

Abstract
Background
Compost habitats sustain a vast ensemble of microbes specializing in the degradation of lignocellulosic plant materials and are thus important both for their roles in the global carbon cycle and as potential sources of biochemical catalysts for advanced biofuels production. Studies have revealed substantial diversity in compost microbiomes, yet how this diversity relates to functions and even to the genes encoding lignocellulolytic enzymes remains obscure. Here, we used a metagenomic analysis of the rice straw-adapted (RSA) microbial consortia enriched from compost ecosystems to decipher the systematic and functional contexts within such a distinctive microbiome.
Results
Analyses of the 16S pyrotag library and 5 Gbp of metagenomic sequence showed that the phylum Actinobacteria was the predominant group among the Bacteria in the RSA consortia, followed by Proteobacteria, Firmicutes, Chloroflexi, and Bacteroidetes. The CAZymes profile revealed that CAZyme genes in the RSA consortia were also widely distributed within these bacterial phyla. Strikingly, about 46.1 % of CAZyme genes were from actinomycetal communities, which harbored a substantially expanded catalog of the cellobiohydrolase, β-glucosidase, acetyl xylan esterase, arabinofuranosidase, pectin lyase, and ligninase genes. Among these communities, a variety of previously unrecognized species was found, which reveals a greater ecological functional diversity of thermophilic Actinobacteria than previously assumed.
Conclusion
These data underline the pivotal role of thermophilic Actinobacteria in lignocellulose biodegradation processes in the compost habitat. Besides revealing a new benchmark for microbial enzymatic deconstruction of lignocelluloses, the results suggest that actinomycetes found in compost ecosystems are potential candidates for mining efficient lignocellulosic enzymes in the biofuel industry.
Electronic supplementary material
The online version of this article (doi:10.1186/s13068-016-0440-2) contains supplementary material, which is available to authorized users.
Keywords: Lignocellulose degradation, Metagenomics, Compost ecosystem, Actinobacteria
Background
The dramatic rate of fossil fuels depletion and the dwindling supplies of geological reserves have spurred the search for alternative renewable energy sources such as biofuels. Utilization of lignocellulosic biomass derived from renewable plant material represents a promising alternative to fossil fuel [1]. However, the effective conversion of plant biomass remains a formidable challenge due to the paucity of highly efficient enzymes. Furthermore, most of the attractive lignocellulosic biomass conversion strategies are insensitive to fluctuations in feedstock and frequently dependent on thermal or chemical pre-treatments of the biomass [2]. As a consequence, search for microbes and enzymes displaying these desired characteristics of stability and efficiency in habitats where lignocellulosic biomass is naturally processed by microbiomes seems promising.
To recycle organic waste sustainably, composting is generally mediated by indigenous microbial communities under the aerobic conditions and in the solid state. Given that composts sustain an immense diversity of microbes for the efficient degradation of lignocellulosic biomass [3, 4], compost habitats are considered as one of the most important bioreactors for renewable bioenergy on the planet [5]. Therefore, various research groups have attempted to characterize these functional microbial communities. A range of thermophilic cellulose-degrading bacteria including, for example, Bacillus spp., Thermus spp., and Streptomycetes spp. have been isolated from compost and cultured in laboratory studies [6]. Moreover, many culture-independent molecular analyses with PCR-DGGE and clone libraries observed that cellulolytic actinomycetes are enriched in compost habitats. These observations suggest that thermophilic Saccharomonospora viridis and Thermobifida fusca as well as mesophilic Micrococcineae jointly contribute to the degradation of lignocellulosic materials [7, 8]. However, because of the complexity of lignocellulosic enzyme systems and compost microbiomes, it has generally been considered unfeasible to characterize the microbial degradation of lignocellulose in compost ecosystems by applying the above-mentioned techniques, which are principally based on isolated pure strains or 16S rRNA gene cloning. This not only inhibits furthering our understanding of the functions, interactions and ecology of microbial communities related to the degradation and transformation of lignocellulosic materials, but also seriously impedes exploiting the repertoire of novel enzymes and proteins that could enhance the efficiency of biomass conversion in compost ecosystems.
To date, the development to exploit ‘-omics’ approaches for the identification of the metabolic functions of multispecies consortia has progressed considerably [9]. These powerful approaches allow the identification of lignocellulosic enzymes and the resulting comprehensive sets of translated gene sequences can be used to infer the physiology and biochemistry of lignocellulose degradation processes. Recent advances in metagenomics generated functional genomic datasets of diverse environmental samples, including bovine rumen [10], guts of termite, panda, and honey bee [11–13], actively fermenting platform communities and natural grassland soil [14, 15]. These studies suggest that metagenomics approaches could provide an overview of the functioning of environmental microbial communities. Furthermore, such metagenomics approaches revealed the cellulolytic capabilities of various complex microbiome samples and substantially expanded the repertoire of genes and genomes participating in the decomposition of lignocellulose, which are of interest for industrial biorefineries and white biotechnology. To the best of our knowledge, to day a few metagenomic analyses have been conducted to analyze the structure and potential function of microbial communities in the compost ecosystem, which is also regarded as a unique and highly efficient system for bio-recycling of lignocellulosic biomass [5, 16]. However, the mechanism by which resident microbial communities synergistically degrade and utilize the lignocellulosic plant biomass under high temperature and solid conditions of the compost habitat is poorly defined.
It is almost universally accepted that the enrichment culture technique is a powerful tool for building microbial consortia with desired properties [17, 18]. A typical enrichment method for generating a desired consortium is to adapt environmental communities to feedstocks under specific conditions, with the expectation that the developing communities will be enriched with microbes displaying the desired functions. For example, Hess et al. [9] successfully obtained an enriched fiber-degrading microbial community on switchgrass incubated within the rumen. Likely, DeAngelis et al. [19] demonstrated that the switchgrass-adapted communities were less complex and more suited to degrading targeted feedstock than the starting inoculum. It should also be noted that the enrichment technique could lower the complexity of metagenomic datasets, subsequently rendering its future assembling more amenable [19].
Hence, in this study, a unique rice straw-adapted (RSA) microbial community was enriched within a nylon bag throughout the cow manure composting process. Owing to the intensive degradation of lignocellulosic biomass occurring during the thermophilic and maturing stages of composting process [7, 20, 21], the nylon bag filled with rice straw was retrieved at the end of maturing stage that is suggested to be the optimal time to fully explore the large repertoire of microbes and genes participating in the lignocellulose decomposition within the compost habitat. We therefore applied 454 pyrosequencing and massive depth metagenomic sequencing to systematically complement the microbial taxonomic analyses and to characterize the microbial functional diversity of the RSA consortia. The specific investigation of carbohydrate-active enzymes repertoire and their phylogenetic distribution in the RSA consortia will contribute to a better understanding of the microbial enzymatic decomposition of lignocelluloses in the compost ecosystem with extreme complexity. Ultimately, this study will provide valuable insights into the compost microbiome for bioprospecting novel lignocellulosic biocatalysts in the biofuel industry.
Results and discussion
Changes in the pre- and post-enrichment rice straw contents and composition
After 30 days of enrichment, substantial degradation of the rice straw in the nylon bag occurred (31.5 % dry mass reduction, Additional file 1: Table S1). Further analysis showed that the mass reduction was largely attributed to the degradation of cellulose and hemicellulose, both of which accounted for 78.9 % of the decrease in dry mass during the enrichment phase. The remaining reduction was likely due to the degradation of other components of the rice straw, such as pectin, lignin, and protein. In addition, the effectiveness of the microbial enrichment was preliminarily verified by our observation of a high abundance of cellulolytic microbes from the post-enrichment rice straw on Congo red agar, while only very few microbes were found from the pre-enrichment rice straw (Additional file 2: Fig. S1). Together, these results demonstrate that the rice straw-adapted microbial consortia enriched from the compost ecosystem were capable of degrading lignocellulosic biomass and support previous observations that compost habitats harbor a large number of cellulolytic microbes [3, 7].
Microbial diversity in RSA consortia enriched from manure compost
Metagenomic sequencing of the RSA consortia yielded total read counts of 59,627,950 after quality filtering. We performed de novo assembly and predicted 174,022 open reading frames (ORFs). The average ORF length was 657 bp, and 39.7 % of the ORFs were predicted to represent full-length genes (Additional file 3: Table S2). To extend the taxonomic analysis, a 16S rRNA amplicon library was additionally generated. A total of 27,644 fragments spanning the V1–V3 variable region were analyzed. Both metagenomic and 16S rRNA amplicon datasets showed that the RSA consortia were primarily comprised bacterial members (>98 %), along with very few archaea and eukarya. Various analysis approaches were then used to accurately estimate the microbial diversity within the RSA consortia, such as MetaPhlAn, mOTUs, Mothur, and SortMeRNA [22–25]. While these analyses showed minor differences in the rank abundance order, the phylum Actinobacteria was the predominant group among the bacteria in the RSA consortia, followed by Proteobacteria, Firmicutes, Chloroflexi, and Bacteroidetes (Fig. 1a). To gain further insight into the diversity within the RSA consortia, the metagenomic dataset was taxonomically profiled using MetaPhlAn [22]. This tool can accurately identify specific microbial clades at the species level or higher taxonomic levels, thereby providing new insights into the complexity of microbial communities. The MetaPhlAn analysis showed that Thermobispora bispora, Rhodothermus marinus, Sphaerobacter thermophilus and Thermomonospora curvata, were extremely abundant, while the thermophile Symbiobacterium, Thermobifida, and Geobacillus species were presented at low abundance in the RSA consortia (Additional file 4: Table S3). Most of the species identified as being abundant in our samples have been detected previously in the environment of hot composts. It was reported that they are proficient degraders of plant biomass, especially the Actinobacteria, including T. bispora,T. curvata, and Thermobifida fusca [26–28]. The predominance of these thermophile species in the RSA consortia may be closely related to the increased biomass degradation in the high-temperature composting phase as has been reported in previous research [5, 29]. It should be also noted that the large proportion of lignocellulolytic species suggests that the enrichment technique we used could contribute to the achievement of the desired property in the RSA consortia.
Fig. 1.
