

Abstract
Background
We assessed the impact of retinoid X receptor (RXR) agonist bexarotene on brain amyloid measured by amyloid imaging in patients with Alzheimer’s disease (AD) in a proof-of-concept trial.
Methods
Twenty patients with AD [Mini Mental State Examination (MMSE) score 10–20 inclusive] with positive florbetapir scans were randomized to receive 300 mg of bexarotene or placebo for 4 weeks. The amyloid imaging result was the primary outcome. Whole-population analyses and prespecified analyses by genotype [apolipoprotein E ε4 (ApoE4) carriers and ApoE4 noncarriers] were conducted. Secondary outcomes included scores on the Alzheimer’s Disease Assessment Scale–Cognitive subscale, Alzheimer’s Disease Cooperative Study–Activities of Daily Living scale, MMSE, Clinical Dementia Rating scale, and Neuropsychiatric Inventory. Serum amyloid-β (Aβ) peptide sequences Aβ1–40 and Aβ1–42 measurements were collected as biomarker outcomes.
Results
There was no change in the composite or regional amyloid burden when all patients were included in the analysis. ApoE4 noncarriers showed a significant reduction in brain amyloid on the composite measure in five of six regional measurements. No change in amyloid burden was observed in ApoE4 carriers. There was a significant association between increased serum Aβ1–42 and reductions in brain amyloid in ApoE4 noncarriers (not in carriers). There were significant elevations in serum triglycerides in bexarotene-treated patients. There was no consistent change in any clinical measure.
Conclusions
The primary outcome of this trial was negative. The data suggest that bexarotene reduced brain amyloid and increased serum Aβ1–42 in ApoE4 noncarriers. Elevated triglycerides could represent a cardiovascular risk, and bexarotene should not be administered outside a research setting. RXR agonists warrant further investigations as AD therapies.
Trial registration
ClinicalTrials.gov identifier NCT01782742. Registered 29 January 2013.
Electronic supplementary material
The online version of this article (doi:10.1186/s13195-016-0173-2) contains supplementary material, which is available to authorized users.
Keywords: Clinical trial, Alzheimer’s disease, MRI, Amyloid, PET, Bexarotene, ApoE genotype
Background
There is an urgent need to develop new treatments for Alzheimer’s disease (AD). AD is currently the third leading cause of death in the United States [1] and costs the economy more annually than cardiovascular disease or cancer [2]. By 2050, the annual cost of AD to the U.S. economy will exceed $1 trillion [3]. All attempts to develop disease-modifying treatments for AD have failed [4].
Much current therapeutic research is focused on the amyloid hypothesis and means of redressing the imbalance between production and clearance of amyloid-β (Aβ) protein that leads to peptide aggregation, neurotoxicity, and formation of neuritic plaques [5]. Pharmaceutical approaches include reducing Aβ production, inhibiting its aggregation, and facilitating its removal [6].
The apolipoprotein E ε4 (ApoE4) allele is among the most potent risk factors for AD, increasing the risk of developing the disease and decreasing its age of onset [7]. ApoE4 has deleterious effects on protein metabolism, enhances Aβ1–42 aggregation, and impairs mitochondrial function [8]. The ApoE4 genotype is associated with greater amyloid burden as measured by amyloid imaging [9]. ApoE4 gene carriers have greater cortical and vascular amyloid deposition than noncarriers [10]. ApoE function and its ability to bind Aβ is influenced by its lipidation status, which is deficient in ApoE4 carriers. Lipidation is determined by the ATP-binding cassette A1 (ABCA1), which is in turn under the control of nuclear retinoid X receptors (RXRs) [9, 11, 12]. RXR agonists induce the expression of ApoE and ABCA1 and increase ApoE lipidation, enhancing its ability to remove Aβ1–42 from the brain [13]. Cramer and colleagues [14] reported a marked effect of bexarotene, an RXR agonist, on Aβ levels in transgenic (Tg) mice. Eleven-month-old amyloid precursor protein-presenilin 1 mice treated with bexarotene for seven days had a 50 % reduction in plaque burden, significantly reduced levels of soluble and insoluble brain Aβ, and restoration of cognitive and memory functions. Attempts to confirm this observation have been only partially successful. Most follow-up studies reproduced effects on soluble Aβ; effects on amyloid plaques and behavioral outcomes were more variable [15–19].
Bexarotene is approved for treatment of cutaneous T-cell lymphoma and can be repurposed for treatment of other indications. It has been used off label for treatment of non–small cell lung cancer, breast cancer, and Kaposi’s sarcoma [20]. The observation that bexarotene may be effective in reducing the pathology and cognitive deficits of Tg animals with Aβ pathology suggested that assessment of bexarotene as a repurposed therapy for AD is warranted [21]. Off-label use of bexarotene in AD based on results in Tg animal systems is controversial because bexarotene is known to have substantial toxicity, commonly elevating triglyceride and cholesterol levels and increasing the risk of hypothyroidism [22–26]. One case report has suggested benefit from treatment of AD with bexarotene [27]. Rigorous evaluation of the potential benefits and harm of bexarotene in controlled trials is the optimal means of providing information on the therapeutic potential of this agent.
Drug development proceeds from preclinical observations in animal model systems to human studies, including proof-of-concept (POC), dose-finding, and large-scale pivotal phase III trials in preparation for regulatory submission. Clinical outcomes for trials of disease-modifying agents typically require large numbers of patients and long-term observations and do not lend themselves to POC investigations [28]. Although no biomarker has gained surrogate status and is known to predict clinical outcomes, biomarkers can be used to develop go–no-go decisions in early stages of drug development [29]. A drug lacking measurable biological effects would not be expected to have clinical benefits in larger trials and would not be advanced for further study. POC studies are exploratory, signal-seeking studies aimed at determining if a meaningful biological effect is present. Informative POC studies derisk drug development programs and increase the likelihood of success [30].
