

Graphical abstract
Keywords: Retinal pigment epithelial cells, Oxidative stress, RTA 408, Nrf2
Abstract
Oxidative stress-induced retinal pigment epithelial (RPE) cell damage is an important factor in the pathogenesis of age-related macular degeneration (AMD). Previous studies have shown that RTA 408, a synthetic triterpenoid compound, potently activates Nrf2. This study aimed to investigate the protective effects of RTA 408 in cultured RPE cells during oxidative stress and to determine the effects of RTA 408 on Nrf2 and its downstream target genes. Primary human RPE cells were pretreated with RTA 408 and then incubated in 200 μM H2O2 for 6 h. Cell viability was measured with the WST-8 assay. Apoptosis was quantitatively measured by annexin V/propidium iodide (PI) double staining and Hoechst 33342 fluorescent staining. Reduced (GSH) and oxidized glutathione (GSSG) were measured using colorimetric assays. Nrf2 activation and its downstream effects on phase II enzymes were examined by Western blot. Treatment of RPE cells with nanomolar ranges (10 and 100 nM) of RTA 408 markedly attenuated H2O2-induced viability loss and apoptosis. RTA 408 pretreatment significantly protected cells from oxidative stress-induced GSH loss, GSSG formation and decreased ROS production. RTA 408 activated Nrf2 and increased the expression of its downstream genes, such as HO-1, NQO1, SOD2, catalase, Grx1, and Trx1. Consequently, the enzyme activities of NQO1, Grx1, and Trx1 were fully protected by RTA 408 pretreatment under oxidative stress. Moreover, knockdown of Nrf2 by siRNA significantly reduced the cytoprotective effects of RTA 408. In conclusion, our data suggest that RTA 408 protect primary human RPE cells from oxidative stress-induced damage by activating Nrf2 and its downstream genes.
Highlights
-
•
Oxidative stress contributes to the pathogenesis of AMD.
-
•
RTA408 in nanomolar concentrations increased survival and restored the redox state in H2O2-damaged RPE cells.
-
•
RTA408 stimulated Nrf2 and downstream antioxidant enzymes Grx1, NQO1, and HO-1.
-
•
Inhibition of Nrf2 abolished RTA408’s protective effects.
-
•
RTA408 may have the potential to treat AMD and other degenerative eye diseases.
1. Introduction
Age related macular degeneration (AMD), a leading cause of blindness in the elderly, is a disease in which there is progressive loss of central vision [1]. AMD occurs in two major forms, the dry form and the wet form [2]. Anti-vascular endothelial growth factor (VEGF) antibody therapy has revolutionized the treatment of wet AMD [3]. However, dry AMD treatment remains a major challenge. Currently, the only treatment for dry AMD is the use of the Age-Related Eye Disease Study (AREDS)-based vitamin formulation, which includes vitamin C, vitamin E, zinc oxide, cupric oxide, lutein, and zeaxanthin [4], [5]. However, this formulation does not reverse vision loss but only lowers the risk of developing advanced stages of AMD in certain patients. Therefore, identifying novel therapeutic targets and development of novel therapeutic molecules for AMD are urgently needed.
Oxidative stress-induced retinal pigment epithelial (RPE) cell death is an early event in the development of AMD [6]. The RPE cells remain in a quiescent state throughout life. RPE cells present at birth are constantly exposed to years of oxidative damage before the onset of AMD. Therefore, RPE are very sensitive to oxidative damage, often induced by external sources like UV light and internal sources like reactive oxygen species (ROS) produced by the electron transport chain. Proteins are the main targets of free radicals due to their high abundance and their high reactivity with ROS. As oxidative stress defense systems deteriorate with age, oxidatively modified proteins gradually accumulate underneath the RPE adjacent to the basement membrane and lead to drusen formation, which is the hallmark of AMD [7]. Thus, understanding the function of antioxidant pathways in the retina is critical for developing new therapies for AMD.
One of the crucial antioxidant pathways involved is the nuclear factor (erythroid-derived-2)-like 2 (Nrf2) pathway. Nrf2 is a 65 kDa molecule with a basic leucine zipper structure. Normally, Nrf2 in its inactive state is kept in the cytoplasm bound to kelch-like ECH-associated protein 1 (Keap1) [8], [9]. With a half-life of only 20 min, Nrf2 is constantly targeted for ubiquitination by Keap1 with consequential degradation via the proteasome. When the cell is in an oxidative stress environment, oxidative stress oxidizes Keap1’s active site cysteine residues, preventing Keap1 from interacting with Nrf2. With the accumulation of Nrf2 in the cytoplasm, Nrf2 moves to the nucleus where it binds to the small Maf protein and the antioxidant response element (ARE). Activation of ARE leads to the transcriptional activation of several other antioxidant enzymes and proteins, such as NADPH dehydrogenase (NQO1), heme oxygenase-1 (HO-1), glutaredoxin 1 (Grx1), and thioredoxin 1 (Trx1) [10]. All these enzymes are distinguished by their ability to reverse oxidative damage and stress. NADPH dehydrogenase transforms enzymes and proteins back into their reduced state by the exchange of electrons between NADPH and NADP [11]. HO-1 may be involved indirectly in the antioxidant system by converting heme to other products such as iron (II), carbon monoxide, and biliverdin [12]. Glutaredoxin and thioredoxin are two distinct yet similar systems. Although they are both involved in reducing oxidized protein thiols and allowing proteins to return to their functional state, Grx1 is considered as a vital antioxidant enzyme, considering its essential locations in both the cytoplasm [13], [14], the intermembrane space of mitochondria [15], and possibly, the nucleus. Therefore, drugs enabling and amplifying the Nrf2 system are thought to be promising therapies for AMD and other degenerative diseases that rely on the delicate balance of oxidative species in the cell.
RTA 408 represents a novel class of therapeutics that has the potential to increase Nrf2 expression and thereby increase expression of antioxidant enzymes. RTA 408 is a member of the synthetic oleanane triterpenoid compounds. It is currently under clinical investigation for the prevention of cataract surgery-induced loss of corneal endothelial cells, prevention of radiation-induced dermatitis in breast cancer patients undergoing radiotherapy, treatment of solid tumors including melanoma and lung cancer, and treatment of Friedreich’s Ataxia and mitochondrial myopathies. Previous studies have demonstrated that RTA 408 has significant cytoprotective effects attributed to the activation of the Nrf2 pathway [16], [17], [18], [19]. The present study investigates the connection between RTA 408 and the Nrf2 pathway as well as multiple antioxidant enzymes in RPE cells. This will help determine whether RTA 408 may serve as a potent therapy for AMD and other degenerative eye diseases.
