

Abstract
TGFβ is a multifunctional cytokine that regulates cell proliferation, cell immortalization, and cell death, acting as a key homeostatic mediator in various cell types and tissues. Autophagy is a programmed mechanism that plays a pivotal role in controlling cell fate and, consequently, many physiological and pathological processes, including carcinogenesis. Although autophagy is often considered a pro-survival mechanism that renders cells viable in stressful conditions and thus might promote tumor growth, emerging evidence suggests that autophagy is also a tumor suppressor pathway. The relationship between TGFβ signaling and autophagy is context-dependent and remains unclear. TGFβ-mediated activation of autophagy has recently been suggested to contribute to the growth inhibitory effect of TGFβ in hepatocarcinoma cells. In the present study, we define a novel process of TGFβ-mediated autophagy in cancer cell lines of various origins. We found that autophagosome initiation and maturation by TGFβ is dependent on the retinoblastoma tumor suppressor protein/E2 promoter binding factor (pRb/E2F1) pathway, which we have previously established as a critical signaling axis leading to various TGFβ tumor suppressive effects. We further determined that TGFβ induces pRb/E2F1-dependent transcriptional activation of several autophagy-related genes. Together, our findings reveal that TGFβ induces autophagy through the pRb/E2F1 pathway and transcriptional activation of autophagy-related genes and further highlight the central relevance of the pRb/E2F1 pathway downstream of TGFβ signaling in tumor suppression.
Keywords: autophagy; cancer; hepatocellular carcinoma; retinoblastoma protein (pRb, RB); transforming growth factor beta (TGF-β); tumor suppressor gene
Introduction
TGFβ is a multifunctional cytokine that is involved in the regulation of numerous diverse fundamental biological processes, including cell proliferation, differentiation, immortalization, and apoptosis, in a context- and cell-specific manner (1–3). TGFβ is a key mediator in the maintenance of homeostasis between cell growth and cell death and the growth inhibitory and pro-apoptotic effects of TGFβ have been described in various cell types, including epithelial cells, hepatocytes, and hematopoietic cells, among others (4–11).
Autophagy is an evolutionarily conserved process by which a cell self-digests its own cytoplasmic materials, including long-lived or malfunctioning proteins and damaged organelles, through a lysosomal degradative pathway in response to various stress conditions (12, 13). Briefly, the general mechanism of autophagy involves the formation of a double membrane-bound vesicle called an autophagosome that envelops and sequesters a targeted region of the cell. The autophagosome then fuses with a lysosome, forming an autolysosome, in which hydrolases digest the sequestered contents to metabolites that are recycled for biosynthesis in the cell (14). As such, autophagy is often considered a pro-survival mechanism, protecting cells and maintaining homeostasis under poor nutrient conditions or cell stress (15). However, autophagy has also been implicated in several vital biological processes, including aging, cellular remodeling, pathogenic infection, and programmed cell death (12, 15).
In addition to normal cell growth and homeostasis, autophagy has been implicated to play a protective role in preventing the progression of a number of human diseases, including muscular disorders, neurodegeneration, and cancer (14, 16–19). Autophagy in fact plays a dual and opposing role in cancer, demonstrating evidence of both tumor-promoting and tumor suppressive functions in a context-dependent manner. Although many studies support a role for autophagy in maintaining tumor cell survival in response to metabolic stress or hypoxia, thus promoting the growth of solid tumors (20–23), mounting evidence suggests that autophagy is intrinsically a tumor suppressor pathway (24–28). Moreover, alterations in numerous key autophagy-related genes have been linked to various human cancers, and mice deficient for these genes are prone to tumor development (29–32). However, the mechanisms underlying how autophagy suppresses cancer have not been well established.
Interestingly, TGFβ has recently been shown to also induce autophagy (33). TGFβ signals through serine/threonine kinase receptors that recruit and phosphorylate the canonical downstream mediators, the Smad proteins, inducing Smad nuclear translocation to regulate target gene expression. Nuclear Smad complexes interact with various co-activators or co-repressors, which are differentially expressed in different cell types and are thus thought to provide a basis for tissue- and cell type-specific functions of TGFβ ligands (34, 35). Alternatively, TGFβ activates other intracellular signaling pathways independently of the Smads. These include the stress-activated kinases p38 and JNK, MAPK, ERK, and PI3K/Akt pathways (36).
In a recent study, we identified the retinoblastoma tumor suppressor protein/E2 promoter binding factor (pRb/E2F1) pathway as a major signaling axis leading to apoptosis downstream of TGFβ in a number of cell types and in both normal and cancer cells (6). We found that TGFβ recruits E2F1 into transcriptionally active pRb-E2F1-p300/CBP-associated factor (P/CAF) complexes, increasing the expression of multiple pro-apoptotic target genes and inducing programmed cell death. Intriguingly, the pRb/E2F pathway appears to also be required for a number of other TGFβ tumor suppressive responses. Indeed, TGFβ-mediated growth inhibition involves E2F4/5 recruitment into repressive pRb family member-E2F-HDAC complexes that target key cell cycle regulators, such as cdc25a (5) and c-myc (4, 37), preventing cell cycle entry. Although E2F4-p130 complexes and E2F4/5-p107 complexes bind to the cdc25a and c-myc promoters, respectively, pRb is not involved in this process. Moreover, we also previously found that TGFβ induces E2F1 recruitment into repressive E2F-HDAC complexes, inhibiting hTERT expression, thereby preventing cell immortalization (38).
Kiyono et al. (33) previously showed that activation of autophagy may in fact contribute to TGFβ-mediated tumor suppressive effects. They found that TGFβ induces autophagy and enhances the degradation of long-lived proteins. Moreover, they showed that induction of autophagy relies on both Smad-dependent and Smad-independent signaling and proceeds via transcriptional activation of a number of autophagic genes in hepatocellular carcinoma cells, potentiating the tumor suppressive effects of TGFβ in these cells. In addition to cancer cells, TGFβ has been shown to induce autophagy in mammary and renal epithelial cells, as well as mesangial cells (39–41). These studies provide emerging evidence for a novel TGFβ-mediated tumor suppressive pathway, although the precise mechanisms and therapeutic implications thereof remain to be fully elucidated.
Having previously identified the pRb/E2F pathway as a critical regulator of TGFβ-mediated tumor suppressor effects in both preventing cell immortalization (38) and inducing apoptosis (6), we investigated the potential role of this pathway in TGFβ-mediated autophagy. We found that TGFβ activates autophagy in various cancer cell lines and that these effects are dependent on E2F1 and pRb. Moreover, our results indicate that TGFβ may regulate autophagy through pRb/E2F1-dependent transcriptional activation of multiple autophagy-related genes that function at various stages in the autophagy process. These data further support the crucial role for pRb/E2F signaling as a potent tumor suppressive pathway downstream of TGFβ.
Experimental Procedures
Cell Culture and Transfections
HuH7, HepG2, and Hep3B cell lines, as well as H1299 cells stably expressing GFP-LC3 (gift from Dr. Gordon Shore, McGill University) were cultured in DMEM (HyClone), and WM278 cells in RPMI 1640 (HyClone). Medium for all cells was supplemented with 10% FBS (HyClone) and 2 mm l-glutamine (Wisent), and the cells were grown at 37 °C in 5% CO2 conditions. To generate HuH7 cell lines stably expressing GFP-LC3, cells were transfected with pEGFP-LC3 (Addgene plasmid 21073) using LipofectamineTM 2000 reagent (Life Technologies), and G418-resistent colonies were screened for expression of GFP-LC3. Prior to treatment, cells were serum-starved for 24 h, and all stimulations were done in serum-free medium containing 100 pm TGFβ1 (Peprotech). Cells were transiently transfected with different siRNAs against E2F1, E2F4 (Ambion), pRb, or P/CAF (Sigma-Aldrich), or with wild-type and mutant E2F1 expression vectors using LipofectamineTM 2000 reagent (Life Technologies), according to the manufacturer's instructions.
