

Abstract
Acetoacetate (AA) is a ketone body and acts as a fuel to supply energy for cellular activity of various tissues. Here, we uncovered a novel function of AA in promoting muscle cell proliferation. Notably, the functional role of AA in regulating muscle cell function is further evidenced by its capability to accelerate muscle regeneration in normal mice, and it ameliorates muscular dystrophy in mdx mice. Mechanistically, our data from multiparameter analyses consistently support the notion that AA plays a non-metabolic role in regulating muscle cell function. Finally, we show that AA exerts its function through activation of the MEK1-ERK1/2-cyclin D1 pathway, revealing a novel mechanism in which AA serves as a signaling metabolite in mediating muscle cell function. Our findings highlight the profound functions of a small metabolite as signaling molecule in mammalian cells.
Keywords: acetoacetate, cyclin D1, extracellular-signal-regulated kinase (ERK), muscle regeneration, muscular dystrophy
Introduction
Satellite cells, which are among the most abundant well defined adult stem cell types in skeletal muscle, play functionally important roles in postnatal growth, repair, and the regeneration of skeletal muscle (1–6). Because of their powerful ability to regenerate in vivo in response to muscle damage and various stimuli, satellite cells represent important targets for the treatment of muscular diseases (7–10). The recent development of stem cell-based regenerative medicine strategies has brought enormous interest in the discovery of regulatory factors capable of controlling satellite cell functions, such as activation, proliferation, differentiation, and self-renewal (11–13). Identification of such factors is expected to not only improve our understanding of the regulatory mechanisms that govern satellite cell functions, but also to facilitate the development of stem cell-based therapies for the treatment of muscular dystrophy or other chronic diseases associated with muscle wasting.
Recent studies demonstrating a close correlation between cell proliferation and metabolic alterations in various tumor types have drawn attention to the significance of intrinsic small metabolites as signaling molecules responsible for regulating various cellular activities (14, 15). Although only a very limited number of such metabolites have been identified to date, accumulating evidence suggests that these metabolites can be oncogenic and alter cell signaling through epigenetic regulation. For example, 2-hydroxyglutarate (2-HG),4 succinate, and fumarate, which are the best characterized small metabolites with oncogenic function, have come to be regarded as oncometabolites (16–19). In tumor cells, 2-HG is generated by mutant forms of isocitrate dehydrogenase (IDH1 and IDH2) (20–23), whereas succinate and fumarate accumulate via mutant forms of succinate dehydrogenase and fumarate hydratase, respectively (24–27). It has been clearly demonstrated that increases in the levels of these oncometabolites play causal roles in tumorigenesis (26–34). Recent studies of the molecular mechanisms underlying their action have revealed that 2-HG and elevated levels of succinate and fumarate exert their oncogenic functions by broadly inhibiting multiple α-ketoglutarate-dependent histone and DNA demethylases, including histone demethylases, prolyl hydroxylases, collagen prolyl-4-hydroxylases, and the TET family of 5-methlycytosine hydroxylases (34–36). In addition, some lipid synthesis intermediates, such as malonyl-CoA, participate in promoting cancer cell proliferation through the transcriptional regulation of growth factor receptors (37). Lysophosphatidic acid, however, signals through lysophosphatidic acid receptors to stimulate cancer cell proliferation and survival (6, 38). Minetti et al. (39) recently reported that lysophosphatidic acid stimulates muscle hypertrophy and differentiation through the activation of Gnai2 (G protein-binding, α-inhibiting 2) in a nuclear factor of activated T-cell-dependent and protein kinase C (PKC)-dependent manner. Interestingly, another recent study reveals that 2-HG plays an epigenetic role in regulating the expression levels of genes involved in the differentiation of non-transformed cells (40), indicating that the signaling role of metabolites is not restricted to cancer development and suggesting that metabolites may exert significant actions through non-metabolic pathways during normal development. However, we are only just beginning to understand the range of these functions in non-cancer cells.
Acetoacetate (AA) and 3-hydroxybutyrate (3HB) are the main ketone bodies. They are produced by the liver and used peripherally as energy sources for various organs, particularly the skeletal muscle and brain, under conditions that include starvation, limited carbohydrate availability, and the neonatal period (41). AA participates in various biological processes that do not involve 3HB, including insulin release in vitro (42), generation of free oxygen radicals (43–45), and lipid peroxidation (46). AA also stimulates chaperone-mediated autophagy (47) and down-regulates the expression of ATP-binding cassette transporter A1 (ABCA1) in vitro (48). AA, but not 3HB, promotes the secretion of interleukin (IL)-6 in cultured U937 monocytic cells (49). Interestingly, in neural cells, AA exerts its protective effect against glutamate-induced oxidative stress on HT22 cells and rat primary hippocampal neurons by decreasing glutamate-induced production of reactive oxygen species (50). Increasing concentrations of AA, but not 3HB, cause a significant up-regulation of ICAM1 (intercellular adhesion molecule 1) in human brain microvascular endothelial cells (51). In myocardial cells, AA is involved in regulating the incorporation of glucose into glycogen and mediating the relative contributions of exogenous glucose and endogenous carbohydrate to myocardial energy metabolism (52). Interestingly, a ketogenic diet has long been known to be beneficial for the treatment of children with intractable seizures by unknown mechanisms (53, 54). These tantalizing reports not only suggest that AA may play important roles in various biological processes but also suggest that it may have regulatory functions in addition to its involvement in energy production. In fact, the function of AA as a signaling molecule in regulating the atoDAEB operon has been well characterized in a bacterial two-component system (55), in which AA generates a signal that mediates short-chain fatty acid (SCFA) catabolism during bacterial cell growth and in response to a wide range of stimuli (56). However, a regulatory function of AA as a signaling molecule has not been reported in a mammalian system.