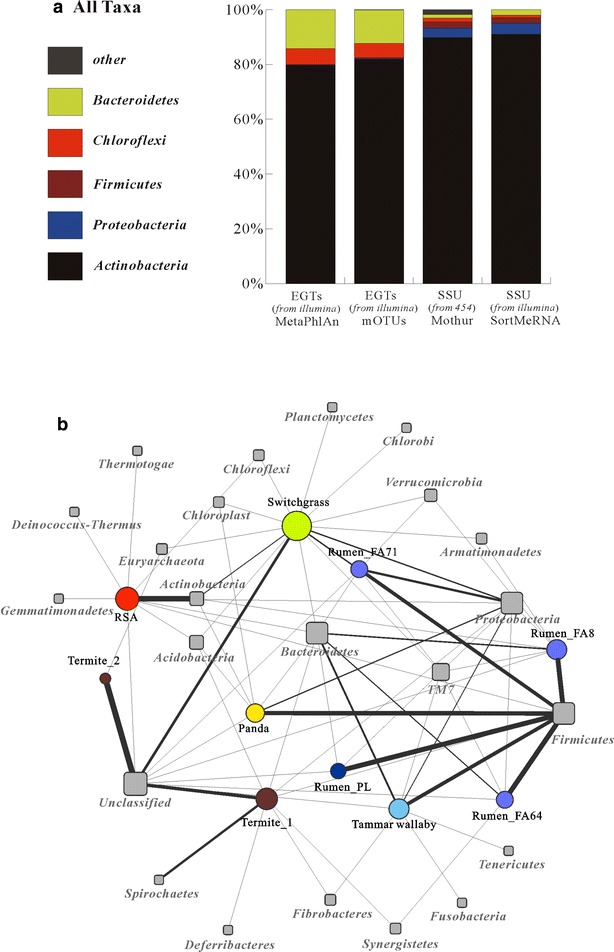
Phylogenetic composition of bacterial phyla from environmental gene tags (EGTs) and pyrosequence 16S rDNA sequences from the RSA consortia (a), and network for metagenome taxonomic profiling from the bovine, termite, panda, tammar wallaby samples as well as the RSA consortia (b)
Obviously, taxonomic profiling distinguished our metagenome from other typical and distinctive lignocellulose-degrading ecosystem metagenomes, which were derived from bovine, termite, panda, and tammar wallaby samples (Fig. 1b). The latter were dominated by the representatives of the Firmicutes and Bacteroidetes [10, 12, 30]. This incisive taxonomic disparity was most likely due to different environmental properties. For example, unlike the other lignocellulose-degrading ecosystems listed above, the compost ecosystem is characterized by high temperature, weak alkaline and aerobic conditions. These could significantly facilitate the growth of actinomycetes, which have been shown to exhibit a high plant biomass-degrading capacity [28, 31–33]. It is also worth noting that the taxonomic profile of our RSA consortia bore the greatest resemblance with that of the switchgrass-adapted compost community: both contained a number of actinomycetal members (Fig. 1b). However, presumably due to the differences in composting conditions and feedstock composition (i.e., rice straw vs. switchgrass), these two consortia substantially differed in the composition of the microbial community at the genus level. The actinobacterial genus Stackebrandtia was dominant in the switchgrass-adapted compost community [5], whereas Thermobispora was extremely abundant in our RSA consortia (Additional file 4: Table S3). Although both actinobacterial genera were reported to effectively degrade cellulose and hemicellulose [34, 35], their physiological adaptability and growth performance were considerably different. One of the largest discrepancies is their ability to grow at various temperatures. It was reported that the growth temperature range for Stackebrandtia nassauensis and T. bispora was 15 ~ 37 and 50 ~ 65 °C, respectively [36, 37]. These figures explain why thermophilic actinomycetes thrive in the RSA consortia but are scarcely detected in bovine, termite, panda, tammar wallaby, as well as switchgrass-adapted compost microbiomes. Based on the unique taxonomic profile of the RSA consortia, we hypothesize that these thermophilic communities may display distinctive functional properties and subsequently may be uniquely adapted to the lignocellulosic materials in the compost habitat.
Microbial gene functions in RSA consortia enriched from manure compost
Given that metagenomics can provide an initial picture of the physiological properties of microbial consortia, we compared the catalog with the COG and KEGG databases to assess the functions prominent in the RSA consortia. BLAST comparisons of all sequences yielded 124,378 hits for the COG database and 48,204 hits for the KEGG pathways. The enrichment of COG categories in Fig. 2a shows that the RSA consortia were enriched for general function (12.7 % in all COG functional categories), amino acid transport and metabolism (9.8 %), transcription (7.5 %), and carbohydrate transport/metabolism (7.3 %). The ontology analysis based on KEGG revealed similar patterns as shown in Fig. 2b, where membrane transport (12.7 % in all KEGG pathways), carbohydrate metabolism (12.1 %), amino acid metabolism (10.4 %), as well as xenobiotics biodegradation and metabolism (8.5 %) were abundant in the RSA consortia. Further, the comparative COG functional analysis of the RSA consortia with eight other well-known lignocelluloses-degrading microbiomes from termite, panda, 55 and 40 degree reactor, switchgrass-adapted compost, honey bee, wallaby and bovine rumen proved that they share similar metabolic patterns, particularly associated with transcription, carbohydrates and amino acids metabolism (Additional file 5: Fig. S2). These metabolic reconstructions suggest that the RSA consortia have enriched several functional capacities, especially, as expected, for carbohydrate metabolism. In order to determine the potential roles of microbial communities in the decomposition of plant polymer, specific COGs involved in carbohydrate transport and metabolism were analyzed in more detail (Additional file 6: Table S4). Notably, the RSA consortia harbored a diversity of genes involved in the metabolisms of the glucose, mannose, arabinose, galactose, glucuronose, xylose, and fructose, all of which accounted for 11.8 % of the COG subcategory G (Carbohydrate transport and metabolism). This suggests that the plant polysaccharides which the compost microbiome consumed were rich in glucan-, xylan-, pectin-, arabinose-containing carbohydrate structures, potentially supporting our previously mentioned observation that the lignocellulosic biomass reduction during the composting phase was mainly attributed to the degradation of cellulose and hemicellulose. Furthermore, the genes related to sugar/polysaccharide transport and phosphotransferase systems in the RSA consortia were more abundant than those encoding for polysaccharide-degrading enzymes (Additional file 6: Table S4). For example, the ATP-binding cassette (ABC) -type sugar transporters (COG1653, COG1175, COG0395, and COG1129), which may be involved in the uptake of carbohydrate [38], accounted for 17.7 % of the COG subcategory G. Another carbohydrate-related function enriched in the RSA consortia was the family of arabinose efflux permease (COG2814, 6.8 % of the COG subcategory G), which is responsible for the export and import of compounds such as sugars, antimicrobials, and amino acids [39]. Again, predicted genes related to phosphotransferase system (COG1925, COG1762, and COG1263) were detected. They might function in the transport and phosphorylation of numerous monosaccharides, disaccharides, amino sugars, polyols, and other sugar derivatives [40]. Altogether, the wide diversity of gene functions in carbohydrate metabolizing and transporting reveals a high potential of the RSA consortia for glycan degradation and sugar uptake and transport in the compost ecosystem.
Fig. 2.
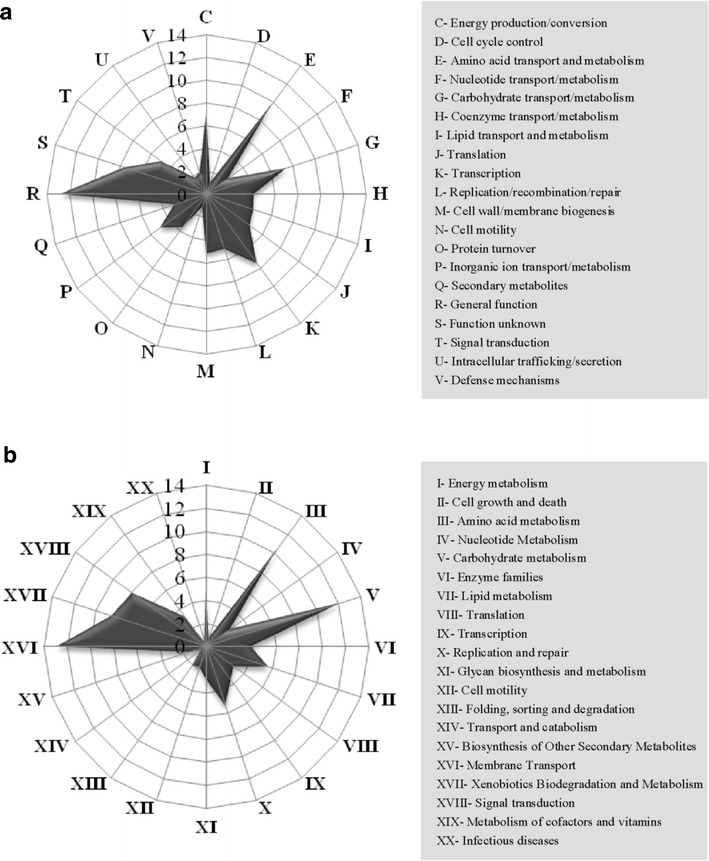
Distribution pattern of a COG-assigned and b KEGG-assigned proteins in the RSA consortia. Genes not assignable to any COGs or KEGGs are not shown in this figure. The percentage of matched gene numbers was assigned to specific COG or KEGG functional categories
The abundance, variety and phylogenetic distribution of CAZymes in the RSA consortia
To better understand the carbohydrate metabolism in the compost ecosystem, where microorganisms synergistically degrade and utilize the cell wall components, we screened our metagenome for genes encoding enzymes that catalyze the hydrolysis of plant polymers. The RSA consortia contained 4810 different CAZyme genes distributed unequally between glycoside hydrolases (GHs, 35 %), carbohydrate-binding modules (CBMs, 13 %), polysaccharide lyases (PLs, 3 %), carbohydrate esterases (CEs, 18 %), and glycosyltransferases (GTs, 31 %). It is noteworthy that the CAZyme profile in the RSA consortia was close to the transcriptional profile of CAZyme genes in a maturing compost sample (unpublished data) (Additional file 7: Fig. S3). This finding suggests that carbohydrate metabolism pathways performed by CAZymes in the RSA consortia may to a large extent be expressed and active within the compost ecosystem at the time when we sampled.