2. Methods
2.1. Materials
The 2-cyano-3,12-dioxooleana-1,9 (11)-dien-28-oic acid (CDDO) derivative RTA 408 was >98% pure (Reata Pharmaceuticals, Inc., Irving, TX, USA). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), penicillin/streptomycin, and 0.05% trypsin and other cell culture reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA). Hydrogen peroxide and other chemicals were obtained from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise stated. Antibodies listed below were used: anti-Nrf2 (Cell Signaling, Beverly, MA, USA, #12721), anti-NQO1 (Cell Signaling, #3187), anti-Bax (Cell Signaling, #2772), anti-Bcl2 (Cell Signaling, #2876), anti-cleaved caspase 3 (17 kDa) (Cell Signaling, #9664), anti-HO-1 (Cell Signaling, #5061), anti-Grx1 (Abcam, Cambridge, MA, USA, ab45953), anti-Trx1 (Abcam, ab86255), anti-PSSG (Virogen, Watertown, MA, USA, 101-A-100), anti-SOD2 (signal, HPA001814), anti-catalase (Abcam, ab16731), anti-GAPDH (Santa Cruz, Santa Cruz, CA, USA, sc-32233), anti-B23 antibodies, and horseradish peroxidase-conjugated secondary antibodies (sc2061, sc2060, sc2030; Santa Cruz Biotechnology, Santa Cruz, CA, USA).
2.2. Human retinal pigment epithelial (RPE) cell culture
Human fetal RPE cells were purchased at passage one from ScienCell™ Research Laboratories (Carlsbad, CA, USA), and all experiments were performed with cells between passages two to eight. The cells were maintained in DMEM supplemented with 10% FBS, 100 μg/ml streptomycin, and 100 U/ml of penicillin. Cell cultures were maintained at 37 °C in a humid atmosphere incubator with 5% CO2 and 95% air. The medium was changed every 3-4 days. For H2O2-induced apoptotic studies, cells were synchronized by gradual serum deprivation with the following procedure: cells were cultured overnight in DMEM with 2% FBS followed by incubation in serum-free medium for 30 min before exposure to a bolus of 200 µM H2O2 for 6 h.
2.3. Cell viability assay
Cell viability was measured by a colorimetric cell viability kit (Promokine, Heidelberg, Germany) with the tetrazolium salt WST-8 (2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt), which can be bioreduced to a water-soluble orange formazan dye by dehydrogenases present in the viable cells. The amount of formazan produced is directly proportional to the number of living cells. Cells were seeded at a density of 5000 cells/well (100 μl total volume/well) in a 96-well assay. Cells were incubated with or without RTA 408 (1–100 nM) for 24 h and then treated with 200 µM of H2O2 for 6 h. After treatment, 10 µl of WST-8 solution was added to each well of the culture plate and incubated for 2 h in the incubator. The absorption was evaluated at 450 nm using a microplate reader (BioTek, Winooski, VT).
2.4. Hoechst 33342 fluorescent staining
Cells were plated on poly-l-lysine coated glass coverslips for overnight and then incubated with or without RTA 408 (1–100 nM) for 24 h followed by 200 μM H2O2 treatment for 6 h. The cells were washed twice with ice cold PBS and then fixed with cold 4% paraformaldehyde for 15 min followed by PBS wash. The fixed cells were then stained with 0.1 µg/mL Hoechst 33342 (Invitrogen, Grand Island, NY) for 10 min and rinsed with PBS. The images were taken using a fluorescence microscope (Olympus, Center Valley, PA).
2.5. Flow cytometry analysis of cell apoptosis
Cells were seeded in 100 mm culture plates and incubated overnight at 37 °C and then incubated with or without RTA 408 (1–100 nM) for 24 h followed by 200 μM H2O2 treatment for 6 h. After that, the cells were trypsinized and stained with annexin V and PI using annexin V apoptosis Kit (Invitrogen, Grand island, NY) according to the manufacturer’s protocol. The stained cells were then analyzed by flow cytometer (FC500, Beckman Colter, Indianapolis, IN) to differentiate among viable (annexin V−/PI−), early apoptotic (annexin V+/PI−), late apoptotic (annexin V+/PI+) cells, and necrotic cells (annexin V−/PI+).
2.6. ROS detection
Intracellular ROS levels were determined using the fluorescent probe CellROX Orange Reagent (Life Technologies, city, state). After pretreatment with RTA 408 for 24 h, RPE cells were exposed to 200 μM H2O2 for another 30 min. After treatment, the cells were loaded with 5 mM CellROX Orange Reagent for 30 min at 37 °C, washed with DPBS and immediately imaged on a fluorescence microscope (Olympus, Center Valley, PA). The cellular fluorescence was quantified using the ImageJ software, after background subtraction for each image.
2.7. GSH and GSSG assays
Cells were collected on ice using cell scrapers and were lysed with RIPA buffer (Sigma, St. Louis, MO, USA). For glutathione (GSH) measurement, an aliquot of fresh cell lysate was treated with an equal volume of 20% trichloroacetic acid followed by centrifugation. The supernatant was used immediately for GSH analysis using Ellman’s (5,5’-dithiobis-(2-nitrobenzoic acid)) reagent, following the method of Lou et al. [20]. GSSG level was measured by using a oxidized glutathione (GSSG) assay kit (Abcam, Cambridge, MA, USA) according to the manufacturer’s instruction.
2.8. Western blot analysis
Protein concentration was determined with a BCA assay kit (Thermo Scientific, Rockford, IL). Equal amounts of protein were boiled in Laemmli buffer (Bio-Rad, Bio-Rad, Richmond, CA, USA) and loaded onto 12% SDS-PAGE gel and transferred to a 0.2 µm polyvinylidene difluoride membrane (GE Healthcare, Boulder, CO). The membranes were incubated with appropriate primary antibodies for overnight at 4 °C and were then incubated with the appropriate secondary antibodies for 1 h. Detection was performed using the ECL Western blotting detection system (Thermo Scientific, Rockford, IL). The immunoblot was analyzed with a Bio-Rad imaging system (Versadoc 5000 MP Imaging System, Bio-Rad, Richmond, CA, USA).
2.9. Nrf2 translocation
Cells were treated without and with RTA 408 for 0.5, 2, and 6 h. The cytoplasmic and nuclear fractions of each sample were obtained using the NE-PER Nuclear and Cytoplasmic Extraction Reagents from Thermo Scientific (Thermo Scientific, Rockford, IL). 60 μg of each sample was loaded onto a 12% SDS-PAGE gel and transferred onto a PVDF membrane. The membranes were incubated with Nrf2 antibody for overnight at 4 °C and were then incubated with the appropriate secondary antibodies for 1 h. β-actin and B23 were used as cytoplasmic and nuclear marker, respectively. Detection was performed using the ECL Western blotting detection system (Thermo Scientific, Rockford, IL). The immunoblot was analyzed with a Bio-Rad imaging system (Versadoc 5000 MP Imaging System, Bio-Rad, Richmond, CA, USA).
2.10. Protein glutathionylation detection
Cells were pretreated with or without 100 nM RTA 408 for 24 h and then incubated with 1 mM H2O2 for 30 min and lysed with RIPA buffer (Sigma). Equal amounts of protein (80 μg) from each group were loaded onto an SDS-PAGE gel under non-reducing (no β-mercaptoethanol) conditions, immunoblotted, and probed with an anti-PSSG antibody, which previously has been shown to selectively recognize glutathionylated proteins [21], [22], [23].