Immunoblotting
Cells were lysed in cold radioimmune precipitation assay buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Triton X-100, 0.1% SDS, 1 mm EDTA), containing 1 mm sodium orthovanadate, 1 mm phenylmethylsulfonyl fluoride, 5 μg/ml aprotinin, 2 μg/ml leupeptin, and 1 μg/ml pepstatin. Lysates were separated by SDS-PAGE, transferred to nitrocellulose, and incubated with the specified antibodies overnight at 4 °C: anti-Beclin1 (Novus Biologicals), anti-LC3 (Novus Biologicals), anti-p62 (Santa Cruz Biotechnology), anti-E2F1 (KH95) (Santa Cruz Biotechnology), and anti-β-tubulin (Sigma). Following primary antibody incubation, membranes were washed twice in TBST (50 mm Tris-HCl, pH 7.6, 200 mm NaCl, 0.05% Tween 20) and incubated with secondary antibody coupled to horseradish peroxidase (Sigma) at 1:10,000 dilution for 1 h at room temperature. Membranes were then washed in TBST four times for 15 min. Immunoreactivity was revealed by chemiluminescence and detected using an Alpha Innotech Fluorochem Imaging system (Packard Canberra).
Subcellular Localization of LC3
Cells expressing EGFP-LC3 were fixed with 4% paraformaldehyde, and the change in the LC3 localization was examined using a Zeiss LSM-510 Meta Axiovert confocal microscope. Autophagy was measured by quantitation of the GFP-LC3 puncta as described by Klionsky et al. (42) The areas of GFP-LC3 puncta were analyzed using ImageJ software (National Institutes of Health), and a minimum of 100 cells from three independent experiments was counted for quantitative analysis.
RNA Isolation and Real Time Quantitative PCR
Total RNA was isolated from cell lines using TRIzol reagent (Invitrogen) and reverse transcribed using random hexamers and Moloney murine leukemia virus reverse transcriptase (Invitrogen), as per the manufacturer's instructions. Subsequently, real time qPCR2 was carried out using SsoFastTM EvaGreen® Supermix (Bio-Rad) in a RotorGene 6000 PCR detection system (Corbett Life Science). Conditions for qPCR were as follows: 95 °C for 30 s, 40 cycles of 95 °C for 5 s, and 60 °C for 20 s. Primer sequences are listed in Table 1.
TABLE 1.
Primer sequences and amplicon sizes
| Primer name | Primer sequence | Amplicon size |
|---|---|---|
| bp | ||
| PCR primer sequences | ||
| Beclin1 (2) forward | 5′-TGTCACCATCCAGGAACTCA-3′ | 180 |
| Beclin1 (2) reverse | 5′-CTGTTGGCACTTTCTGTGGA-3′ | |
| ULK1 forward | 5′-TCGAGTTCTCCCGCAAGG-3′ | 134 |
| ULK1 reverse | 5′-CGTCTGAGACTTGGCGAGGT-3′ | |
| ULK2 forward | 5′-GGCTCTCCTACTAAGACCACAG-3′ | 82 |
| ULK2 reverse | 5′-GACGAGTAACCAAGGCTAACAG-3′ | |
| UVRAG forward | 5′-CTGTTTGGATGGGCTGAAAT-3′ | 167 |
| UVRAG reverse | 5′-TGCGAACACAGTTCTGATCC-3′ | |
| ATG14 forward | 5′-ATGAGCGTCTGGCAAATCTT-3′ | 192 |
| ATG14 reverse | 5′-CCCATCGTCCTGAGAGGTAA-3′ | |
| PIK3C3 forward | 5′-AAGCAGTGCCTGTAGGAGGA-3′ | 199 |
| PIK3C3 forward | 5′-TGTCGATGAGCTTTGGTGAG-3′ | |
| MAP1LC3A forward | 5′-CGTCCTGGACAAGACCAAGT-3′ | 181 |
| MAP1LC3A reverse | 5′-CTCGTCTTTCTCCTGCTCGT-3′ | |
| MAP1LC3B forward | 5′-AGCAGCATCCAACCAAAATC-3′ | 187 |
| MAP1LC3B reverse | 5′-CTGTGTCCGTTCACCAACAG-3′ | |
| ATG4B forward | 5′-GCCGAGATTGGAGGTG-3′ | 143 |
| ATG4B reverse | 5′-GCCTATGGACTTGCCTTC-3′ | |
| GABARAPL1 forward | 5′-TTTGGTGCCCCTTATCTCAC-3′ | 241 |
| GABARAPL1 reverse | 5′-GGCCATCATGTAGCATTCCTT-3′ | |
| ATG12 forward | 5′-AGTAGAGCGAACACGAACCATCC-3′ | 110 |
| ATG12 reverse | 5′-AAGGAGCAAAGGACTGATTCACATA-3′ | |
| BCL2 forward | 5′-GAGTTCGGTGGGGTCATGT-3′ | 97 |
| BCL2 reverse | 5′-GCCGGTTCAGGTACTCAGTC-3′ | |
| DAPK1 forward | 5′-CAGTGTTGTTGCTCTAGGAAG-3′ | 196 |
| DAPK1 reverse | 5′-GGGACTGCCACAAATGATGAG-3′ | |
| c-myc forward | 5′-TTCGGGTAGTGGAAAACCAG-3′ | 203 |
| c-myc reverse | 5′-CAGCAGCTCGAATTTCTTCC-3′ | |
| E2F1 forward | 5′-TGCAGAGCAGATGGTTATGG-3′ | 265 |
| E2F1 reverse | 5′-ATCTGTGGTGAGGGATGAGG-3′ | |
| E2F4 forward | 5′-GTGCCACCACCTGAAGATTT-3′ | 229 |
| E2F4 reverse | 5′-TGAGCTCACCACTGTCCTTG-3′ | |
| GAPDH forward | 5′-GCCTCAAGATCATCAGCAATGCCT-3′ | 104 |
| GAPDH reverse | 5′-TGTGGTCATGAGTCCTTCCACGAT-3′ | |
| ChIP primer sequences | ||
| Beclin1 (2) forward | 5′GATCTCGACTCACTGCAACCTC-3′ | |
| Beclin1 (2) reverse | 5′-GAGGTTGCAGTGAGCCGAGAT-3′ | |
| ATG1/ULK1 forward | 5′-CAGAGAAGAAGCGGGGCGG-3′ | |
| ATG1/ULK1 reverse | 5′-CCGGGCGTGACGAACAGA-3′ | |
| ATG7 forward | 5′-CTTCATCCCACTTCCCATATTC-3′ | |
| ATG7 reverse | 5′-GGCGCCTGTAGTCCCAGCTACTTG-3′ | |
| LC3 forward | 5′-TGGAGGGGAAAGGATGGTCG-3′ | |
| LC3 reverse | 5′-GGGGCGGAGCAGGTGTGTG-3′ | |
Chromatin Immunoprecipitation
Protein complexes were cross-linked to DNA by adding formaldehyde directly to tissue culture medium to a final concentration of 1%. Cross-linking was allowed to proceed for 10 min at room temperature and stopped by the addition of glycine to a final concentration of 0.125 m. Cross-linked cells were harvested, washed with PBS, pelleted by centrifugation for 5 min at 4 °C, and lysed in nuclear lysis buffer (1% SDS, 10 mm EDTA, 50 mm Tris-HCl, pH 8.1, supplemented with 1 mm PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 2 μg/ml pepstatin, for 10 min on ice. The resulting chromatin solution was sonicated for five pulses of 20 s to generate 300–500-bp DNA fragments. After centrifugation for 10 min at 4 °C, the supernatant was immunocleared by incubation with protein A-Sepharose beads for 1 h at 4 °C. Immunocleared chromatin was immunoprecipitated overnight with 2 μg of the indicated antibodies. Antibody-protein-DNA complexes were then isolated by immunoprecipitation with 40 μl of protein A-G-agarose beads (Santa Cruz Biotechnology), overnight with rotation at 4 °C. The beads were washed three times for 10 min each with low salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 150 mm NaCl, 20 mm Tris-HCl, pH 8.1), high salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 500 mm NaCl, 20 mm Tris-HCl, pH 8.1), and LiCl wash buffer (0.25 m LiCl, 1% Nonidet P-40, 1% sodium deoxycholate, 1 mm EDTA, 10 mm Tris-HCl, pH 8.1), and twice in TE buffer. Complexes were then eluted twice in 150 μl of elution buffer (1% SDS, 0.1 m NaHCO3) by incubating at 65 °C for 15 min. To reverse cross-linking, 0.2 m NaCl, and 1 μl of 10 mg/ml RNaseA was added to each sample, and they were incubated at 65 °C overnight. Following this, 5 mm EDTA and 2 μl of 10 mg/ml proteinase K was added, and samples were incubated for at 45 °C for 2 h. DNA was recovered using the QIAquick spin columns (Qiagen) as per the manufacturer's protocol. Quantification was performed by qPCR, as described above.
