperfusion injury in mice
Mice were anesthetized with 5% isoflurane intubation. The adequacy of anesthesia and body temperature were monitored during surgery. The surgery was described previously [22]. In brief, the left coronary artery (LCA) was ligated and induced myocardial ischemia. In sham operated mice, the suture was placed beneath the LCA without ligating. After 30 min of myocardial ischemia, releasing the ligature to initiate the reperfusion, and reperfusion was last for 3 h. Then, the myocardial infarction area was determined. Mice were anesthetized and sacrificed by cervical dislocation.
Myocardial infarction area detection
After 3 h of reperfusion, 4% Evans’ blue dye was injected into cardiac cavity via the thoracic aorta in a retrogradefashion. Ischemia region (area at risk, AAR) could not be dyed, and the non-ischemia regionwas dyed into blue. The heart was immediately explanted, and washed with normal saline. Left ventricle was then sliced into 1 to 2 mm cross sections, which were stained with 2% triphenyltetrazolium-chloride (TCC) at 37°C for 20 min. The viable tissue was stained in red and infarction tissue was not stained. The sections were observed and pictured using microscope. Infarct size (IS) was measured by Image-Pro Plus image analysis software 4.1 and calculated as a percentage of the AAR in total left ventricle.
Cell culture
Mouse atrial-derived cardiac cell line HL-1 was cultured in culture plates, with coating gelatin/fibronectin overnight at 37°C, and maintained in Claycomb medium(Sigma, USA) containing 10% fetal bovine serum, 1% penicillin/streptomycin, 0.1 mM norepinephrine and 2 mM L-glutamine at 37°C in a humid atmosphere of 5% CO2. Cells were incubated with 5.5 mM (normal glucose) or 30 mM glucose (high glucose). Checked the cell status daily and replaced the medium every two days. HL-1 cells were transfected with miR-135a mimic, miR-135a inhibitor, pcDNA-TXNIP, negative control (Pre-NC) of miR-135a mimic, negative control (NC) of miR-135a inhibitor or empty pcDNA (Guangzhou Ribobio Co., LTD, China) using Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer’s protocol.
Hypoxia/reoxygenation in cells
HL-1 cell line was subjected to hypoxia-reperfusion (H/R) as an in vitro model of I/R injury to the heart. The H/R cells were incubated in a humid atmosphere of 0.5% O2 for 15 h, and then treated with normal air atmosphere for another 6 h.
TUNEL assay
Mouse cardiac tissues were fixed with formaldehyde for 24 h, and then routinely paraffin-embedded and sectioned. HL-1 cells were fixed with 4% fresh paraformaldehyde for 30 min at room temperature. Then, the apoptosis of cardiac myocytes from mice and HL-1 cells were measured by Colorimetric TUNEL Apoptosis Assay Kit (Beyotime, China) according to the instructions. Fluorescence microscope showed that the nuclear of normal myocardial cells was dyed in blue, and apoptotic cell was dyed in green (TUNEL positive cells). The TUNEL positive cells were pictured in six fields/section. The average TUNEL positive cell was calculated.
Real-time PCR
Total RNA was isolated from mouse cardiac tissues and HL-1 cells with TRIzol reagent (Invitrogen, USA), and then utilized to synthesize complementary DNA (cDNA) using reverse transcriptional kit (Takara, China) according to the manufacturer’s protocol. The expression levels of miRNAs and TXNIP were quantified by fluorescent quantitative real-time PCR with SYBR® Green PCR Master Mix (Applied Biosystems, USA) on ABIPRISM-7900 Sequence Detection System (Applied Biosystems, USA). An initial denaturation at 94°C for 10 min was followedby 45 cycles of denaturation at 94°C for 15 s, anneal at 60°C for 20 s, and extension at 72°C for 15 s. The relative expression of miRNAs and mRNA wascalculated with 2(-ΔΔCt) methods, with the levels of U6 and β-Actin acting as controls, respectivly.
Western blot
The myocardial tissuesof mice were harvested andwashed with ice-cold normal saline to homogenize. After removing the supernatant, the homogenate was used to abstract the total protein. RIPA lysis buffer was employed to obtain protein from tissue homogenate and HL-1 cells. Protein concentration was determined using a BCA Protein Assay Kit (Beyotime, China). Equal amounts of protein were separated by 10% SDS-PAGE, transferred to PVDF membranes and blocked in 5% nonfat milk. The primary antibodies against TXNIP (1:1000, Abcam, USA) and β-Actin (1:1000, Abcam, USA) were then incubated with membranes at 4°C overnight. After being washed with TBST three times, horseradish peroxidase-conjugated secondary antibody was then incubated for 1 h at room temperature. The bands were visualized using the enhanced chemiluminescent (ECL) substrate, with β-Actin severing as control.
Luciferase reporter assay
HL-1 cells were cultured in a 24-well plate at the concentration of 1×104 cells/well. Fragments of the 3’UTR of TXNIP containing potential miR-135a binding sites were amplified by PCR method. The amplified product was cloned into psiCHECK-2 vector. Psi-TXNIP or empty vector was co-transfected with pre-miR-135a mimic/inhibitor or negative control (pre-NC or NC) into cells using Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer’s protocol. After 24 h of transfection, the luciferase activity was detected with a Dual-LuciferaseReporter Assay system (Promega, USA), and fireflyluciferase was used to normalize the Renilla luciferase.