2.11. Enzyme activity assays
Grx1 was assayed using 2-Hydroxyethyl disulfide (HEDS) as the substrate according to the method described in Raghavachari and Lou [24]. In brief, the cell lysate was mixed with potassium phosphate buffer (200 mM, pH 7.5) containing 0.5 mM glutathione (GSH), 0.4 U/mL glutathione reductase (GR), 0.2 mM β-nicotinamide adenine dinucleotide phosphate, reduced tetra (cyclohexylammonium) salt (NADPH), and 2 mM hydroxyethyl disulfide
(HED). The reaction was carried out at 30 °C, and the decrease in absorbance at 340 nm was monitored for 5 min and used to determine the Grx1 activity. Trx1 activity was determined using the methods of Holmgren and Bjornstedt [25]. The cell lysate was mixed with potassium phosphate buffer (100 mM, pH 7.0, with 2 mM EDTA) containing 0.05 U/mL thioredoxin reductase (TR), 0.2 mM NADPH, and 0.14 mM insulin. The reaction was carried out at 30 °C, and the decrease in absorbance at 340 nm was monitored for 5 min and used to determine the Trx1 activity. NQO1 activity was measured using NQO1 activity assay kit (Abcam) according to manufacturer’s instructions.
2.12. SiRNA transfection
Control and Nrf2 siRNA used in this study were chemically synthesized by Santa Cruz Biotechnology (Santa Cruz, CA, USA). SiRNA duplexes (scrambled siRNA, sc-37007; Nrf2 siRNA, sc-72089) were transfected into primary human RPE cell with siRNA transfection reagent (sc-29528; Santa Cruz, CA, USA) according to the manufacturer’s instructions.
2.13. Statistics
Each experiment was performed at least three times and statistical analyzes were performed using Student’s t-test when comparing between two groups and one-way ANOVA followed by Bonferroni’s test as a post hoc test when comparing among three or more groups with the Prism software (GraphPad, La Jolla, CA). The number of experimental samples used in each group is presented in the. All data are expressed as means±SD and differences were considered significant at P<0.05.
3. Results
3.1. RTA 408 inhibits H2O2-induced cytotoxicity in RPE cells
RTA 408 at concentrations 1 to 100 nM had no apparent cytotoxic effects on RPE cells (Fig. 1A). To examine the cytotoxicity of H2O2, RPE cells were treated with different concentration of H2O2 (100–500 μM) for 6 h followed by a cell viability assay. As shown in Fig. 1B, H2O2 dose-dependently inhibited RPE cell survival. 200 uM of H2O2, which induced around 60% of cell viability loss, was used in the following experiments. To investigate the cytoprotective effects of RTA 408, RPE cells were pretreated with 1–100 nM of RTA 408 for 24 h followed by addition of 200 uM H2O2 for 6 h. Pretreatment with 1, 10, and 100 nM RTA 408 increased cell viability to 52.7±1.2% (mean±SD, n=6, P<0.01), 80.1±3.1% (P<0.01), and 95.4±2.8% (P<0.01), respectively (Fig. 1C).
Fig. 1.

RTA 408 protected primary human RPE cells against H2O2-induced oxidative damage and cell death. (A) Lack of RTA 408 cytotoxicity in RPE cells. Cell viability from different concentrations of RTA 408 treated (1, 10, and 100 nM), non-treated (control), and vehicle (DMSO-treated) RPE cells was measured by WST-8 assay. Each value is expressed as the mean±SD (n=6). (B) Dose-dependent effect of H2O2 on the viability of RPE cells. Cells were exposed to 0, 100, 200, 300, 300, 400, or 500 µM H2O2 for 6 h. Viable cells were quantified by WST-8 assay. *P<0.05, **P<0.01, ***P<0.001 comparing with the 0 μM H2O2 group. (C) Cytoprotective effects of RTA 408. RPE cells were first pretreated with 1 nM, 10 nM, and 100 nM of RTA 408 for 24 h and then incubated in 200 uM H2O2 for 6 h. Control and vehicle (DMSO-treated) groups were also incubated in 200 uM H2O2 for 6 h as well for comparison to RTA 408 treated groups. Cell viability was measured via the WST-8 assay and represented by mean±SD. ***P<0.001 comparing with the H2O2 alone group. (n=6) (D) Cell apoptosis in RTA 408 treated cells. RPE cells were first pretreated with or without 100 nM RTA 408 for 24 h and then incubated in 200 uM H2O2 for 6 h. Cells were then subjected to Hoechst staining. Apoptotic cells are labeled with white arrows as having a nuclear shrinkage or strong fluorescence, (n=3).
3.2. Anti-apoptotic effects of RTA 408
To better understand the anti-apoptotic effect of RTA 408, cell apoptosis was evaluated by two different assays: Hoechst 33342 nuclei staining and Annexin V/PI double staining. As shown in Fig. 1D, the control and RTA 408 alone groups both showed normal nuclear morphology with uniform blue chromatin and organized structure. After adding 200 μM H2O2 for 6 h, RPE cells showed condensed nuclei, increased fluorescent intensity, and fragmented chromatin (labeled with arrows). In contrast, 100 nM RTA 408 effectively prevented H2O2-induced nuclear changes as indicated by less nuclear fragmentation and condensation. To further confirm the anti-apoptotic effects of RTA 408, annexin V/PI double staining and flow cytometry analysis were performed to quantify early (annexin V+/PI-) or late apoptotic (annexin V+/PI+) cells. The representative images for flow cytometry and the summarized data are presented in Fig. 2. Cell apoptosis levels were similarly low in control and RTA 408 treatment alone cells. However, after H2O2 treatment, the rate of early apoptosis increased to 30.5±5.6% but remained very low (4.6±0.5%) in the RTA 408 pretreated group (P<0.05) (Fig. 2B). Consistently, after exposure to H2O2, the number of late apoptotic cells increased to 13.7±3.3%, and pretreatment with RTA 408 reduced the percentage of late apoptosis to 2.1±1.2% (Fig. 2C).
Fig. 2.

RTA 408 decreases apoptotic cell death in H2O2-treated RPE cells. (A) Flow cytometry of annexin/PI double stained of control, RTA 408 treatment only, H2O2-treated only, and RTA 408 and H2O2-treated RPE cells, showing live cells in quadrant A3, early apoptotic cells in quadrant A4, late apoptotic cells in quadrant A2, and necrosis in quadrant A1. Representative figures showing population of viable (annexin V−/PI−), early apoptotic (annexin V+/PI−), late apoptotic (annexin V+/PI+) and necrotic (annexin V−/PI+) cells. Bar graphs showing the quantification of early (B) and late apoptotic (C) cells. Data were mean±SD of three independent experiments. ***P<0.001 compared with H2O2 only group, (n=3).
