

Abstract
Purpose of review
The purpose of this review is to discuss the mechanisms of central and peripheral tolerance in relation to T-cell mediated autoimmunity in rheumatoid arthritis (RA).
Recent findings
The well established association between major histocompatibility complex class II and RA has led us to understand that T cells, and the adaptive immune response, are important in the pathogenesis of disease. In order for autoimmune disease to develop, there is a breach of tolerance to self antigen and the mechanisms of both central and peripheral tolerance aim to prevent this. Here, we review evidence from mouse models indicating that alterations in T-cell receptor signalling thresholds during thymic selection may be linked to the escape of T cells that mediate autoimmune arthritis. In addition, we summarize the role of dendritic cells and Foxp3+ regulatory T cells in both peripheral and thymic tolerance, and highlight their relevance to what we know about the aetiology of RA.
Summary
Mechanisms of central tolerance in the thymus and peripheral tolerance are in place to control autoreactive T cells and to prevent the development of autoimmune disease. We anticipate that a better understanding of these mechanisms will lead to the development of better, antigen-specific therapeutics to restore tolerance.
Keywords: dendritic cell, T cell, thymus, tolerance, Treg
INTRODUCTION
The immune system is heavily implicated in the pathogenesis of rheumatoid arthritis (RA) and there is a generally accepted view that some type of ‘breach of self-tolerance’ underlies this. However, the exact mechanisms involved, whether these relate to differences in central versus peripheral tolerance, and how this relates to the aetiology of the disease remain unclear. Here, we discuss how defects in central and peripheral tolerance may be involved, how they might be linked and how they relate to what we know about RA. The ultimate goal for treatment of autoimmune disease would be the development of therapies that promote and restore self-tolerance in patients. As a result, understanding central and peripheral tolerogenic mechanisms that have gone awry in RA will lead to the development of improved therapeutics that target the restoration of tolerance.
Box 1.

no caption available
CENTRAL TOLERANCE MECHANISMS IN RHEUMATOID ARTHRITIS
The antigen receptors of T cells are randomly generated to provide the potential to recognize a wide array of pathogens. As a consequence, self-reactive T cells are generated. To avoid autoimmunity, regulatory mechanisms must be in place to remove and/or control these cells. This ‘immunological tolerance’ is mediated at several levels and in several cell types. Here, we will focus on tolerance in T cells in the context of RA.
Thymic selection
The thymus supports the development of T cells expressing the αβ form of the T-cell receptor. Following their entry at the corticomedullary junction [1], CD4− CD8− lymphoid precursors randomly rearrange their T-cell receptor (TCR) genes to maximize variation within the developing T-cell repertoire. Following the induction of CD4 and CD8 expression [2], these processes generate a large cohort of CD4+CD8+ thymocytes that express a wide range of αβ TCR specificities. Owing to the random nature of TCR gene rearrangement, such cells undergo selection processes to ensure the thymus is biased toward the generation of self-tolerant T cells capable of self-major histocompatibility complex (MHC) recognition.
During positive selection, recognition of self-peptides bound to MHC II or MHC I molecules on cortical thymic epithelial cells ensures the generation of single positive CD4+ and CD8+ thymocytes, respectively [3]. During this process, newly selected thymocytes undergo Chemokine Receptor 7 (CCR7)-mediated migration to the thymic medulla (Fig. 1), an important site for tolerance induction [4]. Here, CD4+ and CD8+ thymocytes are screened further for their reactivity to self-antigens, including those expressed intrathymically as a result of the Autoimmune regulator (AIRE)-mediated transcription of tissue-restricted antigens in medullary thymic epithelial cells [5,6]. In the medulla, dendritic cells also play a key role in thymic tolerance through both cross-presentation of self-antigens from medullary thymic epithelial cells as well as migration of self-antigen bearing dendritic cells from the periphery [7]. Importantly, the outcome of TCR-MHC interactions within the medulla is highly dependent upon how strongly the TCR recognizes self-antigen. High affinity TCR-mediated interactions result in negative selection through apoptosis induction, whereas medium affinity interactions induce expression of the transcription factor Foxp3, and the emergence of the natural T-regulatory (Treg) lineage [8]. Collectively, T-cell development involves intrathymic selection events based upon self-antigen interaction, to maximize the spectrum of antigen recognition by newly selected T cells and also minimize the risk of generating autoreactive T cells. As a consequence, factors with the potential to alter TCR signal strength in response to self-antigen during intrathymic development can lead to alterations in thymic tolerance and potentially skew the T-cell repertoire, leading to autoimmune disease.