Histone Acetylation
Histone proteins were extracted as previously described (51). Briefly, HuH7 cells were serum-starved and stimulated in the presence or the absence of TGFβ or 1 μm trichostatin A. Cells were harvested and resuspended in cold hypotonic lysis buffer (10 mm Tris-HCl, pH 8.0, 1 mm KCl, 1.5 mm MgCl2, 1 mm DTT, protease inhibitors, 1 μm trichostatin A, and 10 mm sodium butyrate). Cell lysates were centrifuged at 10,000 × g at 4 °C for 10 min, and nuclei pellets were resuspended in 400 ml of 0.4 n H2SO4 and incubated overnight on a rotator at 4 °C. Samples were centrifuged at 16,000 × g for 10 min, and supernatants containing histones were transferred into a fresh tube. 132 ml of trichloroacetic acid was added drop by drop to the histone solution, inverted several times, and then incubated on ice for 30 min. The histone precipitates were centrifuged at 16,000 × g for 10 min, and pellets were washed twice with ice-cold acetone, and the histone pellets were air-dried for 20 min. Total histone proteins were subjected to Western blot analysis using an acetylated lysine antibody (Cell Signaling).
Statistical Analysis
The results are expressed as means ± standard deviation of at least three independent experiments. Statistical differences were determined by two-tailed unpaired t test. p < 0.05 was considered statistically significant.
Results
TGFβ Induces Autophagy in Human Hepatocarcinoma Cell Lines
We first assessed the autophagy response to TGFβ in two human hepatocarcinoma cell lines: HuH7 and HepG2. For this, we initially examined whether TGFβ treatment induced the conversion of cytosolic LC3-I to the lipidated LC3-II form, which is localized in the autophagosome membrane and is widely used as a marker of autophagy in mammalian cells (43). Immunoblotting analyses revealed an up-regulation in the endogenous protein level of LC3-I and a clear accumulation of LC3-II isoform in cells treated with TGFβ for 24 h (Fig. 1A). We also examined the expression of two key autophagic regulators, Beclin-1 and p62, in response to TGFβ in both cell lines. Although Beclin-1 is required for the initiation of autophagosome formation (44), p62 links ubiquitinated proteins to the autophagy machinery and enables their degradation in lysosomes (45). As such, p62 levels are preferentially degraded by autophagy. Accordingly, we found that TGFβ treatment in HuH7 and HepG2 cells resulted in an increase in Beclin-1 and loss of p62 protein expression levels, supporting that the idea that TGFβ induces autophagy in these cells.
FIGURE 1.
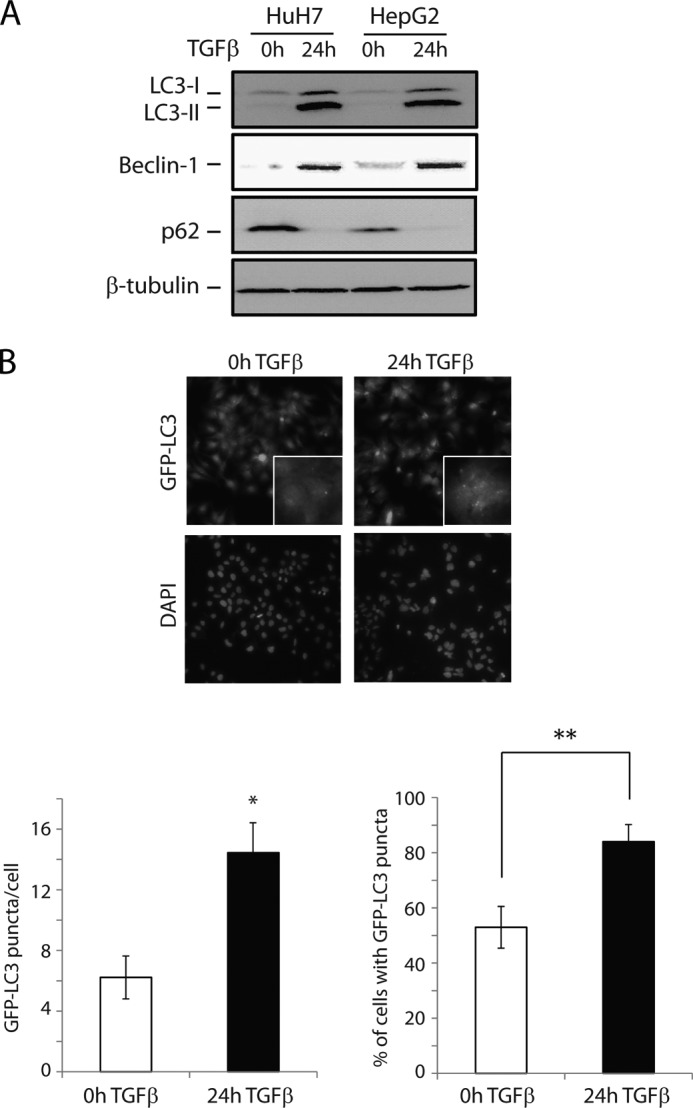
TGFβ induces autophagy in human hepatocarcinoma cell lines. A, immunoblot analysis of the conversion of endogenous LC3 (LC3-I to the more rapidly migrating LC3-II), as well as expression levels of Beclin-1 and p62, in HuH7 and HepG2 cells treated with TGFβ for 24 h. B, HuH7 cells stably expressing GFP-LC3 were treated with TGFβ as indicated, and the relocalization of GFP-LC3 to autophagosomes was detected as punctate formation, visualized by fluorescence microscopy (representative images, left panel). The number of GFP-LC3 puncta per cell and percentage of cells exhibiting more than five GFP-LC3 puncta were quantified (right panel). The data are presented as means ± S.D. **, p < 0.01.
To further corroborate the activation of autophagy, we employed a GFP-tagged LC3 plasmid (pEGFP-LC3) to monitor the cellular localization of LC3. Fluorescent GFP-LC3 is used extensively to measure autophagy because it displays a diffuse cytoplasmic distribution (corresponding to the LC3-I form) under normal conditions, and a distinctly punctate distribution once LC3 is lipidated and integrated into the autophagosome membrane (LC3-II form) under autophagy-inducing conditions (46). For this, HuH7 cells were stably transfected with pEGFP-LC3, and the expression pattern of LC3 was examined before and after TGFβ treatment. In the absence of TGFβ, only occasional puncta were detected, representing a basal level of autophagy. In contrast, TGFβ treatment markedly increased punctate distribution (Fig. 1B). Taken together, these data indicate that TGFβ acts as a potent inducer of autophagy in these cells.