Next, the overall phylogenetic distribution of CAZyme genes in the RSA consortia was analyzed to link compost microbial community composition and lignocellulose decomposition processes. The abundance of CAZyme genes varied across bacterial phyla. The majority of these genes in the RSA consortia were found from Actinobacteria (46.1 %), followed by Proteobacteria (16.7 %), Firmicutes (14.2 %), Chloroflexi (7.7 %), and Bacteroidetes (6.1 %), and more importantly, the members from Actinobacteria were dominant in all carbohydrate-active modules (GHs, GTs, PLs, and CEs) and their associated CBMs (Additional file 8: Fig. S4). Since the content of CAZyme genes was not homogeneously distributed within each phylum, we also investigated the detailed phylogenetic distribution of genes involved in lignocelluloses degradation at lower taxonomic levels. Intriguingly, twelve different species of phylum Actinobacteria were present in the 20 most abundant members possessing CAZyme genes, revealing that the phylum Actinobacteria harbored much more CAZyme genes and exhibited a much wider distribution of CAZyme repertoire in the RSA consortia (Fig. 3). These findings, taken together with the fact that the phylogenetic distributions of CAZyme genes largely distinguished the RSA consortia (predominantly Actinobacteria) from that of the bovine rumen (Clostridiales), the termite hindgut (Fibrobacteres and Spirochetes), the honey bee gut (Proteobacteria), and the Tammar wallaby foregut (Bacteroidetes) [10, 11, 13, 30], strongly emphasized the functional versatility and distinctive adaptation of actinobacterial communities in the processing and metabolizing of carbohydrates within the compost habitat. However, in addition to T. bispora (8.79 %), T. curvata (8.56 %), and Streptosporangium roseum (6.43 %) of the phylum Actinobacteria, the CAZyme genes were also mainly detected in S. thermophilus (7.99 %) of the phylum Chloroflexi as well as R. marinus (7.47 %) of the phylum Bacteroidetes (Fig. 3). There are several implications of this observation: Firstly, the composition of functional members corresponds well to the structure of the ecologically dominant species in the RSA consortia. Potentially, a link exists between microbial taxa and their functional traits. This suggests that polysaccharides within the compost habitat are hydrolyzed by the predominant Actinobacteria in cooperation with Chloroflexi and Bacteroidetes. Further, although high levels of cellulose-degrading enzymes were found previously to be secreted by T. bispora, T. curvata, and R. marinus [41–43], our data provide genetic evidence for both highly cellulolytic and highly transglycosylatic abilities of these well-characterized strains, which harbor an abundant of genes encoding GHs and GTs.
Fig. 3.
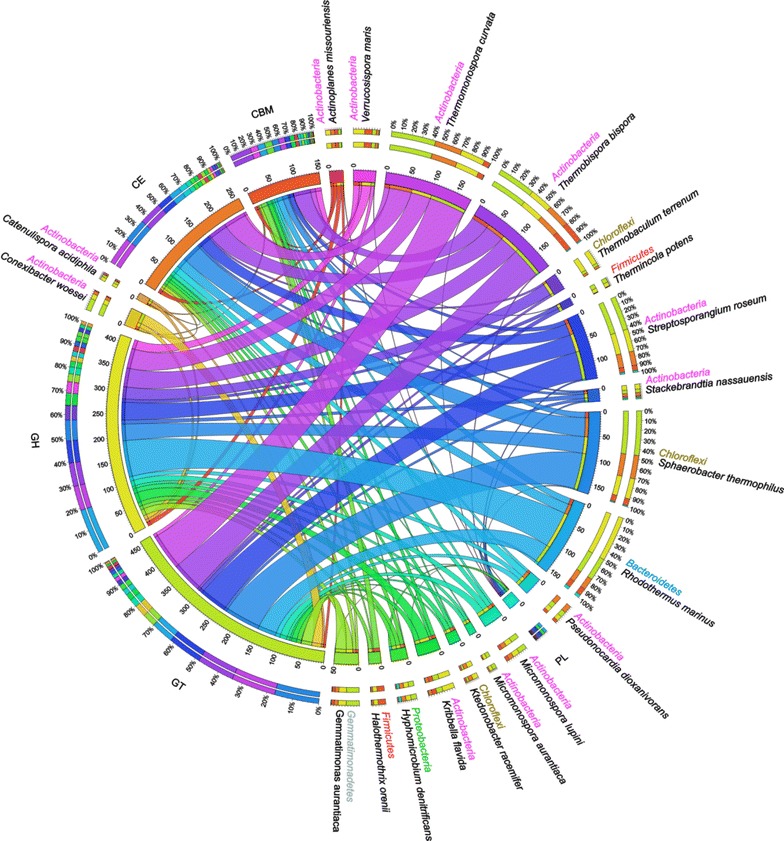
Phylogenetic distributions of carbohydrate-active enzymes in the most abundant members possessing CAZyme genes. The data were visualized via Circos software [84]. The width of bars from each microbial species and functional enzyme family indicates their relative abundance in the RSA consortia
As a widespread group of enzymes that hydrolyze the glycosidic bond between carbohydrates, GH families were most attached to oligosaccharide-degrading enzymes (18.18 %), along with relatively few to cellulases (4.60 %) and endohemicellulases (4.90 %) (Table 1). This vast majority of oligosaccharide-degrading enzymes identified in our metagenome suggests that the RSA consortia were predominantly processing and metabolizing the partially degraded plant materials in the compost ecosystem. In particular, β-glucosidases responsible for the processing of small oligosaccharides in the RSA consortia were mostly present in GH3 and GH1 (8.62 %), whereas the endo- and exo-cellulases were predominantly found in GH5 and GH9 (3.48 %) (Table 1). A range of CAZymes targeting hemicelluloses (such as GH10 xylanases and GH26 mannanase, 3.01 %) and pectic polysaccharides (PL9 pectate lyase) were also abundant (Additional file 9: Table S5). Together with the COG profiles for glycan degradation, the diverse repertoire of CAZymes systems provides the basis for a collaborative system efficient in the processing and metabolizing of carbohydrates in the compost habitat. However, among the total of 85 GHs, the largest two sequence-based families were the families of GH109 and GH13 (Additional file 9: Table S5), known as the dehydrogenase and α-amylase family associated with starch biosynthesis and turnover [44]. The next most abundant is the GH23 family, among which nearly half were derived from Actinobacteria. This family was thought to possess inverting lysozymes and lytic transglycosylases that cleave glycosidic bonds via a water-independent substrate-assisted mechanism [45]. In addition to these putative GHs, we also detected a number of CBM2 and CBM50, in which Actinobacteria accounted for 95.5 and 9.8 %, respectively. These non-catalytic CBMs are contiguous amino acids within the carbohydrate-active enzyme with a discreet fold promoting efficient adherence of the enzyme to insoluble substrates and thus enhancing the catalytic cleavage of the substrate [46–48]. It is plausible that the abundance of GH23 together with CBM2 and CBM50, leads to the efficiency of the dominant populations in the RSA consortia, especially of the Actinobacteria, to degrade the insoluble lignocellulosic plant biomass in the solid condition of the compost habitat.
Table 1.
Glycoside hydrolase profiles targeting plant structural polysaccharides in four metagenomes
| Predominant activity | Termite [11] | Bovine [10] | Macropod [30] | RSA | |
|---|---|---|---|---|---|
| Cellulases | |||||
| GH5 | Cellulases | 7.97 | 1.23 | 4.31 | 2.36 |
| GH6 | Endoglucanases | 0.00 | 0.00 | 0.18 | 0.77 |
| GH7 | Endoglucanases | 0.00 | 0.00 | 0.00 | 0.00 |
| GH9 | Endoglucanases | 1.28 | 0.92 | 0.36 | 1.12 |
| GH44 | Endoglucanases | 0.85 | 0.00 | 1.08 | 0.06 |
| GH45 | Endoglucanases | 0.57 | 0.00 | 0.00 | 0.00 |
| GH48 | Cellobiohydrolases | 0.00 | 0.00 | 0.00 | 0.30 |
| Subtotal (%) | 10.67 | 2.15 | 5.92 | 4.60 | |
| Endohemicellulases | |||||
| GH8 | Endoxylanases | 0.71 | 0.61 | 0.36 | 0.00 |
| GH10 | Endo-1,4-β-xylanases | 6.54 | 1.08 | 2.51 | 2.18 |
| GH11 | Xylanases | 1.99 | 0.15 | 0.00 | 0.47 |
| GH12 | Xyloglucanases | 0.00 | 0.00 | 0.00 | 0.41 |
| GH26 | β-mannanase & xylanases | 2.13 | 0.77 | 2.33 | 0.83 |
| GH28 | Galacturonases | 0.85 | 0.77 | 0.54 | 0.65 |
| GH53 | Endo-1,4-β-galactanases | 1.71 | 2.61 | 1.62 | 0.35 |
| Subtotal (%) | 13.94 | 5.99 | 7.36 | 4.90 | |
| Debranching enzymes | |||||
| GH51 | α-L-arabinofuranosidases | 2.56 | 9.83 | 2.33 | 1.42 |
| GH54 | α-L-arabinofuranosidases | 0.00 | 0.15 | 0.00 | 0.00 |
| GH62 | α-L-arabinofuranosidases | 0.00 | 0.00 | 0.00 | 0.35 |
| GH67 | α-glucuronidases | 1.42 | 0.00 | 0.90 | 0.35 |
| GH78 | α-L-rhamnosidases | 0.00 | 5.22 | 4.49 | 2.30 |
| Subtotal (%) | 3.98 | 15.21 | 7.72 | 4.43 | |
| Oligosaccharide-degrading enzymes | |||||
| GH1 | β-glucosidases | 3.13 | 1.54 | 11.13 | 3.72 |
| GH2 | β-galactosidases | 3.27 | 28.57 | 5.03 | 1.59 |
| GH3 | β-glucosidases | 9.82 | 27.04 | 13.46 | 4.90 |
| GH29 | α-L-fucosidases | 0.00 | 11.37 | 0.54 | 0.89 |
| GH35 | β-galactosidases | 0.43 | 1.84 | 0.72 | 0.41 |
| GH38 | α-mannosidases | 1.56 | 2.61 | 0.54 | 0.77 |
| GH39 | β-xylosidases | 0.43 | 0.31 | 0.18 | 1.89 |
| GH42 | β-galactosidases | 3.41 | 1.69 | 1.62 | 0.83 |
| GH43 | Arabino/xylosidases | 2.28 | 9.37 | 3.41 | 3.01 |
| GH52 | β-xylosidases | 0.43 | 0.00 | 0.00 | 0.18 |
| Subtotal (%) | 24.75 | 84.33 | 36.62 | 18.18 | |
| Total GHs | 703 | 651 | 557 | 1694 | |
| % ORFS | 0.78 | 0.78 | 0.71 | 0.97 | |
The listed value is the population abundance weighted relative abundance (%) of GH families among the total GHs
Although most of these CAZymes profiles of the RSA consortia were similar to those of the termite, bovine and macropod microbiomes, 0.97 % of the sequences assigned to the GH families in the RSA consortia are subtly greater than those in the termite (0.78 %), bovine (0.78 %), and macropod (0.71 %) communities (Table 1). Considering the fact that the numbers of hydrolytic enzymes produced by the anaerobic gut habitats are relatively lower than those observed for their aerobic counterparts [49], the higher percentage of average GH families in the aerobic compost habitat indicates a much more abundant and diverse set of microbial genes directed towards accessing a wide range of polysaccharides. Furthermore, compared to these herbivore microbiomes, the RSA consortia contained a certain amount of genes associated with cellulosome complexes (e.g., GH48, 0.30 %), which facilitate interactions between the insoluble substrate and the bacterial enzymatic machinery [50]. Other differences in the GHs profile of these four metagenomes include the presence of the GH12 (0.41 %) and GH62 (0.35 %) families as well as the absence of the GH8 family in our study. It is important to note that GH12 and GH62 mainly originated from Actinobacteria (75 and 100 %, respectively). The detection of GH12, a thermostable endoglucanase with a high efficiency for rice straw saccharification [51, 52], further confirms the high capacity of Actinobacteria for degrading rice straw in the thermophilic composting environment. Again, the existence of GH62 supports that Actinobacteria possess several accessory enzymes, which could facilitate the arabinoxylan degradation [53]. The diverse repertoire and distinct phylogenetic distribution of CAZymes in the RSA consortia, therefore, further reflect these typical cellulolytic characteristics (thermostability and versatility) of actinomycetal community along with its capacity of attacking the insoluble substrates, hence contributing to the unique adaptation of composts-specific microbial populations to lignocellulosic plant biomass.
