

Abstract
The vascular smooth muscle cell (VSMC) phenotypic switch is considered to be the key pathophysiological change in various cardiovascular diseases, such as aortic dissection, atherosclerosis, and hypertension. The results in this study showed that TGF-β1 promotes the proliferation, migration and morphological changes of VSMC.TGF-β1 promoted the expressions of PI3K, P-PI3K, AKT, P-AKT, ID2, and OPN protein and suppressed the expressions of α-SMA and SM22α protein; the opposite results were observed for TGF-β1 inhibitor group, AKT inhibitor group and Combined inhibitors group. After the stimulation of TGF-β1 signaling, the mRNA levels of PI3K, AKT, ID2, and OPN were the highest, while the mRNA levels of α-SMA and SM22α were the lowest; the opposite results were found in the same groups above. These results suggested the PI3K/AKT/ID2 signaling pathway is involved in TGF-β1-mediated human aortic VSMC phenotypic switching, that is from a contractile to synthetic phenotype, and Combined inhibitors was more effective in inhibiting the phenotypic switch than a single inhibitor. The Combined inhibitors experiments may provide new avenues for the prevention and treatment of thoracic aortic dissection (TAD) that are based on the pathological effects of phenotypic switching.
Keywords: Vascular smooth muscle cell, phenotype switch, TGF-β1, PI3K/AKT, ID2
Introduction
Vascular smooth muscle cells (VSMCs) are major components of the media of aorta and exhibit two different phenotypes, i.e., a contractile phenotype and a synthetic phenotype. Contractile phenotype has a weak ability of proliferation and migration, and exhibit a spindle elongated morphology. Moreover, the cell has a limited ability to synthesize extracellular matrix. Contractile phenotype primarily participates in controlling the diameter of blood vessels, regulating organ blood flow and maintaining a stable blood pressure. In the normal and mature blood vessel wall, VCMCs are predominantly made up of the contractile phenotype [1-4]. Synthetic phenotype has a stronger proliferation and migration ability, and the cell has a hypertrophic appearance and exhibit “hill and valley” growth. Its ability to synthesize extracellular matrix (e.g., collagen, elastin, and proteoglycans) is strong and plays an important role in maintaining the integrity, compliance and compression capability of the normal aorta wall [4,5].
The marker molecules that are specific for contractile VSMC include smooth muscle α-actin (α-SMA) and smooth muscle 22α (SM22α), and those for synthetic VSMC include Osteopontin (OPN) [5-7]. The VSMC switch to the synthetic phenotype leads to the increased expressions of markers for this phenotype. When cardiovascular diseases, such as atherosclerosis, hypertension, and diabetic vascular disease, cause vascular injury, contractile VSMC may switch to synthetic VSMC. This phenotypic switch can repair the injured vessel wall through the migration, proliferation and synthesis of extracellular matrix, having a profound impact on injury repair [5,6,8,9]. Recently, it was found that the release of various growth factors (TGF-β1, PDGF-BB, etc.) and the activation of downstream signaling are involved in this phenotypic switch [4,5,8,10].
The transforming growth factor-β (TGF-β) superfamily is a class of secreted polypeptides. TGF-β plays an important role in the regulation of cell growth, differentiation, and apoptosis, and participates in the occurrence of cardiovascular and fibrous proliferative diseases [11-13]. TGF-β functions by activating downstream signaling pathways, which are divided into two categories: Smad-dependent classic signaling pathways and Smad-independent signaling pathways, such as PI3K/AKT, p38, and ERK [10,14,15]. Of these, Smad-independent signaling pathways may play a regulatory role independently or in cooperation with Smad-dependent signaling pathways [11,16-20]. PI3K/AKT is considered an important signaling pathway that regulates the VSMC phenotype, which is important not only in vascular biology [21] but also in diabetes and obesity. The regulatory effect of PI3K/AKT on VSMC phenotype may be mediated by regulating unidentified downstream transcription factors [4]. Lasorella A et al. [22] found that ID2, as a transcriptional regulatory factor, plays an important regulatory role in muscle cell growth, proliferation and differentiation. George S et al. [23] stated that an important feature of the VSMC phenotypic switch is the enhancement in cell migration and proliferation and that ID2 is very likely to be an important regulatory factor in this switch. Although the above evidence suggests that ID2 may be involved in VSMC phenotypic switch, its specific role and the connection to PI3K/AKT signaling remain unclear.
We hypothesized that TGF-β1 can stimulate ID2 expressions by activating the PI3K/AKT signaling pathway, resulting in VSMCs switch from contractile to synthetic phenotype. The significant changes in this process include the following: 1) morphological changes of the cells, i.e., from spindle elongated to hypertrophic morphology and exhibit “hill and valley” growth; 2) changes in the levels of protein and mRNA markers of the respective types (i.e., a decrease in the protein/mRNA expressions of the contractile marker proteins α-SMA and SM22α, and an increase protein/mRNA expressions of the synthetic marker protein OPN); and 3) enhanced proliferation and migration.
Materials and methods
Reagents and instruments
The main reagents and instruments of this study included the following: human aortic VSMCs (T/G HA-VSMCs, ATCC, US); high-glucose DMEM, trypsin, penicillin-streptomycin, PBS (all from Hyclone, US); fetal bovine serum (FBS, Zhejiang Tianhang Biological Technology Stock Co., Ltd., China); recombinant human TGF-β1 (PeproTech, US); SB525334 and MK2206-2HCL (Selleck, US); an inverted microscope (OLYMPUS CK2-TR, Japan); a biological safety cabinet (Haier HR40-IIA2, China); a CO2 incubator (SANYO MCO-175, Japan); a microplate reader (Rayto RT-6500, US); a real-time PCR system (ABI7900, US); and a fluorescence microscope (OLYMPUS BX51, Japan).
