

Abstract
Reductions in brain glucose metabolism have long been associated with Alzheimer’s disease. A study now demonstrates that the endothelial glucose transporter GLUT1 is vital for maintaining brain energy metabolism and vascular clearance of amyloid-β.
The brain is the most energy-demanding organ of the body and is critically dependent on a daily supply of a quarter of a pound of glucose, its main energy source, to generate the ATP it needs to function1. The job of delivering such a large amount of glucose across the blood-brain barrier (BBB) falls exclusively to endothelial cells lining cerebral blood vessels. Endothelial cells make up less than 1% of brain cells, but are loaded with GLUT1, a specialized transporter protein that helps glucose cross the BBB and enter the brain2. Defects in cerebral glucose metabolism in Alzheimer’s disease (AD), the most frequent cause of dementia, have suggested that ‘brain starvation’ could be involved in the disease process3. However, proof that this mechanism, albeit plausible, could induce brain dysfunction and contribute to AD has been lacking. As reported in this issue of Nature Neuroscience, Winkler et al.4 used mouse models to demonstrate that GLUT1 deficiency leads to profound changes in vascular, BBB and neuronal function that are particularly damaging in the setting of AD pathology. In addition to highlighting a new function of GLUT1 in vascular homeostasis, these findings establish endothelial GLUT1 as critical factor in maintaining brain health and a potential therapeutic target in AD.
Brain scans using [18F]fluoro-2-deoxyglucose first revealed reductions in cerebral glucose transport and utilization in selected brain regions of AD patients5,6. These changes occurred early in the disease course and were also present in cognitively normal individuals at genetic risk for AD5, suggesting a causal involvement in the disease process. Reductions in the endothelial GLUT1 transporter were subsequently found in the brains of AD patients, raising the possibility that the reduced glucose utilization was a consequence of the deficits in glucose transport across the BBB7,8. However, the effect of these alterations on brain function and disease process remained uncertain for over two decades. Winkler et al.4 set out to answer this longstanding question using mice with a deficit in the gene encoding GLUT1 (Slc2a1+/− mice). They found that Slc2a1 haploinsufficiency led to age-dependent reductions in vascular length, cerebral blood flow and glucose uptake, and to an increase in BBB permeability (Fig. 1). These vascular and metabolic alterations were associated with reduced dendritic spines in the CA1 region of the hippocampus, coupled with alterations in recognition of novel objects and nesting behavior, suggesting cognitive dysfunction. With advancing age, Slc2a1-deficent mice also exhibited evidence of neurodegeneration in the cortex and hippocampus. These observations reveal an unexpected effect of endothelial GLUT1 deficiency on the brain and its vessels that results in brain dysfunction and neurodegeneration.
Figure 1.
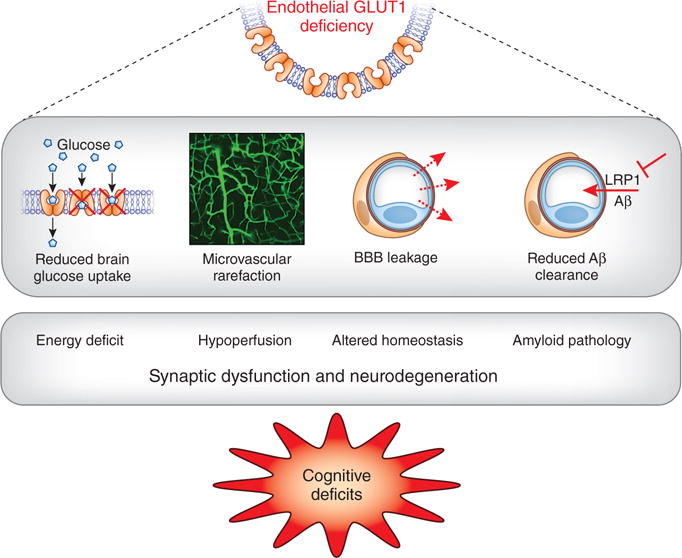
Mechanisms of brain dysfunction and damage caused by GLUT1 deficiency. Endothelial GLUT1 deficiency leads to reduced brain glucose transport, vascular rarefaction and disruption of the BBB, as well as reduced Aβ clearance by suppressing vascular LRP1 expression. These events result in energy deficit, reduced cerebral blood flow (hypoperfusion), altered homeostasis of the brain microenvironment and enhanced amyloid pathology. The resulting synaptic dysfunction and neurodegeneration in turn lead to cognitive deficits.
On the basis of the reported reduction in GLUT1 in AD brains, Winkler et al.4 sought to examine the effect of GLUT1 deficiency on AD pathology. To this end, they crossed Slc2a1-haploinsufficient mice with mice overexpressing the amyloid precursor protein containing the Swedish mutation (APPSw mice), which develop amyloid plaques, a hallmark of AD pathology. They found that Slc2a1 deficiency exacerbated the alterations in vascular structure, function and BBB permeability observed in APPSw mice. The brain accumulation of amyloid-β (Aβ), a pathogenic factor in AD, was enhanced as well, an effect related to reduced brain Aβ clearance caused by suppression of the vascular Aβ transporter lipoprotein receptor-1 (LRP1; Fig. 1). Furthermore, Slc2a1 deficiency worsened the neuronal dysfunction, CA1 spine loss and behavioral deficits in APPSw mice.
At variance with the findings in human disease, endothelial GLUT1 and glucose uptake were not markedly suppressed in APPSw mice, and certainly less so than in Slc2a1 mice. Yet APPSw mice displayed vascular, BBB, neuronal and behavioral alterations similar to those of Slc2a1 mice. Thus, GLUT1 deficiency does not drive the vascular phenotype in APPSw mice, and other factors, such as Aβ, must be responsible for their vascular and cognitive alterations. However, Slc2a1 deficiency greatly exacerbated the dysfunction and damage in APPSw mice. Thus, the reduction in endothelial GLUT1 observed in AD must have the potential to act synergistically with AD pathology to enhance its damaging effects on the brain and promote the progression of the dementia.