A systematic overview of lignocellulose degradation by the RSA consortia
Lignocellulose of plant biomass is chemically complex consisting of some major polymers, such as cellulose, hemicellulose, pectin, and lignin. In nature, the efficient degradation of lignocellulosic biomass involves a cooperative action of various microorganisms producing multiple enzymes that act specifically and synergistically in the degradation of plant biomass. Therefore, in this study, the cultivation-independent metagenomic approach was applied to provide an overview of microbial degradation of primary plant cell wall polysaccharides along with lignin by individual members of the RSA consortia, which were effectively enriched from the compost habitat. Specifically, we examined the carbohydrate-digestive capacity of the RSA consortia, determined the top Blast-hit organism of each identified enzyme at the species level, and interpreted the ecosystem functions of major microorganisms and their potential synergistic action in these complex processes.
Cellulose bioconversion
The classical scheme for cellulose degradation involves at least three different classes of hydrolytic enzymes to participate: endoglucanases for catalyzing random cleavage of internal bonds in the cellulose chain, cellobiohydrolases for attacking the chain ends and hence releasing cellobiose, and β-glucosidases for acting on cello-oligosaccharides and cellobiose and subsequently releasing glucose. Genes encoding for these putative cellulases in the RSA consortia were mainly from the families GH1, GH3, GH5, GH6, GH9, GH12, GH44, GH48, and GH74 (Additional file 9: Table S5). Clearly, cellulolytic species in the RSA consortia were remarkably diverse (Fig. 4a). Endoglucanase was predominantly present in thermotolerant S. thermophilus and R. marinus, while both cellobiohydrolase and β-glucosidase activities were mainly derived from members of the Actinobacteria, such as Verrucosispora maris, T. curvata, and T. bispora. This strongly suggests a synergistic action of multiple members affiliated to Actinobacteria, Chloroflexi, and Bacteroidetes during the degradation of cellulose. The synergistic pattern shown in the compost habitat differs substantially from the rumen microbiome, in which cellulolytic activities were performed by Firmicutes, Fibrobacteres and Bacteroidetes [50] and from the termite hindgut microbiome, in which these processes were dominated by Spirochaetes and Fibrobacteres [11].
Fig. 4.
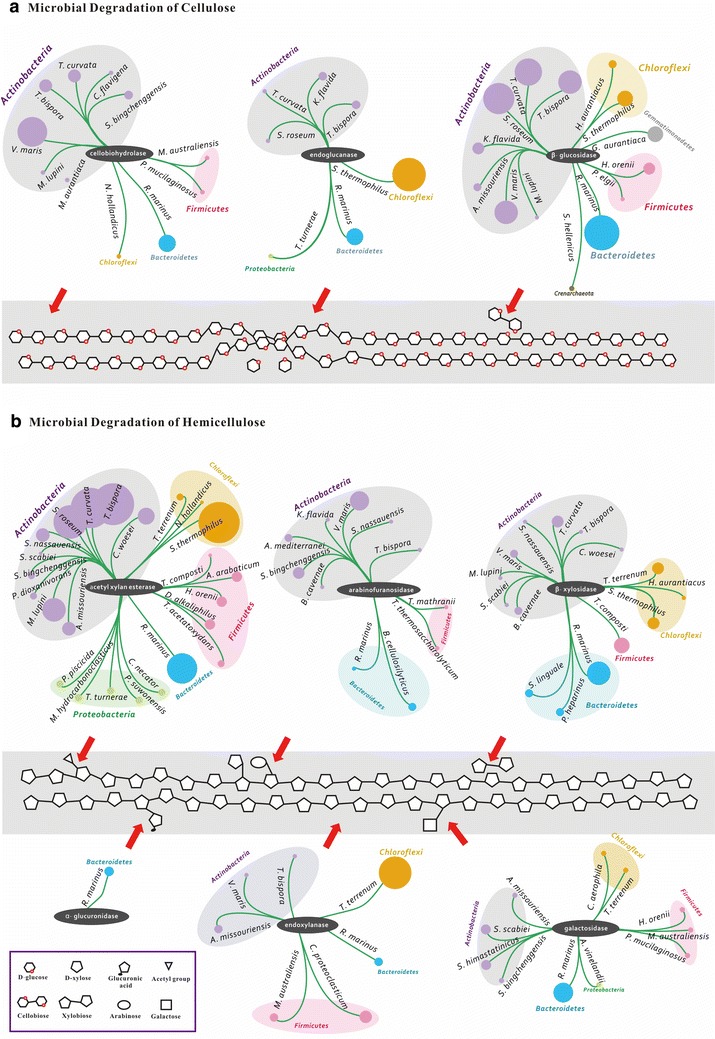
An overview of microbial degradation of cellulose and hemicellulose in the RSA consortia. We show the dominant species that digest the cellulose and hemicellulose by depicting the distribution of genes encoding the CAZymes in the RSA consortia. The purple, pink, green, blue, yellow, gray and brown circles represent the members of phylum Actinobacteria, Firmicutes, Proteobacteria, Bacteroidetes, Chloroflexi, Gemmatimonadetes and Crenarchaeota, respectively. The diameter of each circle is proportional to its relative abundance. A. arabaticum, Acetohalobium arabaticum; A. mediterranei, Amycolatopsis mediterranei; A. missouriensis, Actinoplanes missouriensis; A. vinelandii, Azotobacter vinelandii; B. cellulosilyticus, Bacteroides cellulosilyticus; B. cavernae, Beutenbergia cavernae; C. aerophila, Caldilinea aerophila; C. proteoclasticum, Clostridium proteoclasticum; C. flavigena, Cellulomonas flavigena; C. necator, Cupriavidus necator; C. woesei, Conexibacter woesei; D. alkaliphilus, Dethiobacter alkaliphilus; G. aurantiaca, Gemmatimonas aurantiaca; H. aurantiacus, Herpetosiphon aurantiacus; H. orenii, Halothermothrix orenii; K. flavida, Kribbella flavida; M. aurantiaca, Micromonospora aurantiaca; M. australiensis, Mahella australiensis; M. hydrocarbonoclasticus, Marinobacter hydrocarbonoclasticus; M. lupini, Micromonospora lupini; N. hollandicus, Nitrolancetus hollandicus; P. heparinus, Pedobacter heparinus; P. piscicida, Pseudoalteromonas piscicida; P. dioxanivorans, Pseudonocardia dioxanivorans; P. elgii, Paenibacillu elgii; P. maris, Planctomyces maris; P. mucilaginosus, Paenibacillus mucilaginosus; P. suwonensis, Pseudoxanthomonas suwonensis; R. marinus, Rhodothermus marinus; S. bingchenggensis, Streptomyces bingchenggensis; S. clavuligerus, Streptomyces clavuligerus; S. hellenicus, Staphylothermus hellenicus; S. himastatinicus, Streptomyces himastatinicus; S. linguale, Spirosoma linguale; S. nassauensis, Stackebrandtia nassauensis; S. roseum, Streptosporangium roseum; S. scabiei, Streptomyces scabiei; S. thermophilus, Sphaerobacter thermophilus; T. acetatoxydans, Tepidanaerobacter acetatoxydans; T. curvata, Thermomonospora curvata; T. terrenum, Thermobaculum terrenum; T. turnerae, Teredinibacter turnerae; T. bispora, Thermobispora bispora; T. composti, Thermobacillus composti; T. mathranii, Thermoanaerobacter mathranii; T. potens, Thermincola potens; T. thermosaccharolyticum, Thermoanaerobacterium thermosaccharolyticum; V. maris, Verrucosispora maris. The bubble plot indicates the relative abundances of each microbial species in the RSA consortia
It has been generally accepted that the β-glucosidase activity is a key factor in the cellulolytic process [54]. In this study, the putative β-glucosidase was the most abundant enzyme. Its phylogenetic distribution was the most diverse among the three classes of hydrolytic enzymes, mainly from a variety of Actinobacteria specie including V. maris, T. curvata, T. bispora, S. roseum, and Kribbella flavida. Of these cellulolytic species, S. roseum and K. flavida were firstly detected to participate in the degradation of cellulose in the compost habitat, although V. maris, T. curvata and T. bispora have been frequently identified as primary actinomycetes for cellulose degradation in natural habitats [55]. More importantly, the strictly thermophilic T. bispora, as the predominant member in the RSA consortia, has the potential to carry out the functions of three classes of cellulolytic enzymes as well as those of some xylan-degrading enzymes (Fig. 4a, b). This suggests a greater phylogenetic and functional diversity of actinomycetal communities involved in cellulose degradation than previously assumed.