FIGURE 1.
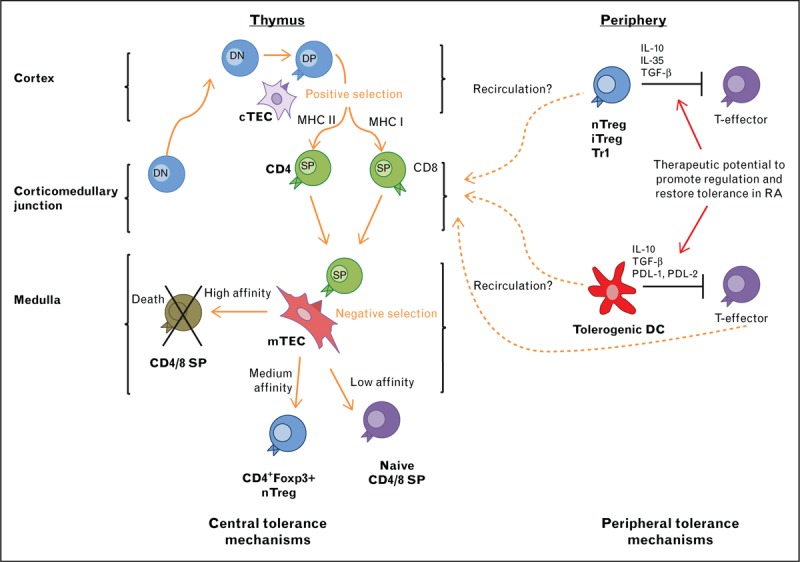
Regulation of central and peripheral tolerance. The organization of the thymus into cortex and medulla ensures that thymic selection events take place in a stepwise fashion. In the medulla, tolerance-inducing events coordinated by medullary thymic epithelial cells and dendritic cells help to remove potentially autoreactive specificities from the newly produced T-cell pool, and also result in the generation of Foxp3+ regulatory T cells. In peripheral tissues, several immune-regulatory products of Dendritic cells and Treg have been linked to the control of effector T cells that mount responses to self-antigens. Perhaps significantly, the thymus also acts as a site that supports T-cell and dendritic cell populations that enter from peripheral tissues, and evidence suggests a role for thymus recirculation in the control of T-cell tolerance. DN: CD4-CD8-, DP: CD4+CD8+; SP CD4+ or CD8+.
Rheumatoid arthritis and altered T-cell selection
As noted above, RA is a common autoimmune disease with the cause being linked to a combination of both genetic and environmental factors [9]. Various murine models have improved our understanding of the genetic aspects of the disease with a particular focus on mutations that alter central and peripheral T-cell tolerance. For instance, under circumstances where mutations dampen the perception of TCR affinity strength during selection, high affinity autoreactive TCRs may escape negative selection. These instead are allowed to enter into the peripheral T-cell pool, where they have a greater propensity to react with self and elicit an autoimmune phenotype [8]. There are a number of models and clinical examples which illustrate this.
The murine SKG model of autoimmune arthritis
Mice with a recessive point mutation in the gene encoding the TCR signalling protein Zeta-chain-associated protein kinase 70 (ZAP- 70), are known as SKG mice and are frequently used as a model for autoimmune arthritis. SKG mice have suppressed TCR signal perception allowing selection of a more autoreactive T-cell repertoire in the thymus that peripherally contributes to the development of autoimmune arthritis [10]. To illustrate this, autologous mixed lymphocyte reactions were used to examine the reactivity of peripheral SKG T cells. These were shown to have high levels of proliferation and activation to autologous Antigen Presenting Cells (APCs) when compared with control T cells, highlighting their autoreactive nature [11]. In addition, analysis of TCR Variable Beta subunit usage showed preferential expression of certain subfamilies with more autoreactive tendencies in SKG mice than controls [12]. When these TCRs were isolated, transfected into bone marrow cells and transferred into Rag2−/− hosts, autoimmune arthritis was induced, reinforcing the idea that SKG mice have a skewed T-cell repertoire toward specificities that are more autoreactive [13▪▪]. Collectively, such observations suggest that alterations in the perception of TCR signal strength may drive the development of arthritis in SKG mice by altering intrathymic selection of the developing T-cell repertoire. In addition, intrathymically generated Treg from SKG mice show a defective suppressive capacity when transferred along with SKG conventional T cells into nude hosts [12,14], again suggesting that altering the strength of TCR signalling during intrathymic T-cell selection events directly impacts upon the T-cell repertoire that is selected, and increases susceptibility to abnormality [15].