Cell culture and intervention
Cell culture
T/G HA-VSMCs were cultured at 37°C and 5% CO2 in high-glucose DMEM medium containing 20% FBS and a penicillin-streptomycin mixture (1:100, containing 10000 U/mL penicillin and 10 mg/mL streptomycin). When the cells reached 90% confluence, they were passaged by digestion with 0.25% trypsin containing EDTA.
Intervention
Thirty thousand cells (1.5 mL) per well were seeded in 6-well plates and cultured using the complete medium containing 10% FBS for 24 hours. The cells were then washed once with an equal volume of PBS to remove the residual serum. After serum-starvation for 24 hours, the cells were washed once with PBS and divided into five groups according to the experimental design: ① Blank control group; ② TGF-β1 group; ③ TGF-β1 inhibitor group; ④ AKT inhibitor group; ⑤ Combined inhibitors group. DMEM was added in groups ① and ②, while groups ③, ④, and ⑤ were pre-treated in an incubator for 1 h with SB525334 (10 ng/mL), MK2206-2HCL (5 ng/mL), or SB525334+MK2206-2HCL, respectively. The inhibitors were removed by washing once with PBS, and TGF-β1 (5 ng/mL) was added to groups ② to ⑤, then all these groups were incubated for 24 h.
Cell counting kit-8 assay (CCK-8)
When the cells reached 90% confluence, they were then counted using a hemocytometer. After the cell concentration was adjusted to 2×105 cells/mL, these cells were added to 60 wells (100 µL per well) in the center of a 96-well plate, while an equal volume of PBS was added to the remaining surrounding wells. After labeling, the cell culture plate was placed in the incubator for 24 hours. This step was followed by serum-starvation with the same volume of serum-free medium for 24 hours, and the cells were then washed once with PBS. The cells were grouped and treated according to the above-described treatments (100 µL was added to each well). Twelve replicates were performed for each of the five treatment groups, and the cells were cultured for 24 h after treatment. A 10% CCK-8 solution was prepared by adding 1.2 mL of CCK-8 into 12 mL of DMEM. Treated cells were washed with PBS, and 100 µL of 10% CCK-8 was added to each well. After incubation for 3 h, the absorbance (OD) at 450 nm was measured using a microplate reader.
Transwell migration assays
The cells were counted using a hemocytometer, and the concentration was adjusted to 7.5×104 cells/ml. In a Transwell plate, 800 µl of DMEM, TGF-β1 (5 ng/ml) (added to four wells), SB525334 (10 ng/ml), MK2206-2HCl (5 ng/ml), or Combined inhibitors solution were respectively added to each of eight wells. A Transwell chamber was placed in each well (only one TGF-β1 well received a chamber at this point), and 200 µL of the above cell suspension was added to each Transwell chamber to react for 1 h. The Transwell chambers in the SB525334, MK2206-2HCl, and Combined inhibitors solution wells were transferred to three of the wells with TGF-β1. After labeling, the cells were cultured for 24 h and then washed once with PBS. The un-migrated cells on the upper chamber surface were wiped with a cotton ball. The migrated cells were fixed with 4% paraformaldehyde for 30 min, stained with crystal violet for 20 min, and washed three times with PBS. The residual dye on both surfaces of the chamber was washed. After air-drying, the filter membrane was cut with a blade and observed under a microscope on a microslide. Cell counting was conducted for three randomly selected fields.
Coomassie blue staining
The cells were seeded on 6-well plates that were pre-set with coverslips, and the culturing and intervention protocols were the same as described in section above. After treatment, the coverslip with the cells were taken from the 6-well plate and washed twice with PBS. The cells were pre-fixed with 2% paraformaldehyde for 5 s, and washed once with PBS. And next were treatment with 1% Triton X-100 for 30 min and washed three times with PBS. The cells on the coverslip were then fixed with 4% paraformaldehyde at room temperature for 20 min and stained with 0.2% Coomassie blue for 30 min. Lastly, the cells were washed three times with distilled water, allowed to air dry, and cleared in xylene. The obtained coverslip was mounted with neutral balsam and observed under a fluorescence microscope. All of the images were saved and analyzed.