3.3. RTA 408 inhibits Bax and caspase 3 activation and upregulates Bcl-2
The pro-apoptotic protein Bax initiates cell apoptosis by directly binding with Bcl-2, thus antagonizing the anti-apoptotic effect of Bcl-2. Under oxidative stress, Bax undergoes a conformational change and translocates from the cytosol to the mitochondrial membrane, leading to the release of cytochrome C and activation of caspase 3 [26]. Therefore, the Bax/Bcl2 ratio and caspase 3 cleavage were measured to further confirm the anti-apoptotic effect of RTA 408. As demonstrated in Fig. 3A and B, protein level of anti-apoptotic factor Bcl-2 was suppressed by ~80% after H2O2 treatment. However, 100 nM RTA 408 could block the oxidative stress mediated reduction in Bcl-2 expression. In contrast, the pro-apoptotic Bax increased to three fold over control after 200 μM H2O2 treatment. The increased level of Bax was significantly inhibited by pretreatment with 100 nM RTA 408 (Fig. 3A and C). Furthermore, caspase 3 protein was not detected in control cells; however, oxidative stress-induced caspase 3 cleavage as seen by the appearance of caspase 3 positive band at 17 kDa in H2O2-treated cells but not in the RTA 408 pretreated group (Fig. 3A and C). These results indicate that RTA 408 treatment strongly protected RPE cells from H2O2-induced apoptosis.
Fig. 3.

Effects of RTA 408 treatment on Bcl-2, Bax, and caspase 3 levels. (A) Whole cell lysates of control (no H2O2 treatment), H2O2-treated, and both RTA 408 and H2O2-treated RPE cells were prepared, and 60 ug of each protein sample was subjected to SDS-PAGE and then blotted onto a PVDF membrane. The blot was incubated with Bcl2, Bax, and cleaved caspase 3 antibodies. GAPDH was used as a reference control for equal protein loading. (B) Quantitative analysis of Bcl2 protein expression levels in all treatment groups are represented as mean±SD. (C) Quantitative analysis of Bax protein levels in all treatment groups are represented on a bar graph as mean±SD. (D) Quantitative analysis of western blot data of cleaved caspase 3 shown in bar graph form, ***P<0.001, comparing H2O2 only group, (n=3).
3.4. Inhibition of ROS production by RTA 408
Intracellular ROS levels were measured by preloading the cells with CellROX Orange followed by stimulating with H2O2 to monitor the resulting ROS sensitive fluorescent product. Fig. 4 shows the fluorescence in the live RPE cells by confocal microscopy 30 min after H2O2 exposure. No fluorescence could be detected at the same time interval in the control cells where no H2O2 was added (Fig. 4A). H2O2 treatment for 30 min induced a massive production of ROS (Fig. 4B). RTA 408 significantly suppressed ROS production in a dose-dependent manner (Fig. 4C–E). Quantitative fluorescence intensities of CellROX Orange in the various groups are shown in Fig. 4F.
Fig. 4.

RTA 408 significantly reduces reactive oxygen species (ROS) production in H2O2-treated RPE cells. RPE cells were pretreated with or without 1 nM, 10 nM, and 100 nM RTA 408 for 24 h followed by 200 µM H2O2 treatment for 30 min. Fluorescence was detected using the probe CellROX Orange reagent for all groups and control (no H2O2 treatment) cells. Fluorescence intensity is quantified and represented as mean±SD via a bar graph, ***P<0.001 compared with H2O2 only group, (n=3).
3.5. Preservation of the redox state in RTA 408 treated RPE cells
The glutathione disulfide-glutathione couple (GSSG/GSH) can serve as an important indicator of cellular redox environment. After H2O2 treatment, the intracellular GSH levels in RPE cells were significantly decreased from 25.6±2.9 to 5.2±2.1 nmol/mg protein (Fig. 5A). However, only less than a 10% decrease in GSH levels was observed in RTA 408 pretreated cells after H2O2 exposure. RTA 408 treatment alone slightly increased GSH level to 35.2±6.5 nmol/mg. On the other hand, the level of GSSG was significantly increased in response to H2O2 but remained relatively low in RTA 408 pretreated cells (without RTA 408 pretreatment: 2.4±0.3 vs RTA 408 treatment: 1.2±0.3 nmol/mg protein, P<0.05) (Fig. 5B). RTA treatment alone had no apparent effect on cellular GSSG levels. Furthermore, H2O2 treatment caused a significant increase in protein glutathionylation, an index of protein cysteine thiol oxidation. Pretreatment with 100 nM RTA 408 significantly inhibited the H2O2-induced protein glutathionylation by ~60% (Fig. 5C,D).
Fig. 5.

Effects of RTA 408 treatment on levels of GSH, GSSG, and protein-GSH mixed disulfide (PSSP) levels. Cells were pretreated with 100 nM RTA 408 for 24 h followed by 200 μM H2O2 challenge for 6 h. GSH levels (A), GSSG levels (B) were measured as described in Materials and Methods. (C) Comparison of protein glutathionylation (PSSG) levels in RTA 408 pretreated cells. The cells were treated with 1 mM H2O2 for 30 min and total proteins were separated on a 12% SDS-gel under non-reducing condition (no β-mercaptoethanol) for western blot analysis using an anti-PSSG antibody. (D) The relative pixel density of all the PSSG bands compared to β-actin. Data are mean±SD of three independent experiments, **P<0.01, and ***P<0.001, comparing with H2O2 only group, (n=3).
3.6. RTA 408 activates Nrf2 and its downstream genes in RPE cells
To investigate the effect of RTA 408 on Nrf2 and its downstream genes, cells were treated with RTA 408 at 1, 10, and 100 nM for 24 h. Western blot analysis indicated a dose-dependent increase of Nrf2, HO-1, NQO1, SOD2, catalase, Grx1, and Trx1 protein expression (Fig. 6A). When cells were treated with 100 nM RTA 408 for various time, Nrf2 and its downstream targets were upregulated as early as 6 h after RTA 408 treatment and lasted for at least 24 h (Fig. 6B). RTA 408 also enhanced Nrf2’s nuclear translocation. In the cytoplasm, Nrf2 expression slowly decreased, as more Nrf2 accumulated and progressively moved into the nucleus (Fig. 6C and D). This data verifies that RTA 408’s main protective mechanism is through Nrf2 activation.
Fig. 6.