As a consequence of altered thymic selection, autoreactive peripheral T cells can respond to self-antigens expressed by dendritic cells, triggering their activation and differentiation into functionally distinct T-cell subsets. In SKG mice, as with human RA, the main effector cells have been shown to be Th17 cells driven by APC-derived IL-6, with IL-6 deficient SKG mice being devoid of IL-17-producing CD4 T cells [11]. Interestingly, Th17 cells can be recruited to joints through CCR6 expression, attracted by the high CCL20 levels found in arthritic joints where they are thought to mediate innate cell activation and joint destruction [16].
Rheumatoid arthritis and PTPN22 variants
Protein tyrosine phosphatase, non-receptor type 22 (PTPN22) encodes lymphocyte tyrosine phosphatase (Lyp) or the mouse ortholog Pep, and is a critical negative regulator of TCR signal transduction upstream of ZAP-70. A single nucleotide polymorphism in the protein tyrosine phosphatase N22 producing a PTPN22 variant (R620W) has been shown to support the progression of autoimmune disease such as type I diabetes and it was further suggested to be the second strongest genetic risk factor for RA, second to HLA variants in humans [17,18]. Interestingly, this PTPN22 variant (R620W) has been suggested to be a gain of function mutation, linked to reduced TCR signalling during intrathymic T-cell selection and high affinity self-reactive T cells in the periphery [17,19]. However, other studies have reported that the consequence of PTPN22 polymorphisms may be a decrease in phosphatase activity and an increase in TCR signalling [20].
Despite this discrepancy, a variant of Lyp (Lyp620W) which is encoded by PTPN22 has been associated with autoimmune disease in humans. Its effect on disease development can be studied using mice expressing the Lyp variant homolog Pep619W. Analysis of these mice carrying OT-II TCR or male H-Y antigen TCR transgenes showed increased CD4+ T-cell positive selection, proliferation of the memory/effector T-cell pool, but no alteration in negative selection indicating a complex requirement for functional Lyp in ensuring appropriate TCR signalling during T-cell selection [20]. In addition, a lack of PTPN22 expression was investigated in mice with regard to T-regulatory cells, which have been shown to express higher levels of PTPN22 than conventional T cells [21]. Interestingly, when compared with WT mice, Ptpn22-/- mice showed significant increases in Treg and their precursor populations [21], suggesting alterations in TCR signalling are able to affect the balance between conventional and Treg development, which might further affect autoimmune status.
PERIPHERAL TOLERANCE MECHANISMS IN RHEUMATOID ARTHRITIS
Although mutations within the thymus can alter T-cell selection and skew the T-cell repertoire toward an autoreactive nature, under normal circumstances, the selection processes that are in place to prevent autoreactive T cells escaping from the thymus are still not 100% effective. As a result, there are always some autoreactive T cells that do escape, undetected, into the periphery. For this reason, the mechanisms of peripheral tolerance are vital to prevent the development of autoimmune disease. In this section, various mechanisms of peripheral tolerance will be discussed. These include regulation via Treg, and tolerogenic dendritic cells as well as their relevance to RA, and how these mechanisms may fail in RA patients, leading to a breakdown of tolerance.
T-regulatory cells
Treg are known to be vital for the maintenance of immunological tolerance (reviewed in [22,23]). Along with natural CD4+CD25+Foxp3+ Treg (nTreg), other regulatory populations include inducible CD4+Foxp3+ Treg (iTreg), which develop in the periphery after induction of Foxp3+ expression, and CD4+Foxp3+ type 1 regulatory T cells (Tr1). Foxp3+ has been shown to be vital for the development and suppressive function of Treg [24] as in its absence aggressive autoimmune disease develops as self-tolerance is broken [25–29]. Treg manifest their suppressive function through various mechanisms including direct cell-to-cell contact, or indirectly through the secretion of anti-inflammatory cytokines (e.g. IL-10, IL-35, or TGF-β) as IL-10 or IL-35 deficient have an impaired suppressive capacity [30,31]. The secretion of TGF-β can induce the development of further Treg from CD4+CD25- naïve T cells [32,33]. Treg can also act by reducing the functions of APCs by inhibiting CD80 and CD86 expression, via a CTLA-4 dependent mechanism [34–36].