Here, we report that AA plays a previously unrecognized signaling role in promoting skeletal muscle cell proliferation in vitro. AA also significantly enhanced muscle regeneration by stimulating muscle satellite cell activation and proliferation in vivo. More remarkably, treatment of muscular dystrophin-deficient mdx mice with AA significantly ameliorates muscular dystrophy characterized by the improved muscle integrity, recovered muscle strength, and enhanced exercise performance. Moreover, we demonstrate that AA, functioning as a signaling molecule, acts through the MEK-ERK-cyclin D1 pathway in a Ras-independent manner. Our findings not only provide a proof of concept that small metabolites can act to couple cell metabolism and muscle stem cell functions in mammals, but also provide a rationale for potentially utilizing this metabolite and maybe its derivatives treating diseases associated with muscle wasting in humans.
Experimental Procedures
Skeletal Muscle Regeneration and AA Treatment in Mice
All animal experiments were performed in C57BL/6 or mdx mice. The care and handling of animals was performed in accordance with the guidelines of the Animal Ethics Committee of Peking Union Medical College, Beijing, China. The mice were anesthetized by intraperitoneal injection of 10 mg/kg ketamine and 1 mg/kg xylazine. For monitoring of muscle regeneration, muscle injury was induced in 8-week-old C57BL/6 mice by injection of CTX (20 μl of 10 μm CTX in PBS; Sigma) into the mid-belly of the right tibialis anterior (TA) muscle. The left TA muscle of each tested mouse was injected with PBS (20 μl) as a control. For AA (Sigma) treatment, TA muscles were injected with 20 μl of different doses of AA (20, 30, and 50 mm) or PBS at 0, 2, and 4 days after CTX injection (n = 5 per group). For the mdx rescue experiment, 3-month-old mdx mice (5–10 mice each) were given daily intraperitoneal injections of AA (220 mg/kg); 0.9% NaCl was used as control.
Cell Culture and Treatments
Mouse C2C12 cells were cultured in growth medium consisting of Dulbecco's modified Eagle's medium (DMEM; Gibco) supplemented with 4.5 g/liter glucose, 10% fetal bovine serum (FBS), 1% antibiotic/antimycotic, and 1% gentamycin at 37 °C in a 5% CO2 atmosphere. After reaching about 20–30% confluence, cells were starved by incubation in serum-free DMEM for 10–12 h, and then treated with different doses (0.1, 1, and 5 mm) of AA in low glucose (1 g/liter glucose) DMEM for different lengths of time. Cells treated with the same amount of PBS were used as controls. Under the same experimental conditions, cells were also treated with 3HB, acetone, acetoacetyl-CoA, acetyl-CoA, succinate (all from Sigma), and IGF1 (R&D). For inhibitor studies, the pharmacological inhibitors, PD98059 (20 μm; Cell Signaling), FTA (manumycin A, 5 μm; Calbiochem), GW5074 (2.5 μm; Calbiochem), Trolox (0.5 mm; Sigma), N-acetyl-l-cysteine (2.5 mm; Sigma), suramin (300 μm; TOCRIS), nystatin (25 μg/ml; Sigma), chlorpromazine (50 μm), and bafilomycin A1 (BFA1; 50 nm) were added to the low glucose DMEM 1 h prior to AA treatment. Cell proliferation was measured by FACS analysis. Each assay was performed in three replicates and was repeated at least three times. For cyclin D1 knockdown, C2C12 cells were transfected with three cyclin D1-specific siRNAs (100 μm) or a control siRNA, using the FuGENE HD transfection reagent (Roche Applied Science). The transfected cells were treated with AA as described above. Acetoacetate sodium was synthesized and purified by HPLC.
Muscle Histology and Hematoxylin and Eosin (H&E) Staining
TA muscles were harvested 2, 3, 5, and 7 days after CTX injury, fixed overnight with 4% paraformaldehyde (Sigma) in PBS, and paraffin-embedded for H&E staining. Newly regenerating (centronucleated) myofibers were quantified by analysis of randomly selected fields from muscle sections, using the ImagePro Plus 5.1 software (Olympus).
Immunofluorescence Staining of Frozen Sections and C2C12 Cells
For Pax7 immunostaining, fresh sections were fixed in 4% paraformaldehyde for 20 min, permeabilized with methanol (−20 °C) for 6 min, and blocked with a solution containing 4% bovine serum albumin (BSA; Jackson ImmunoResearch) in PBS. The sections were then subjected to antigen retrieval with 100 mm sodium citrate, followed by a second round of blocking with goat anti-mouse AffiniPure Fab fragments (Jackson ImmunoResearch, 1:100). For Pax7 immunostaining, sections were incubated overnight at 4 °C with an anti-Pax7 antibody (Developmental Studies Hybridoma Bank; 1:20). The sections were then washed with PBS, and Pax7 signals were visualized by incubation with biotin-conjugated goat anti-mouse IgG1 (Jackson ImmunoResearch; 1:1000) and Cy3-conjugated streptavidin (Jackson ImmunoResearch; 1:2500). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen). For MyoD immunostaining, fresh sections were fixed in 4% paraformaldehyde for 20 min, permeabilized with 0.2% Triton X-100 in PBS for 10 min, and blocked by incubation with 5% BSA, 6% goat serum, 0.2% PBST at room temperature for 2 h. Immunostaining with anti-MyoD (Santa Cruz Biotechnology; 1:50) antibodies was performed by overnight incubations at 4 °C. After the sections were washed, immunoreactive proteins were visualized by incubation with fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit IgG (ZhongShanJinQiao Co.). Nuclei were stained with DAPI (Roche Applied Science). The numbers of Pax7+ and MyoD+ cells in sections (n = 10) were counted. Differentiated C2C12 cells were immunostained with anti-myogenin and anti-MHC as described for MyoD, and the numbers of myogenin+ and MHC+ cells were quantified from more than 20 images obtained from three sections.