Effects of RTA 408 on expression of Nrf2 and downstream genes. (A) Dose-dependent effects of RTA 408 on Nrf2 and downstream gene expression. RPE cells were pretreated with 1–100 nM RTA for 24 h. Control cells were treated identically, but without addition of RTA 408. Whole cell lysates of each group were prepared, and 60 ug of each protein sample was used to detect the levels of Nrf2, SOD2, catalase, NQO1, HO-1, Grx1, and Trx1 by western immunoblotting. GAPDH was used as a loading control. (B) Time-dependent effects of RTA 408 on Nrf2 and its downstream gene expression. Cells were treated with 100 nM RTA for various time (0–24 h). 60 μg of each protein sample was subjected to western immunoblot analysis. The blot was incubated with GAPDH, Nrf2, SOD2, catalase, NQO1, HO-1, Grx1, and Trx1 antibodies. GAPDH was used as a reference for equal protein loading. All experiments were repeated three times with similar results. (C) Nrf2 expression in the cytoplasmic and nuclear fractions. Cells were treated with 100 nM of RTA 408 for various time points (0, 0.5, 2 and 6 h) and then were separated to its nuclear and cytoplasmic portions using a kit. 40 μg of each sample were subjected to western immunoblot analysis and incubated with B23, Nrf2, and β-actin antibodies. All experiments were repeated three times with similar results. (D) Quantitative analysis of time-dependent Nrf2 translocation are depicted as mean±SD, **P<0.01, and ***P<0.001, comparing control (0 h) group (n=3).
3.7. RTA 408 increases NQO1, Grx1, and Trx1 activities
To further investigate the effects of RTA 408 on Nrf2 downstream target genes, the enzyme activities of Grx1, Trx1, and NQO1 were examined. The activities of Grx1 and Trx1 in normal RPE cells were 5.8±1.1 and 4.2±0.4 mU/mg protein, respectively. When cells were exposed to H2O2, Grx1 was slightly decreased to below 35% of the initial activity but was restored up to 95% of its normal level when pretreated with RTA 408 (Fig.7A). Similarly, H2O2 treatment caused a significant decrease in Trx1 activity to ~50% control levels, and RTA 408 pretreatment (100 nM for 24 h) provided a full protection (Fig. 7B). As shown in Fig. 7C, strong protection was also observed for NQO1 activity in RTA 408 pretreated cells. H2O2 treatment inhibited NQO1 enzyme activity by 40%, whereas RTA 408 pretreatment almost completely protected NQO1 from H2O2-induced inactivation.
Fig. 7.

Effects of RTA 408 on Grx1, Trx1, and NQO1 enzyme activities. RPE cells were pretreated with 100 nM RTA 408 for 24 h followed by 200 μM H2O2 for another 6 h. Quantitative analysis of Grx1 (A), Trx1 (B), and (C) NQO1 activity of all treatment groups (control, H2O2-treated, RTA 408 and H2O2-treated, and 100 nM RTA 408 only RPE cells) are depicted as mean±SD, **P<0.01, and ***P<0.001, comparing H2O2 only group (n=3).
3.8. Nrf2 siRNA abolishes the cytoprotective effects of RTA 408
SiRNA was used to confirm that RTA 408-induced Nrf2 activation protects cells from H2O2-induced cell injury. Nrf2 siRNA-treated groups showed lower levels of Nrf2 as compared to scrambled siRNA transfected and non-transfected control cells (Fig. 8A and B). Under non-stressed condition, Nrf2 inhibition slightly lowered RPE cell viability. When cells were challenged with H2O2, Nrf2 siRNA transfected cells had a much lower survival rate compared with that of scrambled siRNA transfected cells. Furthermore, as shown in Fig. 8C, the cytoprotective effect of RTA 408 was significantly blocked by the Nrf2 siRNA, indicating RTA 408 promotes cell survival through Nrf2 activation.
Fig. 8.

Nrf2 knockdown abolished the cytoprotective effects of RTA 408 in RPE cells. RPE cells were transfected with or without scrambled or Nrf2 siRNA for 48 h, then incubated with or without 100 nM RTA 408 for 24 h, followed by 200 μM H2O2 treatment for another 6 h. (A) Western blot analysis of Nrf2 protein levels. Total 60 µg of protein of each sample was subjected to Western blot with Nrf2 and GAPDH antibodies. (B) Bar graph of western blot data showing Nrf2 protein expression normalized to GAPDH expression. Values are the mean±SD, ***P<0.001 comparing scramble siRNA group, (n=6). (C) Cell viability was measured in RPE cells transfected with or without Nrf2 siRNA. Values are the mean±SD, **P<0.01, and ***P<0.001, comparing scramble siRNA group (n=6).
4. Discussion
Age-related macular degeneration (AMD) is a leading cause of irreversible blindness and loss of central vision. Dry AMD has no known current treatment and can account for nearly 90% of all AMD cases, highlighting the importance of finding a good therapy for treating this form of AMD [1]. Because oxidative stress plays a significant role in AMD pathogenesis, antioxidant therapy has been a novel proposed solution in treating AMD [6]. Specifically, reinforcing the cellular defense systems that protect the retina and RPE cells against oxidative stress is a viable option for preventing and/or reducing the progression of macular degeneration. Our data show that RTA 408 protects RPE cells from oxidative stress-induced cell death, and this protective mechanism is related to Nrf2 activation. The present study demonstrated that: (1) RTA 408 protected primary human RPE cells from H2O2-induced cell death in a dose- and time-dependent manner. (2) RTA 408 inhibited pro-apoptotic factors like Bax and caspase 3 and promoted anti-apoptotic factors like Bcl-2. (3) RTA 408 pretreatment restored the redox state in oxidatively damaged cells as indicated by less ROS production, higher GSH levels, and less protein glutathionylation. (4) In nanomolar ranges, RTA 408 potently activated the expression of Nrf2 and its downstream targets SOD2, catalase, NQO1, HO-1, Grx1, and Trx1.
Nrf2 is a basic leucine zipper transcription factor which binds to a promoter sequence known as the antioxidant-responsive element (ARE), leading to coordinated induction of a battery of cytoprotective and antioxidant genes [10]. Increased Nrf2 activity has often correlated with cellular survival and protection. Johnson and others have shown previously that Nrf2 activation can protect against oxidative injury to the RPE [27], [28]. Conversely, mice that are deficient of Nrf2 developed pathological features similar to human AMD [29]. These data strongly suggest that Nrf2-mediated signaling pathways are essential in protecting the RPE cells against aging and age-related degeneration. As such, Nrf2-activating drugs are of current interest in the treatment of AMD.
In particular, RTA 408 is a triterpenoid CDDO derivative that reversibly and covalently modifies reactive cysteine residues on multiple proteins including Keap1, the negative regulator of Nrf2. Binding of RTA 408 to Cys151 in Keap1 results in Keap1 inhibition, thereby activating the Nrf2 signaling pathway and its downstream target genes to protect many different tissues from various toxic insults [30]. For example, Reisman et al. reported that topical application of RTA 408 protected mice from radiation-induced dermatitis by a Nrf2-induced cytoprotective response [17], [18]. They also found that RTA 408 provided robust radiation protection to the murine gastrointestinal and hematopoietic system and improved overall survival of irradiated mice [18]. As well as inducing protection of normal tissues, RTA 408 was also shown to inhibit human prostate cancer xenograft growth in mice [19]. More recently, it has been proposed that RTA 408 may address and treat ophthalmic disorders. For example, a phase II clinical trial is ongoing to test an RTA 408 ophthalmic suspension for the prevention of corneal endothelial cell loss following cataract surgery. Although several studies have indicated that RTA 408 is a potent cytoprotective agent in multiple different cell types, our experiment is the first to show its protective function in human RPE cells against oxidative stress.