Murine studies, using collagen-induced arthritis as a model of RA, have shown that the depletion of Treg using anti-CD25 resulted in increased disease severity [37], and reduced disease severity was observed when Treg were introduced by adoptive transfer [38]. Treg from RA patients have been shown to be defective in their ability to suppress proinflammatory cytokine production by CD4+ T cells, although they can efficiently suppress their proliferation; this defect in their suppressive abilities may be because of a defect in CTLA-4 expression and function [39–41]. B cells are known to have a key pathogenic role in the aetiology of RA, as evidenced by the efficacy of rituximab treatment in RA patients [42,43]. In relation to this, Treg have been shown to regulate the pathogenic function of B cells during inflammation both in murine models and in RA itself [44,45].
Tr1 cells mediate their suppressive function through the secretion of IL-10 and TGF-β and are known to suppress both immune and autoimmune responses [46]. A population of antigen-specific IL-10-producing Tr1 cells has been identified in the blood of RA patients [47] indicating they may have a role in maintenance of peripheral tolerance in RA. The transfer of Tr1 cells in murine models of RA was found to be beneficial, reducing the incidence and severity of arthritis when administered before and after induction of disease [48]. The Tr1 cell population could, therefore, be a promising target for the restoration of tolerance in autoimmune diseases, including RA.
Role of dendritic cells in the regulation of peripheral tolerance
Dendritic cells are vital for the induction of the inflammatory immune response against, for example, invading pathogens. In addition to this, dendritic cells are also central to immune regulation and inducing and maintaining tolerance (Fig. 1), as demonstrated by the development of fatal spontaneous autoimmune disease following dendritic cell depletion [49].
‘Tolerogenic’ dendritic cells are generated through the incomplete maturation of dendritic cells, as occurs during the steady state. These tolerogenic dendritic cells present antigen to T cells in the absence of adequate costimulatory signals and result in the maintenance of peripheral tolerance through mechanisms including T-cell deletion, unresponsiveness or anergy [50], and the induction of regulatory T cells [51]. These effects are mainly attributed to the production of anti-inflammatory cytokines, for example, IL-10 and TGF-β, and the expression of downregulatory/inhibitory markers, for example, Programmed death-ligand 1 and 2 (PDL-1, PDL-2) (reviewed in [52,53]).
Restoring tolerance through the use of immunomodulatory tolerogenic dendritic cells in RA has become an exciting line of therapeutic potential. Studies in murine CIA have shown tolerogenic dendritic cells to be highly effective. Introducing in-vitro derived type-II collagen-pulsed tolerogenic dendritic cells into arthritic CIA mice reduced the severity of the disease, reduced the inflammatory environment (lower levels of Th17 cells) and increased the anti-inflammatory environment through increased levels of IL-10 producing T cells [54]. The generation of tolerogenic dendritic cells from RA patients has been investigated [55] and their safety and efficacy as a therapy are currently being determined in clinical trials [56]. Cellular therapies like tolerogenic dendritic cells or the manipulation of dendritic cells in vivo to induce a more tolerogenic population could be a potential and feasible therapy for RA patients in the future.
In addition to tolerogenic dendritic cells being able to regulate immune responses, different subsets of dendritic cells may have different roles in autoimmune disease. Using a novel breach of self-tolerance murine model of arthritis [57], we have shown that plasmacytoid dendritic cells have an anti-inflammatory role [58]. By contrast, conventional dendritic cells have a more proinflammatory role. Their depletion resulted in reduced severity of disease as well as reduced anticollagen responses [59]. Interestingly, peripheral dendritic cell homing to the thymus provides a source of peripheral antigens for tolerance induction in the steady state [60,61]. Whether the onset of autoimmune reactions alters this process, and so impacts on intrathymic tolerance mechanisms is not known.