TGFβ Transcriptionally Mediates Autophagy in an E2F1-dependent Manner
Autophagy is regulated by numerous autophagy-related genes (ATGs) that function collaboratively in autophagy initiation and progression. We then investigated whether TGFβ transcriptionally modulates the expression levels of various genes involved in different steps of the autophagy process. Our results indicate that TGFβ regulates multiple autophagy-related genes in HuH7 cells (Fig. 2A, upper panel) but that there is in fact no functional distinction in the genes that are regulated. This suggests that TGFβ plays a more general role in promoting autophagy rather than specifically targeting one phase or function. Importantly, in addition to assessing known TGFβ target genes (Beclin-1, DAPK, ATG5, ATG7, and MAP1LC3), we uncovered a number of novel TGFβ-targeted autophagy regulators (PIK3C3, ULK1, ULK2, GABARAP, ATG4b, ATG12, and ATG14). Many of these genes were also transcriptionally induced by TGFβ in HepG2 cells, as well as human non-small cell lung carcinoma cells (H1299) (Fig. 2A, middle and lower panels). These results indicate that the autophagy response to TGFβ is not limited to hepatocarcinoma but applies to other types of human cancer cell types. In addition, we measured the expression levels of B-cell lymphoma 2 (Bcl-2), an anti-apoptotic protein that also inhibits autophagy by binding and sequestering Beclin-1 (29). We found that TGFβ treatment transcriptionally repressed Bcl-2 expression, thus further contributing to the activation of autophagy in these cells (Fig. 2B).
FIGURE 2.
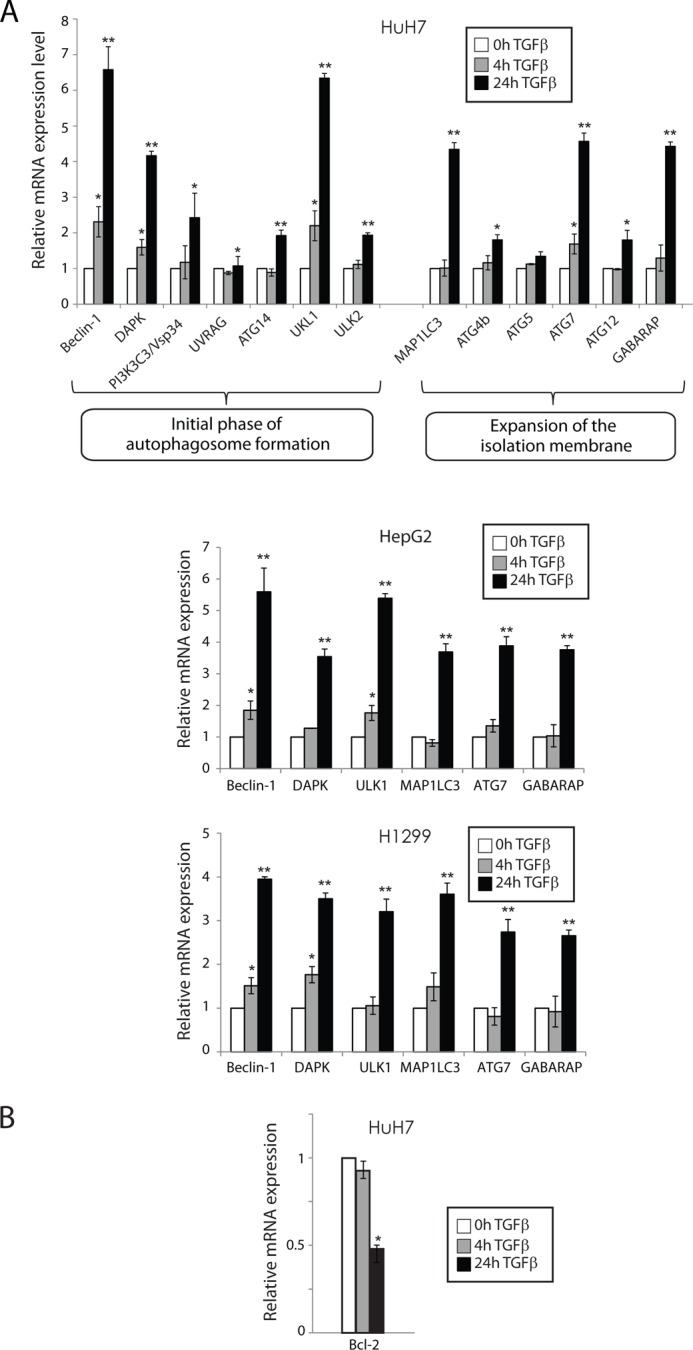
TGFβ transcriptionally mediates autophagy in an E2F1-dependent manner. A, HuH7, HepG2, and H1299 cells were stimulated with TGFβ as indicated, and mRNA levels for the specified autophagy genes were measured by real time qPCR analysis. The results are normalized to GAPDH and shown relative to levels observed in untreated cells (set to 1). The data are represented as means ± S.D. *, p < 0.05; **, p < 0.01. B, HuH7 cells were stimulated with TGFβ as indicated, and mRNA levels for Bcl-2 gene were measured by real time qPCR analysis. The results are normalized to GAPDH and shown relative to levels observed in untreated cells (set to 1). The data are represented as means ± S.D. *, p < 0.05.
In previous studies from our laboratory, we identified E2F1 as a critical transcriptional co-regulator of multiple TGFβ-targeted genes involved in tumor suppression, leading to apoptosis as well as inhibition of cell immortalization (6, 38). We therefore assessed whether E2F1 could also contribute to the transcriptional regulation of autophagy-related genes in response to TGFβ. Quite remarkably, loss of E2F1 expression by siRNA significantly impaired the TGFβ-mediated regulation of most of these target genes in both HuH7 and H1299 cells (Fig. 3A). Efficiency of the E2F1 knockdown was assessed by real time qPCR and Western blot (Fig. 3B). These data indicate that E2F1 plays a central role downstream of TGFβ-mediated transcriptional activation of autophagy regulatory genes. In addition to providing novel TGFβ targets, these data also highlight a novel pathway by which TGFβ regulates autophagy regulatory genes and potentially the autophagy process itself. To further address this, we silenced E2F1 expression and examined the activation of autophagy by TGFβ, as assessed previously. As shown in (Fig. 3C), loss of E2F1 expression strongly attenuated the autophagy response to TGFβ, as measured by the induction of Beclin-1, degradation of p62, and accumulation of LC3-II, at the protein level. Moreover, E2F1 knockdown markedly and significantly reduced the number of GFP-LC3 puncta observed in response to TGFβ treatment (Fig. 3D). Our results here suggest that E2F1 regulates TGFβ-mediated autophagy through the transcriptional control of autophagy-related genes.
FIGURE 3.

TGFβ activation of autophagy acts through E2F1. A, HuH7 and H1299 cells were transiently transfected with siRNA against E2F1 or a control nonsilencing siRNA and stimulated with TGFβ for 24 h. mRNA levels for the specified autophagy genes were measured by real time qPCR analysis. The results are normalized to GAPDH and shown relative to levels observed in untreated cells (set to 1). The data are represented as means ± S.D. *, p < 0.05; **, p < 0.01. B, HuH7 cells were transiently transfected with a control nonsilencing siRNA or siRNA against E2F1 and E2F1 mRNA levels (left panel), and protein levels (right panel) were determined by real time qPCR and Western blot analysis, respectively. The data are represented as means ± S.D. ***, p < 0.001. C and D, following siRNA transfection, HuH7 cells were treated or not with TGFβ and LC3 conversion (C) and GFP-LC3 puncta formation (D) were analyzed by immunoblotting and fluorescence microscopy, respectively. The effect of E2F1 knockdown on Beclin-1 and p62 expression was also evaluated by immunoblotting (C). E and F, HuH7 cells were transiently transfected with siRNA against E2F4 or a control nonsilencing siRNA and stimulated with TGFβ for 24 h. The mRNA levels for the indicated genes were measured as in A. **, p < 0.01; (***, p < 0.001. E, middle and right panels, E2F4 mRNA levels (middle panel) and protein levels (right panel) were determined by real time qPCR and Western blot analysis.