Activation of Nrf2 by RTA 408 upregulates multiple enzymes and proteins capable of detoxifying electrophiles and ROS, which simultaneously prevents oxidative damage to critical macromolecules like proteins, DNA, or lipids. RTA 408 also decreases ROS-mediated activation of pro-inflammatory transcription factors. Nanomolar concentrations of RTA 408 promoted cellular protection and survival and eliminated oxidative stress-induced RPE cell injury demonstrating high potency and efficacy in these assays. Compared to other RPE protective agents such as sulforaphane [31], [32], lipoic acid [33], [34], and sulindac [35], RTA 408 required much lower doses to exert cytoprotective effects. Furthermore, Nrf2 activation can also cause other ROS scavenging activities and upregulation of key cytoprotective molecules such as GSH and the glutaredoxin systems [8], [36]. As the most abundant low molecular weight thiol-containing molecule, GSH takes an active part in scavenging ROS, both by direct reaction with ROS and as the cofactor of other antioxidant enzymes, including glutathione peroxidase and Grx1 [14]. Once oxidized, GSH is converted into GSSG and GSH/GSSG ratio decreases, which further leads to protein glutathionylation. Protein glutathionylation, defined as the reversible formation of a mixed disulfide (PSSG) between protein thiols and glutathione (GSH), appears to be the most important mode of thiol oxidation [37]. In high oxidative stress environments, protein glutathionylation accumulates, causing protein inactivation, damage, and eventually cell death [13], [14]. However, RTA 408 treatment diminished protein glutathionylation and increased GSH levels, enhancing the redox state of RPE cells. Therefore, RTA 408 may have the potential to prevent or reverse oxidative damage commonly found in AMD and other retinopathies.
Our data in Fig. 6C strongly supports RTA 408’s ability to stimulate the Nrf2 pathway, as RTA 408 treatment significantly increased Nrf2 accumulation and translocation into the nucleus. This is the primary mechanism for how RTA 408 exerts its powerful antioxidant effects. Following the treatment of primary RPE cells with RTA 408, we also observed significant increases in phase II enzymes including SOD2, catalase, NQO1, HO-1, Grx1, and Trx1. Grx1 belongs to the oxidoreductase family and is a component of the endogenous antioxidant defense system [38]. Grx1 is critical for protecting human RPE cells against oxidative damage, and our previous study has shown that Grx1 prevented oxidative stress-induced protein glutathionylation [14]. The protective effect of Grx1 is also associated with its ability to prevent oxidative stress-induced AKT glutathionylation and thus stimulate AKT activation, a pathway involved in the transcription of several survival and protective genes [14]. Another powerful and crucial oxidoreductase that functions similarly to Grx1, Trx1 can reverse protein oxidation that results in disulfide bonds and cause cellular protection. Trx1 upregulation has corresponded with anti-apoptotic effects and cell survival [39], [40]. Working closely together, SOD2 and catalase are vital enzymes involved in the detoxification of hydrogen peroxide and superoxide, both extremely damaging molecules involved in AMD [41], [42], [43], [44]. Another Nrf2 downstream target and powerful indirect antioxidant enzyme, HO-1 can also convert heme to beneficial byproducts such as carbon monoxide and bilirubin that can directly scavenge free radicals and repair DNA damage caused by oxidative stress [12], [28]. RTA 408 also stimulates NQO1 expression, another extremely important enzyme that reduces oxidized proteins and promotes a better cellular redox state.
The Nrf2 activation capabilities of RTA 408 also affect the apoptotic pathway by decreasing the expression of pro-apoptotic Bax and stimulating anti-apoptotic Bcl2 during oxidative insults. The cytoprotective effects of RTA 408 was also seen by Annexin V/PI double staining. These data clearly show that H2O2 mediated RPE apoptosis and necrosis was significantly decreased by treatment with RTA 408. Overall, RTA 408 enhanced antioxidant and survival proteins’ expression by stimulating Nrf2 activation, leading to the prevention and reversal of damage generated by oxidative insults.
To specifically show that RTA 408 was involved in the Nrf2 signaling pathway, we decided to observe the effect of RTA 408 on cell viability and Nrf2 downstream proteins in Nrf2-deficient cells. Therefore, we knocked down Nrf2 using a specific siRNA and found that the induction of HO-1 and NQO1 by RTA 408 was inhibited; this result confirmed previous conclusions regarding the regulation of phase II enzymes by Nrf2. In an oxidative stress environment, Nrf2-deficient RPE cells died despite RTA 408 treatment. Considering that we did not use siRNA to inhibit other Nrf2 downstream genes or GSH synthesis enzymes and view how it affected RTA 408's effectiveness, we cannot rule out other possible pathways but will make sure to address these limitations in our future studies. Overall, these data significantly support our hypothesis that RTA 408 is a potent Nrf2 activator that can enhance RPE survival and protection.
In conclusion, as summarized in Fig. 9, our results strongly suggest that RTA 408 can activate the Nrf2 pathway and induce phase II enzyme expression, thereby protecting RPE cells from oxidative stress-induced cell apoptosis, preventing ROS over production, and inhibiting protein oxidation. The potency and efficacy of RTA 408 in protecting RPE cells makes it an attractive candidate for potential clinical use in treating oxidative stress-related retinal degenerative disease. In our future studies, we hope to replicate the success of RTA 408 in an in vivo AMD disease model to further inspire the use of Nrf2-activating drugs to treat oxidative stress-related degenerative eye diseases.
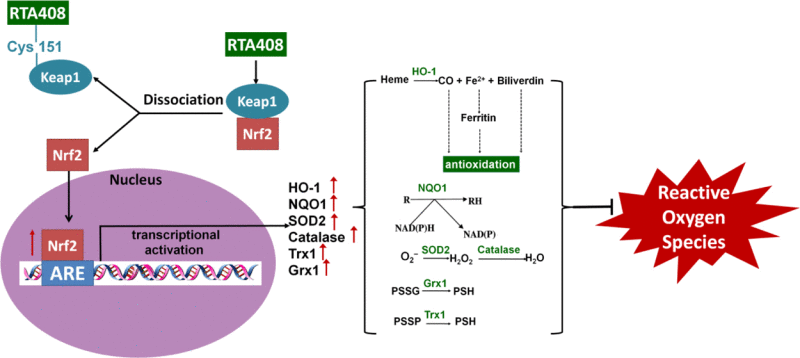
Fig. 9.