We conducted a biomarker-driven POC trial of bexarotene to determine if the provocative preclinical observations predict human biology and if studies with clinical outcomes should be pursued. We conducted a double-blind, randomized, placebo-controlled, parallel-group study of a single dose (300 mg/day) of bexarotene. We treated patients for 4 weeks in the double-blind portion of the study and continued observation for 4 additional weeks with all patients on treatment. The primary outcome was the effect of bexarotene on brain amyloid imaging after 1 month of treatment. We report the primary outcomes of the study.
Methods
Standard protocol, approvals, registrations, and patient consents
The study was approved by the Cleveland Clinic Institutional Review Board. Written informed consent was obtained from all patients (or guardians of patients) participating in the study. The trial is registered with ClinicalTrials.gov under the identifier NCT01782742.
Trial design
The BExarotene Amyloid Treatment for Alzheimer’s Disease (BEAT AD) trial was a double-blind, placebo-controlled, parallel-group, single-site study with an allocation ratio in the 4 weeks of four bexarotene-treated patients to one placebo-treated patient (Additional File 1). In weeks 4–8, all patients received treatment with bexarotene. There were no changes in methodology after trial initiation.
Participants
Patients were men women ages 50–90 years who met National Institute of Neurological Disorders and Stroke/Alzheimer’s Disease and Related Disorders Association criteria for AD dementia [31]. Each patient was required to have a positive florbetapir positron emission tomogram (PET) and a Mini Mental State Examination (MMSE) [32] score between 10 and 20 (inclusive) to be included in the trial. Positive amyloid PET was determined visually by two readers; if they disagreed, a third rater was invoked, and the majority reading was accepted. Subjects were required to have a study partner who could comply with all required study procedures. Subjects had at least 8 years of education, were capable of communicating effectively with the trial team, and had no uncontrolled medical illnesses. Treatment with any pharmacologic agent, including antidementia agents (cholinesterase inhibitors and/or memantine), had to be stable for at least 1 month before randomization. Patients were required to consent to ApoE genotyping. Patients were not stratified by ApoE genotype, and genotypes were not known until after unblinding of the trial.
Interventions
Patients received bexarotene 75 mg or matching placebo twice daily for days 1–7. The dose was increased to 150 mg twice daily or matching placebo for days 8–28. Patients were treated with atorvastatin for elevated cholesterol levels and clofibrate for elevated triglyceride levels when these emerged in the course of the trial. These were not implemented until the end of week 4.
Outcomes
The primary endpoint was the drug–placebo difference in change from baseline to week 4 in the composite Aβ burden of the brain as measured by standardized uptake value ratio (SUVr) with a florbetapir PET. A white matter standard for comparison was prespecified for the primary endpoint [33]. The effect of ApoE genotype was hypothesized to be influential, and change from baseline on treatment compared with placebo at week 4 on composite and regional Aβ burden according to ApoE genotype (ApoE4 carriers compared with ApoE4 noncarriers) was a declared primary outcome (Additional File 2). Determination of SUVr values using a cerebellar standard was included as a secondary outcome. Secondary clinical outcomes addressed change from baseline in the active treatment group compared with change from baseline in the placebo group at week 4. Changes in scores on the MMSE, Alzheimer’s Disease Assessment Scale–Cognitive subscale (ADAS-Cog) [34], Clinical Dementia Rating Sum of Boxes (CDR-SOB) [35], Neuropsychiatric Inventory (NPI) [36], and Alzheimer’s Disease Cooperative Study–Activities of Daily Living (ADCS-ADL) scale [37] were recorded. Change in Aβ1–40 and Aβ1–42 serum levels from baseline to week 4 were secondary biomarker outcomes. Safety and tolerability were measured by incidence of adverse events (AEs) or serious AEs, clinical laboratory data, vital signs, electrocardiograms, and magnetic resonance imaging (MRI) scans.
Blinding and randomization
Patients were randomized in a 4:1 ratio of bexarotene to placebo as determined by a randomization sequence generated by Cleveland Clinic Department of Quantitative Health Sciences. Patients received either the active agent or identical appearing placebos. Patients were enrolled at the Cleveland Clinic Lou Ruvo Center for Brain Health, Las Vegas, NV, USA, and all study personnel, patients, and caregivers were blinded to allocation sequence and treatment status. Among trial personnel, only the Cleveland Clinic pharmacist had access to the randomization details.
Sample size determination
There was no previous history of the use of bexarotene in AD to guide effect size estimates or standard deviations. The effects observed in Tg mice were dramatic and occurred within a short time frame. We hypothesized that a marked effect of the type seen in Tg animals could be observed in a sample size of 10 subjects with AD; thus, we planned recruitment of 20 subjects, with 4 assigned to placebo.
ApoE genotyping
Whole blood was collected at baseline into PAXgene Blood DNA Tubes (PreAnalytiX, Hombrechtikon, Switzerland). DNA was extracted from the whole-blood samples using the PAXgene Blood DNA Kit according to kit specifications. DNA concentration and purity were assessed using a NanoDrop spectrophotometer (Thermo Scientific, Wilmington, DE, USA). Samples were genotyped for ApoE using the method and reported by Calero et al. [38].
Serum amyloid analysis using enzyme-linked immunosorbent assays
Plasma was collected at baseline, 4 weeks, and 8 weeks. Plasma samples from each time point were analyzed using enzyme-linked immunosorbent assays for Aβ1–40 and Aβ1–42 (Wako Pure Chemical Industries, Osaka, Japan) according to kit instructions. All samples were run in triplicate. The samples were read on a SpectraMax Plus 384 spectrophotometer and plate reader (Molecular Devices, Sunnyvale, CA, USA).
Amyloid imaging analysis
PET data were acquired from all 20 subjects after injection of 370 MBq (±10 %) of florbetapir at baseline and at week 4 for the primary analysis. PETs at all the sessions were acquired for 10 minutes at 50 minutes after injection. A Biograph mCT scanner (Siemens Healthcare, Malvern, PA, USA) was used for acquiring all PET data, and the images were reconstructed using three-dimensional ordered-subset expectation maximization to an image size of 128 × 128 pixels, slice thickness of 2 mm, and postreconstruction Gaussian filter of 2 mm. SUVr values at baseline were calculated using anatomically predefined, atlas-based cortical regions—medial orbital frontal, precuneus, parietal, temporal, anterior cingulate, and posterior cingulate [39]. SUVr values for week 4 and week 8 images were calculated using the prespecified primary method, implementing white matter as the internal standard [40] for measuring change (baseline to week 4).