Detection of reactive oxygen species
The reactive oxygen species (ROS) level of myocardial cells was determined by Reactive Oxygen Species Assay Kit (Beyotime, China) containing 2’,7’-dichlorofluorescin diacetate (DCFH-DA), which can freely pass through the cell membrane. Non-fluorescent molecule DCFH-DA can be oxidized by ROS to fluorescent 2’,7’-dichlorofluorescin (DCF). The intracellular ROS level can be reflected by the fluorescence intensity of DCF.
Statistical analysis
All analysis were conducted using SPSS17.0. Data were represented as mean ± SEM. Differences in data between the groups were compared with independent-samples T test. A P value less than 0.05 was considered statistical significant.
Results
Diabetes aggravates the myocardial ischemia/reperfusion injury
Both nondiabetic and diabeticmice were subjected to 30 min of LCA ischemia followed by reperfusion, and the myocardial functions were evaluated at 3 h of reperfusion. As shown in Figure 1, the infarct size and apoptosis of myocardial cells were significantly elevated by mI/R in diabetic mice compared with nondiabetic ones (P<0.01).
Figure 1.
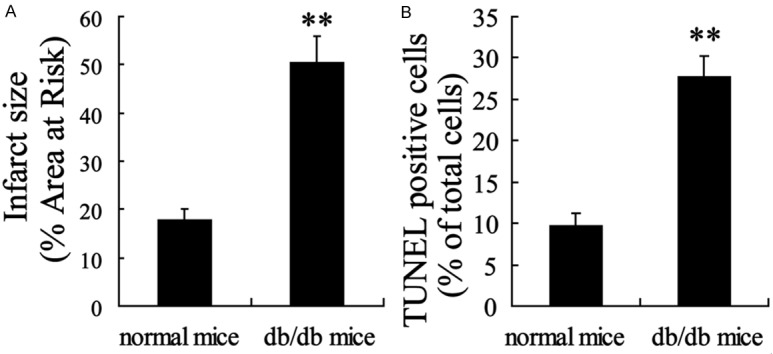
Diabetic state promoted myocardial injury in myocardial ischemia/reperfusion (mI/R) mice. A: Infarct size was measured by TCC staining. B: The apoptosis of myocardial cells was detected with TUNEL assay. Values are expressed as means ± SEM. Ten mice were investigated in each group. **P<0.01 vs. normal mice.
TXNIP expression is up-regulated in myocardialischemia/reperfusion injury
TXNIP has been reported to link hyperglycemia and mI/R injury [20]. Therefore, we evaluated its expression in native mice with sham operation and then investigated in mI/R mice. Compared with Sham group, myocardial TXNIP expression in mRNA and protein levels was dramatically up-regulated in I/R mice (Figure 2). Moreover, the effect was more obvious in db/db mice.
Figure 2.
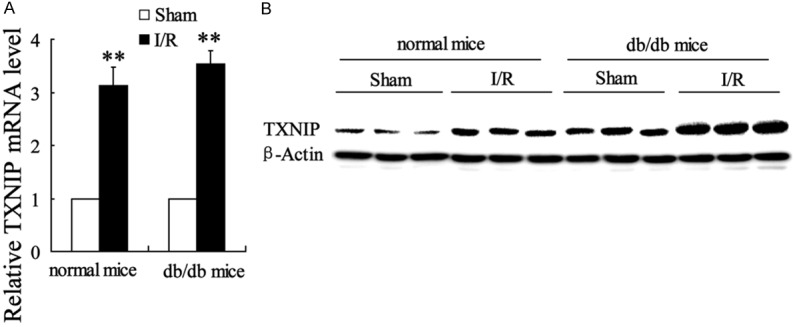
TXNIP expression in mRNA and protein levels was up-regulated by myocardial ischemiareperfusion (mI/R) injury both in normal and db/db mice. A: The relative TXNIP mRNA expression level was quantified with real-time PCR. B: The TXNIP protein level was detected by western blot. Values are expressed as means ± SEM. Ten mice were investigated in each group. **P<0.01 vs. mice with sham operation.
miR-135 expression is down-regulated in myocardial ischemia/reperfusion injury in vivo
Then, the expression of miRNAs including miR-34a, miR-155, miR-153, miR-143 and miR-135a was detected in myocardial cells. In normal or db/db mice, mI/R had no significant effect on the expression of miR-34a, miR-155, miR-153 and miR-143 levels. However, miR-135a expression was markedly decreased by mI/R (Figure 3A and 3B). In addition, compared with normal mice, diabetic mice had the reduction of miR-135a level regardless of mI/R treatment or not (Figure 3C and 3D).
Figure 3.