Recirculation of peripheral T cells back to the thymus
It has been known for some time now that peripheral T cells, including both conventional and Foxp3+ Treg, can home back to the thymus, which challenges the view that movement out of the thymus is unidirectional (Fig. 1). The first evidence showed labelled lymph node cells transferred into syngeneic hosts could be found within the thymus in both adult and neonatal hosts [62]. In the mouse, mature peripheral T cells that migrate into the thymus resemble activated, or previously activated, CD44hi T cells which appear to preferentially enter over naïve T cells [63–65] (reviewed in detail [66]). The question remains as to what function these recirculating peripheral lymphocytes have in the thymus. There is emerging evidence that these cells are able to alter central tolerance and induce the deletion of thymic APC populations in an antigen-specific manner [67]. In addition, very recently it has been shown that peripheral Treg can also recirculate back to the thymus and once there they suppress the development of new Treg through the inhibition of IL-2 [68▪▪]. In the same study, evidence of the reentry of mature T cells and Treg into the human thymus was also found. In the setting of autoimmune disease and RA, this could be an interesting mechanism for silencing autoreactive T cells. Moreover, despite having sufficient numbers of progenitor cells [69–71], RA patients exhibit impaired thymic function as indicated by fewer recent thymic emigrants. Whether this is linked to changes in peripheral T-cell recirculation back to the thymus caused by ageing and/or RA is not clear.
CONCLUSION
The thymus represents a key site for the generation of αβT cells that play an essential role in immune responses. However, the removal of autoreactive T cells from the developing TCR repertoire via intrathymic selection mechanisms is incomplete, which is a significant factor in relation to the onset of T-cell mediated autoimmune diseases. To combat this, peripheral tolerance mechanisms involving modulation of dendritic cell function and Foxp3+ Treg are in place. In RA, evidence suggests that a breakdown in T-cell tolerance takes place. However, whether this maps to altered T-cell responses in either the thymus or within peripheral tissues is not clear. Perhaps significantly, both sites are linked not only by the conventional T cells, Foxp3+ Treg and dendritic cell subsets they contain, but also by trafficking of these cell types between each site. How such processes impact on the maintenance of tolerance, and its breakdown, is not understood. We propose that adopting an overarching approach to studying tolerance regulation at sites of T-cell production and effector function will provide new opportunities to better understand tolerance maintenance and breakdown, and inform future strategies for immune intervention.
Acknowledgements
This work was funded by the ARUK Rheumatoid Arthritis Pathogenesis Centre for Excellence (RACE). G.A. is supported by an MRC Programme Grant; P.G. is supported by an ARUK Programme Grant.
Financial support and sponsorship
None.
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
REFERENCES
- 1.Forster R, Davalos-Misslitz AC, Rot A. CCR7 and its ligands: balancing immunity and tolerance. Nat Rev Immunol 2008; 8:362–371. [DOI] [PubMed] [Google Scholar]
- 2.Anderson G, Takahama Y. Thymic epithelial cells: working class heroes for T cell development and repertoire selection. Trends Immunol 2012; 33:256–263. [DOI] [PubMed] [Google Scholar]
- 3.Singer A, Adoro S, Park JH. Lineage fate and intense debate: myths, models and mechanisms of CD4- versus CD8-lineage choice. Nat Rev Immunol 2008; 8:788–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kurobe H, Liu C, Ueno T, et al. CCR7-dependent cortex-to-medulla migration of positively selected thymocytes is essential for establishing central tolerance. Immunity 2006; 24:165–177. [DOI] [PubMed] [Google Scholar]
- 5.Derbinski J, Gabler J, Brors B, et al. Promiscuous gene expression in thymic epithelial cells is regulated at multiple levels. J Exp Med 2005; 202:33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rossi SW, Kim MY, Leibbrandt A, et al. RANK signals from CD4(+)3(−) inducer cells regulate development of Aire-expressing epithelial cells in the thymic medulla. J Exp Med 2007; 204:1267–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klein L, Hinterberger M, von Rohrscheidt J, Aichinger M. Autonomous versus dendritic cell-dependent contributions of medullary thymic epithelial cells to central tolerance. Trends Immunol 2011; 32:188–193. [DOI] [PubMed] [Google Scholar]
- 8.Starr TK, Jameson SC, Hogquist KA. Positive and negative selection of T cells. Ann Rev Immunol 2003; 21:139–176. [DOI] [PubMed] [Google Scholar]
- 9.Viatte S, Plant D, Raychaudhuri S. Genetics and epigenetics of rheumatoid arthritis. Nat Rev Rheumatol 2013; 9:141–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakaguchi N, Takahashi T, Hata H, et al. Altered thymic T-cell selection due to a mutation of the ZAP-70 gene causes autoimmune arthritis in mice. Nature 2003; 426:454–460. [DOI] [PubMed] [Google Scholar]
- 11.Hirota K, Hashimoto M, Yoshitomi H, et al. T cell self-reactivity forms a cytokine milieu for spontaneous development of IL-17+ Th cells that cause autoimmune arthritis. J Exp Med 2007; 204:41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanaka S, Maeda S, Hashimoto M, et al. Graded attenuation of TCR signaling elicits distinct autoimmune diseases by altering thymic T cell selection and regulatory T cell function. J Immunol 2010; 185:2295–2305. [DOI] [PubMed] [Google Scholar]
- 13▪▪.Ito Y, Hashimoto M, Hirota K, et al. Detection of T cell responses to a ubiquitous cellular protein in autoimmune disease. Science 2014; 346:363–368. [DOI] [PMC free article] [PubMed] [Google Scholar]; Identifies the antigen specificity of TCRs which cause autoimmune arthritis.