Statistical methods
Analyses were performed on the basis of the intention-to-treat principle (Additional File 2). Categorical factors were summarized using frequencies and percentages, while continuous measures were described using means and standard deviations. Comparisons of the changes between groups at 4 weeks were performed using linear models with change as the outcome. Results are presented as the mean and 95 % confidence interval. Spearman correlations were used to evaluate associations between changes in clinical measures. Each measure was evaluated for outliers. The small sample size limits the ability to detect departures from normality on which the results rely. Only very large differences between groups can be detected as significant, and larger samples would be needed to confirm any statistically significant findings. Analyses were performed using SAS software (version 9.3; SAS Institute, Cary, NC, USA) with a significance level of 0.05. In this exploratory POC trial, no adjustment for multiplicity of testing was performed.
Results
Participant flow and baseline features
Figure 1 shows the study disposition. Forty-nine subjects were screened, twenty-nine subjects did not meet the inclusion criteria and were not assessed further. Twenty patients had amyloid imaging and all twenty met the criteria for an elevated amyloid burden at baseline. One patient required three readers to reach a consensus on the presence of an abnormal amyloid signal. Twenty subjects were randomized, and all twenty completed the 4-week assessment. One subject was not continued into the weeks 4–8 open-label treatment period at the principal investigator’s request because of a greater than tenfold elevation in serum triglycerides.
Fig. 1.
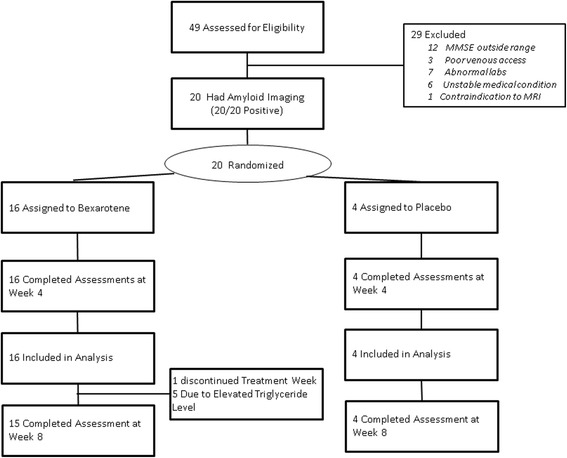
Disposition of subjects in the Bexarotene Amyloid Treatment for Alzheimer’s Disease trial. MMSE Mini Mental State Examination, MRI magnetic resonance imaging
Study disposition
Patient recruitment began on 18 February 2013 and was completed on 5 June 2014. Recruitment was stopped when the prespecified number of subjects was reached, and the trial was completed 8 weeks after the last patient entered the trial.
Table 1 summarizes the baseline characteristics of the overall population and by treatment group. Patients were well matched for age; sex; years of education; and MMSE, ADAS-Cog, CDR-SOB, NPI, and ADCS-ADL scores. On average, the impairment of patients assigned to placebo was somewhat less severe than that of patients randomized to bexarotene. The recruited population included seven ApoE4 noncarriers, seven ApoE4 heterozygotes, and six ApoE4 homozygotes. Composite SUVr amyloid levels ranged from 1.37 to 1.41.
Table 1.
Baseline characteristics, overall and by group
| Characteristic | Total (N = 20) | Bexarotene (n = 16) | Placebo (n = 4) |
|---|---|---|---|
| Age at screening, yr | 75.5 ± 6.8 | 74.9 ± 6.6 | 78.1 ± 8.0 |
| Male sex | 7 (35.0) | 6 (37.5) | 1 (25.0) |
| White race | 19 (95.0) | 15 (93.8) | 4 (100.0) |
| Years of education | 14.2 ± 4.7 | 14.7 ± 4.9 | 12.3 ± 3.3 |
| Years of cognitive symptoms | 4.3 ± 1.9 | 4.6 ± 1.9 | 2.8 ± 0.96 |
| MMSE total score | 14.4 ± 3.8 | 13.7 ± 3.7 | 17.0 ± 3.6 |
| ADAS-Cog scorea | 48.0 ± 9.8 | 49.9 ± 8.9 | 40.3 ± 10.7 |
| CDS-SOB scorea | 1.4 ± 0.56 | 1.4 ± 0.55 | 1.1 ± 0.63 |
| NPI scorea | 8.4 ± 8.1 | 8.7 ± 8.6 | 7.0 ± 6.4 |
| ADCS-ADL scorea | 55.9 ± 12.9 | 53.7 ± 13.1 | 64.5 ± 8.2 |
| Composite SUVr | 1.40 (0.11) | 1.41 (0.09) | 1.37 (0.16) |
| ApoE4 genotype | |||
| Noncarriers | 7 (35.0) | 4 (25.0) | 3 (75.0) |
| Heterozygotes | 7 (35.0) | 6 (37.5) | 1 (25.0) |
| Homozygotes | 6 (30.0) | 6 (37.5) | 0 (0.0) |
ADAS-Cog Alzheimer’s Disease Assessment Scale–Cognitive subscale, ADCS-ADL Alzheimer’s Disease Cooperative Study–Activities of Daily Living scale, CDR-SOB Clinical Dementia Rating Sum of Boxes, MMSE Mini Mental State Examination, NPI Neuropsychiatric Inventory, SUVr standardized uptake value ratio
Values presented as mean ± standard deviation, or count (%)
a Data not available for all subjects; missing values: ADAS-Cog score = 1, CDS-SOB score = 3, NPI score = 4, NPI distress score = 4, ADCS-ADL score = 1
Outcomes
The composite SUVr for all subjects showed no difference in change between the bexarotene-treated subjects and those receiving placebo (Table 2) following 4 weeks of treatment. The prespecified comparison of all subjects for specified brain regions also showed no drug–placebo difference.
Table 2.