Western blotting
The cells of five groups were washed with PBS and transferred to five Eppendorf tubes. After centrifugation at 12000 rpm, the supernatant was discarded. The appropriate amount RIPA lysis buffer was added to extract the proteins from the cells. The protein concentration was measured, and 50 µg of total protein from each sample was added to 10% PAGE electrophoresis gels that were prepared according to the molecular weight of the protein of interest. The electrophoresis separation was stopped after the target proteins were fully separated based on the pre-stained marker. The target bands were cut from the gel using the pre-stained marker as a reference and washed with distilled water. The PVDF membrane and filter paper were cut in the same size of the PAGE gel. The PVDF membrane was soaked with methanol for a few seconds and then soaked in the transfer buffer together with the filter paper. The protein transfer sandwich was assembled in the following order: black panel - fiber pad - filter paper - gel - PVDF membrane - filter paper - fiber pad - white panel. After closing the clamp of the cassette, the protein transfer sandwich was placed into the transmembrane tank, with the black panel facing the black cathode. The transmembrane conditions were as follows: PI3K with P-PI3K - 00 mA for 120 min, plus 300 mA for 15 min; AKT, P-AKT and OPN - 200 mA for 120 min; α-SMA - 200 mA for 80 min; ID2 and sm22α - 200 mA for 60 min. The obtained PVDF membrane was blocked by soaking in TBST containing 5% nonfat dry milk, with shaking at room temperature for 2 h. After the corresponding primary antibodies were diluted in the blocking buffer, the PVDF membrane was soaked in the primary antibody solution and incubated at 4°C overnight. The corresponding dilutions were as follows: PI3K - 1:1000, P-PI3K - 1:1000, AKT - 1:1000, P-AKT - 1:2000, α-SMA - 1:300, OPN - 1:500, ID2 and sm22α - 1:1000. The PVDF membrane was thoroughly washed with TBST for 5-6 times, with 5 min/wash. The corresponding HRP-labeled secondary antibody was diluted with the blocking buffer to a dilution of 1:50,000. The PVDF membrane was then soaked in the secondary antibody solution, followed by incubation at room temperature with shaking for 2 h. The PVDF membrane was then thoroughly washed with TBST for 5-6 times. An appropriate amount of ECL substrate solution was added dropwise onto each membrane and incubated for several minutes. After the fluorescence bands were visible, the excess substrate solution was absorbed using filter paper. The membrane was then covered with plastic wrap, processed with an X-ray film, developed and fixed.
Quantitative real-time polymerase chain reaction (RT-PCR)
1 mL of Trizol was added to the well of each group, and 200 µL of chloroform was added. After mixing by gently inverting several times, the samples were placed at room temperature for 5 min and then centrifuged at 12000 rpm at 4°C for 15 min. The aqueous supernatant (approximately 400 µL) was transferred into a new 1.5-ml Eppendorf tube, and 400 µL of isopropanol was added. After mixing, the tube was placed at room temperature for 10 min. This step was followed by centrifugation at 12000 rpm at 4°C for 10 min, and the supernatant was removed. The precipitated pellet was washed with cold 70% ethanol three times, air-dried for 5-10 min, and then dissolved in 20 µL of DEPC water. The concentration of RNA was determined with a spectrophotometer. The same amount of RNA from each group was used for the reverse transcription, and the cDNA obtained from the reverse transcription served as a template for RT-PCR to amplify the target gene. β-Actin served as the internal control. The reverse transcription reaction conditions were as follows: 42°C for 60 min, followed by 95°C for 5 min. The reaction conditions for β-actin were 94°C for 4 min, 94°C for 30 s, 56°C for 30 s, 72°C for 25 s, 72°C for 4 min, and 4°C for 4 min, with 30 cycle counts. Then, real time-PCR was performed for β-actin and the target cDNA in 10-fold diluted reaction. The reaction conditions were as follows: 50°C for 2 min, 95°C for 10 min, 95°C for 30 s, and 60°C for 30 s, with 40 cycle counts. The PCR products were subjected to agarose electrophoresis. After processing with a gel imaging system, the grayscale was obtained by scanning, and the expressions levels was determined using integrated optical density (IOD). The ratio of the target gene and β-actin was analyzed, and the relative expression levels of the target genes were calculated using the 2-ΔΔt method (Table 1).
Table 1.
The sequences of the primers and the length of the corresponding PCR products
| Gene | Forward 5’→3’ | Reverse 5’→3’ | Productsize (bp) |
|---|---|---|---|
| PI3K | TGCTGTTCGGTGCTTGGA | CATCCCACATGCACGACA | 250 |
| AKT | TGGGCAAGGGCACTTTCGG | CGGGACAGGTGGAAGAACAGC | 256 |
| ID2 | CCCCAGAACAAGAAGGTGA | ATCCGTGTTGAGGGTGGT | 168 |
| OPN | TGATGCTACAGACGAGGAC | ACTATCAATCACATCGGAAT | 244 |
| α-SMA | TCATGGTCGGTATGGGTCAG | CGTTGTAGAAGGTGTGGTGC | 150 |
| SM22α | TGGCAGTGACCAAGAATGAT | GGTCGTCCGTAGCCTGTC | 181 |
| β-Actin | CACGATGGAGGGGCCGGACTCATC | TAAAGACCTCTATGCCAACACAGT | 240 |
Statistical analysis
Statistical analysis was performed with SPSS 20.0 (SPSS Inc, USA). Data are presented as a mean ± standard deviation (x̅±SD). Comparisons among groups were assessed with one-way analysis of variance (ANOVA) followed by Student-Newman-Keuls test. A value of P<0.05 was considered statistically significant.
Results
TGF-β1 stimulates proliferations in human aortic VSMCs
CCK-8 test showed that the OD value of TGF-β1 group was increased, while the OD values in AKT inhibitor group and Combined inhibitors group were decreased compared with Blank control group; these differences were statistically significant (P<0.01). The OD values showed no significant difference between TGF-β1 inhibitor group and Blank control group nor were there differences between AKT inhibitor group and Combined inhibitor group (Figure 1). These results indicated that treatment with TGF-β1 for 24 hours significantly promoted proliferations of HA-VSMCs, whereas AKT inhibitor and Combined inhibitors significantly inhibited proliferations, while there were no significant differences between these two groups. The TGF-β1 inhibitor could not effectively promote or inhibit cell proliferations.
Figure 1.