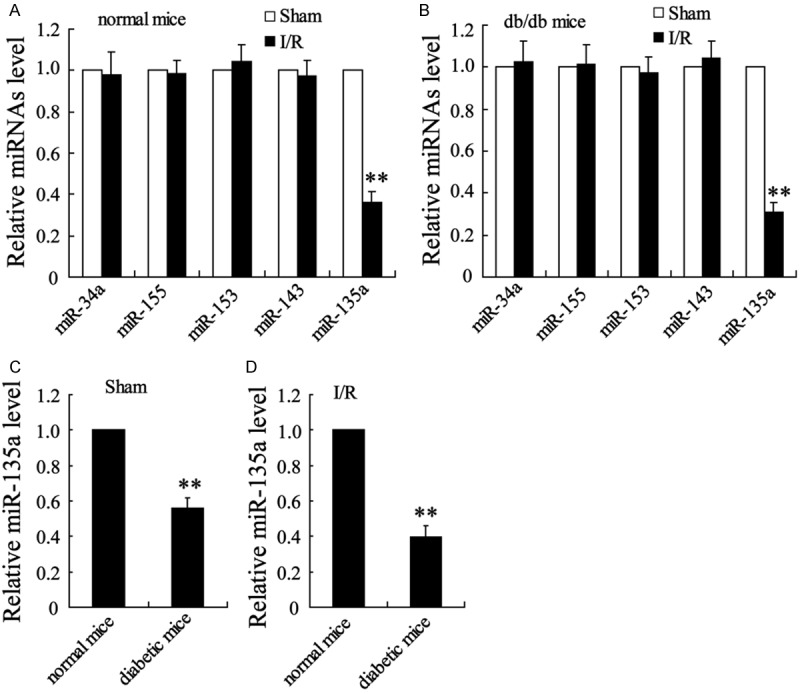
diabetic state aggravatedthe down-regulation of miR-135a inmyocardialischemia/reperfusion (mI/R) injured mice. (A) miRNAs expression was detected with real-time PCR in normal mice with or without the treatment of I/R. (B) miRNAs expression in db/db mice with or without the treatment of I/R. (C) miR-135a expression in normal and db/db mice with sham operation. (D) miR-135a expression in normal and db/db mice with I/R treatment. Values are expressed as means ± SEM. Ten mice were investigated in each group. **P<0.01 vs. Sham group in (A and B) or normal mice in (C and D).
The effects ofhypoxia/reoxygenationon expression of TXNIP and miR-135a in HL-1 cells exposed to high glucose
HL-1 cell line was subjected to H/R as in vitro model of I/R injury to the heart. The TXNIP and miR-135a expression was evaluated in this model. As shown in Figure 4A and 4B, the H/R cells had a decrease of miR-135a expression and an increase of TXNIP mRNA and protein expression both in cells exposed to 5.5 mM and 30 mM glucose. In the other hand, high glucose contributed to a more significant down-regulation of miR-135a and up-regulation of TXNIP regardless of the treatment of H/R or not (Figure 4C and 4D).
Figure 4.
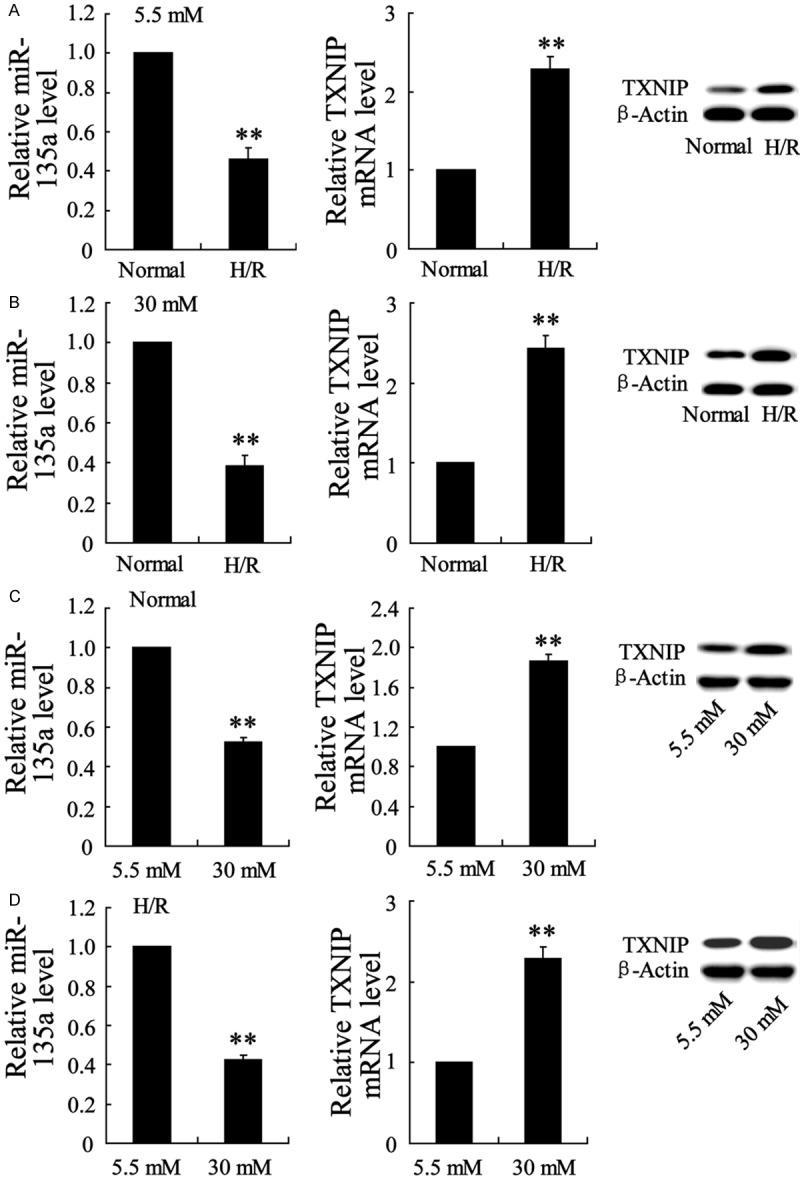
High glucosepromoted the down-regulation of miR-135a and up-regulation of TXNIP in HL-1 cellsexposed to hypoxia/reoxygenation (H/R). (A) The miR-135a and TXNIP expression levels were detected by Real-time PCR and western blot in H/R injured HL-1 cells exposed to normal glucose (5.5 mM). (B) The miR-135a and TXNIP expression levels in H/R injured HL-1 cells exposed to high glucose (30 mM). (C) The miR-135a and TXNIP expression levels in normal HL-1 cells exposed to 5.5 mM or 30 mM glucose. (D) The miR-135a and TXNIP expression levels in H/R injured HL-1 cells exposed to 5.5 mM or 30 mM glucose. Values are expressed as means ± SEM. Each experiment was repeated three times. **P<0.01 vs. Normal group in (A and B) or 5.5 mM group in (C and D).