- 14.Takahashi T, Kuniyasu Y, Toda M, et al. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int Immunol 1998; 10:1969–1980. [DOI] [PubMed] [Google Scholar]
- 15.Vang T, Congia M, Macis MD, et al. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat Genet 2005; 37:1317–1319. [DOI] [PubMed] [Google Scholar]
- 16.Hirota K, Yoshitomi H, Hashimoto M, et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med 2007; 204:2803–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rawlings DJ, Dai X, Buckner JH. The role of PTPN22 risk variant in the development of autoimmunity: finding common ground between mouse and human. J Immunol 2015; 194:2977–2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Messemaker TC, Huizinga TW, Kurreeman F. Immunogenetics of rheumatoid arthritis: understanding functional implications. J Autoimmun 2015; 64:74–81. [DOI] [PubMed] [Google Scholar]
- 19.Burn GL, Svensson L, Sanchez-Blanco C, et al. Why is PTPN22 a good candidate susceptibility gene for autoimmune disease? FEBS Lett 2011; 585:3689–3698. [DOI] [PubMed] [Google Scholar]
- 20.Zhang J, Zahir N, Jiang Q, et al. The autoimmune disease-associated PTPN22 variant promotes calpain-mediated Lyp/Pep degradation associated with lymphocyte and dendritic cell hyperresponsiveness. Nat Genet 2011; 43:902–907. [DOI] [PubMed] [Google Scholar]
- 21.Maine CJ, Hamilton-Williams EE, Cheung J, et al. PTPN22 alters the development of regulatory T cells in the thymus. J Immunol 2012; 188:5267–5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sakaguchi S. Regulatory T cells: key controllers of immunologic self-tolerance. Cell 2000; 101:455–458. [DOI] [PubMed] [Google Scholar]
- 23.Schwartz RH. Natural regulatory T cells and self-tolerance. Nat Immunol 2005; 6:327–330. [DOI] [PubMed] [Google Scholar]
- 24.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 2003; 4:330–336. [DOI] [PubMed] [Google Scholar]
- 25.Kanangat S, Blair P, Reddy R, et al. Disease in the scurfy (sf) mouse is associated with overexpression of cytokine genes. Eur J Immunol 1996; 26:161–165. [DOI] [PubMed] [Google Scholar]
- 26.Clark LB, Appleby MW, Brunkow ME, et al. Cellular and molecular characterization of the scurfy mouse mutant. J Immunol 1999; 162:2546–2554. [PubMed] [Google Scholar]
- 27.Bennett CL, Christie J, Ramsdell F, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet 2001; 27:20–21. [DOI] [PubMed] [Google Scholar]
- 28.Gambineri E, Torgerson TR, Ochs HD. Immune dysregulation, polyendocrinopathy, enteropathy, and X-linked inheritance (IPEX), a syndrome of systemic autoimmunity caused by mutations of FOXP3, a critical regulator of T-cell homeostasis. Curr Opin Rheumatol 2003; 15:430–435. [DOI] [PubMed] [Google Scholar]
- 29.Wildin RS, Ramsdell F, Peake J, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet 2001; 27:18–20. [DOI] [PubMed] [Google Scholar]
- 30.Asseman C, Mauze S, Leach MW, et al. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J Exp Med 1999; 190:995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Collison LW, Workman CJ, Kuo TT, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 2007; 450:566–569. [DOI] [PubMed] [Google Scholar]
- 32.Chen W, Jin W, Hardegen N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med 2003; 198:1875–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andersson J, Tran DQ, Pesu M, et al. CD4+ FoxP3+ regulatory T cells confer infectious tolerance in a TGF-beta-dependent manner. J Exp Med 2008; 205:1975–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Serra P, Amrani A, Yamanouchi J, et al. CD40 ligation releases immature dendritic cells from the control of regulatory CD4+CD25+ T cells. Immunity 2003; 19:877–889. [DOI] [PubMed] [Google Scholar]