BrdU Incorporation Assay
For in vivo BrdU incorporation assays, 125 mg/kg BrdU (Sigma) was injected into TA muscles 2 h prior to harvesting. Frozen sections were fixed with cold acetone (−20 °C) for 10 min, incubated in 2 n HCl at 37 °C for 30 min for DNA denaturation, and then immersed twice in 0.1 m borate buffer (5 min each) for neutralization of the acid. After three washes with PBS, the sections were blocked with 5% goat serum for 30 min and then incubated with anti-BrdU (Abcam) at 4 °C overnight. The sections were then washed with PBS and incubated with FITC-conjugated goat anti-rat IgG for visualization of the BrdU signals. The BrdU-positive cells were quantified in 50 sections from five mice.
Cell Proliferation Assay
For FACS assays, cells were digested with trypsin, transferred to a 15-ml tube, and collected by centrifugation at 1100 × g for 3.5 min. The harvested cells were washed with 1 ml of PBS and fixed by incubation overnight at 4 °C with 4 ml of 75% ethanol in PBS. The fixed cells were harvested and washed with PBS, and then incubated with 100 μl of 1 mg/ml RNase A (AMRESCO) at 37 °C for 30 min. Finally, the cells were stained with 400 μl of propidium iodide (50 μg/μl; Calbiochem), and the ratio of cells in S-phase was analyzed by FACS. For analysis of [3H]thymidine incorporation, 5 μCi of [3H]thymidine was added to the culture medium. Six hours later, the cells were harvested, washed three times with PBS, and trypsinized, and radioactivity was measured using a scintillation counter.
Treadmill Test
The treadmill test was performed using an Exer3/6 (Columbus Instruments). Mice were acclimated to treadmill running three times (every other day) before the test was performed. Mice ran on the treadmill at 15° downhill, starting at a speed of 10 m/min. After 3 min, the speed was increased by 1 m/min to a final speed of 20 m/min. Exhaustion was defined as the inability of the animal to remain on the treadmill despite electrical prodding.
Evan's Blue Assay
Evan's blue dye was dissolved in PBS (0.15 m NaCl, 10 mm phosphate buffer, pH 7.4) and sterilized by passage through membrane filters (0.2-μm pore size). Animals were injected intravenously with 50 μl of dye solution (0.5 mg of Evan's blue dye, 0.05 ml PBS) per 10 g of body weight and then run for 30 min every day; 72 h later, they were killed by cervical dislocation. The extensor longus digitorum (EDL) muscles were embedded in OCT and snap-frozen in liquid nitrogen. For visualization, the muscle sections were incubated in ice-cold acetone at −20 °C for 10 min, washed three times with PBS (10 min each), and mounted. Fluorescence microscopy showed Evan's blue dye staining as a bright red emission.
Force Measurements
EDL muscles were constantly immersed in physiological saline solution containing 118.5 mm NaCl, 4.7 mm KCl, 2.4 mm CaCl2, 3.1 mm MgCl2, 25 mm NaHCO3, 2 mm NaH2PO4, and 5.5 mm d-glucose. All solutions were continuously bubbled with 95% O2, 5% CO2 (v/v) and maintained at a pH of 7.4. All experiments were carried out at 25 °C. Contractions were elicited by passing a current between two platinum electrodes located on opposite sides of the muscle. Twitch contractions were elicited with a single 0.3-ms square pulse of 10 V at 200 Hz, whereas tetanic contractions were elicited with a 200-ms train of the same pulse at frequencies of 200 Hz. Muscle length was adjusted to obtain maximum tetanic force, and a 15-min equilibrium period was allowed before any force-frequency measurements. Contractions were elicited every 2 min during the experiment. Force was measured with a dual-mode muscle lever system (ADInstruments Pty Ltd. and GRASS S88X) and digitized at 4 kHz with an analog-digital board (ADInstruments LabChart). Peak twitch and tetanic force were calculated as the difference between the maximum force during contraction, and the force was measured at 5 ms before the contraction.
Creatine Kinase Assay
Creatine kinase concentration was measured using a commercially available kit (Abnova).
Single Fiber Isolation and AA Treatment
Single myofibers were isolated from the gastrocnemius muscles of 8-week-old mice by collagenase I (Sigma; C-0130) digestion (57). Briefly, each harvested gastrocnemius muscle was incubated in 3 ml of 0.2% collagenase I in serum-free DMEM, in a shaking water bath at 35 °C for 60–90 min. Digestion was considered complete when the muscle looked less defined and slightly swollen, with hair-like single fibers appearing to come away from the edges. The digested muscles were placed in a Petri dish, and myofibers were isolated under a microscope. Single fibers were placed in 6-well plates pre-coated with Matrigel (1:3; BD Biosciences) and allowed to attach for 3 min. Then, 2 ml of fiber medium (DMEM supplemented with 10% horse serum, 0.5% chick embryo extract, 1% antibiotic/antimycotic, and 5 mm AA) was added. The fibers were cultured for 48 h at 37 °C in a 5% CO2 atmosphere, fixed with 4% paraformaldehyde, and stained for Pax7 and MyoD as described below. The numbers of Pax7+ and MyoD+ cells determined from counts of at least 40 single fibers were used for statistical analyses.