TXNIP is a potential target gene of miR-135a
We then investigated the relationship between miR-135a and TXNIP. Luciferase reporter assay demonstrated that co-transfection of miR-135a mimic decreased the TXNIP expression and its 3’UTR activity (Figure 5A), while miR-135a inhibitor increased these levels (Figure 5B), which suggesting TXNIP was a potential target gene of miR-135a.
Figure 5.
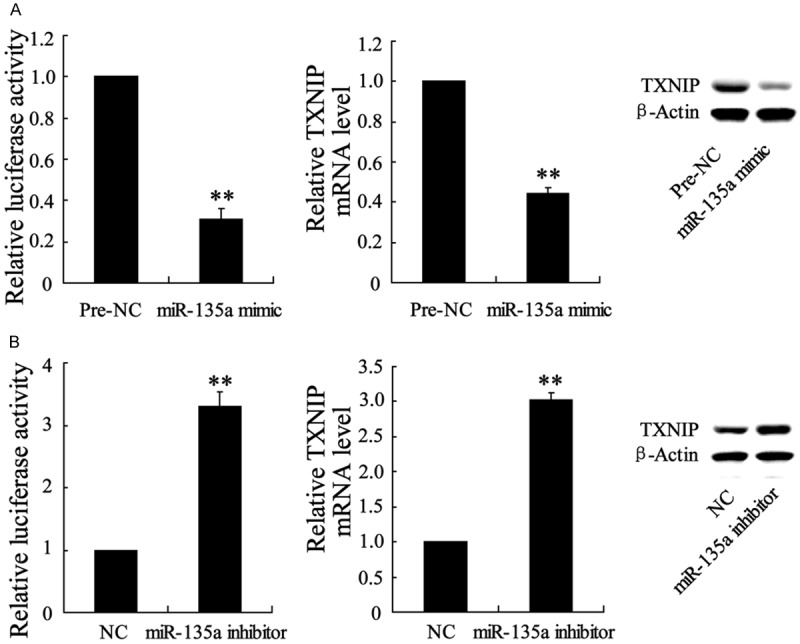
Alteration of miR-135a expression led to the regulation in 3’UTR activity of TXNIP in HL-1 cells. (A) Co-transfected with psiCHECK-2-TXNIP vector and miR-135a mimic (or its negative control-Pre-NC) into cells, 3’UTR activity of TXNIPwas detected using luciferase reporter assay. (B) Co-transfected with psiCHECK-2-TXNIP vector and miR-135a inhibitor (or its negative control-NC) into cells, 3’UTR activity of TXNIP was measured. Values are expressed as means ± SEM. Each experiment was repeated three times. **P<0.01 vs. Pre-NC group in (A) or NC group in (B).
miR-135a overexpression protects against hypoxia/reoxygenation injury in HL-1 cells exposed to high glucose
The protective effects of miR-135a on H/R injured myocardialcells were illustrated. As shown in Figure 6, H/R led to the increased levels of ROS production and apoptosis in HL-1 cells exposed to 5.5 mM or 30 mM glucose. Transfected cells with miR-135a mimic to up-regulate its expression. The results showed that miR-135a overexpression protected HL-1 cells against H/R induced increase of ROS and apoptosis in cells exposed to high glucose but not normal glucose. Moreover, up-regulated TXNIP reversed the protective effects of miR-135a on H/R injured HL-1 cells.
Figure 6.