Hemicellulose bioconversion
During the enzymatic hydrolysis of hemicellulose, endoxylanase, β-xylosidase, α-glucuronidase, arabinofuranosidase, galactosidase, and acetylxylan esterase all act on the different heteropolymers available in lignocellulosic biomass (Fig. 4b). Genes encoding for these putative hemicellulases in the RSA consortia were mainly from the families GH1–GH4, GH10, GH11, GH27, GH35, GH36, GH39, GH43, GH51, GH52, GH62, GH67, GH127, CE1, CE3, CE4, and CE7 (Additional file 9: Table S5). Strikingly, the dominant members associated with hemicellulose bioconversion in the RSA consortia were also thermophilic. Thermobaculum terrenum of the phylum Chloroflexi was predominantly responsible for the hydrolysis of the internal glycosidic linkages of the heteroxylan backbone (endoxylanase), whereas Rhodothermus marinus of the phylum Bacteroidetes was mainly responsible for degrading short molecules such as xylooligosaccharide, xylobiose, galactolipid, and glucuronide. These findings indicate that the members mainly from Bacteroidetes along with Chloroflexi probably had a crucial role in the degradation of hemicellulose, specializing towards its glycosidic linkage hydrolysis under the thermophilic conditions of the compost habitat. It should also be noted that the key role of Bacteroidetes in degrading hemicellulose polymers was confirmed by a previous report on the metagenome of the mesophilic fermenting platform community [15]. However, unlike this community, the dominant species of the phylum Bacteroidetes functioning in the RSA consortia is R. marinus, which is an extreme thermophilic bacterium thriving in various thermal environments and displaying diverse inherent enzyme activities in degrading lignocellulosic biomass [56].
Of note, however, is that the actinomycetal population containing genes for encoding endoxylanase and β-xylosidase was less well represented but more diverse, including the following species T. bispora, T. curvata, S. roseum, Micromonospora lupini, V. maris, Conexibacter woesei, Stackebrandtia nassauensis, Beutenbergia cavernae, Streptomyces scabiei, and Actinoplanes missouriensis. Most of these actinomycetal species were reported to be able to simultaneously produce a number of acetyl xylan esterases and arabinofuranosidases, which hydrolyze the acetyl substitutions on xylose moieties and arabinofuranosyl linkages in arabinoxylan, respectively [57]. Considering the synergisms between esterases/arabinofuranosidases and xylanases in the enzymatic degradation of xylan [57, 58], we suggest that the prominent actions of actinomycetal esterase and arabinofuranosidase may greatly facilitate the efficient and complete the breakdown of xylan during the bioconversion of hemicelluloses.
Pectin bioconversion
Pectin, a family of covalently linked galacturonic acid-rich polysaccharides, is the most structurally and functionally complex polysaccharide of plant cell walls [59]. Complete hydrolysis of pectins involves a set of diverse enzymes (Fig. 5), including pectin lyase, polygalacturonase, galactosidase, arabinofuranosidase, and rhamnosidase. Genes encoding for these putative pectinases in the RSA consortia were mainly from the families PL1, PL3, PL4, PL9, PL10, GH2, GH4, GH27, GH28, GH35, GH36, GH51, GH62, GH78, GH106, and GH127 (Additional file 9: Table S5). Among these known pectinases, pectin lyase has an essential role in the pectin bioconversion since it is the only enzyme that can cleave the α-1, 4 bonds of highly esterified pectins without prior actions of other enzymes. It has been generally accepted that the microbial sources of pectin lyase are specific fungi and bacteria, such as Aspergillus niger, Bacillus spp., and Pseudomonas spp. [60–63]. However, in our study, a range of rare actinomycetes was the major producer of this important enzyme, especially Stackebrandtia nassauensis, Actinoplanes missouriensis, T. bispora, and T. curvata. While pectin lyase has been previously detected in Streptomyces viridochromogenes and Thermomonospora flisca [64, 65], so far very limited information is available on the pectinolytic enzyme system of the class Actinobacteria. This first report on pectin lyase production by rare actinomycetes highlights their importance in the degradation of pectin in the compost ecosystem.
Fig. 5.
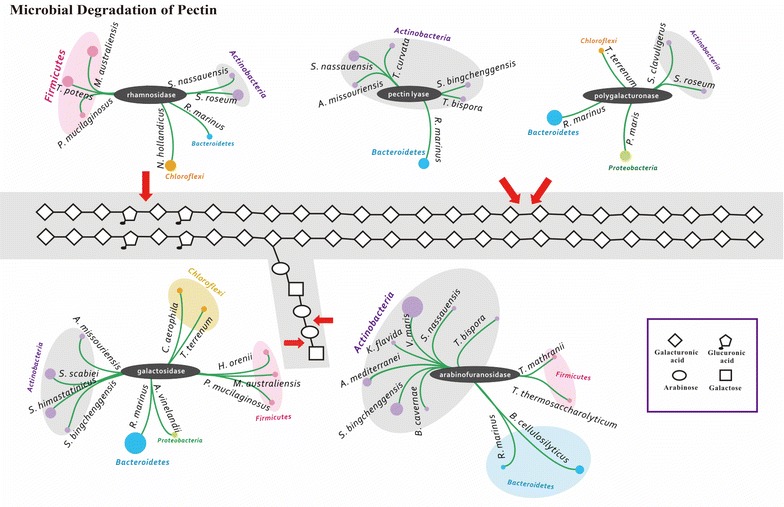
As in Fig. 4 but for microbial degradation of pectin in the RSA consortia
Lignin bioconversion
As the main non-carbohydrate structural component of the plant cell wall, lignin is a highly complex aromatic heterogeneous polymer that is covalently cross-linked with polysaccharide fibers by covalent bonds [66]. Lignin attack is an oxidative process and requires a suite of oxidative enzymes, such as lignin peroxidase (LiP), manganese peroxidase (MnP), versatile peroxidase (VP), and laccase [67]. Genes encoding for these putative ligninases were from the family AA2 in the “Auxiliary Activities” (AAs) groups, and they accounted for 8.7 % of all AAs (Additional file 10: Table S6). After examining the phylogenetic distribution of genes in the family AA2, we found that in addition to Symbiobacterium thermophilum of the phylum Firmicutes, the dominant members involved in the degradation of lignin compounds in the RSA consortia were mainly from a variety of Actinobacteria species, including T. curvata, Mycobacterium xenopi, Amycolicicoccus subflavus, and Mycobacterium thermoresistibile. The diversity of these previously unrecognized members greatly increases the inventory of ligninases since actinomycetes identified by in vitro assays for lignin degradation were affiliated within a limited number of genera, including Streptomyces, Rhodococcus and Nocardia [68].
Implications
In recent years, thermostable lignocellulases have been a target for metabolic engineering related to biomass conversion and biofuel production [69]. The compost habitat, characterized by high temperature (~70 °C) and abundant lignocellulosic materials (e.g., rice straw, dry leaves, sawdust), is potentially a site for prospecting thermophilic biomass-degradation enzymes [70]. As expected, our pyrosequencing and metagenomic analyses of the RSA consortia revealed that the numerically dominant and deeply branched thermophilic members from Actinobacteria have the potential to effectively degrade the lignocellulosic biomass in the compost habitat. Properties of the GHs and CBMs found in this study indicated that lignocellulosic enzymes produced by Actinobacteria in composts might be thermostable, which is supported by the previous finding that cellulase derived from Streptomyces sp. was active at 37 ~ 75 °C [71]. Therefore, from an applied point of view, the compost-specific lignocellulases operating stable under a range of high temperatures would have advantages in industrial lignocellulose processing compared to the most commonly used commercial enzyme cocktails produced by the fungus Trichoderma reesei with an optimum temperature range between 40 and 50 °C [72].
To our knowledge, the present study is the first research illustrating a synergistic action of the predominant Actinobacteria along with Chloroflexi and Bacteroidetes and Firmicutes in the hydrolysis of polysaccharides within the compost habitat. It is also the first study that provides genetic evidence supporting that compost microbiome contained a variety of previously known and so far unrecognized actinomycetal members potentially displaying versatile lignocelluloses-degrading abilities, especially in the β-glucosidase, acetyl xylan esterase, arabinofuranosidase, pectin lyase and ligninase activity. These data reveal a greater ecological functional diversity of thermophilic Actinobacteria in carbohydrate metabolism in the compost habitat than previously assumed, underlining that there is still a broad swath of actinomycetes to be explored as potential candidates for mining efficient lignocellulosic enzymes in the biofuel industry.
It should be also noted that lignocellulose degradation within the compost habitat is a continuous process of dynamic interactions between complex microbial community and recalcitrant substances. It would be desirable to apply broad sampling and adopt a broad array of approaches, including metatranscriptomics, proteomics and metabolomics, so as to temporally identify and quantify the expression of genes and metabolic functions associated with the bioconversion of the major components of plant biomass during the composting period, such as cellulose, hemicellulose, pectin, lignin as well as silica, although pioneering our metagenomic study provided a first glimpse into the microbial enzymatic decomposition of lignocelluloses within the compost ecosystem.
Conclusions
Pyrosequencing and metagenomic analyses revealed that the RSA consortia enriched from composting ecosystems contained a distinctive set of microorganisms, including Actinobacteria, Proteobacteria, Firmicutes, Chloroflexi, and Bacteroidetes. The diverse and distinct repertoire of CAZyme genes in the RSA consortia were also primarily distributed within these bacteria phyla, which suggests a synergistic system efficient in the processing and metabolizing of carbohydrates in the compost habitat. Interestingly, 46.1 % of the CAZyme genes were from Actinobacteria with an expanded catalog of cellobiohydrolase, β-glucosidase, acetyl xylan esterase, arabinofuranosidase, pectin lyase, and ligninase genes. The versatile functions illustrate the importance of compost actinomycetal communities in the bioconversion of lignocellulosic biomass. In addition, the detection of a variety of previously known and so far unrecognized actinomycetal members in the RSA consortia highlights the power of moving beyond surveys of microbial diversity and mining enzymes from microbial communities for biofuel industry by functional-driven metagenomics.