Polager et al. (47) established a role for E2F1 in DNA damage-induced autophagy, identifying ATG5, MAP1LC3, ULK1, and DRAM (DNA damage-regulated autophagy modulator) as E2F1 transcriptional targets. Intriguingly, they found not only E2F1 but also E2F4 to be associated with these gene promoters (47). Given their results, and because E2F4 also acts downstream of TGFβ signaling to regulate cell growth arrest through transcriptional modulation of cell cycle genes (4, 48), we investigated whether E2F4 could also regulate these autophagy genes in response to TGFβ. Interestingly, we found that silencing E2F4 expression by means of siRNA had no significant effect on the regulation of these genes by TGFβ (Fig. 3E, left panel). As a control, we also measured c-myc expression levels and, as expected, E2F4 knockdown partially impeded the down-regulation of c-myc by TGFβ (Fig. 3E, right panel). Taken together, these data indicate that E2F1, but not E2F4, plays a role in TGFβ-mediated induction of autophagy-related genes, suggesting that the autophagy response to TGFβ is not only dependent on but also specific to E2F1. This also indicates that TGFβ-mediated and DNA damaged-induced autophagy processes occur through distinct mechanisms.
TGFβ-induced Activation of Autophagy Acts through pRb/E2F1 Signaling
Having previously established that TGFβ regulates apoptotic gene expression via a transcriptionally active pRb-E2F1-P/CAF complex (6), we sought to determine whether these components could also be involved in regulating autophagy genes in response to TGFβ. In support of this, a recent study showed that pRb overexpression could induce autophagy (26). Using pRb restoration in pRb-deficient cells, Jiang et al. (26) demonstrated that pRb activates the autophagy response and that pRb binding to E2F1 is required for autophagy induction. Accordingly, we first speculated whether an E2F1 mutant that is deficient for pRb binding (E2F1 Y411C) would lose the capability to induce autophagy in response to TGFβ, by acting as a dominant negative when overexpressed. Interestingly, we found that ectopic expression of E2F1 Y411C suppressed the transcriptional activation of multiple autophagy genes in response to TGFβ (Fig. 4A), suggesting that pRb binding to E2F1 is indeed required for TGFβ-induced autophagy. This parallels our previous study in which we found pRb-E2F1 association to also be required for TGFβ-induced apoptosis (6).
FIGURE 4.

TGFβ activation of autophagy acts through pRb/E2F1 signaling. A, HuH7 cells transiently transfected with empty vector or an E2F1 mutant deficient for pRb binding (E2F1 Y411C) treated with TGFβ for 24 h. Levels of mRNA expression of the specified genes were determined by real time qPCR analysis. The results are normalized to GAPDH and shown relative to levels observed in untreated cells (set to 1). B, GFP-LC3 puncta formation was analyzed by fluorescence microscopy and quantified as described previously. C, HuH7 cells were transiently transfected a control nonsilencing siRNA or siRNA against pRb and stimulated with TGFβ for 24 h. The mRNA levels for the indicated genes were determined by real time qPCR analysis. The data are represented as mean ± S.D. *, p < 0.05; **, p < 0.01. D, HuH7 cells were transiently transfected with a control nonsilencing siRNA or siRNA against pRb and pRb mRNA levels (left panel) and protein levels (right panel) were determined by real time qPCR and Western blot analysis.
We subsequently addressed the contribution of pRb to TGFβ-mediated autophagy. As shown in (Fig. 4B), knockdown of pRb expression significantly reduced the number of GFP-LC3 puncta observed in response to TGFβ, indicating impaired autophagosome formation by TGFβ in the absence of pRb. Moreover, the transcriptional induction of a number of autophagic genes in response to TGFβ was also notably impaired when pRb expression was silenced by RNA interference (Fig. 4C). Efficiency of pRb knockdown was assessed by real time qPCR and Western blot (Fig. 4D).
TGFβ-induced Activation of Autophagy Requires the Histone Acetyltransferase P/CAF
Gene transcription downstream of TGFβ signaling may be co-regulated by histone acetyltransferases such as p300/CBP and P/CAF (1, 2, 6, 49–51). In a recent study, we showed that P/CAF is involved in the transcriptional induction of numerous apoptotic genes in response to TGFβ (6). We thus assessed the requirement for this histone acetyltransferase P/CAF to TGFβ-mediated autophagy. As shown in (Fig. 5A), loss of P/CAF expression similarly suppressed the induction of multiple autophagy genes by TGFβ. Efficiency of the P/CAF siRNA was assessed by qPCR and Western blot (Fig. 5B). TGFβ-induced protein histone acetylation requires P/CAF. Indeed, as shown in Fig. 5C, when P/CAF gene expression is knockdown by means of siRNA, TGFβ no longer induces histone acetylation, further emphasizing the role and contribution of P/CAF to TGFβ-mediated transcriptional activation of the autophagy genes. The Smad pathway is central to TGFβ-mediated autophagy. As shown in Fig. 5D, knocking down Smad3 with a specific siRNA drastically impaired the TGFβ effect on Beclin-1, DAPK, ATG1/ULK1, and MAP1LC3 gene expression. Efficiency of the Smad3 siRNA knockdown was assessed by qPCR and Western blot (Fig. 5E).
FIGURE 5.

TGFβ activation of autophagy acts through pRb/E2F1 signaling and requires the histone acetyltransferase P/CAF. A, HuH7 cells were transiently transfected with a control nonsilencing siRNA or siRNA against P/CAF and stimulated with TGFβ for 24 h. The mRNA levels for the indicated genes were determined by real time qPCR analysis. The data are represented as means ± S.D. *, p < 0.05; **, p < 0.01. B, HuH7 cells were transiently transfected with a control nonsilencing siRNA or siRNA against P/CAF. P/CAF mRNA levels (left panel) and protein levels (right panel) were determined by real time qPCR and Western blot analysis. C, HuH7 cells, transfected or not with a scrambled or P/CAF siRNA, were serum-starved and stimulated in the presence or the absence of TGFβ or 1 μm trichostatin A (TSA). Protein histones were precipitated from nuclear pellets and subjected to Western blot analysis using an acetylated lysine antibody. D, HuH7 cells were transiently transfected a control nonsilencing siRNA or siRNA against Smad3 and stimulated with TGFβ for 24 h. The mRNA levels for the indicated genes were determined by real time qPCR analysis. The data are represented as means ± S.D. *, p < 0.05; **, p < 0.01. E, Smad3 mRNA levels (left panel) and protein levels (right panel) were determined by real time qPCR and Western blot analysis.
To further address the role and contribution of P/CAF to TGFβ-mediated autophagy, we then assessed the functional relevance of the TGFβ induced pRb-E2F1-P/CAF complex in regulating TGFβ transcriptional responses. For this, we performed chromatin immunoprecipitation assays to determine whether this complex is recruited to the autophagy target gene promoters in response to TGFβ. As shown in Fig. 6, TGFβ treatment markedly induced recruitment of all three partners (E2F1, pRb, and P/CAF) to the ATG1/ULK1, LC3, Beclin-1, and ATG7 gene promoters, concurring with the TGFβ-mediated increase in the mRNA levels of these autophagy genes and further activation of the autophagy program. As negative controls, ChIP assays using control IgG and E2F4 antibodies showed no TGFβ effect on the autophagy gene promoters. As shown in Fig. 6 (bottom panel), the β-actin promoter was used as a negative control locus.
FIGURE 6.

TGFβ induces recruitment of all three partners (E2F1, pRb, and P/CAF) to autophagy gene promoters. HuH7 cells were untreated or treated with TGFβ for the indicated times and the binding of E2F1, pRb, and P/CAF to the LC3, Beclin-1, ATG1/ULK1, and ATG7 gene promoters was determined by ChIP. For negative controls, ChIP assays using control IgG and E2F4 antibodies, as well as a negative control locus (β-actin promoter) were performed.
Collectively, these findings support a role for pRb and P/CAF downstream of TGFβ in the transcriptional activation of autophagy genes and the mediation of autophagy and indicate that a transcriptionally active pRb-E2F1 complex plays a role in mediating the TGFβ autophagy response.