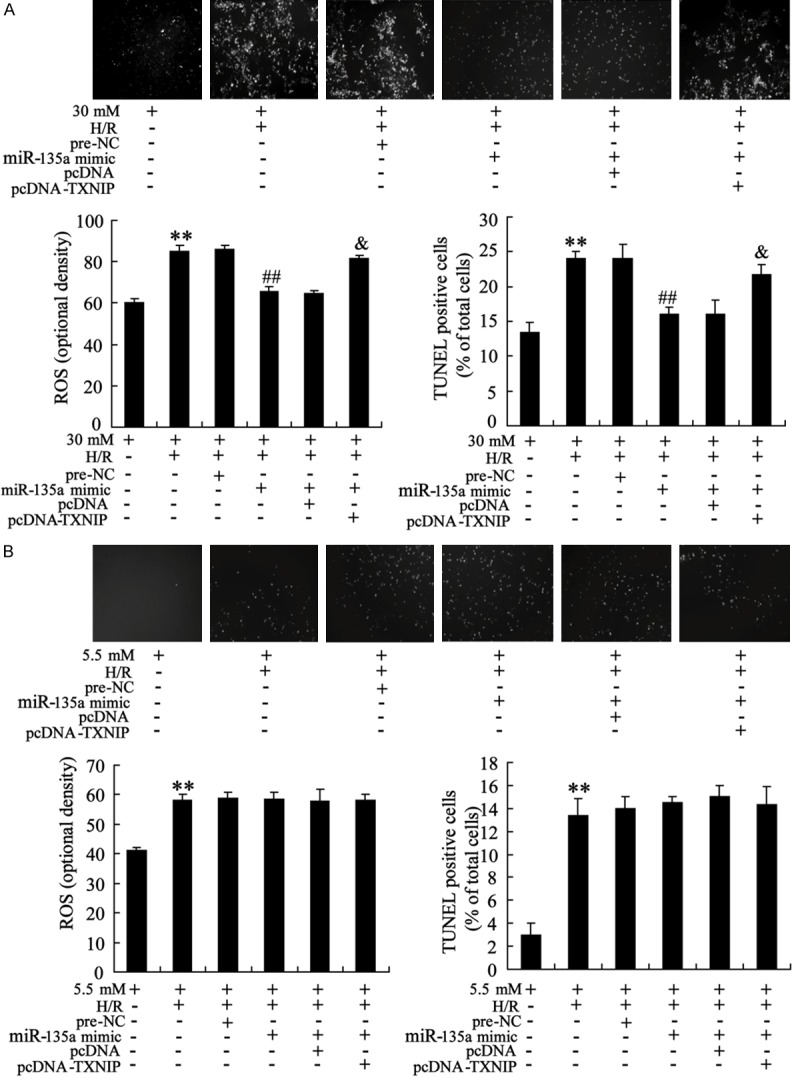
miR-135a overexpression protects against hypoxia/reoxygenation (H/R) induced increase of ROS production and apoptosis levels in HL-1 cells exposed to high glucose. A: HL-1 cells with the treatment of H/R were cultured in 30 mM glucose. MiR-135a mimic (or pre-NC) and pcDNA-TXNIP (or pcDNA) were transfected into cells for 24 h. The intracellular ROS level and cell apoptosis were measured by DCFH-DA method and TUNEL assay, respectively. B: HL-1 cells with the treatment of H/R were cultured in 5.5 mM glucose. The intracellular ROS level and cell apoptosis were detected in HL-1 cells transfected with miR-135a mimic (or pre-NC) and pcDNA-TXNIP (or pcDNA). Values are expressed as means ± SEM. Each experiment was repeated three times. **P<0.01 vs. cells exposed to 5 mM or 30 mM glucose. ##P<0.01 vs. cells treated with H/R and pre-NC. &P<0.05 vs. cells treated with H/R, miR-135a mimic and pcDNA.
miR-135a overexpression protects myocardial cells from ischemia/reperfusion injury in diabetic mice
Toconfirm whether miR-135a has a myocardial protective role in the process of mI/R in vivo, miR-135a mimic was injected into mice. The myocardial infarct size and apoptosis were significantly elevated in mI/R, while injection of miR-135a mimic could prominently reduce these levels (Figure 7A and 7B).
Figure 7.
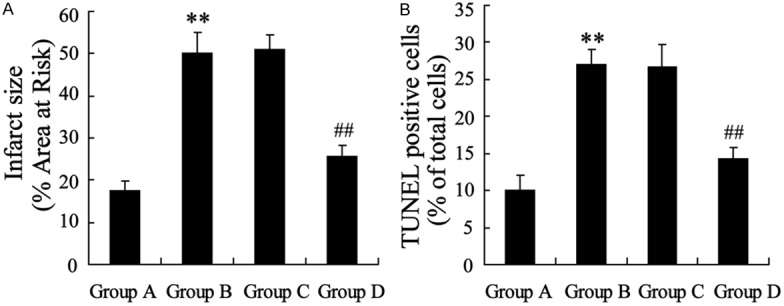
miR-135a overexpression reduced myocardial infarct size and apoptosis in myocardial ischemia/reperfusion (mI/R) diabetic mice. Group A: diabetic mice with sham operation. Group B: diabetic mice with I/R injury. Group C: mI/R injured diabetic mice were injected with pre-NC. Group D: mI/R injured diabetic mice were administrated with miR-135a mimic by tail intravenous injection. A: Infarct size was detected with TCC staining. B: Myocardial apoptosis was measured by TUNEL assay. Values are expressed as means ± SEM. Ten mice were in each group. **P<0.01 vs. Group A. ##P<0.01 vs. Group C.