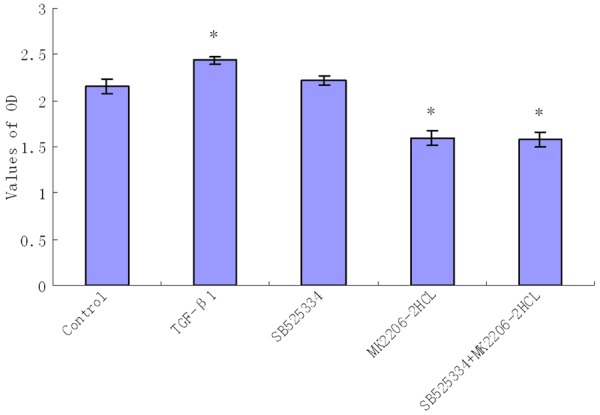
Comparison of the OD value in the different groups with CCK-8 test (The resulting data were represented as x̅±SD, *P<0.01 vs. Blank control group).
TGF-β1 stimulates migrations in human aortic VSMCs
The number of the migrating cells of TGF-β1 group was increased, while the numbers of migrating cells in AKT inhibitor group and combined inhibitors group were decreased. Compared with Blank control group, these differences were statistically significant (P<0.05). The numbers of migrating cells showed no significant differences between TGF-β1 inhibitor group, AKT inhibitor group and Combined inhibitors group (P>0.05) (Figure 2). These results indicated that TGF-β1 significantly promoted the migration of HA-VSMCs, while the TGF-β1 inhibitor, AKT inhibitor and combined inhibitors significantly inhibited cell migrations; there were no significant differences between these three inhibitor groups.
Figure 2.
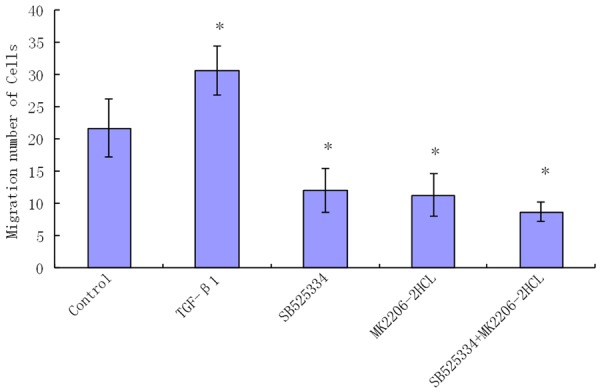
Comparison of the number of migrating cells in the different groups with transwell migration assay. (The resulting data were represented as x̅±SD, *P<0.01 vs. Blank control group).
TGF-β1 induces morphological changes in human aortic VSMCs
In the above five groups of cells, the TGF-β1 group exhibited more hypertrophic appearances in three randomly selected observation fields (×400), with a larger proportion compared with the other four groups. These cells exhibited “hill and valley” growth. The other four groups had fewer hypertrophic cells, prim arily exhibiting a spindle elongated morphology; the smallest proportion of hypertrophic cells were found in Combined inhibitors group (Figure 3). These results indicated that TGF-β1 promoted the morphological changes of HA-VSMCs from a spindle elongated morphology to a hypertrophic appearance and exhibit “hill and valley” growth, while the cells in Combined inhibitors group essentially maintained a spindle elongated morphology, showing no significant morphological changes. At high magnification, some gemistocytic cells were also observed in Blank control group, so it is possible that a certain degree of spontaneous phenotypic switching occurred.
Figure 3.
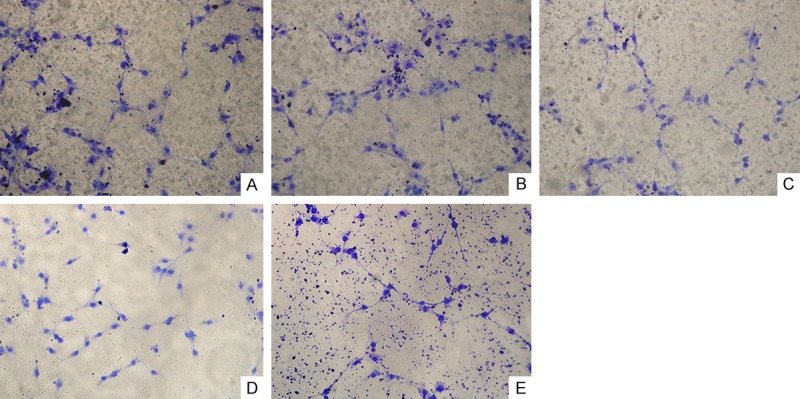
Cell morphology by Coomassie blue staining (×400). A. Blank control group; B. TGF-β1 group; C. TGF-β1 inhibitor group (SB525334); D. AKT inhibitor group (MK2206-2HCL); E. Combined inhibitors group (SB525334+MK2206-2HCL).
TGF-β1 induces PI3K, P-PI3K, AKT, P-AKT, and ID2 expressions in human aortic VSMCs
The PI3K, P-PI3K, AKT, P-AKT, and ID2 levels in TGF-β1 group was increased, while their levels in other three inhibitor groups were decreased and Combined inhibitors groups was the lowest. Compared with Blank control group, these differences were statistically significant (P<0.01), while the PI3K and P-PI3K levels in AKT inhibitor group showed no statistically significant differences with Blank control group (Figure 4). These results suggested that TGF-β1 stimulated PI3K, P-PI3K, AKT, P-AKT, and ID2 expressions, while Combined inhibitors significantly suppress their expressions compared with single inhabitor.
Figure 4.