Comparisons of mean changes from baseline to 4 weeks in amyloid burden between treatment groups (white matter standard)
| Bexarotene | Placebo | Difference | ||||
|---|---|---|---|---|---|---|
| Factor | N | Mean (95 % CI) | N | Mean (95 % CI) | Mean (95 % CI) | p Value |
| All subjects | ||||||
| Composite | 16 | −0.028 (−0.064, 0.008) | 4 | 0.023 (−0.049, 0.096) | −0.052 (−0.133, 0.030) | 0.22 |
| Frontal medial orbital | 16 | −0.043 (−0.081, −0.006) | 4 | −0.021 (−0.096, 0.054) | −0.022 (−0.106, 0.062) | 0.61 |
| Anterior cingulate | 16 | −0.040 (−0.080, 0.000) | 4 | 0.018 (−0.061, 0.098) | −0.059 (−0.147, 0.030) | 0.20 |
| Parietal | 16 | −0.003 (−0.034, 0.027) | 4 | 0.044 (−0.018, 0.105) | −0.047 (−0.116, 0.022) | 0.19 |
| Posterior cingulate | 16 | −0.017 (−0.065, 0.031) | 4 | 0.044 (−0.052, 0.141) | −0.061 (−0.169, 0.047) | 0.27 |
| Precuneus | 16 | −0.027 (−0.069, 0.014) | 4 | 0.040 (−0.043, 0.122) | −0.067 (−0.159, 0.025) | 0.16 |
| Temporal | 16 | −0.038 (−0.075, 0.000) | 4 | 0.016 (−0.059, 0.090) | −0.053 (−0.136, 0.030) | 0.22 |
| ApoE4 noncarriers | ||||||
| Composite | 4 | −0.097 (−0.155, −0.040) | 3 | 0.047 (−0.019, 0.114) | −0.145 (−0.232, −0.057) | 0.012 |
| Frontal medial orbital | 4 | −0.076 (−0.146, −0.007) | 3 | 0.005 (−0.075, 0.085) | −0.081 (−0.187, 0.025) | 0.16 |
| Anterior cingulate | 4 | −0.096 (−0.166, −0.026) | 3 | 0.048 (−0.034, 0.129) | −0.143 (−0.251, −0.036) | 0.029 |
| Parietal | 4 | −0.068 (−0.107, −0.029) | 3 | 0.065 (0.020, 0.110) | −0.133 (−0.193, −0.073) | 0.002 |
| Posterior cingulate | 4 | −0.113 (−0.180, −0.046) | 3 | 0.074 (−0.004, 0.151) | −0.187 (−0.289, −0.084) | 0.007 |
| Precuneus | 4 | −0.127 (−0.188, −0.066) | 3 | 0.062 (−0.008, 0.132) | −0.189 (−0.282, −0.096) | 0.004 |
| Temporal | 4 | −0.104 (−0.162, −0.045) | 3 | 0.031 (−0.037, 0.098) | −0.134 (−0.224, −0.045) | 0.018 |
| ApoE4 carriers | ||||||
| Composite | 12 | −0.005 (−0.041, 0.031) | 1 | −0.048 (NA) | NA | NA |
| Frontal medial orbital | 12 | −0.033 (−0.074, 0.009) | 1 | −0.099 (NA) | NA | NA |
| Anterior cingulate | 12 | −0.022 (−0.062, 0.019) | 1 | −0.069 (NA) | NA | NA |
| Parietal | 12 | 0.018 (−0.013, 0.050) | 1 | −0.019 (NA) | NA | NA |
| Posterior cingulate | 12 | 0.015 (−0.035, 0.065) | 1 | −0.044 (NA) | NA | NA |
| Precuneus | 12 | 0.006 (−0.031, 0.043) | 1 | −0.027 (NA) | NA | NA |
| Temporal | 12 | −0.015 (−0.055, 0.024) | 1 | −0.030 (NA) | NA | NA |
ApoE4 apolipoprotein E ε4, CI confidence interval, NA not applicable
The prespecified analysis of drug–placebo difference in change from baseline of amyloid burden by ApoE genotype demonstrated a significant reduction of Aβ on the composite measure and a reduction of Aβ in anterior cingulate cortex, parietal cortex, posterior cingulate cortex, precuneus, and temporal cortex in ApoE4 noncarriers using the declared white matter standard (Table 2; Figure 2). There was no significant difference in change from baseline to 4 weeks in amyloid measures among ApoE4 carriers receiving bexarotene (heterozygotes, homozygotes, or combined) compared with those on placebo. Analysis of changes at week 4 for composite and regional changes in Aβ using a cerebellar comparison standard showed no differences between drug and placebo for all subjects, including ApoE4 noncarriers (data not shown).
Analysis of MMSE scores showed a significant difference between change from baseline with bexarotene compared with placebo in favor of placebo in ApoE4 noncarriers (p = 0.026) at week 4. This was due to an improvement in MMSE scores in the placebo group. There was no drug-placebo difference between the drug and placebo groups, considered together or by genotype on ADAS-Cog, NPI, and ADCS-ADL (Table 3).
Table 3.