RTA 408 activates the Nrf2 pathway which leads to an upregulation of antioxidant enzymes and protects the cell from oxidative stress. Binding of RTA 408 to Cys151 in Keap1, the negative regulator of Nrf2, results in Keap1 inhibition. This promotes Nrf2 movement into the nucleus where it binds to the antioxidant response element (ARE). With the activation of ARE, transcriptional activation of antioxidant enzymes heme oxygenase-1 (HO-1), NADPH dehydrogenase (NQO1), superoxide dismutase 2 (SOD2), catalase, thioredoxin 1 (Trx1), and glutaredoxin 1 (Grx1) occurs. HO-1 converts heme to carbon monoxide, iron (II), and biliverdin, which all indirectly scavenge ROS. NQO1 converts enzymes and other proteins (R) back to their reduced form (RH) through the electron transfer between NADPH and NADP. Superoxide (O2−) can be converted to hydrogen peroxide using SOD2 and then further processed into water by catalase. Grx1 and Trx1 work together to reduce protein-glutathione mixed disulfide (PSSG) and protein-protein disulfide (PSSP) to protect protein thiols from oxidation. Overall, RTA 408 induction of phase II antioxidant enzymes such as HO-1, NQO1, SOD2, catalase, Grx1, and Trx1 via activation of Nrf2 promote RPE cell survival during oxidative stress.
Disclosure of potential conflicts of interest
This study was supported by REATA Pharmaceuticals, Inc. (Irving, TX). Keith Ward is employed by and has a financial interest in REATA Pharmaceuticals, Inc. Abbot F. Clark and Hongli Wu received a commercial research Grant from REATA Pharmaceuticals, Inc. (RP0162). No potential conflicts of interest were disclosed by the other authors.
References
- 1.de Jong P.T. Age-related macular degeneration. N. Engl. J. Med. 2006;355:1474–1485. doi: 10.1056/NEJMra062326. [DOI] [PubMed] [Google Scholar]
- 2.Fritsche L.G., Fariss R.N., Stambolian D., Abecasis G.R., Curcio C.A., Swaroop A. Age-related macular degeneration: genetics and biology coming together. Annu. Rev. Genom. Hum. Genet. 2014;15:151–171. doi: 10.1146/annurev-genom-090413-025610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lai K., Landa G. Current choice of treatments for neovascular AMD. Expert Rev. Clin. Pharmacol. 2015;8:135–140. doi: 10.1586/17512433.2015.990379. [DOI] [PubMed] [Google Scholar]
- 4.Age-Related Eye Disease Study 2 Research G. Lutein+zeaxanthin and omega-3 fatty acids for age-related macular degeneration: the Age-Related Eye Disease Study 2 (AREDS2) randomized clinical trial. JAMA. 2013;309:2005–2015. doi: 10.1001/jama.2013.4997. [DOI] [PubMed] [Google Scholar]
- 5.Seddon J.M., Ajani U.A., Sperduto R.D., Hiller R., Blair N., Burton T.C., Farber M.D., Gragoudas E.S., Haller J., Miller D.T. Dietary carotenoids, vitamins A, C, and E, and advanced age-related macular degeneration. Eye Disease Case-Control Study Group. JAMA. 1994;272:1413–1420. [PubMed] [Google Scholar]
- 6.Cai J., Nelson K.C., Wu M., Sternberg P., Jr, Jones D.P. Oxidative damage and protection of the RPE. Prog. Retininal Eye Res. 2000;19:205–221. doi: 10.1016/s1350-9462(99)00009-9. [DOI] [PubMed] [Google Scholar]
- 7.Khandhadia S., Lotery A. Oxidation and age-related macular degeneration: insights from molecular biology. Expert. Rev. Mol. Med. 2010;12:e34. doi: 10.1017/S146239941000164X. [DOI] [PubMed] [Google Scholar]
- 8.Howden R. Nrf2 and cardiovascular defense. Oxid. Med. Cell. Longev. 2013;2013:104308. doi: 10.1155/2013/104308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suzuki T., Motohashi H., Yamamoto M. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol. Sci. 2013;34:340–346. doi: 10.1016/j.tips.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 10.Kaspar J.W., Niture S.K., Jaiswal A.K. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free. Radic. Biol. Med. 2009;47:1304–1309. doi: 10.1016/j.freeradbiomed.2009.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salgado D., Forrer R.S., Spiess B.M. Activities of NADPH-dependent reductases and sorbitol dehydrogenase in canine and feline lenses. Am. J. Vet. Res. 2000;61:1322–1324. doi: 10.2460/ajvr.2000.61.1322. [DOI] [PubMed] [Google Scholar]
- 12.He M., Pan H., Chang R.C., So K.F., Brecha N.C., Pu M. Activation of the Nrf2/HO-1 antioxidant pathway contributes to the protective effects of Lycium barbarum polysaccharides in the rodent retina after ischemia-reperfusion-induced damage. PloS One. 2014;9:e84800. doi: 10.1371/journal.pone.0084800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chai Y.C., Hoppe G., Sears J. Reversal of protein S-glutathiolation by glutaredoxin in the retinal pigment epithelium. Exp. Eye Res. 2003;76:155–159. doi: 10.1016/s0014-4835(02)00309-3. [DOI] [PubMed] [Google Scholar]
- 14.Liu X., Jann J., Xavier C., Wu H. Glutaredoxin 1 (Grx1) protects human retinal pigment epithelial cells from oxidative damage by preventing AKT glutathionylation. Invest Ophthalmol. Vis. Sci. 2015;56:2821–2832. doi: 10.1167/iovs.14-15876. [DOI] [PubMed] [Google Scholar]
- 15.Pai H.V., Starke D.W., Lesnefsky E.J., Hoppel C.L., Mieyal J.J. What is the functional significance of the unique location of glutaredoxin 1 (GRx1) in the intermembrane space of mitochondria? Antioxid. Redox Signal. 2007;9:2027–2033. doi: 10.1089/ars.2007.1642. [DOI] [PubMed] [Google Scholar]
- 16.Goldman D.C., Alexeev V., Lash E., Guha C., Rodeck U., Fleming W.H. The triterpenoid RTA 408 is a robust mitigator of hematopoietic acute radiation syndrome in mice. Radiat. Res. 2015;183:338–344. doi: 10.1667/RR13900.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reisman S.A., Lee C.Y., Meyer C.J., Proksch J.W., Sonis S.T., Ward K.W. Topical application of the synthetic triterpenoid RTA 408 protects mice from radiation-induced dermatitis. Radiat. Res. 2014;181:512–520. doi: 10.1667/RR13578.1. [DOI] [PubMed] [Google Scholar]
- 18.Reisman S.A., Lee C.Y., Meyer C.J., Proksch J.W., Ward K.W. Topical application of the synthetic triterpenoid RTA 408 activates Nrf2 and induces cytoprotective genes in rat skin. Arch. Dermatol. Res. 2014;306:447–454. doi: 10.1007/s00403-013-1433-7. [DOI] [PubMed] [Google Scholar]