Methods
Enrichment of rice straw-adapted microbial consortia in a compost habitat
We chose rice (Oryza sativa L.) straw as the feedstock to be degraded since it is more resistant to microbial degradation than the straw from other protein-rich plants such as wheat [73]. Air-dried rice straw was ground to pass a 2-mm sieve using a Retsch ZM200 rotor mill. The ground material (14.81 g) was weighed into a nylon bag with a mesh size of 50 μm. This nylon bag was placed in the middle of an aerobic windrow compost pile of cow manure, which was set up in a suburb of Hangzhou city in China. Aeration of the compost pile occurred via natural ventilation and turning, and the pile was periodically remixed and turned over with a wheel loader. When the pile temperature (29.2 °C) dropped to ambient level towards the end of the maturing stage of composting (day 30), the nylon bag was retrieved from the compost. Rice straw samples of 10.15 g were immediately collected from the bag and shipped to the laboratory on dry ice. To determine the extent of degradation of this substrate incubated in compost ecosystems, the chemical composition (cellulose, hemicellulose and lignin) of pre- and post- enrichment rice straw was determined according to the method described by Liu [74].
Nucleic acid extraction
High molecular weight DNA was extracted from the microbial consortia adherent to the post-enrichment rice straw samples using the MoBio UltraClean Soil DNA isolation kit (MoBio Laboratories, Solana Beach, CA, USA). The extraction was performed according to the manufacturer’s instructions. The extracted DNA was 225 ng/ml and had a 260/280 ratio of 1.91 (Nanodrop, Thermo Scientific, Wilmington, DE, USA). The DNA was eluted in a final volume of 100 μL and the eluents were stored at −20 °C until analysis.
High-throughput 16S rRNA gene pyrosequencing and phylogenetic classification
An amplicon library was constructed for 454 pyrosequencing using bacterial primers 8F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 533R (5′-TTACCGCGGCTGCTGGCAC-3′) for the V1–V3 region of the 16S rRNA gene. The length of the amplicon, including the barcode and 454 primers, was ~596 nt. The PCR reaction was performed in a 25 μL mixture containing 22.5 μL of Platinum PCR SuperMix (Invitrogen, Shanghai, China), 0.5 μL of each primer at 30 μmol L−1, and 1.0–1.5 μL template DNA. The amplification conditions were as follows: 94 °C for 5 min, 28 cycles of 94 °C for 45 s, 55 °C for 30 s, and 72 °C for 45 s followed by extension at 72 °C for 5 min. The PCR products were purified using a 2.0 % agarose gel and quantified using Picogreen (Invitrogen, Shanghai, China). Our sample was subjected to a single pyrosequence run by a Roche massively parallel 454 GS-FLX according to standard protocols [75]. To minimize the effects of random sequencing errors, low-quality sequences were removed by eliminating those without an exact match to the forward primer, without a recognizable reverse primer, length shorter than 200 nucleotides and contained any ambiguous base calls (Ns). After trimming the barcodes and primers from the resulting sequences, we obtained 21,670 high-quality V1–V3 tags of the 16S rRNA gene. Sequences were clustered into an operational taxonomic unit (OTU) at a 0.03 cut-off using the MOTHUR program. The taxonomy of the high-quality reads was further classified using the RDP classifier with a confidence threshold of 80 % and the RDP taxonomic nomenclature [76]. All sequences have been deposited in GenBank short-read archive SRR1292608.
Metagenome processing: DNA library construction and sequencing
DNA library construction and sequencing were performed by BGI (Shenzhen, China) and followed BGI’s previous work on human gut microbe metagenomic sequencing [77]. A library with 180-bp clone insert size was constructed for our sample. Sequence reads were quality trimmed to an accuracy of 99.4 % and duplicate reads were identified and removed prior to assembly. Nearly 59.6 million high-quality reads were generated, and the total data volume of high-quality reads was approximately 5 Gbp (Additional file 3: Table S2).
Illumina genome analyzer short reads de novo assembly
High-quality short reads of the DNA sample were assembled by the SOAPdenovo assembler [78], as used in human gut microbe metagenomic analyses. After assessing different Kmer sizes, we found that Kmer 41 provided the longest contig length and highest read utilization rate. Consequently, we used the contigs of Kmer 41 for our sample as the final assembly result (Additional file 11: Table S7).
Taxonomic profiling and functional classification in metagenomic database
The RSA consortia metagenomic dataset was taxonomically profiled using MetaPhlAn and mOTU [22, 23]. In order to further confirm the composition of microbial community in the RSA consortia, rRNA sequences of the small subunit (SSU) were extracted from the Illumina data using SortMeRNA [25]. Annotations by the MG-RAST pipeline [79] showed that our metagenome protein coding sequences contained 0.4 % RNA genes and 99.6 % protein coding genes, of which 63.3 % have been assigned a putative function. We performed predicted gene functional classification by querying protein sequences of the genes against the evolutionary genealogy of genes: non-supervised orthologous groups (eggNOG) database and the Kyoto Encyclopedia of Genes and the Genomes (KEGG) database using BLASTP with E-value <1 × 10−5. We used the “function comparison” tool of integrated microbial genomes with microbiome samples (IMG/M, http://img.jgi.doe.gov/cgi-bin/m/main.cgi, a data-management and analysis platform for genomic and metagenomic data based on IMG [80]) to compare the clusters of orthologous groups (COG) category of the RSA consortia against eight other microbiomes available on IMG/M including termite, panda, 55 and 40 degree reactor, switchgrass-adapted compost, honey bee, wallaby, and bovine rumen [5, 10–13, 15, 30].
Carbohydrate-active enzymes (CAZymes): annotation and phylogenetic analysis
Searches for CAZymes were performed as described in Liu et al. [81]. Briefly, protein coding genes identified in the RSA consortia metagenome were searched (E < 10−4) against the set of profile hidden markov models (HMMs) representing the signature domain of each CAZy family defined by Yin et al. [82], as deposited on dbCAN (http://csbl.bmb.uga.edu/dbCAN/). The phylogenetic origins of CAZymes-encoding genes were determined using MEGAN [83].
Sequence data submission
Datasets are publicly available on the integrated microbial genomes with microbiome samples (IMG/M, project ID 12014) and the metagenomics RAST server (MG-RAST, ID 4513787.3).
Authors’ contributions
CW, DD, KM, HLW, and WXW designed the experiments and drafted this manuscript and revised it critically for important intellectual content. CW, HSW, and YQ carried out the composting and performed the enrichment of our RSA consortia and the chemical composition test. DD, HSW, and YQ performed the sequencing. KM and HLW participated in the design of the study and helped to draft the manuscript. CW, DD, HSW, and KM performed all data analyses. WXW led and coordinated the overall project. All authors read and approved the final manuscript.
Acknowledgements
This research was supported by the China National Critical Project for Science and Technology on Water Pollution Prevention and Control (No. 2014ZX0710-012) and the International Postdoctoral Exchange Fellowship Program 2015.
Competing interests
The authors declare that they have no competing interests.
Abbreviations
- ABC
ATP-binding cassette
- bp
base pairs
- CAZy
carbohydrate-active enzymes
- CBM
carbohydrate-binding module
- CE
carbohydrate esterase
- COGs
clusters of orthologous groups
- eggNOG
non-supervised orthologous groups
- EGTs
environmental gene tags
- GH
glycoside hydrolase
- GT
glycosyltransferase
- HMMs
hidden markov models
- IMG/M
integrated microbial genomes and metagenomes
- KEGG
kyoto encyclopedia of genes and genomes
- MG-RAST
metagenomics RAST
- ORF
open reading frame
- OTU
operational taxonomic unit
- PL
polysaccharide lyases
- RDP
ribosomal database project
- RSA
rice straw-adapted
- SSU
small subunit
Additional files
10.1186/s13068-016-0440-2 Comparison of the content of pre- and post- enrichment rice straw and its components (34 k).
10.1186/s13068-016-0440-2 Cultures grown on Congo red agar plates from post-enrichment rice straw (left) and pre-enrichment rice straw (right). About 1 g sample was added into 100 mL of sterile water and shaken at 150 rpm for 1 h. Then 100 μL of the resultant solution was inoculated on Congo red agar plates at 28 °C for 7 days. The Congo red agar medium contained K2HPO4 (0.5 g), MgSO4 (0.25 g), CMC-Na (1.88 g), yeast extract (1.0 g), Congo-red (0.4 g), agar (14.0 g), gelatin (2.0 g), pH 7.0 (per liter) (89 k).
10.1186/s13068-016-0440-2 Summary of illumina GA reads and ORF prediction (33 k).
10.1186/s13068-016-0440-2 MetaPhlAn species abundances in the RSA consortia (41 k).
10.1186/s13068-016-0440-2 Metabolic clustering of RSA consortia, termite, panda, reactors, switchgrass-adapted compost, honey bee, wallaby, and bovine rumen metagenomes. The different color represents relative percentage of the metabolic classes within each sample (112 k).
10.1186/s13068-016-0440-2 Glycan metabolism COGs enriched in rice straw-adapted microbial consortia enriched from manure compost (89 k).
10.1186/s13068-016-0440-2 A heat map cluster representing CAZyme profiles in the RSA consortia Metagenome and four typical sample Metatranscriptomes (including samples from the mesophilic stage, thermophilic stage, cooling stage and maturing stage of the composting process; unpublished data). The relative abundances of gene and transcript in Metegenome and Metatranscriptome, respectively, were standardized by z-score transformation (236 k).
10.1186/s13068-016-0440-2 Phylogenetic distributions of carbohydrate-active enzymes at the phylum level in the RSA consortia. The data were visualized via Circos software (427 k).
10.1186/s13068-016-0440-2 Summary of carbohydrate-active enzymes found in the rice straw-adapted consortia enriched from manure compost (262 k).
10.1186/s13068-016-0440-2 Summary of Auxiliary Activities (AA) families found in the rice straw-adapted consortia enriched from manure compost (39 k).
10.1186/s13068-016-0440-2 Summary of de novo assembly results (37 k).
Contributor Information
Cheng Wang, Email: wangcheng0530@126.com.
Da Dong, Email: 562900873@qq.com.
Haoshu Wang, Email: 877478602@qq.com.
Karin Müller, Email: karin.mueller@plantandfood.co.nz.
Yong Qin, Email: 405481949@qq.com.
Hailong Wang, Email: nzhailongwang@gmail.com.
Weixiang Wu, Email: weixiang@zju.edu.cn.