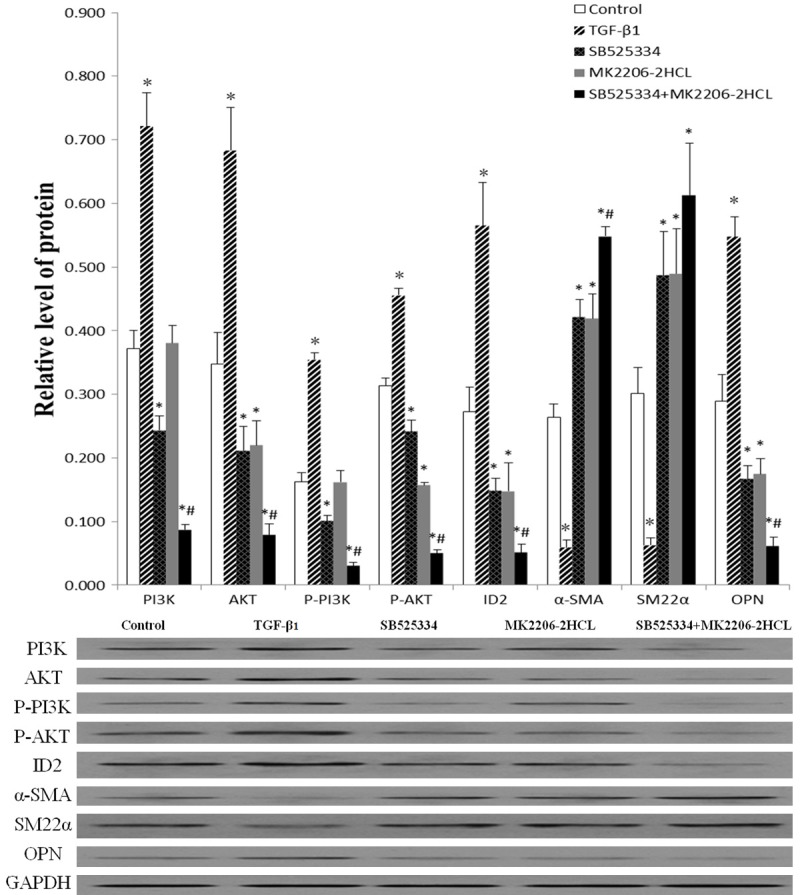
The relative expressions levels of various proteins in the different groups with western blot detection (The resulting datas were represented as x̅±SD, *P<0.01 vs. Blank control group; #P<0.01 vs. TGF-β1 inhibitor group and AKT inhibitor group).
TGF-β1 induces synthetic marker expression (OPN) and suppresses contractile marker expressions (α-SMA, SM22α) in human aortic VSMCs
The OPN levels in TGF-β1 group was increased, while the α-SMA, SM22α levels was decreased, while these three markers’ levels in TGF-β1 inhibitor group, AKT inhibitor group and Combined inhibitors group were opposite. Compared with Blank control group, these differences were statistically significant (P<0.01) (Figure 4). These results suggested that TGF-β1 induces synthetic marker expressions and suppress contractile marker expressions while inhibitor groups have an opposite effect. The effect of Combined inhibitors was the strongest except SM22α, whose effect on SM22α was no significant difference with other single inhibitors.
Effect of TGF-β1 on the expressions of PI3K, AKT and ID2 mRNA in human aortic VSMCs
The PI3K, AKT, and ID2 mRNA levels in the TGF-β1 group was increased, while the levels of PI3K mRNA in TGF-β1 inhibitor group and Combined inhibitors group were decreased, the levels of AKT and ID2 mRNA in all three inhibitor groups were decreased. Compared with Blank control group, these differences of PI3K mRNA levels were statistically significant except AKT inhibitor group (P<0.01), and differences of AKT mRNA levels were statistically significant (P<0.05) while differences between TGF-β1 inhibitor group and Blank control group was relatively weak (P=0.046), and differences of ID2 mRNA levels were statistically significant (P<0.01) (Figure 5). The PI3K mRNA levels in AKT inhibitor group showed no statistically significant difference with Blank control group. These results suggested that TGF-β1 stimulated expressions of PI3K, AKT, and ID2 mRNA, while TGF-β1 inhibitor and combined inhibitors suppressed expressions of PI3K mRNA and all three inhibitor can suppressed expressions of AKT, ID2 mRNA. The suppression effect of combined inhibitors was the strongest, while AKT inhibitor had no effect on expressions of PI3K mRNA.
Figure 5.
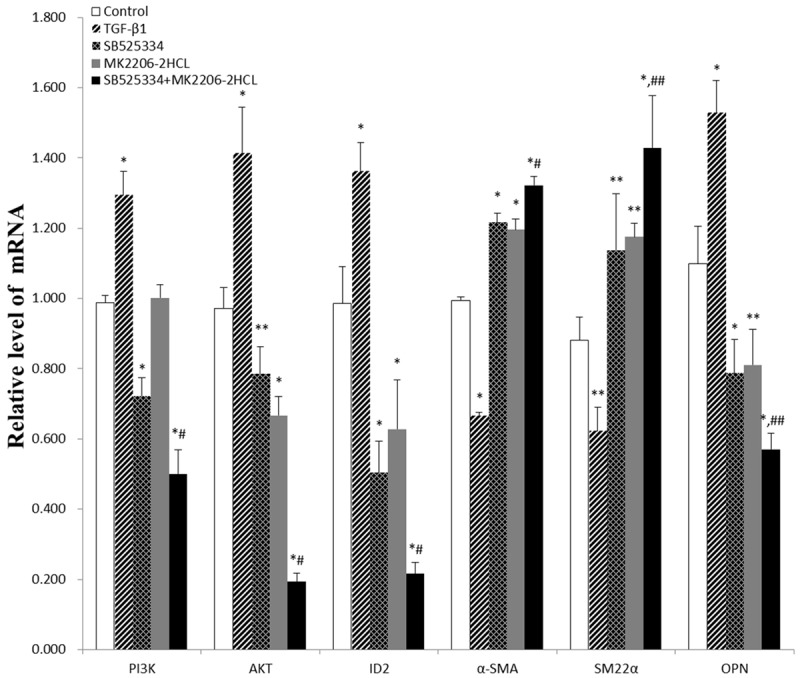
The relative expressions levels of various mRNAs in the different groups with RT-PCR (The resulting datas were represented as x̅±SD, *P<0.01 vs. Blank control group; **0.01≤P<0.05 vs. Blank control group; # P<0.01 vs. TGF-β1 inhibitor group and AKT inhibitor group; ## 0.01<P<0.02 vs. TGF-β1 inhibitor group and AKT inhibitor group).