Comparisons of changes from baseline to 4 weeks in clinical measures between treatment groups
| Bexarotene | Placebo | Difference | ||||
|---|---|---|---|---|---|---|
| Factor | Number of subjects | Mean (95 % CI) | Number of subjects | Mean (95 % CI) | Mean (95 % CI) | p Value |
| All subjects | ||||||
| MMSE | 16 | 0.75 (−0.78, 2.28) | 4 | 1.75 (−1.32, 4.82) | −1.00 (−4.43, 2.43) | 0.57 |
| ADAS-Cog | 16 | 0.38 (−2.15, 2.90) | 4 | −0.25 (−5.31, 4.81) | 0.63 (−5.03, 6.28) | 0.83 |
| CDR-SOB | 16 | 0.00 (0.00, 0.00) | 4 | 0.00 (0.00, 0.00) | 0.00 (0.00, 0.00) | NAa |
| NPI | 16 | −2.63 (−6.78, 1.53) | 4 | −2.25 (−10.57, 6.07) | −0.38 (−9.67, 8.92) | 0.94 |
| ADCS-ADL | 16 | −1.94 (−4.86, 0.99) | 4 | −6.50 (−12.35, −0.65) | 4.56 (−1.98, 11.10) | 0.18 |
| ApoE4 noncarriers | ||||||
| MMSE | 4 | −0.25 (−2.12, 1.62) | 3 | 3.67 (1.51, 5.82) | −3.92 (−6.77, −1.06) | 0.026 |
| ADAS-Cog | 4 | −3.00 (−7.04, 1.04) | 3 | −0.33 (−4.99, 4.33) | −2.67 (−8.83, 3.50) | 0.41 |
| CDR-SOB | 4 | 0.00 (0.00, 0.00) | 3 | 0.00 (0.00, 0.00) | 0.00 (0.00, 0.00) | NAa |
| NPI | 4 | −1.25 (−12.43, 9.93) | 3 | −3.33 (−16.25, 9.58) | 2.08 (−15.00, 19.16) | 0.81 |
| ADCS-ADL | 4 | −4.75 (−10.69, 1.19) | 3 | −7.67 (−14.53, −0.81) | 2.92 (−6.16, 11.99) | 0.53 |
ADAS-Cog Alzheimer’s Disease Assessment Scale–Cognitive subscale, ADCS-ADL Alzheimer’s Disease Cooperative Study–Activities of Daily Living scale, ApoE4 apolipoprotein E ε4, CDR-SOB Clinical Dementia Rating Sum of Boxes, CI confidence interval, MMSE Mini Mental State Examination, NPI Neuropsychiatric Inventory
aNot applicable (NA); p value was not calculated, because no change was observed in any patient at 4 weeks.
A significant increase in serum Aβ1–42 was seen at week 4 in the bexarotene group compared with those on placebo when all subjects were included. Increases in serum Aβ1–42 correlated with decreased cortical amyloid in treated ApoE4 noncarriers (Table 4), not in treated ApoE4 carriers (data not shown). There were no correlations between Aβ1–40 serum level changes and cortical amyloid changes.
Table 4.
Spearman correlations between changes from baseline to 4 weeks (ApoE4 noncarriers)
| Measure | Brain region | r Value | 95 % CI | p Value |
|---|---|---|---|---|
| Cholesterol change | Composite SUVr | −0.66 | (−1.00, 0.39) | 0.16 |
| Frontal medial orbital | −0.54 | (−1.00, 0.62) | 0.27 | |
| Anterior cingulate | −0.66 | (−1.00, 0.39) | 0.16 | |
| Parietal | −0.89 | (−1.00, −0.24) | 0.019 | |
| Posterior cingulate | −0.66 | (−1.00, 0.39) | 0.16 | |
| Precuneus | −0.66 | (−1.00, 0.39) | 0.16 | |
| Temporal | −0.66 | (−1.00, 0.39) | 0.16 | |
| Triglycerides change | Composite SUVr | −0.89 | (−1.00, −0.24) | 0.019 |
| Frontal medial orbital | −0.77 | (−1.00, 0.11) | 0.072 | |
| Anterior cingulate | −0.83 | (−1.00, −0.05) | 0.042 | |
| Parietal | −1.00 | (−1.00, −1.00) | ||
| Posterior cingulate | −0.89 | (−1.00, −0.24) | 0.019 | |
| Precuneus | −0.89 | (−1.00, −0.24) | 0.019 | |
| Temporal | −0.89 | (−1.00, −0.24) | 0.019 | |
| Aβ1–42 change | Composite SUVr | −0.83 | (−1.00, −0.05) | 0.042 |
| Frontal medial orbital | −0.71 | (−1.00, 0.26) | 0.11 | |
| Anterior cingulate | −0.94 | (−1.00, −0.48) | 0.005 | |
| Parietal | −0.54 | (−1.00, 0.62) | 0.27 | |
| Posterior cingulate | −0.83 | (−1.00, −0.05) | 0.042 | |
| Precuneus | −0.83 | (−1.00, −0.05) | 0.042 | |
| Temporal | −0.83 | (−1.00, −0.05) | 0.042 | |
| Aβ1–40 change | Composite SUVr | −0.37 | (−1.00, 0.92) | 0.47 |
| Frontal medial orbital | −0.31 | (−1.00, 1.00) | 0.54 | |
| Anterior cingulate | −0.26 | (−1.00, 1.00) | 0.62 | |
| Parietal | 0.20 | (−1.00, 1.00) | 0.70 | |
| Posterior cingulate | −0.37 | (−1.00, 0.92) | 0.47 | |
| Precuneus | −0.37 | (−1.00, 0.92) | 0.47 | |
| Temporal | −0.37 | (−1.00, 0.92) | 0.47 |
CI confidence interval, SUVr standardized uptake value ratio
Correlational analyses showed significant correlations between elevations in cholesterol and reduced amyloid levels in one cortical region (parietal) and between elevations of triglycerides and reduced cortical amyloid in four of the six cortical regions and in the composite cortical amyloid measure in ApoE4 noncarriers (Table 4). There were no significant correlations between serum cholesterol and triglyceride levels and cortical amyloid change measures in ApoE4 carriers.
Safety
Fifteen of twenty subjects had increases in triglyceride levels to greater than 200 mg/dl. Eleven subjects had increases in cholesterol levels to greater than 300 mg/dl. Other AEs included delusions, dizziness, toe blister, dry cough, and diverticulitis (one each). No amyloid-related imaging abnormalities of either the effusion or hemorrhagic type were seen on MRI scans at week 4 or week 8.
Discussion
This POC trial had a small number of participants, and ApoE genotype subgroups were also small, limiting the conclusions that can be drawn. Patients were excluded if they had abnormal lipid levels at baseline (six subjects were excluded on this basis) or if they had any medical illness that was not adequately controlled (six were excluded on this basis). We do not know the effects of bexarotene in patients who were in excluded categories, and the results should be generalized with caution. SUVr may be susceptible to flow effects, and any impact of bexarotene on blood flow could affect interpretation of the results. Bexarotene is not known to alter blood flow. We chose a brief exposure period in this POC study to explore potential acute effects of bexarotene. Longer exposures are necessary to assess effects on clinical outcomes and to determine if removal of amyloid in ApoE4 carriers is observed with longer exposures.