References
- 1.Lynd LR, Laser MS, Bransby D, Dale BE, Davison B, Hamilton R, et al. How biotech can transform biofuels. Nat Biotechnol. 2008;26(2):169–172. doi: 10.1038/nbt0208-169. [DOI] [PubMed] [Google Scholar]
- 2.Kumar P, Barrett DM, Delwiche MJ, Stroeve P. Methods for pretreatment of lignocellulosic biomass for efficient hydrolysis and biofuel production. Ind Eng Chem Res. 2009;48(8):3713–3729. doi: 10.1021/ie801542g. [DOI] [Google Scholar]
- 3.Hassen A, Belguith K, Jedidi N, Cherif A, Cherif M, Boudabous A. Microbial characterization during composting of municipal solid waste. Bioresour Technol. 2001;80(3):217–225. doi: 10.1016/S0960-8524(01)00065-7. [DOI] [PubMed] [Google Scholar]
- 4.Martinez D, Larrondo LF, Putnam N, Gelpke MDS, Huang K, Chapman J, et al. Genome sequence of the lignocellulose degrading fungus Phanerochaete chrysosporium strain RP78. Nat Biotechnol. 2004;22(6):695–700. doi: 10.1038/nbt967. [DOI] [PubMed] [Google Scholar]
- 5.Allgaier M, Reddy A, Park JI, Ivanova N, D’haeseleer P, Lowry S, et al. Targeted discovery of glycoside hydrolases from a switchgrass-adapted compost community. PLoS One. 2010;5(1):e8812. doi: 10.1371/journal.pone.0008812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mayende L, Wilhelmi BS, Pletschke BI. Cellulases (CMCases) and polyphenol oxidases from thermophilic Bacillus spp. isolated from compost. Soil Biol Biochem. 2006;38(9):2963–2966. doi: 10.1016/j.soilbio.2006.03.019. [DOI] [Google Scholar]
- 7.Wang C, Guo X, Deng H, Dong D, Tu Q, Wu W. New insights into the structure and dynamics of actinomycetal community during manure composting. Appl Microbiol Biotechnol. 2014;98(7):3327–3337. doi: 10.1007/s00253-013-5424-6. [DOI] [PubMed] [Google Scholar]
- 8.Amner W, McCarthy AJ, Edwards C. Quantitative assessment of factors affecting the recovery of indigenous and released thermophilic bacteria from compost. Appl Environ Microb. 1988;54(12):3107–3112. doi: 10.1128/aem.54.12.3107-3112.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hess M, Sczyrba A, Egan R, Kim T-W, Chokhawala H, Schroth G, et al. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science. 2011;331(6016):463–467. doi: 10.1126/science.1200387. [DOI] [PubMed] [Google Scholar]
- 10.Brulc JM, Antonopoulos DA, Miller MEB, Wilson MK, Yannarell AC, Dinsdale EA, et al. Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases. Proc Natl Acad Sci USA. 2009;106(6):1948–1953. doi: 10.1073/pnas.0806191105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Warnecke F, Luginbühl P, Ivanova N, Ghassemian M, Richardson TH, Stege JT, et al. Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature. 2007;450(7169):560–565. doi: 10.1038/nature06269. [DOI] [PubMed] [Google Scholar]
- 12.Zhu L, Wu Q, Dai J, Zhang S, Wei F. Evidence of cellulose metabolism by the giant panda gut microbiome. Proc Natl Acad Sci USA. 2011;108(43):17714–17719. doi: 10.1073/pnas.1017956108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engel P, Martinson VG, Moran NA. Functional diversity within the simple gut microbiota of the honey bee. Proc Natl Acad Sci USA. 2012;109(27):11002–11007. doi: 10.1073/pnas.1202970109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delmont TO, Prestat E, Keegan KP, Faubladier M, Robe P, Clark IM, et al. Structure, fluctuation and magnitude of a natural grassland soil metagenome. ISME J. 2012;6(9):1677–1687. doi: 10.1038/ismej.2011.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hollister EB, Forrest AK, Wilkinson HH, Ebbole DJ, Tringe SG, Malfatti SA, et al. Mesophilic and thermophilic conditions select for unique but highly parallel microbial communities to perform carboxylate platform biomass conversion. PLoS One. 2012;7(6):e39689. doi: 10.1371/journal.pone.0039689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martins LF, Antunes LP, Pascon RC, de Oliveira JCF, Digiampietri LA, Barbosa D, et al. Metagenomic analysis of a tropical composting operation at the Sao Paulo zoo park reveals diversity of biomass degradation functions and organisms. PLoS One. 2013;8(4):e61928. doi: 10.1371/journal.pone.0061928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng YF, Edwards JE, Allison GG, Zhu WY, Theodorou MK. Diversity and activity of enriched ruminal cultures of anaerobic fungi and methanogens grown together on lignocellulose in consecutive batch culture. Bioresour Technol. 2009;100(20):4821–4828. doi: 10.1016/j.biortech.2009.04.031. [DOI] [PubMed] [Google Scholar]
- 18.Lu YT, Chen XB, Zhou P, Li ZH. Screening on oil-decomposing microorganisms and application in organic waste treatment machine. J Environ Sci. 2005;17(3):440–444. [PubMed] [Google Scholar]
- 19.DeAngelis KM, Gladden JM, Allgaier M, D’haeseleer P, Fortney JL, Reddy A, et al. Strategies for enhancing the effectiveness of metagenomic-based enzyme discovery in lignocellulolytic microbial communities. Bioenergy Res. 2010;3(2):146–158. doi: 10.1007/s12155-010-9089-z. [DOI] [Google Scholar]
- 20.Chen Y, Inbar Y, Hadar Y, Malcolm R. Chemical properties and solid-state CPMAS 13C-NMR of composted organic matter. Sci Total Environ. 1989;81:201–208. doi: 10.1016/0048-9697(89)90126-5. [DOI] [Google Scholar]
- 21.Tang JC, Maie N, Tadab Y, Katayama A. Characterization of the maturing process of cattle manure compost. Process Biochem. 2006;41:380–389. doi: 10.1016/j.procbio.2005.06.022. [DOI] [Google Scholar]
- 22.Segata N, Waldron L, Ballarini A, Narasimhan V, Jousson O, Huttenhower C. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat Methods. 2012;9(8):811–814. doi: 10.1038/nmeth.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sunagawa S, Mende DR, Zeller G, Izquierdo-Carrasco F, Berger SA, Kultima JR, et al. Metagenomic species profiling using universal phylogenetic marker genes. Nat Methods. 2013;10(12):1196–1199. doi: 10.1038/nmeth.2693. [DOI] [PubMed] [Google Scholar]
- 24.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75(23):7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kopylova E, Noé L, Touzet H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. 2012;28(24):3211–3217. doi: 10.1093/bioinformatics/bts611. [DOI] [PubMed] [Google Scholar]
- 26.Rastogi G, Bhalla A, Adhikari A, Bischoff KM, Hughes SR, Christopher LP, et al. Characterization of thermostable cellulases produced by Bacillus and Geobacillus strains. Bioresource Technol. 2010;101(22):8798–8806. doi: 10.1016/j.biortech.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 27.Amass W, Amass A, Tighe B. A review of biodegradable polymers: uses, current developments in the synthesis and characterization of biodegradable polyesters, blends of biodegradable polymers and recent advances in biodegradation studies. Polym Int. 1998;47:89–144. doi: 10.1002/(SICI)1097-0126(1998100)47:2<89::AID-PI86>3.0.CO;2-F. [DOI] [Google Scholar]
- 28.Anderson I, Abt B, Lykidis A, Klenk HP, Kyrpides N, Ivanova N. Genomics of aerobic cellulose utilization systems in actinobacteria. PLoS One. 2012;7(6):e39331. doi: 10.1371/journal.pone.0039331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dougherty MJ, D’haeseleer P, Hazen TC, Simmons BA, Adams PD, Hadi MZ. Glycoside hydrolases from a targeted compost metagenome, activity-screening and functional characterization. BMC Biotechnol. 2012;12(1):38. doi: 10.1186/1472-6750-12-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pope P, Denman S, Jones M, Tringe S, Barry K, Malfatti S, et al. Adaptation to herbivory by the Tammar wallaby includes bacterial and glycoside hydrolase profiles different from other herbivores. Proc Natl Acad Sci USA. 2010;107(33):14793–14798. doi: 10.1073/pnas.1005297107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ryckeboer J, Mergaert J, Vaes K, Klammer S, De Clercq D, Coosemans J, et al. A survey of bacteria and fungi occurring during composting and self-heating processes. Ann Microbiol. 2003;53(4):349–410. [Google Scholar]
- 32.Zhang X, Zhong YH, Yang SD, Zhang WX, Xu MQ, Ma AZ, et al. Diversity and dynamics of the microbial community on decomposing wheat straw during mushroom compost production. Bioresour Technol. 2014;170:183–195. doi: 10.1016/j.biortech.2014.07.093. [DOI] [PubMed] [Google Scholar]
- 33.Tuomela M, Vikman M, Hatakka A, Itävaara M. Biodegradation of lignin in a compost environment: a review. Bioresour Technol. 2000;72(2):169–183. doi: 10.1016/S0960-8524(99)00104-2. [DOI] [Google Scholar]
- 34.Munk C, Lapidus A, Copeland A, Jando M, Mayilraj S, Del Rio TG, et al. Complete genome sequence of Stackebrandtia nassauensis type strain (LLR-40 K-21T) Stand Genomic Sci. 2009;1(3):234. doi: 10.4056/sigs.47643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rastogi G, Bhalla A, Adhikari A, Bischoff KM, Hughes SR, Christopher LP, et al. Characterization of thermostable cellulases produced by Bacillus and Geobacillus strains. Bioresour Technol. 2010;101(22):8798–8806. doi: 10.1016/j.biortech.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 36.Labeda D, Kroppenstedt R. Stackebrandtia nassauensis gen. nov., sp. nov. and emended description of the family Glycomycetaceae. Int J Syst Evol Microbiol. 2005;55(4):1687–1691. doi: 10.1099/ijs.0.63496-0. [DOI] [PubMed] [Google Scholar]
- 37.Wang Y, Zhang Z, Ruan J. A proposal to transfer Microbispora bispora (Lechevalier 1965) to a new genus, Thermobispora gen. nov., as Thermobispora bispora comb. nov. Int J Syst Bacteriol. 1996;46(4):933–938. doi: 10.1099/00207713-46-4-933. [DOI] [PubMed] [Google Scholar]