TGF-β1 suppresses the expressions of α-SMA, SM22α mRNA and induces OPN mRNA expressions in human aortic VSMCs
The α-SMA and SM22α mRNA levels in TGF-β1 group were decreased, while their levels in all three inhibitor groups were increased. The OPN mRNA levels in TGF-β1 group were increased, while its levels in all three inhibitor groups were decreased. Compared with Blank control group, these differences of α-SMA, SM22α mRNA levels were statistically significant (P<0.05), and these differences of OPN mRNA levels were statistically significant (P≤0.01) (Figure 5). These results suggested that TGF-β1 suppressed the expressions of α-SMA and SM22α mRNA and stimulated the expressions of OPN mRNA, while TGF-β1 inhibitor, AKT inhibitor and combined inhibitors stimulated the expressions of α-SMA and SM22α mRNA and suppressed OPN mRNA expressions. Combined inhibitor significantly stimulated α-SMA, SM22α mRNA expressions and suppressed OPN mRNA expressions compared with single inhibitor.
Discussion
Most mature and healthy VSMCs are of the contractile phenotype, exhibiting relatively high contractile capability and low proliferation. However, different physiological and pathological stimuli may cause the VSMC phenotypic switch, leading to proliferation, migration and synthesis of excessive extracellular matrix [24]. These effects result in pathological changes to tissues and organs. It is currently known that the morphological changes of TAD are primarily in the media of the vessel wall and that VSMCs form the main structure of the media of aorta. Under normal circumstances, contractile VSMCs can maintain the vascular tone and homeostasis of the blood vessel wall. However, in TAD, the number and proportion of synthetic VSMCs are significantly increased, leading to reduced aortic elasticity and a propensity to rupture [14,25,26]. These findings suggest that the phenotypic switch may be an important factor in the occurrence of TAD.
Studies have shown that TGF-β1 is an important factor that leads to the VSMC phenotypic switch. Various signaling pathways are involved in TGF-β1-induced switching, including Smad-dependent and Smad-independent signaling pathways. The role of Smad-independent signals, such as PI3K/AKT, in this process has received increasing attention in recent years. PI3K is an intracellular signaling protein with catalytic activities and can regulate the level of AKT phosphorylation to initiate the PI3K/AKT signaling pathway [27,28]. By transducing stimulatory extracellular signals to the nucleus, the PI3K/AKT signaling pathway can trigger a series of biological reactions, including cell proliferation, differentiation and apoptosis and plays an important regulatory role in these processes. Recent studies have shown that the PI3K/AKT signaling pathway is related to the contractile function of vascular smooth muscle, contributing to VSMC dysfunction, vasoconstriction and vascular remodeling [21,29]. Recently, it was reported that PI3K/AKT is involved in regulating the VSMC phenotypic switch, acting through an unidentified downstream transcription factor [4]. Recent data showed that ID2, ZEB1 and other transcription factors play an important role in the phenotype transformation of smooth muscle cells [23]. Moreover, our previous study (data not shown) found that the expressions of ID2 was significantly up-regulated in the aortic wall of patients with TAD compared to unaffected individuals and the VSMCs of aortic wall in patients with TAD showed clear signs of phenotypic switching. These data suggest that ID2 is involved in the phenotypic switch. In addition, in our previous study, different concentrations of human TGF-β1 were used to stimulate human aortic VSMCs, and the results showed that the treatment of 5 ng/mL TGF-β1 for 24 hours led to the highest levels of proliferation and migration (unpublished data).