Western Blot Analysis
Muscle tissues and C2C12 cells were lysed in a buffer containing 50 mm Tris, pH 7.5, 150 mm NaCl, 0.5% Nonidet P-40, and protease and phosphatase inhibitors. The protein lysates were resolved by SDS-PAGE and transferred to a polyvinylidene fluoride membrane. Immunoblotting was performed with primary antibodies against Pax7 (from Developmental Studies Hybridoma Bank); MyoD, VDAC1, and Tim23 were from BD Biosciences; cyclin D1, cyclin E, Cdk2, and Cdk4 were from Santa Cruz Biotechnology; Ras, phospho-Raf, Raf, phospho-MEK, MEK, phospho-ERK, ERK, phospho-Akt, Akt, phospho-p38, p38, phospho-AMPKα, AMPKα, phospho-AMPKβ, and AMPKβ were from Cell Signaling Technology; Oxct1, embryonic MHC, and β-actin were from Sigma; and GAPD was from Millipore. Membranes were washed for 30 min, incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (Zhongshanjinqiao Corp.) for 1 h at room temperature, and then washed with PBS for 30 min. Each membrane was then placed into Detection Solution (Thermo), incubated for 1 min at room temperature, and subsequently exposed to x-ray film. The bands were quantified using ImageJ software. Each assay was performed in three replicates and was repeated at least three times.
Quantitative PCR Analysis
Total RNA was extracted using the TRIzol reagent (Life Technologies, Inc.) and reverse-transcribed using RevertAid reverse transcriptase (Thermo Scientific). Quantitative PCR analyses were performed using an iQ5 Multicolor Real Time PCR Detection System (Bio-Rad). The primer sequences were as follows: HKII-F, 5′-AAC CTCAAA GTG ACG GTG GG-3′, and HKII-R, 5′-TCA CATTTC GGA GCC AGA TCT-3′; PFKI-F, 5′-GAA CTA CGCACA CTT GAC CAT-3′, and PFKI-R, 5′-CTC CAA AAC AAA GGT CCT CTG G-3′; Cyto-c-F, 5′-GAG TTT TGG GCT GAT GGG TA-3′, and Cyto-c-R, 5′-ATC CCG CTG TA ACA CCA GTC-3′; TFAM-F, AGC CAG GTC CAG CTC ACT AA-3′, and TFAM-R, 5′-AAA CCC AAG AAA GCA TGT GG-3′; and β-ATP synthase-F, 5′-GAG GGA TTA CCA CCC ATC CT-3′, and β-ATP synthase-R, 5′-CAT GAT TCT GCC CAAGGT CT-3′.
SCOT Enzymatic Activity Assay
C2C12 cells treated with different dosages of AA were trypsinized, harvested, washed twice with PBS, and then lysed in 50 mm sodium phosphate, pH 8.0, and 0.1% Triton X-100. SCOT activity was measured in the direction of succinyl-CoA formation by spectrophotometricomeasurement of acetoacetyl-CoA disappearance, as described by Williamson et al. (58) with some modifications. The detection mixture (final volume, 1 ml) contained 50 mm Tris-HCl buffer, pH 8.5, 10 mm MgCl2, 4 mm iodoacetamide, 30 μm acetoacetyl-CoA, and 50 mm succinate. After the 30 μg of protein sample was added, the rate of increase in absorption at 303 nm was measured for 4 min. The millimolar extinction coefficient used for acetoacetyl-CoA was 21.0 (303 nm).
Measurement of Lactate Release
After treatment with AA or 3HB for 12 or 24 h, the conditioned media of C2C12 cells were collected, and the amount of lactate released into the medium was determined using a commercially available kit (Jiancheng Corp.), as described previously (59).
Luciferase Reporter Assay
To assess cyclin D1 promoter activity, a cyclin D1 promoter-luciferase reporter plasmid was transiently transfected into C2C12 cells using the FuGENE HD transfection reagent (Roche Applied Science). Twenty four hours later, transfected cells were stimulated with AA, and the luciferase activity in cell lysates was determined using the Dual-Luciferase reporter assay system (Promega) according to the manufacturer's instructions.
Metabolic Measurement
Metabolic measurements were obtained by using an Oxymax Lab Animal Monitoring System (Columbus Instruments, Columbus, OH). The system was calibrated against a standard gas mixture to measure O2 consumed (VO2, ml/kg/h) and CO2 generated (VCO2, ml/kg/h). These measurements were taken on animals that had received AA treatment for 2 months. The first 12 h were a period of adaptation for the animals, and then metabolic rate (VO2) and food intake were evaluated for a 24-h period. Energy expenditure (or heat production) was calculated as the product of the calorific value of oxygen (3.815 + 1.232 × respiratory quotient) and the volume of O2 consumed.
RNA-Seq Data Analysis
Raw sequencing data were mapped to mouse genome mm9 assembly via BWA algorithm with default parameters. DEGSeq was used to count the read coverage for each gene. The differentially expressed genes were filtered with the criterion of more than 1.5-fold changes via SAM. GOTERMFINDER, a tool for finding significant GO terms among a group of genes, was used for GO enrichment analysis. Gene validation was performed using a iQ5 Multicolor real time PCR detection system (Bio-Rad). The primer sequences were designed with DNAMAN.
Statistical Analysis
Results are presented as means ± S.E. Statistical significance of the difference between two means was calculated using the Student's t test. p < 0.05 was considered to represent a statistically significant difference.