Discussion
The balance between cell proliferation and cell death is central to many fundamental physiological processes. As such, deregulation of this balance has been linked to the pathogenesis of multiple diseases. Mounting evidence indicates that autophagy is a key programmed mechanism that controls cell survival and cell death, thus playing a pivotal role in maintaining cellular homeostasis and, consequently, in many biological and pathological processes, including carcinogenesis (21, 29, 32).
Autophagy has been implicated in promoting tumor development by enabling tumor cells to temporarily survive stressful environmental conditions. Paradoxically, several studies have also demonstrated a tumor suppressive function for autophagy. Indeed, several genetic links have emerged between perturbations of autophagy and cancer development. For instance, monoallelic deletion of Beclin-1 is frequently observed in human breast, ovarian, and prostate cancers (29, 32). Correspondingly, heterozygous disruption of Beclin-1 in mice has been shown to promote spontaneous tumor formation (30, 52). Another recent study indicated that Beclin-1 expression levels were significantly decreased in hepatocarcinoma tissues compared with adjacent normal tissues (21). Moreover, deletion of the Beclin-1-binding protein UVRAG (53) or other autophagy-related genes, such as ATG4C (31), ATG5, and ATG7 (54), also resulted in accelerated tumorigenesis.
TGFβ is a multifunctional cytokine that regulates cell proliferation as well as cell death, acting as an essential homeostatic mediator in various cell types and tissues. Similar to autophagy, TGFβ plays bidirectional and paradoxical roles in tumor suppression and tumor progression. Further adding to this dichotomy, TGFβ-mediated induction of autophagy may lead to cell survival or cell death in a context-dependent manner. The mechanisms by which TGFβ regulates autophagy are not well established. TGFβ generally signals in a cell type- and context-specific manner, and its regulation of autophagy is no exception. In primary mouse mesangial cells, TGFβ induces autophagy through activation of the TAK1-MKK3-p38 signaling axis, acting as a cytoprotective mechanism against serum deprivation-induced apoptosis (39, 55). Conversely, TGFβ induces autophagy in bovine mammary gland epithelial cells and neonatal piglet gut epithelium, leading to autophagic cell death (PCD type II) (40, 56). Moreover, autophagy activation by TGFβ in hepatocellular carcinoma cells is mediated through the Smad and JNK pathways and contributes to the growth inhibitory effect of TGFβ in these cells (33). Clearly, the underlying mechanisms induced by TGFβ to regulate cell fate remain to be fully elucidated.
In the present study, we uncovered a novel pathway of TGFβ-mediated autophagy in various cancer cell lines. We found that autophagosome initiation and maturation by TGFβ is dependent on the pRb/E2F1 pathway. We further determined that TGFβ potentially regulates autophagy through transcriptional activation of numerous autophagy-related genes in a pRb/E2F1-dependent manner and that this transcriptional effect also relies on the histone acetyltransferase P/CAF.
To identify novel features of TGFβ-induced autophagy, we investigated the transcriptional regulation of a wide range of autophagy-related genes in response to TGFβ treatment. In addition to known TGFβ target genes, our analysis revealed a number of novel and potent TGFβ transcriptional targets, namely PIK3C3, ULK1, ULK2, GABARAP, ATG4b, ATG12, and ATG14. These autophagy-related gene products function cooperatively to facilitate autophagy initiation and maturation, indicating that the transcriptional regulation of autophagy by TGFβ is multifaceted. Moreover, our data provide further support for the premise that autophagy is regulated at the transcriptional level.
In the present study, we demonstrate that the transcriptional regulation of autophagy genes and induction of autophagy by TGFβ is E2F1- and pRb-dependent. Our data provide strong evidence for the importance and specificity of E2F1 in the autophagy response to TGFβ and propose a novel pathway by which TGFβ regulates autophagy activation. The E2F family of transcription factors has been reported to play essential roles in autophagy activation. The transcriptional activity of E2F1 was previously shown to be required for DNA damage-induced autophagy in osteosarcoma cells by upregulating the expression of ATG5, MAP1LC3, ULK1, and DRAM (47). Moreover, BNIP3 (BCL2/adenovirus E1B 19-kDa protein-interacting protein 3) was identified as a direct transcriptional target of E2F1 that is involved in hypoxia-induced autophagy cell death (57). Interestingly, Beclin-1 appears to be regulated by multiple E2F members, because most E2F members occupy the Beclin-1 promoter (58), and both E2F1 and E2F3 have been shown to transactivate the Beclin-1 promoter by reporter assay (59). Recently, Kusama et al. (60) amplified and cloned the putative promoter regions of 23 human autophagy genes, more than half of which were found to be regulated by E2F1 in HeLa cells. These studies strongly support that the E2F transcription factors potentially act as major autophagy regulators. Because our data implicate E2F signaling as an essential regulatory axis through which TGFβ mediates autophagy, this suggests that TGFβ may also represent a major player in autophagy regulation. Although our data suggest that E2F4 is not involved in the induction of autophagy-related genes by TGFβ, further investigation of the contribution of other E2F members to TGFβ-mediated autophagy would be of interest.
The results of our current study, taken together with our previously established role for pRb/E2F1 in TGFβ-mediated apoptosis, suggest that the pRb/E2F1 pathway regulates both apoptosis and autophagy in response to TGFβ. Thus, it is conceivable that autophagy and apoptosis are co-regulated at the transcriptional level downstream of TGFβ signaling. Accordingly, an increasing number of reports suggest that there is indeed a complex interplay between autophagy and apoptosis, evidenced by the extensive molecular cross-talk between autophagy-related and apoptosis-related proteins. In this regard, several ATG proteins have been reported to engage the apoptotic pathway (61–63), whereas depletion of key autophagy regulators or pharmacological interference with autophagy has been shown to mitigate apoptosis (64). Conversely, several apoptotic proteins have been shown to regulate the autophagy process. As mentioned previously, anti-apoptotic Bcl-2 directly binds Beclin-1, inhibiting its autophagy activity (28). Interestingly, a number of apoptotic proteins may in fact promote autophagy induction by enabling the dissociation of Beclin-1 from Bcl-2. This is mediated through competition with Beclin-1 for Bcl-2 binding by various BH3-only proteins (65, 66) or through post-translational modifications of Beclin-1 or Bcl-2 mediated by DAPK, JNK, or TRAF6 (67–69). Although autophagy and apoptosis are correlated in a number of contexts, the causal relationships often remain unclear. It will be important, in future studies, to further characterize the mechanisms by which the autophagy process might be coupled to apoptotic cell death and to identify candidate molecules linking autophagy to apoptosis downstream of TGFβ signaling.
In summary, our data indicate that TGFβ signaling and the pRb/E2F1 pathway induce autophagy through transcriptional activation of multiple autophagy-related genes, acting at different stages of the autophagy process. These findings provide new insights for mechanisms of autophagy regulation downstream of TGFβ signaling and, combined with our previous studies, further highlight the central role played by the pRb/E2F1 pathway downstream of the TGFβ tumor suppressive responses. Both TGFβ and autophagy are involved in numerous diverse physiological effects that may influence multiple important fields beyond cancer research, such as immune regulation and neurodegeneration. Accordingly, autophagy might contribute to various TGFβ-mediated biological functions. Further elucidation of the molecular mechanisms by which TGFβ regulates cell fate through autophagy may thus provide a better understanding of the physiological and pathological roles of TGFβ signaling pathways.
Author Contributions
J. K., L. C., and J.-J. L. designed the study and wrote the paper. J. K. and L. C. did the experiments and contributed to figure preparation. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Gordon Shore for generously providing the H1299 GFP-LC3 cells and Dr. Kristian Helin for kindly providing the mutant E2F1 expression vectors.
This work was supported by Canadian Institutes of Health Research Grant MOP-114904 (to J.-J. L.). The authors declare that they have no conflicts of interest with the contents of this article.
- qPCR
- quantitative PCR
- ATG
- autophagy-related gene.