Subjects recruited into this study were typical of patients with moderate AD with MMSE scores in the range of 10–20. They received the usual standard of care with regard to antidementia therapies. All patients were determined by amyloid imaging to have an elevated brain amyloid burden at baseline meeting clinical and biomarker data for AD dementia [41].
The primary outcome of this study for all subjects was negative; no difference was seen between treatment with bexarotene and placebo when all patients were included in the analysis. Using the prespecified ApoE genotype-based analyses, we observed a highly significant reduction in brain amyloid on the composite measure and in five of six cortical regions of interest among ApoE4 noncarriers. The ApoE4 noncarriers had reductions in amyloid burden in all measured cortical regions after 4 weeks of treatment with bexarotene, and those on placebo had slight increases in burden (Table 2; Figure 2).
Fig. 2.
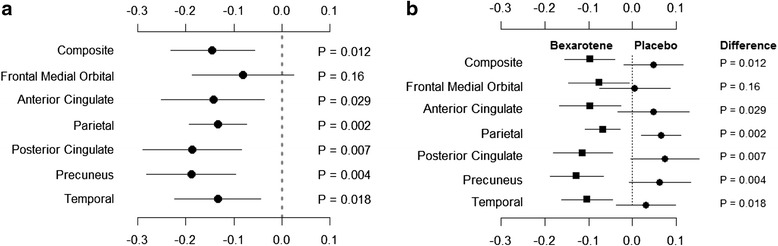
Panel a shows the difference in mean amyloid burden and 95 % confidence intervals for changes from baseline at 4 weeks between bexarotene and placebo groups. Panel b shows mean group changes and 95 % confidence intervals at 4 weeks. P-values reflect the comparisons of mean change between groups
There was a significant correlation between reduced cortical amyloid and elevated serum levels of Aβ1–42, suggesting that soluble forms of amyloid were transferred from brain to blood, although a direct effect of bexarotene on serum Aβ1–42 binding has not been excluded. In ApoE4 noncarriers, there were significant correlations between elevated serum triglyceride levels and decreased cortical amyloid. Bexarotene is an RXR/liver X receptor agonist; the latter mechanism is linked to elevated triglyceride levels.
Reductions in brain amyloid and correlations of reduced brain amyloid with serum Aβ1–42 were seen only in ApoE4 noncarriers. In Tg mice, ApoE4 presence results in more compact plaques; ApoE3 and ApoE2 mice have more diffuse plaques, which may result in more ready mobilization of amyloid with therapy [42]. ApoE4 has a deficient lipidation status, and increased lipidation by bexarotene may be specific to noncarriers [43]. Alternatively, the apparently genotype-specific effects of bexarotene may relate to a potential different time course of response in ApoE4 carriers and noncarriers.
There were no significant changes in cognitive measures. The treatment was brief, the period with relatively decreased amyloid was correspondingly brief, and the trial duration may not have been long enough to observe cognitive benefit. The trial was not powered to observe cognitive change.
Bexarotene has effects on serum lipids that may increase the risk of stroke and heart attack, and this agent should not be used off label for clinical treatment until cognitive efficacy is demonstrated in a clinical trial.
Conclusions
The results of the BEAT AD trial suggest that bexarotene may lower brain amyloid levels in patients with mild to moderate AD who do not carry the ApoE4 gene. This biomarker-based POC trial supports a mechanism-based biological effect of bexarotene on targets relevant to AD pathogenesis. The potential clinical and biological effects of RXR therapeutics in AD and in biologically derived subtypes of AD warrant further investigation.
Acknowledgments
Sources of funding
This study was funded by generous philanthropists and Keep Memory Alive, a 501(c)(3) nonprofit organization in support of the Cleveland Clinic. Avid Pharmaceuticals provided the florbetapir ligand for amyloid imaging. The funding sources had no influence on trial design or interpretation.
Abbreviations
- ABCA1
ATP-binding cassette A1
- AD
Alzheimer’s disease
- ADAS-Cog
Alzheimer’s Disease Assessment Scale–Cognitive subscale
- ADCS-ADL
Alzheimer’s Disease Cooperative Study–Activities of Daily Living scale
- AE
adverse event
- ApoE4
apolipoprotein E ε4
- ATP
adenosine triphosphate
- Aβ
amyloid-β
- BEAT AD trial
Bexarotene Amyloid Treatment for Alzheimer’s Disease trial
- CDR-SOB
Clinical Dementia Rating Sum of Boxes
- CI
confidence interval
- MMSE
Mini Mental State Examination
- MRI
magnetic resonance imaging
- NPI
Neuropsychiatric Inventory
- PET
positron emission tomography
- POC
proof of concept
- RXR
retinoid X receptor
- SUVr
standardized uptake value ratio
- Tg
transgenic
Additional files
BexProtocol-Ver#2-8-1-12. (DOC 299 kb)
BEATADStatisticalPlanFinal_1 3_20141209. (DOC 467 kb)
Footnotes
Competing interests
JLC has received research support from Avid Radiopharmaceuticals, Teva Pharmaceuticals, and Cogstate. JLC has provided consultation services to AbbVie, ACADIA Pharmaceuticals, Actinogen Medical, Adamas Pharmaceuticals, Alzheon, Anavex Life Sciences, Astellas Pharma, Avanir Pharmaceuticals, Boehinger-Ingelheim, Eisai, FORUM Pharmaceuticals, GE Healthcare, Genentech, Intra-Cellular Therapies, Ionis Pharmaceuticals, Eli Lilly and Co., Lundbeck, MedAvante, Merck & Co., Neurim Pharmaceuticals, Neurotrope BioScience, Novartis, Orion, Otsuka Pharmaceutical Co., Pfizer, Piramal, QR Pharma, Resverlogix, Roche, Roivant Sciences, Suven Life Sciences, Takeda Pharmaceutical Co., and Toyama Chemical Co. JLC owns the copyright of the Neuropsychiatric Inventory (NPI). JLC has stock options in Prana Biotechnology, Neurokos, Adamas Pharmaceuticals, MedAvante, and QR Pharma. AJ, MP, and MD are all employed by Avid Pharmaceuticals. The other authors declare that they have no competing interests.