Results
AA Promotes Skeletal Muscle Cell Proliferation
To identify intrinsic metabolites with the potential to function as therapeutics to enhance muscle regeneration and cure diseases involving skeletal muscle degeneration, we examined the functional role of ketone bodies, including AA, 3HB, and acetone, in promoting myogenic C2C12 cell proliferation in vitro. Interestingly, we found that only AA promoted C2C12 cell proliferation evidenced by an increase of cells in S-phase (Fig. 1a). The functional role of AA in promoting C2C12 cell proliferation was further corroborated by treating cells with different doses of AA for the indicated times. AA concentration- and time-dependently increased the percentage of cells in the S-phase of the cell cycle (Fig. 1, b and c). Moreover, the efficiency of AA in promoting cell proliferation was similar to that of IGF1 (insulin-like growth factor 1), a cytokine well known for its ability to stimulate C2C12 cell proliferation. Notably, we observed that AA potentiated the stimulatory effect of IGF1 on C2C12 cell proliferation and partially antagonized the inhibitory effect of myostatin (Fig. 1d). Together, the results indicated that AA promoted C2C12 cell progression from G1-phase to S-phase.
FIGURE 1.

AA promotes C2C12 cell proliferation. a, C2C12 cells were treated with 5 mm AA, 3HB, or acetone for 24 h, and cell proliferation was measured by FACS analysis. b, proliferation of C2C12 cells treated with increasing amounts of AA, recombinant IGF1, or PBS for 24 h was analyzed by FACS and [3H]thymidine incorporation assay. c, proliferation of C2C12 cells treated with PBS or 5 mm AA for different times (24, 36, and 48 h) was analyzed by FACS and [3H]thymidine incorporation assay. d, proliferation of C2C12 cells stimulated by combined treatment with 5 mm AA and IGF1 (or myostatin) was assessed by FACS. Similar results were obtained in three separate experiments. Data are presented as means ± S.E. (error bars; *, p < 0.05; **, p < 0.01; ***, p < 0.001).
AA Accelerates Muscle Regeneration by Stimulating Satellite Cell Activation, Proliferation, and Differentiation in Mice
The promotion of C2C12 cell proliferation in vitro by AA prompted us to investigate whether AA could accelerate skeletal muscle regeneration in vivo. To this end, we induced muscle damage in B6 mice by injecting CTX into the TA muscles of mice, and we examined the effect of different concentrations of AA on the regeneration of damaged TA muscles at the indicated time points. PBS-injected muscles were used as controls. As shown in Fig. 2a, regeneration of damaged muscle was time- and dose-dependently accelerated by AA treatment, as evidenced by the increased numbers of regenerating myofibers with centronucleated fibers compared with PBS-treated muscles. Regenerated myofibers in AA-treated mice were significantly larger than those of control mice (Fig. 2b). To further confirm the muscle regeneration-enhancing function of AA, we evaluated the influence of AA on satellite cell activation in the regenerating muscles by immunostaining sections with antibodies against Pax7 and MyoD (Fig. 2c) and counting the number of Pax7+ and MyoD+ cells (Fig. 2d). The transcription factor Pax7 is expressed in quiescent and activated satellite cells, whereas MyoD expression is considered to be an indicator of satellite cell activation during muscle regeneration (60–62). We found that up to 30 mmol/liter of AA induced a concentration-dependent increase in the numbers of Pax7+ and MyoD+ cells (Fig. 2d). To quantify the effect of AA on satellite cell activation, we counted the number of Pax7+ and MyoD+ cells in injured muscles treated with 30 mmol/liter of AA 1 day post-injury. The number of MyoD+ cells in the Pax7+ cell population was significantly greater in the AA-treated muscle than in PBS controls 1 day after CTX damage (Fig. 2e), indicating that AA accelerated muscle regeneration by promoting satellite cell activation at an early stage of muscle regeneration. Furthermore, AA dramatically increased the number of Pax7+ and MyoD+ cells at each stage during muscle regeneration relative to PBS-treated controls (Fig. 2f). This increase peaked 3 days post-injury, suggesting that AA promotes proliferation of activated myogenic cells during the proliferative phase of muscle regeneration. To further confirm this, we collected AA-treated and control TA muscles 3 days after CTX damage, immunostained the samples with antibodies against the proliferation marker, bromodeoxyuridine (BrdU), and MyoD, and quantified the number of proliferative myogenic cells (BrdU+/MyoD+) in the regenerating muscle. As shown in Fig. 2g, the number of BrdU+/MyoD+ cells was significantly increased in AA-treated regenerating muscle compared with PBS controls. We further validated the AA-stimulated activation and proliferation of satellite cells by treating isolated single fibers with AA. As shown in Fig. 2h, more Pax7+ and MyoD+ satellite cells were detected in AA-treated single fibers than in PBS controls. Consistent with this, AA induced considerable concentration- and time-dependent increases in the protein levels of Pax7 and MyoD in the damaged TA muscles (Fig. 2i). In addition, AA also significantly increased expression of embryonic MHC (Fig. 2i), indicating a functional role of AA in promoting de novo myogenesis during the regeneration. Collectively, these data provide in vivo experimental evidence that AA remarkably accelerates the activation of muscle satellite cells and notably stimulates the proliferation of the activated myogenic cells during muscle regeneration in mice.
FIGURE 2.