References
- 1. Massagué J. (1990) The transforming growth factor-β family. Annu. Rev. Cell Biol. 6, 597–641 [DOI] [PubMed] [Google Scholar]
- 2. Siegel P. M., and Massagué J. (2003) Cytostatic and apoptotic actions of TGF-β in homeostasis and cancer. Nat. Rev. Cancer 3, 807–821 [DOI] [PubMed] [Google Scholar]
- 3. Lebrun J. J. (2012) The dual role of TGF in human cancer: From tumor suppression to cancer metastasis. ISRN Mol. Biol. 2012, 1–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen C. R., Kang Y., Siegel P. M., and Massagué J. (2002) E2F4/5 and p107 as Smad cofactors linking the TGFβ receptor to c-myc repression. Cell 110, 19–32 [DOI] [PubMed] [Google Scholar]
- 5. Iavarone A., and Massagué J. (1999) E2F and histone deacetylase mediate transforming growth factor β repression of cdc25A during keratinocyte cell cycle arrest. Mol. Cell. Biol. 19, 916–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Korah J., Falah N., Lacerte A., and Lebrun J. J. (2012) A transcriptionally active pRb-E2F1-P/CAF signaling pathway is central to TGFβ-mediated apoptosis. Cell Death Dis. 3, e407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ohgushi M., Kuroki S, Fukamachi H, O'Reilly L. A., Kuida K, Strasser A, Yonehara S. (2005) Transforming growth factor β-dependent sequential activation of Smad, Bim, and caspase-9 mediates physiological apoptosis in gastric epithelial cells. Mol. Cell. Biol. 25, 10017–10028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jang C. W., Chen C. H., Chen C. C., Chen J. Y., Su Y. H., Chen R. H. (2002) TGF-β induces apoptosis through Smad-mediated expression of DAP-kinase. Nat. Cell Biol. 4, 51–58 [DOI] [PubMed] [Google Scholar]
- 9. Valderrama-Carvajal H., Cocolakis E, Lacerte A, Lee E. H., Krystal G, Ali S, Lebrun J. J. (2002) Activin/TGF-β induce apoptosis through Smad-dependent expression of the lipid phosphatase SHIP. Nat. Cell Biol. 4, 963–969 [DOI] [PubMed] [Google Scholar]
- 10. Francis J. M., Heyworth C. M., Spooncer E, Pierce A, Dexter T. M., Whetton A. (2000) Transforming growth factor-β1 induces apoptosis independently of p53 and selectively reduces expression of Bcl-2 in multipotent hematopoietic cells. J. Biol. Chem. 275, 39137–39145 [DOI] [PubMed] [Google Scholar]
- 11. Wildey G. M., Patil S., and Howe P. H. (2003) Smad3 potentiates transforming growth factor β (TGFβ)-induced apoptosis and expression of the BH3-only protein Bim in WEHI 231 B lymphocytes. J. Biol. Chem. 278, 18069–18077 [DOI] [PubMed] [Google Scholar]
- 12. Levine B., and Klionsky D. J. (2004) Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 6, 463–477 [DOI] [PubMed] [Google Scholar]
- 13. Xie Z., and Klionsky D. J. (2007) Autophagosome formation: core machinery and adaptations. Nat. Cell Biol. 9, 1102–1109 [DOI] [PubMed] [Google Scholar]
- 14. Mizushima N. (2007) Autophagy: process and function. Genes Dev. 21, 2861–2873 [DOI] [PubMed] [Google Scholar]
- 15. Levine B., and Kroemer G. (2008) Autophagy in the pathogenesis of disease. Cell 132, 27–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shintani T., and Klionsky D. J. (2004) Autophagy in health and disease: a double-edged sword. Science 306, 990–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Baehrecke E. H. (2005) Autophagy: dual roles in life and death? Nat. Rev. Mol. Cell Biol. 6, 505–510 [DOI] [PubMed] [Google Scholar]
- 18. Cecconi F., and Levine B. (2008) The role of autophagy in mammalian development: cell makeover rather than cell death. Dev. Cell 15, 344–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mizushima N., Levine B., Cuervo A. M., and Klionsky D. J. (2008) Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Song J., Guo X., Xie X., Zhao X., Li D., Deng W., Song Y., Shen F., Wu M., and Wei L. (2011) Autophagy in hypoxia protects cancer cells against apoptosis induced by nutrient deprivation through a Beclin1-dependent way in hepatocellular carcinoma. J. Cell. Biochem. 112, 3406–3420 [DOI] [PubMed] [Google Scholar]
- 21. White E. J., Martin V., Liu J. L., Klein S. R., Piya S., Gomez-Manzano C., Fueyo J., and Jiang H. (2011) Autophagy regulation in cancer development and therapy. Am. J. Cancer Res. 1, 362–372 [PMC free article] [PubMed] [Google Scholar]
- 22. Wu W. K., Coffelt S. B., Cho C. H., Wang X. J., Lee C. W., Chan F. K., Yu J., and Sung J. J. (2012) The autophagic paradox in cancer therapy. Oncogene 31, 939–953 [DOI] [PubMed] [Google Scholar]
- 23. Guo J. Y., Chen H. Y., Mathew R., Fan J., Strohecker A. M., Karsli-Uzunbas G., Kamphorst J. J., Chen G., Lemons J. M., Karantza V., Coller H. A., Dipaola R. S., Gelinas C., Rabinowitz J. D., and White E. (2011) Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 25, 460–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maiuri M. C., Malik S. A., Morselli E., Kepp O., Criollo A., Mouchel P. L., Carnuccio R., and Kroemer G. (2009) Stimulation of autophagy by the p53 target gene Sestrin2. Cell Cycle 8, 1571–1576 [DOI] [PubMed] [Google Scholar]
- 25. Maiuri M. C., Tasdemir E., Criollo A., Morselli E., Vicencio J. M., Carnuccio R., and Kroemer G. (2009) Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ 16, 87–93 [DOI] [PubMed] [Google Scholar]
- 26. Jiang H., Martin V., Gomez-Manzano C., Johnson D. G., Alonso M., White E., Xu J., McDonnell T. J., Shinojima N., and Fueyo J. (2010) The RB-E2F1 pathway regulates autophagy. Cancer Res. 70, 7882–7893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ueno T., Sato W., Horie Y., Komatsu M., Tanida I., Yoshida M., Ohshima S., Mak T. W., Watanabe S., and Kominami E. (2008) Loss of Pten, a tumor suppressor, causes the strong inhibition of autophagy without affecting LC3 lipidation. Autophagy 4, 692–700 [DOI] [PubMed] [Google Scholar]
- 28. Pattingre S., Tassa A., Qu X., Garuti R., Liang X. H., Mizushima N., Packer M., Schneider M. D., and Levine B. (2005) Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122, 927–939 [DOI] [PubMed] [Google Scholar]
- 29. Liang X. H., Jackson S., Seaman M., Brown K., Kempkes B., Hibshoosh H., and Levine B. (1999) Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402, 672–676 [DOI] [PubMed] [Google Scholar]
- 30. Qu X., Yu J., Bhagat G., Furuya N., Hibshoosh H., Troxel A., Rosen J., Eskelinen E. L., Mizushima N., Ohsumi Y., Cattoretti G., and Levine B. (2003) Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Invest. 112, 1809–1820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mariño G., Salvador-Montoliu N., Fueyo A., Knecht E., Mizushima N., and López-Otín C. (2007) Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J. Biol. Chem. 282, 18573–18583 [DOI] [PubMed] [Google Scholar]
- 32. Wirawan E., Vanden Berghe T., Lippens S., Agostinis P., and Vandenabeele P. (2012) Autophagy: for better or for worse. Cell Res 22, 43–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kiyono K., Suzuki H. I., Matsuyama H., Morishita Y., Komuro A., Kano M. R., Sugimoto K., and Miyazono K. (2009) Autophagy is activated by TGF-β and potentiates TGF-β-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res. 69, 8844–8852 [DOI] [PubMed] [Google Scholar]
- 34. Massagué J., and Wotton D. (2000) Transcriptional control by the TGF-β/Smad signaling system. EMBO J. 19, 1745–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Feng X. H., and Derynck R. (2005) Specificity and versatility in TGF-β signaling through Smads. Annu. Rev. Cell Dev. Biol. 21, 659–693 [DOI] [PubMed] [Google Scholar]