Discussion
Diabetes not only drives myocardial vulnerability to infarction but also abolishes the effectiveness ofcardioprotective interventions [23]. In the present study, mI/R injury resulted in the increased infarct size and apoptosis of myocardial cells in type 2 diabetic obesedb/db mice compared with in control db/m mice, suggesting heart in the setting of a diabetic state is more sensitive to mI/R injury, which is in consistent with previous experimental and clinical studies [8,24,25]. We also found that miR-135a was down-regulated and targeted TXNIP inmI/R injured myocardial cells, which was a potential mechanism fordeveloping the therapeutic strategies.
MiRNAs have been considered to be an important regulator in various diseases such as cancers, immunological disorders and cardiac-cerebral vascular disease [9,12,26]. Some miRNAs have been reported in diabetes and I/R injury. Boon et al found suppression of miR-34a participates in decreasing cell death and fibrosis in acute myocardial infarction by inducing DNA damage responses andtelomere attrition [27]. The recent study showed that miR-155 promotesI/Rinjury by the alterations in recruitment of inflammatory cell and respiratory oxidative burst ininflammatory sequelae of I/R injury [28]. MiR-153 and miR-143 have been also demonstrated to play the role in the modulation of VSMC proliferation andcardiac chamber morphogenesis [15,16], which are potential molecules related to myocardial injury. MiR-135 is identified as a critical regulator ofmyogenesis, and the increase of miR-135 expression is observed in thediabetic gastrocnemius skeletal muscle [17]. The role of miR-135 in diabetic state is proposed to be mediated through inhibiting insulin signaling and glucose uptake by targeting IRS2. Here, we determined the expression of miR-34a, miR-155, miR-153, miR-143 and miR-135a levels in mI/R injury in vivo. Real-time PCR results showed that only miR-135a was significantly down-regulated by mI/R injury both in diabetic andnondiabetic mice. In vitro, HL-1 cell line was subjected to H/R asmodel of I/R injury to the heart. A decrease of miR-135a expression was also observed in myocardialH/R cells. Moreover, miR-135a overexpression protected against H/R induced ROS and apoptosis increase only in cells incubated with high glucose but not normal glucose. Therefore, we speculated that miR-135a down-regulation might contribute partly to the mI/R injury in the diabetic state. In diabetic db/db mice, we confirmed that the injection of miR-135a mimic could prominently reduce the myocardial infarct size and apoptosis following mI/R injury. Additionally, the previous studies havealso indicated that miR-135a plays an important role in renal fibrosis [29], proliferation of cancer cells [30], and hypoxia-mediated expression of 5-lipoxygenase-activating protein [31].
TXNIP is not only a regulator of cellular redox state, but also a critical modulator in cellular metabolism including glucose uptake/homeostasis [32,33]. Research has shown that TXNIP is implicated in insulin transcription [34], glycolytic activity [32] andbeta-cell apoptosis [35], which links to the pathophysiologic process of diabetes. In the other hand, TXNIP acts as a novel mechanism in mI/R injury by mediating the activation NLRP3 inflammasome in ischemic heart [21]. Su et al found that hyperglycemiapromotes the TXNIP expression and leads to the aggravation of cardiac injury following mI/R injury possibly through modulating p38 MAPK and Akt activation [20]. However, the exact mechanism of TXNIP alterations in mI/R injury remains unclear. In the present study, we demonstrated that TXNIP was a target gene of miR-135a. MiR-135a mimic decreased the TXNIP expression and its 3’UTR activity in HL-1 cells, while miR-135a inhibitor had the reverse result. Even more important, TXNIP upregulation reversed the protective effects of miR-135a on H/L injured HL-1 cells in high glucose condition. Therefore, it is proposed that miR-135a functions as protective effects on myocardial cells against mI/R injury possibly by mediating TXNIP expression.
Taken all together, the main findings of this study have demonstrated that diabetic state aggravated myocardial infarct following mI/R injury, and miR-135a down-regulation modulating TXNIP expression might contribute to this process. This study sheds new light onthe underlying mechanisms by which high glucose condition promotes mI/R injury, highlighting the significance of blood glucose control on cardiovascular disease.
Acknowledgements
This work was funded by National Natural Science Foundation of China (81200142).
Disclosure of conflict of interest
None.