Based on our previous studies, we hypothesized that ID2 was the key downstream factor involved in mediating these effects. After the inhibitor pretreatment, the VSMCs were stimulated using that TGF-β1 concentrations and incubation time described above. Subsequently, the expressions levels of the downstream target proteins and mRNA were determined, and VSMC morphology, proliferation and migration were assayed. It was found that PI3K and AKT were activated through their phosphorylated form. The application of human TGF-β1 and TGF-β1 inhibitor up- and down-regulatedthe expression of PI3K protein and its mRNA, respectively. This result indicates that PI3K is downstream of TGF-β1 in this context and directly regulates the levels of other proteins and genes. Following the application of human TGF-β1 and of AKT inhibitor, the expressions of ID2 protein and mRNA was up-regulated and down-regulated, respectively. These results indicate that ID2 is maybe a direct downstream protein of PI3K/AKT signaling pathway. We also found that the protein levels of the downstream proteins PI3K, P-PI3K, AKT, P-AKT, ID2, and OPN were highest in the TGF-β1 group, while the protein levels of α-SMA and SM22α were the lowest. In addition, the mRNA levels observed above were consistent with their proteins levels. Moreover, the VSMCs showed higher proliferation, increased migration, and exhibited a hypertrophic appearance and exhibit “hill and valley” growth. These data indicate the occurrence of phenotypic switching that was regulated by upstream protein signaling pathways and support the hypothesis that ID2 is an important transcriptional regulatory factor in this process. Moreover, these results explain how PI3K/AKT signaling is involved in TGF-β1-induced human aortic VSMCs phenotypic switching. In Combined inhibitors group, the changes observed in protein and mRNA levels were in the opposite direction of the alteration observed in TGF-β1 group, and no clear phenotypic switch was detected. This observation indicates that Combined inhibitors treatment blocked phenotypic switching. Consistent with the findings of Liu [30], the activation of PI3K/AKT pathway is characterized by increased levels of the phosphorylated versions of these proteins, and inhibitors of this pathway can inhibit the interaction between PI3K and AKT. In the present study, TGF-β1 inhibitor decreased the levels of both P-PI3K and P-AKT, while AKT inhibition only decreased P-AKT levels, confirming that TGF-β1 is an important upstream regulatory factor of the PI3K/AKT pathway. Therefore We speculated that TGF-β1 activates PI3K via phosphorylation, leading to the activation of AKT phosphorylation. As a result, intracellular levels of ID2 are increased, reducing the expressions of the contractile VSMC marker proteins α-SMA and SM22α and increasing levels of the synthetic marker protein OPN. These changes in protein levels mark the occurrence of the switch from a contractile to synthetic phenotypic. These data indicate that PI3K/AKT/ID2 signaling is involved in TGF-β1-induced human aortic VSMCs phenotypic switch at the protein level. The RT-PCR results were consistent with the above changes in protein levels, verifying the above conclusion at a genetic level. Application with either TGF-β1 or AKT inhibitor blocked the phenotypic switch, and there was no significant difference with respect to their inhibition of phenotype marker proteins, although combined inhibitors treatment was more effective than single inhibitor in blocking the phenotypic switch. These data indicate that Combined inhibitors treatment significantly blocked the TGF-β1-induced PI3K/AKT/ID2 signaling pathway, and thereby the phenotypic switching of human aortic VSMCs. We observed a certain degree of phenotypic switching to synthetic cells in un-treated VSMCs by analyzing cell morphology, proliferation, migration, as well as protein and mRNA expressions levels. We speculate that spontaneous phenotypic switching may occur in VSMCs under normal growth conditions. However, few cells exhibited phenotypic switch, and the extent of the switch was minor, having no significant influence on the results. The secretion of extracellular matrix increased based on the characteristics of synthetic VSMCs. This study did not measure the levels of collagen, elastin or other matrix proteins; therefore, we could not determine whether the VSMCs secreted high levels of extracellular matrix proteins after the phenotypic switch. This question will need further investigation. Because the VSMC phenotypic switch is closely connected to the development of TAD and other cardiovascular diseases, analyzing the VSMC phenotype in terms of the pathogenic potential of these cells will be important for the prevention and treatment of several cardiovascular conditions.
In summary, this study showed that the PI3K/AKT/ID2 signaling pathway may be involved in TGF-β1-induced human aortic VSMCs phenotypic switch. In addition, it was observed that the inhibitors of this signaling pathway blocked phenotypic switch. It was also found that treatment with combined inhibitors was more efficient than a single inhibitor in suppressing the phenotypic switch. These results indicated that the use of combined inhibitors may be worthy of further study in the development of targeted drugs for regulating the VSMC phenotypic switch. The prevention the phenotypic switch may provide new advances in combating TAD, and this possibility requires further investigation.
Acknowledgements
This experimental study was sponsored by the Hubei Province Natural Science Foundation of China (No.2014CFB252) and the Fund for Fostering the Key Young Medical Personnel in Wuhan.