What causes the reductions in glucose utilization in AD? This has emerged as a sensitive biomarker of AD and has become an important metric for staging disease progression9. Reductions in glucose utilization in the posterior cingulate and parietal-temporal cortices occur relatively early in the disease and are associated with increased cerebrospinal fluid tau, a neurofilament protein that is abnormally phosphorylated in AD, and hippocampal atrophy10. One possibility is that the hypometabolism is secondary to reduced neuronal activity and, consequently, reduced energy expenditure. However, the report by Winkler et al.4 suggests an alternative explanation. The GLUT1 reduction could reduce glucose uptake and limit the brain’s supply of energy, which, akin to a kink in the fuel line of a combustion engine, could impair neuronal activity and, in the long run, result in neurodegeneration. Deficits in neuronal energy metabolism have long been implicated in AD11, and efforts to provide alternative energy substrates such as dietary ketones to the brain or to enhance neuronal glucose uptake by intranasal insulin have met with some success3,12,13. This scenario raises the possibility that, in AD, the brain is starved of glucose and generates ATP from oxidation of ketone bodies, as suggested by early studies of cerebral metabolism1. A similar situation is found in patients with SLC2A1 deficiency, a rare genetic disease associated with prominent neurological deficits, in whom a ketogenic diet is highly beneficial14.
Could GLUT1 be used to develop new therapeutic interventions in AD? If a reduction in endothelial GLUT1 enhances AD pathology, it is conceivable that restoring GLUT1 levels could ameliorate brain dysfunction and damage in AD. To begin to address this question, Winkler et al.4 performed adenoviral gene transfer in APPSw mice deficient in Slc2a1. They found that restoration of GLUT1 in the hippocampus greatly reduced local Aβ levels. Similar results were obtained with viral gene transfer of LRP1, the Aβ vascular transport protein suppressed by GLUT1 deficiency. Although the authors did not demonstrate rescue of neuronal function and behavior, the findings provide proof of principle that upregulation of GLUT1 clears the brain of amyloid and could have beneficial effects.
Little is known about the mechanism causing GLUT1 dysregulation in AD. GLUT1 expression is controlled by hypoxia-inducible factor 1 (HIF1α,β). Given that HIF1α is down-regulated in AD15, it is conceivable that HIF1α suppression leads to reduced GLUT1 expression. However, earlier studies have indicated that SLC2A1 mRNA is not reduced in AD, implicating post-translational mechanisms8. Thus, further studies on the molecular bases of GLUT1 reduction are needed to provide some indication of how to counteract it.
Irrespective of the many questions outstanding, the data of Winkler et al.4 demonstrate a multifaceted role of glucose transport in the maintenance of brain structure and function, and unveil a damaging interaction with AD pathology. This may open new therapeutic avenues for this devastating neurodegenerative disease.
Footnotes
COMPETING FINANCIAL INTERESTS
The author declares no competing financial interests.
References
- 1.Hoyer S, Oesterreich K, Wagner O. J Neurol. 1988;235:143–148. doi: 10.1007/BF00314304. [DOI] [PubMed] [Google Scholar]
- 2.Harik SI, Kalaria RN, Andersson L, Lundahl P, Perry G. J Neurosci. 1990;10:3862–3872. doi: 10.1523/JNEUROSCI.10-12-03862.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mamelak M. J Alzheimers Dis. 2012;31:459–474. doi: 10.3233/JAD-2012-120370. [DOI] [PubMed] [Google Scholar]
- 4.Winkler EA, et al. Nat Neurosci. 2015;18:521–530. doi: 10.1038/nn.3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nordberg A, Rinne JO, Kadir A, Långström B. Nat Rev Neurol. 2010;6:78–87. doi: 10.1038/nrneurol.2009.217. [DOI] [PubMed] [Google Scholar]
- 6.Jagust WJ, et al. J Cereb Blood Flow Metab. 1991;11:323–330. doi: 10.1038/jcbfm.1991.65. [DOI] [PubMed] [Google Scholar]
- 7.Kalaria RN, Harik S. J Neurochem. 1989;53:1083–1088. doi: 10.1111/j.1471-4159.1989.tb07399.x. [DOI] [PubMed] [Google Scholar]
- 8.Mooradian AD, Chung HC, Shah GN. Neurobiol Aging. 1997;18:469–474. doi: 10.1016/s0197-4580(97)00111-5. [DOI] [PubMed] [Google Scholar]
- 9.Jack CR, et al. Lancet Neurol. 2013;12:207–216. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bateman RJ, et al. N Engl J Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de le Monte SM, Tong M. Biochem Pharmacol. 2014;88:548–559. doi: 10.1016/j.bcp.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krikorian R, et al. Neurobiol Aging. 2012;33:425.e19–425.e27. doi: 10.1016/j.neurobiolaging.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reger MA, et al. Neurobiol Aging. 2004;25:311–314. doi: 10.1016/S0197-4580(03)00087-3. [DOI] [PubMed] [Google Scholar]
- 14.Pearson TS, Akman C, Hinton VJ, Engelstad K, De Vivo DC. Curr Neurol Neurosci Rep. 2013;13:342. doi: 10.1007/s11910-013-0342-7. [DOI] [PubMed] [Google Scholar]
- 15.Liu Y, Liu F, Iqbal K, Grundke-Iqbal I, Gong CX. FEBS Lett. 2008;582:359–364. doi: 10.1016/j.febslet.2007.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]