- 35.Misra N, Bayry J, Lacroix-Desmazes S, et al. Cutting edge: human CD4+CD25+ T cells restrain the maturation and antigen-presenting function of dendritic cells. J Immunol 2004; 172:4676–4680. [DOI] [PubMed] [Google Scholar]
- 36.Wing K, Onishi Y, Prieto-Martin P, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008; 322:271–275. [DOI] [PubMed] [Google Scholar]
- 37.Morgan ME, Sutmuller RP, Witteveen HJ, et al. CD25+ cell depletion hastens the onset of severe disease in collagen-induced arthritis. Arthritis Rheum 2003; 48:1452–1460. [DOI] [PubMed] [Google Scholar]
- 38.Morgan ME, Flierman R, van Duivenvoorde LM, et al. Effective treatment of collagen-induced arthritis by adoptive transfer of CD25+ regulatory T cells. Arthritis Rheum 2005; 52:2212–2221. [DOI] [PubMed] [Google Scholar]
- 39.Ehrenstein MR, Evans JG, Singh A, et al. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med 2004; 200:277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Flores-Borja F, Jury EC, Mauri C, Ehrenstein MR. Defects in CTLA-4 are associated with abnormal regulatory T cell function in rheumatoid arthritis. Proc Natl Acad Sci U S A 2008; 105:19396–19401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cribbs AP, Kennedy A, Penn H, et al. Treg cell function in rheumatoid arthritis is compromised by ctla-4 promoter methylation resulting in a failure to activate the indoleamine 2,3-dioxygenase pathway. Arthritis Rheum 2014; 66:2344–2354. [DOI] [PubMed] [Google Scholar]
- 42.Edwards JC, Szczepanski L, Szechinski J, et al. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med 2004; 350:2572–2581. [DOI] [PubMed] [Google Scholar]
- 43.Emery P, Fleischmann R, Filipowicz-Sosnowska A, et al. The efficacy and safety of rituximab in patients with active rheumatoid arthritis despite methotrexate treatment: results of a phase IIB randomized, double-blind, placebo-controlled, dose-ranging trial. Arthritis Rheum 2006; 54:1390–1400. [DOI] [PubMed] [Google Scholar]
- 44.Jang E, Cho WS, Cho ML, et al. Foxp3+ regulatory T cells control humoral autoimmunity by suppressing the development of long-lived plasma cells. J Immunol 2011; 186:1546–1553. [DOI] [PubMed] [Google Scholar]
- 45.Rapetti L, Chavele KM, Evans CM, Ehrenstein MR. B cell resistance to Fas-mediated apoptosis contributes to their ineffective control by regulatory T cells in rheumatoid arthritis. Ann Rheum Dis 2015; 74:294–302. [DOI] [PubMed] [Google Scholar]
- 46.Groux H, O’Garra A, Bigler M, et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature 1997; 389:737–742. [DOI] [PubMed] [Google Scholar]
- 47.Brun V, Neveu V, Pers YM, et al. Isolation of functional autologous collagen-II specific IL-10 producing Tr1 cell clones from rheumatoid arthritis blood. Int Immunopharmacol 2011; 11:1074–1078. [DOI] [PubMed] [Google Scholar]
- 48.Asnagli H, Martire D, Belmonte N, et al. Type 1 regulatory T cells specific for collagen type II as an efficient cell-based therapy in arthritis. Arthritis Res Ther 2014; 16:R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ohnmacht C, Pullner A, King SB, et al. Constitutive ablation of dendritic cells breaks self-tolerance of CD4 T cells and results in spontaneous fatal autoimmunity. J Exp Med 2009; 206:549–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hawiger D, Inaba K, Dorsett Y, et al. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med 2001; 194:769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang H, Dawicki W, Zhang X, et al. Tolerogenic dendritic cells induce CD4+CD25hiFoxp3+ regulatory T cell differentiation from CD4+CD25-/loFoxp3- effector T cells. J Immunol 2010; 185:5003–5010. [DOI] [PubMed] [Google Scholar]
- 52.Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Ann Rev Immunol 2003; 21:685–711. [DOI] [PubMed] [Google Scholar]