Authors’ contributions
JLC and KZ were responsible for study conception and design. KZ, JWK, CH, and JMT acquired data. JLC, KZ, AJ, MP, MD, AT, and JB analyzed and interpreted data. JLC, KZ, JWK, CH, AJ, MP, AT, and JB drafted the manuscript. JLC, KZ, JWK, CH, JMT, AJ, MP, MD, AT, and JB revised the manuscript critically. AT and JB performed statistical analysis. JLC, KZ, AJ, MP, and MD obtained funding. JLC, KZ, JWK, JMT, AJ, MP, MD, AT, and JB provided administrative, technical, and material support. All authors read and approved the final manuscript.
Contributor Information
Jeffrey L. Cummings, Phone: (702) 483-6029, Email: cumminj@ccf.org
Kate Zhong, Email: zhongk@ccf.org.
Jefferson W. Kinney, Email: jefferson.kinney@unlv.edu
Chelcie Heaney, Email: chelcie.heaney@gmail.com.
Joanne Moll-Tudla, Email: mollj@ccf.org.
Abhinay Joshi, Email: joshi@avidrp.com.
Michael Pontecorvo, Email: pontecorvo@avidrp.com.
Michael Devous, Email: devous@avidrp.com.
Anne Tang, Email: tanga@ccf.org.
James Bena, Email: benaj@ccf.org.
References
- 1.James BD, Leurgans SE, Hebert LE, Scherr PA, Yaffe K, Bennett DA. Contribution of Alzheimer disease to mortality in the United States. Neurology. 2014;82:1045–50. doi: 10.1212/WNL.0000000000000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hurd MD, Martorell P, Delavande A, Mullen KJ, Langa KM. Monetary costs of dementia in the United States. N Engl J Med. 2013;368:1326–34. doi: 10.1056/NEJMsa1204629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Association A’s. 2015 Alzheimer’s disease facts and figures. Alzheimers Dement. 2015;11:332–84. doi: 10.1016/j.jalz.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 4.Cummings JL, Morstorf T, Zhong K. Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther. 2014;6:37. doi: 10.1186/alzrt269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bohm C, Chen F, Sevalle J, Qamar S, Dodd R, Li Y, et al. Current and future implications of basic and translational research on amyloid-β peptide production and removal pathways. Mol Cell Neurosci. 2015;66:3–11. doi: 10.1016/j.mcn.2015.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiang K, Koo EH. Emerging therapeutics for Alzheimer’s disease. Annu Rev Pharmacol Toxicol. 2014;54:381–405. doi: 10.1146/annurev-pharmtox-011613-135932. [DOI] [PubMed] [Google Scholar]
- 7.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–3. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 8.Huang Y. Aβ-independent roles of apolipoprotein E4 in the pathogenesis of Alzheimer’s disease. Trends Mol Med. 2010;16:287–94. doi: 10.1016/j.molmed.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 9.Drzezga A, Grimmer T, Henriksen G, Mühlau M, Perneczky R, Miederer I, et al. Effect of APOE genotype on amyloid plaque load and gray matter volume in Alzheimer disease. Neurology. 2009;72:1487–94. doi: 10.1212/WNL.0b013e3181a2e8d0. [DOI] [PubMed] [Google Scholar]
- 10.Peuralinna T, Tanskanen M, Mäkelä M, Polvikoski T, Paetau A, Kalimo H, et al. APOE and AβPP gene variation in cortical and cerebrovascular amyloid-β pathology and Alzheimer’s disease: a population-based analysis. J Alzheimers Dis. 2011;26:377–85. doi: 10.3233/JAD-2011-102049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koldamova R, Fitz NF, Lefterov I. ATP-binding cassette transporter A1: from metabolism to neurodegeneration. Neurobiol Dis. 2014;72:13–21. doi: 10.1016/j.nbd.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tai LM, Koster KP, Luo J, Lee SH, Wang YT, Collins NC, et al. Amyloid-β pathology and APOE genotype modulate retinoid X receptor agonist activity in vivo. J Biol Chem. 2014;289:30538–55. doi: 10.1074/jbc.M114.600833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao J, Fu Y, Liu CC, Shinohara M, Nielsen HM, Dong Q, et al. Retinoic acid isomers facilitate apolipoprotein E production and lipidation in astrocytes through the retinoid X receptor/retinoic acid receptor pathway. J Biol Chem. 2014;289:11282–92. doi: 10.1074/jbc.M113.526095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, et al. ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–6. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Balducci C, Paladini A, Micotti E, Tolomeo D, La Vitola P, Grigoli E, et al. The continuing failure of bexarotene in Alzheimer’s disease mice. J Alzheimers Dis. 2015;46:471–82. doi: 10.3233/JAD-150029. [DOI] [PubMed] [Google Scholar]
- 16.Veeraraghavalu K, Zhang C, Miller S, Hefendehl JK, Rajapaksha TW, Ulrich J, et al. Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science. 2013;340:924. doi: 10.1126/science.1235505. [DOI] [PubMed] [Google Scholar]
- 17.Fitz NF, Cronican AA, Lefterov I, Koldamova R. Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science. 2013;340:924. doi: 10.1126/science.1235809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Price AR, Xu G, Siemienski ZB, Smithson LA, Borchelt DR, Golde TE, et al. Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science. 2013;340:924. doi: 10.1126/science.1234089. [DOI] [PubMed] [Google Scholar]
- 19.Tesseur I, Lo AC, Roberfroid A, Dietvorst S, Van Broeck B, Borgers M, et al. Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science. 2013;340:924. doi: 10.1126/science.1233937. [DOI] [PubMed] [Google Scholar]