- 38.Nanavati DM, Thirangoon K, Noll KM. Several archaeal homologs of putative oligopeptide-binding proteins encoded by Thermotoga maritima bind sugars. Appl Environ Microb. 2006;72(2):1336–1345. doi: 10.1128/AEM.72.2.1336-1345.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Law CJ, Maloney PC, Wang DN. Ins and outs of major facilitator superfamily antiporters. Annu Rev Microbiol. 2008;62:289. doi: 10.1146/annurev.micro.61.080706.093329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deutscher J, Francke C, Postma PW. How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol Mol Biol R. 2006;70(4):939–1031. doi: 10.1128/MMBR.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng X, Hiras J, Deng K, Bowen B, Simmons BA, Adams PD, et al. High throughput nanostructure-initiator mass spectrometry screening of microbial growth conditions for maximal β-glucosidase production. Front Microbiol. 2013;4:365. doi: 10.3389/fmicb.2013.00365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bernier R, Stutzenberger F. Extracellular and cell-associated forms of beta-glucosidase in Thermomonospora curvata. Lett Appl Microbiol. 1988;7(4):103–107. doi: 10.1111/j.1472-765X.1988.tb01263.x. [DOI] [Google Scholar]
- 43.Gladden JM, Park JI, Bergmann J, Reyes-Ortiz V, Patrik D, Quirino BF, et al. Discovery and characterization of ionic liquid-tolerant thermophilic cellulases from a swithchgrass-adapted microbial community. Biotechnol Biofuels. 2014;7(1):15. doi: 10.1186/1754-6834-7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cayuela M, Sánchez-Monedero M, Roig A. Evaluation of two different aeration systems for composting two-phase olive mill wastes. Process Biochem. 2006;41(3):616–623. doi: 10.1016/j.procbio.2005.08.007. [DOI] [Google Scholar]
- 45.Brás JL, Cartmell A, Carvalho ALM, Verzé G, Bayer EA, Vazana Y, et al. Structural insights into a unique cellulase fold and mechanism of cellulose hydrolysis. Proc Natl Acad Sci USA. 2011;108(13):5237–5242. doi: 10.1073/pnas.1015006108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buist G, Steen A, Kok J, Kuipers OP. LysM, a widely distributed protein motif for binding to (peptido) glycans. Mol Microbiol. 2008;68(4):838–847. doi: 10.1111/j.1365-2958.2008.06211.x. [DOI] [PubMed] [Google Scholar]
- 47.Reddy AP, Simmons CW, D’haeseleer P, Khudyakov J, Burd H, Hadi M, et al. Discovery of microorganisms and enzymes involved in high-solids decomposition of rice straw using metagenomic analyses. PLoS One. 2013;8(10):e77985. doi: 10.1371/journal.pone.0077985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boraston A, Bolam D, Gilbert H, Davies G. Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem J. 2004;382:769–781. doi: 10.1042/BJ20040892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morrison M, Pope PB, Denman SE, McSweeney CS. Plant biomass degradation by gut microbiomes: more of the same or something new? Curr Opin Biotechnol. 2009;20(3):358–363. doi: 10.1016/j.copbio.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 50.Flint HJ, Bayer EA, Rincon MT, Lamed R, White BA. Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nat Rev Microbiol. 2008;6(2):121–131. doi: 10.1038/nrmicro1817. [DOI] [PubMed] [Google Scholar]
- 51.Narra M, Dixit G, Divecha J, Kumar K, Madamwar D, Shah AR. Production, purification and characterization of a novel GH 12 family endoglucanase from Aspergillus terreus and its application in enzymatic degradation of delignified rice straw. Int Biodeter Biodegr. 2014;88:150–161. doi: 10.1016/j.ibiod.2013.12.016. [DOI] [Google Scholar]
- 52.Turner P, Mamo G, Karlsson EN. Potential and utilization of thermophiles and thermostable enzymes in biorefining. Microb Cell Fact. 2007;6(9):1–23. doi: 10.1186/1475-2859-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hashimoto K, Yoshida M, Hasumi K. Isolation and characterization of CcAbf62A, a GH62 α-L-arabinofuranosidase, from the basidiomycete Coprinopsis cinerea. Biosci Biotechnol Biochem. 2011;75(2):342–345. doi: 10.1271/bbb.100434. [DOI] [PubMed] [Google Scholar]
- 54.Berlemont R, Martiny AC. Phylogenetic distribution of potential cellulases in bacteria. Appl Environ Microb. 2013;79(5):1545–1554. doi: 10.1128/AEM.03305-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ball AS, Mccarthy AJ. Sacchariiication of straw by actinomycete enzymes. J Gen Microbiol. 1988;134(8):2139–2147. [Google Scholar]
- 56.Dahlberg L, Holst O, Kristjansson JK. Thermostable xylanolytic enzymes from Rhodothermus marinus grown on xylan. Appl Microbiol Biotechnol. 1993;40(1):63–68. [Google Scholar]
- 57.Sunna A, Antranikian G. Xylanolytic enzymes from fungi and bacteria. Crit Rev Biotechnol. 1997;17(1):39–67. doi: 10.3109/07388559709146606. [DOI] [PubMed] [Google Scholar]
- 58.Biely P, MacKenzie C, Puls J, Schneider H. Cooperativity of esterases and xylanases in the enzymatic degradation of acetyl xylan. Nat Biotechnol. 1986;4(8):731–733. doi: 10.1038/nbt0886-731. [DOI] [Google Scholar]
- 59.Mohnen D. Pectin structure and biosynthesis. Curr Opin Plant Biol. 2008;11(3):266–277. doi: 10.1016/j.pbi.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 60.Hayashi K, Inoue Y, Shiga M, Sato SI, Takano R, Hirayae K, et al. Pectinolytic enzymes from Pseudomonas marginalis maff 03-01173. Phytochemistry. 1997;45(7):1359–1363. doi: 10.1016/S0031-9422(97)00191-X. [DOI] [PubMed] [Google Scholar]
- 61.Raj Kashyap D, Kumar Soni S, Tewari R. Enhanced production of pectinase by Bacillus sp. DT7 using solid state fermentation. Bioresour Technol. 2003;88(3):251–254. doi: 10.1016/S0960-8524(02)00206-7. [DOI] [PubMed] [Google Scholar]
- 62.Prade RA, Zhan D, Ayoubi P, Mort AJ. Pectins, pectinases and plant-microbe interactions. Biotechnol Genet Eng Rev. 1999;16(1):361–392. doi: 10.1080/02648725.1999.10647984. [DOI] [PubMed] [Google Scholar]
- 63.Gysler C, Harmsen J, Kester H, Visser J, Heim J. Isolation and structure of the pectin lyase D-encoding gene from Aspergillus niger. Gene. 1990;89(1):101–108. doi: 10.1016/0378-1119(90)90211-9. [DOI] [PubMed] [Google Scholar]
- 64.Stutzenberger FJ. Inducible thermoalkalophilic polygalacturonate lyase from Thermomonospora fusca. J Bacteriol. 1987;169(6):2774–2780. doi: 10.1128/jb.169.6.2774-2780.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Spooner F, Jr, Hammerschmidt R. Characterization of extracellular pectic enzymes produced by Streptomyces species. Phytopathology. 1989;79:1190. [Google Scholar]
- 66.Sarkanen KV, Ludwig CH. Lignins: occurrence, formation, structure and reactions. New York: John Wiley & Sons Inc; 1971. [Google Scholar]
- 67.Welinder KE. Superfamily of plant, fungal and bacterial peroxidases. Curr Opin Struck Biol. 1992;2(3):388–393. doi: 10.1016/0959-440X(92)90230-5. [DOI] [Google Scholar]
- 68.Bugg TD, Ahmad M, Hardiman EM, Singh R. The emerging role for bacteria in lignin degradation and bio-product formation. Curr Opin Biotechnol. 2011;22:394–400. doi: 10.1016/j.copbio.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 69.Graham JE, Clark ME, Nadler DC, Huffer S, Chokhawala HA, Rowland SE, et al. Identification and characterization of a multidomain hyperthermophilic cellulase from an archaeal enrichment. Nat Commun. 2011;2:375. doi: 10.1038/ncomms1373. [DOI] [PubMed] [Google Scholar]
- 70.Wongwilaiwalin S, Rattanachomsri U, Laothanachareon T, Eurwilaichitr L, Igarashi Y, Champreda V. Analysis of a thermophilic lignocellulose degrading microbial consortium and multi-species lignocellulolytic enzyme system. Enzyme Microb Tech. 2010;47(6):283–290. doi: 10.1016/j.enzmictec.2010.07.013. [DOI] [Google Scholar]
- 71.Jang HD, Chen KS. Production and characterization of thermostable cellulases from Streptomyces transformant T3-1. World J Microb Biotechnol. 2003;19(3):263–268. doi: 10.1023/A:1023641806194. [DOI] [Google Scholar]
- 72.Horn SJ, Vaaje-Kolstad G, Westereng B, Eijsink VG. Novel enzymes for the degradation of cellulose. Biotechnol Biofuels. 2012;5(1):1–13. doi: 10.1186/1754-6834-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Parr J, Papendick R, Hornick S, Meyer R. Soil quality: attributes and relationship to alternative and sustainable agriculture. Am J Altern Agr. 1992;7(1–2):5–11. doi: 10.1017/S0889189300004367. [DOI] [Google Scholar]
- 74.Liu S. Analysis and measurement in papermaking industry. Beijing: China Chemical Industry Press; 2004. [Google Scholar]
- 75.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437(7057):376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, et al. The ribosomal database project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37:D141–D145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464(7285):59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20(2):265–272. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Meyer F, Paarmann D, D’Souza M, Olson R, Glass EM, Kubal M, et al. The metagenomics RAST server—a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008;9(1):386. doi: 10.1186/1471-2105-9-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Markowitz VM, Kyrpides NC. Comparative genome analysis in the integrated microbial genomes (IMG) system. Methods Mol Biol. 2008;395:35–56. doi: 10.1007/978-1-59745-514-5_3. [DOI] [PubMed] [Google Scholar]
- 81.Liu N, Zhang L, Zhou H, Zhang M, Yan X, Wang Q, et al. Metagenomic insights into metabolic capacities of the gut microbiota in a fungus-cultivating termite (Odontotermes yunnanensis) PLoS One. 2013;8(7):e69184. doi: 10.1371/journal.pone.0069184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yin Y, Mao X, Yang J, Chen X, Mao F, Xu Y. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012;40(W1):W445–W451. doi: 10.1093/nar/gks479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Huson DH, Auch AF, Qi J, Schuster SC. MEGAN analysis of metagenomic data. Genome Res. 2007;17(3):377–386. doi: 10.1101/gr.5969107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Krzywinski M, Schein J, Birol İ, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]