References
- 1.Moens AL, Claeys MJ, Timmermans JP, Vrints CJ. Myocardial ischemia/reperfusion-injury, a clinical view on a complex pathophysiological process. Int J Cardiol. 2005;100:179–190. doi: 10.1016/j.ijcard.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 2.Nichols GA, Gullion CM, Koro CE, Ephross SA, Brown JB. The incidence of congestive heart failure in type 2 diabetes: an update. Diabetes Care. 2004;27:1879–1884. doi: 10.2337/diacare.27.8.1879. [DOI] [PubMed] [Google Scholar]
- 3.Almdal T, Scharling H, Jensen JS, Vestergaard H. The independent effect of type 2 diabetes mellitus on ischemic heart disease, stroke, and death: a population-based study of 13 000 men and women with 20 years of follow-up. Arch Intern Med. 2004;164:1422–1426. doi: 10.1001/archinte.164.13.1422. [DOI] [PubMed] [Google Scholar]
- 4.Partamian JO, Bradley RF. Acute myocardial infarction in 258 cases of diabetes. Immediate mortality and five-year survival. N Engl J Med. 1965;273:455–461. doi: 10.1056/NEJM196508262730901. [DOI] [PubMed] [Google Scholar]
- 5.Barthelemy O, Le Feuvre C, Timsit J. Silent myocardial ischemia screening in patients with diabetes mellitus. Arq Bras Endocrinol Metabol. 2007;51:285–293. doi: 10.1590/s0004-27302007000200018. [DOI] [PubMed] [Google Scholar]
- 6.Kapur A, De Palma R. Mortality after myocardial infarction in patients with diabetes mellitus. Heart. 2007;93:1504–1506. doi: 10.1136/hrt.2006.112656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aasum E, Hafstad AD, Severson DL, Larsen TS. Age-dependent changes in metabolism, contractile function, and ischemic sensitivity in hearts from db/db mice. Diabetes. 2003;52:434–441. doi: 10.2337/diabetes.52.2.434. [DOI] [PubMed] [Google Scholar]
- 8.Devereux RB, Roman MJ, Paranicas M, O’Grady MJ, Lee ET, Welty TK, Fabsitz RR, Robbins D, Rhoades ER, Howard BV. Impact of diabetes on cardiac structure and function: the strong heart study. Circulation. 2000;101:2271–2276. doi: 10.1161/01.cir.101.19.2271. [DOI] [PubMed] [Google Scholar]
- 9.Lin S, Gregory RI. MicroRNA biogenesis pathways in cancer. Nat Rev Cancer. 2015;15:321–333. doi: 10.1038/nrc3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sonkoly E, Pivarcsi A. microRNAs in inflammation. Int Rev Immunol. 2009;28:535–561. doi: 10.3109/08830180903208303. [DOI] [PubMed] [Google Scholar]
- 11.van Rooij E, Olson EN. MicroRNA therapeutics for cardiovascular disease: opportunities and obstacles. Nat Rev Drug Discov. 2012;11:860–872. doi: 10.1038/nrd3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quiat D, Olson EN. MicroRNAs in cardiovascular disease: from pathogenesis to prevention and treatment. J Clin Invest. 2013;123:11–18. doi: 10.1172/JCI62876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Y, Xing Y, Liang C, Hu L, Xu F, Chen Y. Crucial microRNAs and genes of human primary breast cancer explored by microRNA-mRNA integrated analysis. Tumour Biol. 2015;36:5571–5579. doi: 10.1007/s13277-015-3227-3. [DOI] [PubMed] [Google Scholar]
- 14.Corsten MF, Papageorgiou A, Verhesen W, Carai P, Lindow M, Obad S, Summer G, Coort SL, Hazebroek M, van Leeuwen R, Gijbels MJ, Wijnands E, Biessen EA, De Winther MP, Stassen FR, Carmeliet P, Kauppinen S, Schroen B, Heymans S. MicroRNA profiling identifies microRNA-155 as an adverse mediator of cardiac injury and dysfunction during acute viral myocarditis. Circ Res. 2012;111:415–425. doi: 10.1161/CIRCRESAHA.112.267443. [DOI] [PubMed] [Google Scholar]
- 15.Song L, Duan P, Guo P, Li D, Li S, Xu Y, Zhou Q. Downregulation of miR-223 and miR-153 mediates mechanical stretch-stimulated proliferation of venous smooth muscle cells via activation of the insulin-like growth factor-1 receptor. Arch Biochem Biophys. 2012;528:204–211. doi: 10.1016/j.abb.2012.08.015. [DOI] [PubMed] [Google Scholar]
- 16.Deacon DC, Nevis KR, Cashman TJ, Zhou Y, Zhao L, Washko D, Guner-Ataman B, Burns CG, Burns CE. The miR-143-adducin3 pathway is essential for cardiac chamber morphogenesis. Development. 2010;137:1887–1896. doi: 10.1242/dev.050526. [DOI] [PubMed] [Google Scholar]
- 17.Agarwal P, Srivastava R, Srivastava AK, Ali S, Datta M. miR-135a targets IRS2 and regulates insulin signaling and glucose uptake in the diabetic gastrocnemius skeletal muscle. Biochim Biophys Acta. 2013;1832:1294–1303. doi: 10.1016/j.bbadis.2013.03.021. [DOI] [PubMed] [Google Scholar]
- 18.Ye Y, Perez-Polo JR, Qian J, Birnbaum Y. The role of microRNA in modulating myocardial ischemia-reperfusion injury. Physiol Genomics. 2011;43:534–542. doi: 10.1152/physiolgenomics.00130.2010. [DOI] [PubMed] [Google Scholar]
- 19.Junn E, Han SH, Im JY, Yang Y, Cho EW, Um HD, Kim DK, Lee KW, Han PL, Rhee SG, Choi I. Vitamin D3 up-regulated protein 1 mediates oxidative stress via suppressing the thioredoxin function. J Immunol. 2000;164:6287–6295. doi: 10.4049/jimmunol.164.12.6287. [DOI] [PubMed] [Google Scholar]