- 19.Alexeev V., Lash E., Aguillard A., Corsini L., Bitterman A., Ward K., Dicker A.P., Linnenbach A., Rodeck U. Radiation protection of the gastrointestinal tract and growth inhibition of prostate cancer xenografts by a single compound. Mol. Cancer Ther. 2014;13:2968–2977. doi: 10.1158/1535-7163.MCT-14-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lou M.F., Dickerson J.E., Jr, Garadi R., York B.M., Jr. Glutathione depletion in the lens of galactosemic and diabetic rats. Exp. Eye Res. 1988;46:517–530. doi: 10.1016/s0014-4835(88)80009-5. [DOI] [PubMed] [Google Scholar]
- 21.Hill B.G., Higdon A.N., Dranka B.P., Darley-Usmar V.M. Regulation of vascular smooth muscle cell bioenergetic function by protein glutathiolation. Biochim. Biophys. Acta. 1797;285–295:2010. doi: 10.1016/j.bbabio.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.West M.B., Hill B.G., Xuan Y.T., Bhatnagar A. Protein glutathiolation by nitric oxide: an intracellular mechanism regulating redox protein modification. FASEB J. 2006;20:1715–1717. doi: 10.1096/fj.06-5843fje. [DOI] [PubMed] [Google Scholar]
- 23.Wu H., Yu Y., David L., Ho Y.S., Lou M.F. Glutaredoxin 2 (Grx2) gene deletion induces early onset of age-dependent cataracts in mice. J. Biol. Chem. 2014;289:36125–36139. doi: 10.1074/jbc.M114.620047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gladyshev V.N., Liu A., Novoselov S.V., Krysan K., Sun Q.A., Kryukov V.M., Kryukov G.V., Lou M.F. Identification and characterization of a new mammalian glutaredoxin (thioltransferase), Grx2. J. Biol. Chem. 2001;276:30374–30380. doi: 10.1074/jbc.M100020200. [DOI] [PubMed] [Google Scholar]
- 25.Holmgren A., Bjornstedt M. Thioredoxin and thioredoxin reductase. Methods Enzymol. 1995;252:199–208. doi: 10.1016/0076-6879(95)52023-6. [DOI] [PubMed] [Google Scholar]
- 26.Finucane D.M., Bossy-Wetzel E., Waterhouse N.J., Cotter T.G., Green D.R. Bax-induced caspase activation and apoptosis via cytochrome c release from mitochondria is inhibitable by Bcl-xL. J. Biol. Chem. 1999;274:2225–2233. doi: 10.1074/jbc.274.4.2225. [DOI] [PubMed] [Google Scholar]
- 27.Johnson J., Maher P., Hanneken A. The flavonoid, eriodictyol, induces long-term protection in ARPE-19 cells through its effects on Nrf2 activation and phase 2 gene expression. Invest Ophthalmol. Vis. Sci. 2009;50:2398–2406. doi: 10.1167/iovs.08-2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang H., Liu Y.Y., Jiang Q., Li K.R., Zhao Y.X., Cao C., Yao J. Salvianolic acid A protects RPE cells against oxidative stress through activation of Nrf2/HO-1 signaling. Free Radic. Biol. Med. 2014;69:219–228. doi: 10.1016/j.freeradbiomed.2014.01.025. [DOI] [PubMed] [Google Scholar]
- 29.Zhao Z., Chen Y., Wang J., Sternberg P., Freeman M.L., Grossniklaus H.E., Cai J. Age-related retinopathy in NRF2-deficient mice. PloS One. 2011;6:e19456. doi: 10.1371/journal.pone.0019456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Probst B.L., Trevino I., McCauley L., Bumeister R., Dulubova I., Wigley W.C., Ferguson D.A. RTA 408, a novel synthetic triterpenoid with broad anticancer and anti-inflammatory activity. PloS One. 2015;10:e0122942. doi: 10.1371/journal.pone.0122942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao X., Talalay P. Induction of phase 2 genes by sulforaphane protects retinal pigment epithelial cells against photooxidative damage. Proc. Natl. Acad. Sci. USA. 2004;101:10446–10451. doi: 10.1073/pnas.0403886101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao X., Dinkova-Kostova A.T., Talalay P. Powerful and prolonged protection of human retinal pigment epithelial cells, keratinocytes, and mouse leukemia cells against oxidative damage: the indirect antioxidant effects of sulforaphane. Proc. Natl. Acad. Sci. USA. 2001;98:15221–15226. doi: 10.1073/pnas.261572998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li X., Liu Z., Luo C., Jia H., Sun L., Hou B., Shen W., Packer L., Cotman C.W., Liu J. Lipoamide protects retinal pigment epithelial cells from oxidative stress and mitochondrial dysfunction. Free Radic. Biol. Med. 2008;44:1465–1474. doi: 10.1016/j.freeradbiomed.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Voloboueva L.A., Liu J., Suh J.H., Ames B.N., Miller S.S. (R)-alpha-lipoic acid protects retinal pigment epithelial cells from oxidative damage. Invest Ophthalmol. Vis. Sci. 2005;46:4302–4310. doi: 10.1167/iovs.04-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sur A., Kesaraju S., Prentice H., Ayyanathan K., Baronas-Lowell D., Zhu D., Hinton D.R., Blanks J., Weissbach H. Pharmacological protection of retinal pigmented epithelial cells by sulindac involves PPAR-alpha. Proc. Natl. Acad. Sci. USA. 2014;111:16754–16759. doi: 10.1073/pnas.1419576111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kensler T.W., Wakabayashi N., Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 37.Lou M.F. Redox regulation in the lens. Prog. Retinal Eye Res. 2003;22:657–682. doi: 10.1016/s1350-9462(03)00050-8. [DOI] [PubMed] [Google Scholar]
- 38.Lillig C.H., Berndt C., Holmgren A. Glutaredoxin systems. Biochim. Biophys. Acta. 2008;1780:1304–1317. doi: 10.1016/j.bbagen.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 39.Lu J., Holmgren A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014;66:75–87. doi: 10.1016/j.freeradbiomed.2013.07.036. [DOI] [PubMed] [Google Scholar]
- 40.Koharyova M., Kollarova M. Thioredoxin system-a novel therapeutic target. Gen. Physiol. Biophys. 2015 doi: 10.4149/gpb_2015006. [DOI] [PubMed] [Google Scholar]
- 41.Chen B., Tang L. Protective effects of catalase on retinal ischemia/reperfusion injury in rats. Exp. Eye Res. 2011;93:599–606. doi: 10.1016/j.exer.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 42.Lewden O., Garcher C., Morales C., Javouhey A., Rochette L., Bron A.M. Changes of catalase activity after ischemia-reperfusion in rat retina. Ophthalmic Res. 1996;28:331–335. doi: 10.1159/000267924. [DOI] [PubMed] [Google Scholar]
- 43.Justilien V., Pang J.J., Renganathan K., Zhan X., Crabb J.W., Kim S.R., Sparrow J.R., Hauswirth W.W., Lewin A.S. SOD2 knockdown mouse model of early AMD. Invest Ophthalmol. Vis. Sci. 2007;48:4407–4420. doi: 10.1167/iovs.07-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sandbach J.M., Coscun P.E., Grossniklaus H.E., Kokoszka J.E., Newman N.J., Wallace D.C. Ocular pathology in mitochondrial superoxide dismutase (Sod2)-deficient mice. Invest Ophthalmol. Vis. Sci. 2001;42:2173–2178. [PubMed] [Google Scholar]