AA treatment stimulates satellite cell activation and proliferation during muscle regeneration. The data are representative of those obtained in experiments on TA muscles from five mice. a, H&E-stained serial cross-sections from mouse TA muscle treated with different dosages of AA at post-injury day 3. b, cross-sectional areas of regenerated myofibers in PBS control (Con) and 30 mm AA-treated TA muscle 7 days after CTX injury were measured by Image-Pro Plus based on H&E staining of CTX-injured TA muscle sections. c, representative image of sections from TA muscles, injected CTX and treated with AA for 3 days, immunostained for Pax7 (red), MyoD (green), and nuclei (DAPI; blue). Right panel shows merged image with DAPI. d, quantification of Pax7+ and MyoD+ cells. The histogram represents the percentage of cells retaining Pax7+ and MyoD+ in each subpopulation (≥2000 cells/condition). e, representative image of Pax7 and MyoD staining and quantification of Pax7+/MyoD+ cells 1 day post-injury with or without 30 mm AA treatment (≥300 cells each). f, quantification of Pax7+ and MyoD+ cells in sections of 30 mm AA-injected TA muscles 2, 3, and 5 days (d) after muscle damage. g, representative images of MyoD (red) and BrdU (green) immunostaining 3 days post-injury showing the percentage of BrdU+ cells in the MyoD+ cell population (≥300 cells each). h, double staining for Pax7 and MyoD in expanded satellite cells from single fibers isolated from gastrocnemius muscle and cultured for 48 h. i, 1st panel shows the expression of Pax7 and MyoD in TA muscles damaged for 3 days treated with different dosages of AA. The other two panels show the expression level of Pax7, MyoD, and embryonic MHC (eMHC) protein levels in 30 mm AA-treated TA muscles 2, 3, and 5 days post-injury. GAPDH protein served as a loading control. Scale bar, 50 μm. More than five pairs of mice were used for each experiment. Data are presented as mean ± S.E. (error bars; *, p < 0.05; **, p < 0.01; ***, p < 0.001).
AA Treatment Improves the Dystrophic Muscles of Mdx Mice
Duchenne muscular dystrophy is a devastating genetic muscular disorder of childhood; it is characterized by progressive debilitating muscle weakness and wasting, followed ultimately by death in the 2nd or 3rd decades of life. mdx mice, a model of human Duchenne muscular dystrophy, have been widely used to study the development of the disease and identify potential candidate drugs (63, 64). Therefore, we investigated the therapeutic potential of AA in ameliorating muscular dystrophy in mdx mice. Prior to treating the mdx mice with AA, we first assessed the possible side effects of long term AA administration in B6 mice for 2 months. The AA-treated mice did not show overt physiological abnormality as evidenced by unaffected body weight and 3HB levels, normal blood glucose, and glucose tolerance (data not shown). In addition, metabolic chamber analysis indicated that AA-treated mice showed no change of food intake, O2 consumption, and energy expenditure (data not shown). Based on the assessment results, the mdx mice were treated with AA for 2 months (starting at 12 weeks of age), and the morphological, pathological, and functional recovery of dystrophic muscles were examined. Although histological examination of skeletal muscles from AA-treated mdx mice demonstrated normal muscle architecture (Fig. 3a), we observed significant increases in the size of the myofibers from AA-treated mice (Fig. 3b). Next, we examined the uptake of Evans blue dye as a measure of muscle damage and observed the post-contraction integrity of the sarcolemma of EDL. The amount of damaged fibers, as quantified by Evans blue-positive area, was significantly lower in the AA-treated muscles compared with untreated muscles (Fig. 3, c and d). The improvement of muscle integrity in the AA-treated muscles of mdx mice was further supported by the decreased serum concentration of muscle creatine kinase (Fig. 3e), a biomarker for the severity of muscle damage, indicating that AA plays a significant role in protecting dystrophic muscles from contraction-induced injury. To further assess whether the improved integrity of muscle by AA treatment could functionally enhance muscle performance in mdx mice, we measured the strength of the EDL muscle in mdx mice after intraperitoneal injection with AA or PBS. AA significantly increased both peak tetanic and twitch forces of the muscles in mdx mice (Fig. 3, f and g), indicating the functional recovery of dystrophic muscle treated with AA in the mdx mice. Consistent with the above observations, we also demonstrated that AA treatment notably improved exercise performance of mdx mice (Fig. 3h). Together, these results reveal that AA treatment can partially rescue the dystrophic muscles of mdx mice morphologically and functionally, suggesting that AA could be a potential pharmacological drug for treating human muscular dystrophies.
FIGURE 3.