- 53.Morelli AE, Thomson AW. Tolerogenic dendritic cells and the quest for transplant tolerance. Nat Rev Immunol 2007; 7:610–621. [DOI] [PubMed] [Google Scholar]
- 54.Stoop JN, Harry RA, von Delwig A, et al. Therapeutic effect of tolerogenic dendritic cells in established collagen-induced arthritis is associated with a reduction in Th17 responses. Arthritis Rheum 2010; 62:3656–3665. [DOI] [PubMed] [Google Scholar]
- 55.Harry RA, Anderson AE, Isaacs JD, Hilkens CM. Generation and characterisation of therapeutic tolerogenic dendritic cells for rheumatoid arthritis. Ann Rheum Dis 2010; 69:2042–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hilkens CM, Isaacs JD. Tolerogenic dendritic cell therapy for rheumatoid arthritis: where are we now? Clin Exp Immunol 2013; 172:148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maffia P, Brewer JM, Gracie JA, et al. Inducing experimental arthritis and breaking self-tolerance to joint-specific antigens with trackable, ovalbumin-specific T cells. J Immunol 2004; 173:151–156. [DOI] [PubMed] [Google Scholar]
- 58.Jongbloed SL, Benson RA, Nickdel MB, et al. Plasmacytoid dendritic cells regulate breach of self-tolerance in autoimmune arthritis. J Immunol 2009; 182:963–968. [DOI] [PubMed] [Google Scholar]
- 59.Benson RA, Patakas A, Conigliaro P, et al. Identifying the cells breaching self-tolerance in autoimmunity. J Immunol 2010; 184:6378–6385. [DOI] [PubMed] [Google Scholar]
- 60.Baba T, Nakamoto Y, Mukaida N. Crucial contribution of thymic Sirp alpha+ conventional dendritic cells to central tolerance against blood-borne antigens in a CCR2-dependent manner. J Immunol 2009; 183:3053–3063. [DOI] [PubMed] [Google Scholar]
- 61.Hadeiba H, Lahl K, Edalati A, et al. Plasmacytoid dendritic cells transport peripheral antigens to the thymus to promote central tolerance. Immunity 2012; 36:438–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Galton M, Reed PB. Entry of lymph node cells into the normal thymus. Transplantation 1966; 4:168–177. [DOI] [PubMed] [Google Scholar]
- 63.Michie SA, Kirkpatrick EA, Rouse RV. Rare peripheral T cells migrate to and persist in normal mouse thymus. J Exp Med 1988; 168:1929–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Agus DB, Surh CD, Sprent J. Reentry of T cells to the adult thymus is restricted to activated T cells. J Exp Med 1991; 173:1039–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reinhardt RL, Khoruts A, Merica R, et al. Visualizing the generation of memory CD4 T cells in the whole body. Nature 2001; 410:101–105. [DOI] [PubMed] [Google Scholar]
- 66.Hale JS, Fink PJ. Back to the thymus: peripheral T cells come home. Immunol Cell Biol 2009; 87:58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Edelmann SL, Marconi P, Brocker T. Peripheral T cells re-enter the thymus and interfere with central tolerance induction. J Immunol 2011; 186:5612–5619. [DOI] [PubMed] [Google Scholar]
- 68▪▪.Thiault N, Darrigues J, Adoue V, et al. Peripheral regulatory T lymphocytes recirculating to the thymus suppress the development of their precursors. Nat Immunol 2015; 16:628–634. [DOI] [PubMed] [Google Scholar]; Provides evidence that thymus homing T-Reg influence de novo T-Reg production.
- 69.Koetz K, Bryl E, Spickschen K, et al. T cell homeostasis in patients with rheumatoid arthritis. Proc Natl Acad Sci U S A 2000; 97:9203–9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ponchel F, Morgan AW, Bingham SJ, et al. Dysregulated lymphocyte proliferation and differentiation in patients with rheumatoid arthritis. Blood 2002; 100:4550–4556. [DOI] [PubMed] [Google Scholar]
- 71.Wagner U, Schatz A, Baerwald C, Rossol M. Brief report: deficient thymic output in rheumatoid arthritis despite abundance of prethymic progenitors. Arthritis Rheum 2013; 65:2567–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]