References
- 1.Fisher SA. Vascular smooth muscle phenotypic diversity and function. Physiol Genomics. 2010;42A:169–187. doi: 10.1152/physiolgenomics.00111.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ogut O, Brozovich FV. Regulation of force in vascular smooth muscle. J Mol Cell Cardiol. 2003;35:347–355. doi: 10.1016/s0022-2828(03)00045-2. [DOI] [PubMed] [Google Scholar]
- 3.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 4.Rzucidlo EM, Martin KA, Powell RJ. Regulation of vascular smooth muscle cell differentiation. J Vasc Surg. 2007;45(Suppl A):A25–32. doi: 10.1016/j.jvs.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 5.Rodríguez AI, Csányi G, Ranayhossaini DJ, Feck DM, Blose KJ, Assatourian L, Vorp DA, Pagano PJ. MEF2B-Nox1 signaling is critical for stretchinduced phenotypic modulation of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2015;35:430–8. doi: 10.1161/ATVBAHA.114.304936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mack CP. Signaling mechanisms that regulate smooth muscle cell differentiation. Arterioscler Thromb Vasc Biol. 2011;31:1495–1505. doi: 10.1161/ATVBAHA.110.221135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuan SM, Wang J, Huang HR, Jing H. Osteopontin expression and its possible functions in the aortic disorders and coronary artery disease. Rev Bras Cir Cardiovasc. 2011;26:173–182. doi: 10.1590/s0102-76382011000200006. [DOI] [PubMed] [Google Scholar]
- 8.Shi N, Chen SY. Mechanisms simultaneously regulate smooth muscle proliferation and differentiation. J Biomed Res. 2014;28:40–6. doi: 10.7555/JBR.28.20130130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 10.Pardali E, Goumans MJ, ten Dijke P. Signaling by members of the TGF-beta family in vascular morphogenesis and disease. Trends Cell Biol. 2010;20:556–67. doi: 10.1016/j.tcb.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 11.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 12.Weiss A, Attisano L. The TGF beta superfamily signaling pathway. Wiley Interdiscip Rev Dev Biol. 2013;2:47–63. doi: 10.1002/wdev.86. [DOI] [PubMed] [Google Scholar]
- 13.Choi JC, LeMaire SA. Thoracic aortic dissection: genes, molecules, and the knife. Tex Heart Inst J. 2012;39:838–839. [PMC free article] [PubMed] [Google Scholar]
- 14.EI-Hamamsy I, Yacoub MH. Cellular and molecular mechanisms of thoracic aortic aneurysms. Nat Rev Cardiol. 2009;6:771–786. doi: 10.1038/nrcardio.2009.191. [DOI] [PubMed] [Google Scholar]
- 15.Derynck R. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 16.Lee MK, Pardoux C, Hall MC, Lee PS, Warburton D, Qing J, Smith SM, Derynck R. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007;26:3957–3967. doi: 10.1038/sj.emboj.7601818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hayashida T, Decaestecker M, Schnaper HW. Cross-talk between ERK MAP kinase and Smad signaling pathways enhances TGF-betadependent responses in human mesangial cells. FASEB J. 2003;17:1576–1578. doi: 10.1096/fj.03-0037fje. [DOI] [PubMed] [Google Scholar]
- 18.Conery AR, Cao Y, Thompson EA, Townsend CM Jr, Ko TC, Luo K. Akt interacts directly with Smad3 to regulate the sensitivity to TGF-beta induced apoptosis. Nat Cell Biol. 2004;6:366–72. doi: 10.1038/ncb1117. [DOI] [PubMed] [Google Scholar]
- 19.Remy I, Montmarquette A, Michnick SW. PKB/Akt modulates TGF-beta signalling through a direct interaction with Smad3. Nat Cell Biol. 2004;6:358–365. doi: 10.1038/ncb1113. [DOI] [PubMed] [Google Scholar]
- 20.Kato M, Putta S, Wang M, Yuan H, Lanting L, Nair I, Gunn A, Nakagawa Y, Shimano H, Todorov I, Rossi JJ, Natarajan R. TGF-beta activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN. Nat Cell Biol. 2009;11:881–889. doi: 10.1038/ncb1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morello F, Perino A, Hirsch E. Phosphoinositide 3-kinase signalling in the vascular system. Cardiovasc Res. 2009;82:261–271. doi: 10.1093/cvr/cvn325. [DOI] [PubMed] [Google Scholar]
- 22.Lasorella A, Noseda M, Beyna M, Yokota Y, Iavarone A. Id2 is a retinoblastoma protein target and mediates signaling by Myc oncoproteins. Nature. 2000;407:592–598. doi: 10.1038/35036504. [DOI] [PubMed] [Google Scholar]
- 23.Karagiannis GS, Weile J, Bader GD, Minta J. Integrative pathway dissection of molecular mechanisms of moxLDL-induced vascular smooth muscle phenotype transformation. BMC Cardiovasc Disord. 2013;13:4. doi: 10.1186/1471-2261-13-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kudryavtseva O, Aalkjaer C, Matchkov VV. Vascular smooth muscle cell phenotype is defined by Ca2+-dependent transcription factors. FEBS J. 2013;280:5488–99. doi: 10.1111/febs.12414. [DOI] [PubMed] [Google Scholar]
- 25.Alexander MR, Owens GK. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu Rev Physiol. 2012;74:13–40. doi: 10.1146/annurev-physiol-012110-142315. [DOI] [PubMed] [Google Scholar]
- 26.Lacolley P, Regnault V, Nicoletti A, Li Z, Michel JB. The vascular smooth muscle cell in arterial pathology: a cell that can take on multiple roles. Cardiovasc Res. 2012;95:194–204. doi: 10.1093/cvr/cvs135. [DOI] [PubMed] [Google Scholar]
- 27.Williams R, Berndt A, Miller S, Hon WC, Zhang X. Form and flexibility in phosphoinositide 3-kinases. Biochem Soc Trans. 2009;37:615–626. doi: 10.1042/BST0370615. [DOI] [PubMed] [Google Scholar]
- 28.Shukla S, Maclennan GT, Hartman DJ, Fu P, Resnick MI, Gupta S. Activation of PI3K/Akt signaling pathway promotes prostate cancer cell invasion. Int J Cancer. 2007;121:1424–1432. doi: 10.1002/ijc.22862. [DOI] [PubMed] [Google Scholar]
- 29.Matsuura E, Kobayashi K, Inoue K, Lopez LR, Shoenfeld Y. Oxidized LDL/beta2-glycoprotein I complexes: new aspects in atherosclerosis. Lupus. 2005;14:736–741. doi: 10.1191/0961203305lu2211oa. [DOI] [PubMed] [Google Scholar]
- 30.Liu C, Su T, Li F, Li L, Qin X, Pan W, Feng F, Chen F, Liao D, Chen L. PI3K/Akt signaling transduction pathway is involved in rat vascular smooth muscle cell proliferation induced by apelin-13. Acta Biochim Biophys Sin. 2010;42:396–402. doi: 10.1093/abbs/gmq035. [DOI] [PubMed] [Google Scholar]