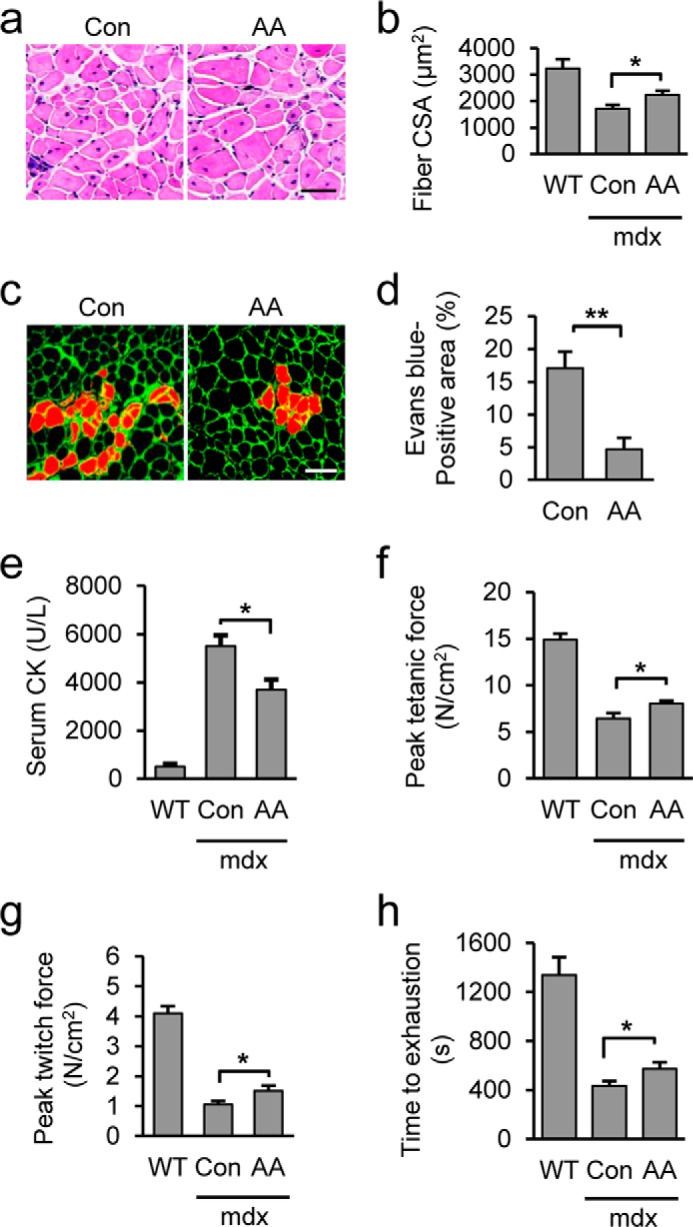
AA treatment restores muscle function and morphology in mdx mice. a, H&E staining of TA muscles from mdx mice treated with AA or PBS control (Con) for 60 days. b, fiber cross-sectional fiber area (CSA) was calculated on the sections in a. C57BL/6 normal mice (B6) served as control. c, Evans blue dye uptake in EDL muscles. d, quantification of the percentage of Evans blue positive fiber area in AA-treated versus control mdx muscle. e, quantitation of serum creatine kinase (CK) levels in B6 and mdx mice treated with AA or PBS control. f, EDL muscles isolated from B6 and mdx mice treated with AA or PBS control were electrically stimulated in vitro to elicit tetanic contractions. g, peak twitch force was measured on EDL muscles isolated from B6 and mdx mice treated with AA or PBS control. h, treadmill test. Scale bar, 50 μm (a) and 100 μm (c). More than five mice were used for each group. Data are presented as mean ± S.E. (error bars; *, p < 0.05; **, p < 0.01).
AA Acts in a Non-metabolic Capacity when Regulating Muscle Cell Function
Because AA and 3HB are produced by the liver and used peripherally as energy sources for various organs, including skeletal muscle, we next asked whether AA promotes C2C12 cell proliferation in an energy-dependent manner. Based on the biochemical reactions of ketolysis, we hypothesized that if AA regulates muscle stem cell function by acting as an energy source, it would need to be converted into the energy-rich compounds, acetoacetyl-CoA or acetyl-CoA, in a reaction catalyzed by the mitochondrial enzyme, succinyl-CoA:3-oxo-acid CoA-transferase (SCOT; EC 2.8.3.5), the rate-limiting enzyme of ketolysis. According to this scenario, the function of AA should be reduced or eliminated in SCOT-deficient cells. However, the AA-mediated promotion of cell proliferation was retained in SCOT knocking down C2C12 cells (Fig. 4a). The ability of AA to stimulate cell proliferation in a SCOT-independent manner was further corroborated by showing that AA also dose-dependently promoted proliferation of HepG2 hepatocellular cells, which is known not to express SCOT protein (Fig. 4b) (65). Previous studies found that SCOT activity was inhibited by AA at concentrations greater than 5 mm (66, 67). This suggested that the enzymatic activity of SCOT should be inhibited by 5 mm AA in C2C12 cells, and the proliferation-promoting role of AA at these physiological levels should be independent of SCOT activity. We tested this by assessing the catalytic activity of SCOT in C2C12 cells treated with increasing doses of AA (0, 1, 3, and 5 mm) for 12 h. As reported previously, SCOT activity was increased by AA at concentrations 1 and 3 mm; however, it was greatly reduced by 5 mm AA (Fig. 4c). Together, these results clearly demonstrate that this function of AA appears to be SCOT-independent. In the reaction catalyzed by SCOT, succinate is generated as a product. Because of succinate functions in various cellular processes, we tested whether AA might act through this intermediate metabolite. However, we found that succinate had no effect on C2C12 cell proliferation (Fig. 4d). We also assessed whether an AA-induced change in mitochondrial biogenesis might stimulate C2C12 cells to proliferate by analyzing the expression levels of the genes encoding Cyto-c, β-ATP synthase, and TFAM. As shown in Fig. 4e, expression of those genes is time-dependently increased in control (PBS-treated)-proliferating C2C12 cells; this was consistent with increased rates of mitochondrial biogenesis, as recently reported by Garedew (68). However, AA treatment did not significantly enhance the expression of Cyto-c, β-ATP synthase, or TFAM as cell proliferation progressed (Fig. 4e). We also used Western blotting in AA-treated proliferating C2C12 cells to assess mitochondrial biogenesis by measuring the levels of the representative mitochondrial proteins, VDAC1 (voltage-dependent anion channel 1) and Tim23 (translocase of inner mitochondrial membrane 23 homolog), and the phosphorylation (activation) status of the key energy sensor AMPK. Consistent with the above results, we found that AA did not increase the levels of VDAC1, Tim23, or phosphorylated AMPKα/β (data not shown). Furthermore, we did not find an obvious effect of AA on mitochondrial morphology in the cells treated with AA compared with PBS (data not shown). Consistently, AA did not alter the mitochondrial metabolic state of the AA-treated mice measured with a metabolic chamber (data not shown). Together, these data indicate that mitochondrial biogenesis is not altered by AA stimulation during cell proliferation. Because proliferating cells are known to have a high rate of glycolysis, we next examined the involvement of glycolysis in AA function by analyzing expression of the genes encoding two key glycolytic enzymes essential for executing glycolysis, hexokinase II (HKII) and phosphofructokinase I (PFKI), and by measuring the released lactate in medium (all in C2C12 cells). As shown in Fig. 4f, AA treatment had almost no effect on expression of HKII and PFKI, or lactate level, indicating that AA did not alter glycolytic activity in C2C12 cells. Interestingly, although 3HB did not promote muscle cell proliferation (Fig. 1a), it increased both mitochondrial activity and glycolysis in C2C12 cells (Fig. 4, e and f). Moreover, we also examined the effect of AA on fatty acid metabolism by assessing expression of genes (cpt1b, cs, acc2, and fasn) required for fatty acid metabolism. Consistently, expression of these genes did not differ in the C2C12 cells in the presence or absence of AA (data not shown). Thus, AA also had no influence on fatty acid metabolism. Collectively, our findings convincingly demonstrate that the specific role of AA in regulating muscle cell function is not directly associated with energy production.