- 20.Farol LT, Hymes KB. Bexarotene: a clinical review. Expert Rev Anticancer Ther. 2004;4:180–8. doi: 10.1586/14737140.4.2.180. [DOI] [PubMed] [Google Scholar]
- 21.Appleby BS, Nacopoulos D, Milano N, Zhong K, Cummings JL. A review: treatment of Alzheimer’s disease discovered in repurposed agents. Dement Geriatr Cogn Disord. 2013;35:1–22. doi: 10.1159/000345791. [DOI] [PubMed] [Google Scholar]
- 22.Graeppi-Dulac J, Vlaeminck-Guillem V, Perier-Muzet M, Dalle S, Orgiazzi J. Endocrine side-effects of anti-cancer drugs: the impact of retinoids on the thyroid axis. Eur J Endocrinol. 2014;170:R253–62. doi: 10.1530/EJE-13-0920. [DOI] [PubMed] [Google Scholar]
- 23.LaFerla FM. Preclinical success against Alzheimer’s disease with an old drug. N Engl J Med. 2012;367:570–2. doi: 10.1056/NEJMcibr1204890. [DOI] [PubMed] [Google Scholar]
- 24.Lowenthal J, Hull SC, Pearson SD. The ethics of early evidence—preparing for a possible breakthrough in Alzheimer’s disease. N Engl J Med. 2012;367:488–90. doi: 10.1056/NEJMp1203104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strittmatter WJ. Old drug, new hope for Alzheimer’s disease. Science. 2012;335:1447–8. doi: 10.1126/science.1220725. [DOI] [PubMed] [Google Scholar]
- 26.de Vries-van der Weij J, de Haan W, Hu L, Kuif M, Oei HL, van der Hoorn JW, et al. Bexarotene induces dyslipidemia by increased very low-density lipoprotein production and cholesteryl ester transfer protein-mediated reduction of high-density lipoprotein. Endocrinology. 2009;150:2368–75. doi: 10.1210/en.2008-1540. [DOI] [PubMed] [Google Scholar]
- 27.Pierrot N, Lhommel R, Quenon L, Hanseeuw B, Dricot L, Sindic C, et al. Targretin improves cognitive and biological markers in a patient with Alzheimer’s disease. J Alzheimers Dis. 2015;49:271–6. doi: 10.3233/JAD-150405. [DOI] [PubMed] [Google Scholar]
- 28.Cummings JL. Optimizing phase II of drug development for disease-modifying compounds. Alzheimers Dement. 2008;4:S15–20. doi: 10.1016/j.jalz.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 29.Cummings JL. Biomarkers in Alzheimer’s disease drug development. Alzheimers Dement. 2011;7:e13–44. doi: 10.1016/j.jalz.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 30.Cook D, Brown D, Alexander R, March R, Morgan P, Satterthwaite G, et al. Lessons learned from the fate of AstraZeneca’s drug pipeline: a five-dimensional framework. Nat Rev Drug Discov. 2014;13:419–31. doi: 10.1038/nrd4309. [DOI] [PubMed] [Google Scholar]
- 31.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–44. doi: 10.1212/WNL.34.7.939. [DOI] [PubMed] [Google Scholar]
- 32.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–98. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 33.Chen K, Roontiva A, Thiyyagura P, Lee W, Liu X, Ayutyanont N, et al. Improved power to characterize longitudinal amyloid-β PET changes and evaluate amyloid-modifying treatments using a cerebral white matter reference region. J Nucl Med. 2015;56:560–6. doi: 10.2967/jnumed.114.149732. [DOI] [PubMed] [Google Scholar]
- 34.Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer’s disease. Am J Psychiatry. 1984;141:1356–64. doi: 10.1176/ajp.141.11.1356. [DOI] [PubMed] [Google Scholar]
- 35.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412–4. doi: 10.1212/WNL.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- 36.Cummings JL, Mega M, Gray K, Rosenberg-Thompson S, Carusi DA, Gornbein J. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology. 1994;44:2308–14. doi: 10.1212/WNL.44.12.2308. [DOI] [PubMed] [Google Scholar]
- 37.Galasko D, Bennett D, Sano M, Ernesto C, Thomas R, Grundman M, et al. An inventory to assess activities of daily living for clinical trials in Alzheimer’s disease: the Alzheimer’s Disease Cooperative Study. Alzheimer Dis Assoc Disord. 1997;11:S33–9. doi: 10.1097/00002093-199700112-00005. [DOI] [PubMed] [Google Scholar]
- 38.Calero O, Hortiguela R, Bullido MJ, Calero M. Apolipoprotein E genotyping method by real time PCR, a fast and cost-effective alternative to the TaqMan and FRET assays. J Neurosci Methods. 2009;183:238–40. doi: 10.1016/j.jneumeth.2009.06.033. [DOI] [PubMed] [Google Scholar]
- 39.Clark CM, Schneider JA, Bedell BJ, Beach TG, Bilker WB, Mintun MA, et al. Use of florbetapir-PET for imaging β-amyloid pathology. JAMA. 2011;305:275–83. doi: 10.1001/jama.2010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Landau SM, Fero A, Baker SL, Koeppe R, Mintun M, Chen K, et al. Measurement of longitudinal β-amyloid change with 18F-florbetapir PET and standardized uptake value ratios. J Nucl Med. 2015;56:567–74. doi: 10.2967/jnumed.114.148981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dubois B, Feldman HH, Jacova C, Hampel H, Molinuevo JL, Blennow K, et al. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol. 2014;13:614–29. doi: 10.1016/S1474-4422(14)70090-0. [DOI] [PubMed] [Google Scholar]
- 42.Youmans KL, Tai LM, Nwabuisi-Heath E, Jungbauer L, Kanekiyo T, Gan M, et al. APOE4-specific changes in Aβ accumulation in a new transgenic mouse model of Alzheimer disease. J Biol Chem. 2012;287:41774–86. doi: 10.1074/jbc.M112.407957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Michaelson DM. APOE ε4: the most prevalent yet understudied risk factor for Alzheimer’s disease. Alzheimers Dement. 2014;10:861–8. doi: 10.1016/j.jalz.2014.06.015. [DOI] [PubMed] [Google Scholar]