- 36. Zhang Y. E. (2009) Non-Smad pathways in TGF-β signaling. Cell Res. 19, 128–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Frederick J. P., Liberati N. T., Waddell D. S., Shi Y., and Wang X. F. (2004) Transforming growth factor β-mediated transcriptional repression of c-myc is dependent on direct binding of Smad3 to a novel repressive Smad binding element. Mol. Cell. Biol. 24, 2546–2559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lacerte A., Korah J., Roy M., Yang X. J., Lemay S., and Lebrun J. J. (2008) Transforming growth factor-β inhibits telomerase through SMAD3 and E2F transcription factors. Cell. Signal. 20, 50–59 [DOI] [PubMed] [Google Scholar]
- 39. Ding Y., Kim J. K., Kim S. I., Na H. J., Jun S. Y., Lee S. J., and Choi M. E. (2010) TGF-β1 protects against mesangial cell apoptosis via induction of autophagy. J. Biol. Chem. 285, 37909–37919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gajewska M., Gajkowska B., and Motyl T. (2005) Apoptosis and autophagy induced by TGF-B1 in bovine mammary epithelial BME-UV1 cells. J. Physiol. Pharmacol. 56, 143–157 [PubMed] [Google Scholar]
- 41. Koesters R., Kaissling B., Lehir M., Picard N., Theilig F., Gebhardt R., Glick A. B., Hähnel B., Hosser H., Gröne H. J., and Kriz W. (2010) Tubular overexpression of transforming growth factor-β1 induces autophagy and fibrosis but not mesenchymal transition of renal epithelial cells. Am. J. Pathol. 177, 632–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Klionsky D. J., et al. (2008) Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4, 151–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tasdemir E., Galluzzi L., Maiuri M. C., Criollo A., Vitale I., Hangen E., Modjtahedi N., and Kroemer G. (2008) Methods for assessing autophagy and autophagic cell death. Methods Mol. Biol. 445, 29–76 [DOI] [PubMed] [Google Scholar]
- 44. Wirawan E., Lippens S., Vanden Berghe T., Romagnoli A., Fimia G. M., Piacentini M., and Vandenabeele P. (2012) Beclin1: a role in membrane dynamics and beyond. Autophagy 8, 6–17 [DOI] [PubMed] [Google Scholar]
- 45. Pankiv S., Clausen T. H., Lamark T., Brech A., Bruun J. A., Outzen H., Øvervatn A., Bjørkøy G., and Johansen T. (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131–24145 [DOI] [PubMed] [Google Scholar]
- 46. Mizushima N., Yoshimori T., and Levine B. (2010) Methods in mammalian autophagy research. Cell 140, 313–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Polager S., Ofir M., and Ginsberg D. (2008) E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene 27, 4860–4864 [DOI] [PubMed] [Google Scholar]
- 48. Li J. M., Hu P. P., Shen X., Yu Y., and Wang X. F. (1997) E2F4-RB and E2F4-p107 complexes suppress gene expression by transforming growth factor β through E2F binding sites. Proc. Natl. Acad. Sci. U.S.A. 94, 4948–4953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Feng X. H., Zhang Y., Wu R. Y., and Derynck R. (1998) The tumor suppressor Smad4/DPC4 and transcriptional adaptor CBP/p300 are coactivators for smad3 in TGF-β-induced transcriptional activation. Genes Dev. 12, 2153–2163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Itoh S., Ericsson J., Nishikawa J., Heldin C. H., and ten Dijke P. (2000) The transcriptional co-activator P/CAF potentiates TGF-β/Smad signaling. Nucleic Acids Res. 28, 4291–4298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dai M., Al-Odaini AA, Arakelian A., Rabbani SA, Ali S., and Lebrun J. J. (2012) A novel function for p21Cip1 and acetyltransferase p/CAF as critical transcriptional regulators of TGFβ-mediated breast cancer cell migration and invasion. Breast Cancer Res. 14, R127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yue Z., Jin S., Yang C., Levine A. J., and Heintz N. (2003) Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. U.S.A. 100, 15077–15082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Liang C., Feng P., Ku B., Dotan I., Canaani D., Oh B. H., and Jung J. U. (2006) Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Biol. 8, 688–699 [DOI] [PubMed] [Google Scholar]
- 54. Takamura A., Komatsu M., Hara T., Sakamoto A., Kishi C., Waguri S., Eishi Y., Hino O., Tanaka K., and Mizushima N. (2011) Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 25, 795–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kim S. I., Na H. J., Ding Y., Wang Z., Lee S. J., and Choi M. E. (2012) Autophagy promotes intracellular degradation of type I collagen induced by transforming growth factor (TGF)-β1. J. Biol. Chem. 287, 11677–11688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Godlewski M. M., Hallay N., Bierla J. B., and Zabielski R. (2007) Molecular mechanism of programmed cell death in the gut epithelium of neonatal piglets. J. Physiol. Pharmacol. 58, 97–113 [PubMed] [Google Scholar]
- 57. Tracy K., Dibling B. C., Spike B. T., Knabb J. R., Schumacker P., and Macleod K. F. (2007) BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol. Cell. Biol. 27, 6229–6242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Weinmann A. S., Bartley S. M., Zhang T., Zhang M. Q., and Farnham P. J. (2001) Use of chromatin immunoprecipitation to clone novel E2F target promoters. Mol. Cell. Biol. 21, 6820–6832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang B., Ling S., and Lin W. C. (2010) 14-3-3Tau regulates Beclin 1 and is required for autophagy. PLoS One 5, e10409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kusama Y., Sato K., Kimura N., Mitamura J., Ohdaira H., and Yoshida K. (2009) Comprehensive analysis of expression pattern and promoter regulation of human autophagy-related genes. Apoptosis 14, 1165–1175 [DOI] [PubMed] [Google Scholar]
- 61. Pyo J. O., Jang M. H., Kwon Y. K., Lee H. J., Jun J. I., Woo H. N., Cho D. H., Choi B., Lee H., Kim J. H., Mizushima N., Oshumi Y., and Jung Y. K. (2005) Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J. Biol. Chem. 280, 20722–20729 [DOI] [PubMed] [Google Scholar]
- 62. Heidari N., Hicks M. A., and Harada H. (2010) GX15–070 (obatoclax) overcomes glucocorticoid resistance in acute lymphoblastic leukemia through induction of apoptosis and autophagy. Cell Death Dis. 1, e76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Radoshevich L., Murrow L., Chen N., Fernandez E., Roy S., Fung C., and Debnath J. (2010) ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell 142, 590–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Crighton D., Wilkinson S., O'Prey J., Syed N., Smith P., Harrison P. R., Gasco M., Garrone O., Crook T., and Ryan K. M. (2006) DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 126, 121–134 [DOI] [PubMed] [Google Scholar]
- 65. Kang R., Zeh H. J., Lotze M. T., and Tang D. (2011) The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 18, 571–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Marquez R. T., and Xu L. (2012) Bcl-2:Beclin 1 complex: multiple, mechanisms regulating autophagy/apoptosis toggle switch. Am. J. Cancer Res. 2, 214–221 [PMC free article] [PubMed] [Google Scholar]
- 67. Zalckvar E., Berissi H., Eisenstein M., and Kimchi A. (2009) Phosphorylation of Beclin 1 by DAP-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-XL. Autophagy 5, 720–722 [DOI] [PubMed] [Google Scholar]
- 68. Wei Y., Pattingre S., Sinha S., Bassik M., and Levine B. (2008) JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 30, 678–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Shi C. S., and Kehrl J. H. (2010) Traf6 and A20 differentially regulate TLR4-induced autophagy by affecting the ubiquitination of Beclin 1. Autophagy 6, 986–987 [DOI] [PMC free article] [PubMed] [Google Scholar]