- 20.Su H, Ji L, Xing W, Zhang W, Zhou H, Qian X, Wang X, Gao F, Sun X, Zhang H. Acute hyperglycaemia enhances oxidative stress and aggravates myocardial ischaemia/reperfusion injury: role of thioredoxin-interacting protein. J Cell Mol Med. 2013;17:181–191. doi: 10.1111/j.1582-4934.2012.01661.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y, Lian K, Zhang L, Wang R, Yi F, Gao C, Xin C, Zhu D, Li Y, Yan W, Xiong L, Gao E, Wang H, Tao L. TXNIP mediates NLRP3 inflammasome activation in cardiac microvascular endothelial cells as a novel mechanism in myocardial ischemia/reperfusion injury. Basic Res Cardiol. 2014;109:415. doi: 10.1007/s00395-014-0415-z. [DOI] [PubMed] [Google Scholar]
- 22.Arslan F, Lai RC, Smeets MB, Akeroyd L, Choo A, Aguor EN, Timmers L, van Rijen HV, Doevendans PA, Pasterkamp G, Lim SK, de Kleijn DP. Mesenchymal stem cell-derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res. 2013;10:301–312. doi: 10.1016/j.scr.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 23.Przyklenk K, Maynard M, Greiner DL, Whittaker P. Cardioprotection with postconditioning: loss of efficacy in murine models of type-2 and type-1 diabetes. Antioxid Redox Signal. 2011;14:781–790. doi: 10.1089/ars.2010.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H, Liu Z, Wang J, Wong GT, Cheung CW, Zhang L, Chen C, Xia Z, Irwin MG. Susceptibility to myocardial ischemia reperfusion injury at early stage of type 1 diabetes in rats. Cardiovasc Diabetol. 2013;12:133. doi: 10.1186/1475-2840-12-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Engbersen R, Riksen NP, Mol MJ, Bravenboer B, Boerman OC, Meijer P, Oyen WJ, Tack C, Rongen GA, Smits P. Improved resistance to ischemia and reperfusion, but impaired protection by ischemic preconditioning in patients with type 1 diabetes mellitus: a pilot study. Cardiovasc Diabetol. 2012;11:124. doi: 10.1186/1475-2840-11-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pauley KM, Chan EK. MicroRNAs and their emerging roles in immunology. Ann N Y Acad Sci. 2008;1143:226–239. doi: 10.1196/annals.1443.009. [DOI] [PubMed] [Google Scholar]
- 27.Boon RA, Iekushi K, Lechner S, Seeger T, Fischer A, Heydt S, Kaluza D, Treguer K, Carmona G, Bonauer A, Horrevoets AJ, Didier N, Girmatsion Z, Biliczki P, Ehrlich JR, Katus HA, Muller OJ, Potente M, Zeiher AM, Hermeking H, Dimmeler S. MicroRNA-34a regulates cardiac ageing and function. Nature. 2013;495:107–110. doi: 10.1038/nature11919. [DOI] [PubMed] [Google Scholar]
- 28.Eisenhardt SU, Weiss JB, Smolka C, Maxeiner J, Pankratz F, Bemtgen X, Kustermann M, Thiele JR, Schmidt Y, Bjoern Stark G, Moser M, Bode C, Grundmann S. MicroRNA-155 aggravates ischemia-reperfusion injury by modulation of inflammatory cell recruitment and the respiratory oxidative burst. Basic Res Cardiol. 2015;110:32. doi: 10.1007/s00395-015-0490-9. [DOI] [PubMed] [Google Scholar]
- 29.He F, Peng F, Xia X, Zhao C, Luo Q, Guan W, Li Z, Yu X, Huang F. MiR-135a promotes renal fibrosis in diabetic nephropathy by regulating TRPC1. Diabetologia. 2014;57:1726–1736. doi: 10.1007/s00125-014-3282-0. [DOI] [PubMed] [Google Scholar]
- 30.Mao XP, Zhang LS, Huang B, Zhou SY, Liao J, Chen LW, Qiu SP, Chen JX. Mir-135a enhances cellular proliferation through post-transcriptionally regulating PHLPP2 and FOXO1 in human bladder cancer. J Transl Med. 2015;13:86. doi: 10.1186/s12967-015-0438-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gonsalves CS, Kalra VK. Hypoxia-mediated expression of 5-lipoxygenase-activating protein involves HIF-1alpha and NF-kappaB and microRNAs 135a and 199a-5p. J Immunol. 2010;184:3878–3888. doi: 10.4049/jimmunol.0902594. [DOI] [PubMed] [Google Scholar]
- 32.Yu FX, Chai TF, He H, Hagen T, Luo Y. Thioredoxin-interacting protein (Txnip) gene expression: sensing oxidative phosphorylation status and glycolytic rate. J Biol Chem. 2010;285:25822–25830. doi: 10.1074/jbc.M110.108290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perrone L, Devi TS, Hosoya KI, Terasaki T, Singh LP. Inhibition of TXNIP expression in vivo blocks early pathologies of diabetic retinopathy. Cell Death Dis. 2010;1:e65. doi: 10.1038/cddis.2010.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu G, Chen J, Jing G, Shalev A. Thioredoxininteracting protein regulates insulin transcription through microRNA-204. Nat Med. 2013;19:1141–1146. doi: 10.1038/nm.3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen J, Saxena G, Mungrue IN, Lusis AJ, Shalev A. Thioredoxin-interacting protein: a critical link between glucose toxicity and betacell apoptosis. Diabetes. 2008;57:938–944. doi: 10.2337/db07-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]