FIGURE 4.

AA plays a non-metabolic role in ERK activation and muscle cell proliferation. a, cell proliferation was analyzed in Oxct1-knockdown C2C12 cells treated with 5 mm AA or PBS for 24 h. The efficiency of Oxct1 knockdown was examined by Western blotting. b, proliferation of HepG2 cells treated with PBS or AA (5 mm) was analyzed by FACS. c, SCOT catalytic activity was measured in the C2C12 cells treated with different dosages of AA (0, 1, 3, and 5 mm) for 24 h. d, cell proliferation was analyzed in C2C12 cells treated with 5 mm AA or succinate for 24 h. e, normalized Cyto-c, β-ATP synthase, and TFAM mRNA levels in C2C12 cells treated with 5 mm AA or 3HB for 6 or 24 h were determined by quantitative RT-PCR. f, normalized phosphofructokinase I (PFK1) and hexokinase II (HK2) mRNA levels in C2C12 cells treated with 5 mm AA or 3HB for 6 or 24 h were determined by quantitative RT-PCR. Lactate concentration in cell media was measured using a commercial kit. Similar results were obtained in three separate experiments. Data are presented as means ± S.E. (error bars; *, p < 0.05). NS, not significant.
To provide more molecular insight into the AA signaling role in regulating the muscle cell proliferation, we performed RNA-Seq analyses to identify AA-regulated genes in proliferating C2C12 myoblasts in the present or absent of AA. Differentially regulated genes and gene set enrichment analysis revealed that altered expression of the AA-mediated genes in growth medium was highly enriched in the signaling pathways associated with cell proliferation (data not shown); however, the genes encoding enzymes in biochemical pathways of ketolysis for energy production were not transcriptionally changed in the cells cultured in growth medium in response to AA (data not shown). This indicates that AA-mediated myogenic cells function by transcriptional regulation of the genes required for cell proliferation. Taken together, the data from our multiparameter analyses consistently support the notion that AA plays a non-metabolic role in regulating muscle cell function.
Signaling Function of AA in Promoting Muscle Cell Proliferation by Increasing Expression of Cyclin D1 through MEK-ERK1/2 Activation
The energy-independent function of AA in promoting cell proliferation was further substantiated by exploring the molecular mechanism underlying its action. Because AA was found to promote cell cycle progression from the G1- to S-phase, we assessed the protein expression levels of various G1 regulators in AA-treated C2C12 myoblasts. AA significantly increased the steady-state levels of cyclin D1 protein (Fig. 5a) in a concentration-dependent manner (Fig. 5b). AA also stimulated cyclin D1 expression at the mRNA level, as demonstrated by quantitative real time RT-PCR analyses and assessment of cyclin D1 promoter-driven luciferase reporter activity (data not shown). To directly test whether cyclin D1 up-regulation was a determining factor in AA-induced cell proliferation, we knocked down cyclin D1 in proliferating C2C12 cells using small interfering RNA (siRNA) (Fig. 5c). The AA-induced promotion of myoblast cell proliferation was greatly attenuated in cyclin D1 knockdown cells compared with control cells (Fig. 5d), demonstrating that cyclin D1 is required for AA-mediated enhancement of cell proliferation.
FIGURE 5.

MEK-ERK1/2 activation is required for AA-induced C2C12 cell proliferation through up-regulation of cyclin D1 expression. a, C2C12 cells were treated with 5 mm AA for 24 h. Cell lysates were used for Western blot detection of cyclin D1, cyclin E, Cdk2, and Cdk4. β-Actin served as a loading control. PBS treatment served as control (Con). b, proteins in total lysates of C2C12 cells treated with different concentrations of AA (0.1, 0.5, 1, and 5 mm) and IGF1 (100 ng/ml) for 24 h were resolved by SDS-PAGE and analyzed by Western blotting for cyclin D1, Cdk2, and β-actin. c, efficiency of cyclin D1 knockdown in C2C12 cells was examined by Western blotting. d, cell proliferation was analyzed in cyclin D1-knockdown C2C12 cells treated with 5 mm AA or PBS for 24 h. e, expression levels of phospho (p)-MEK, total (t)-MEK, p-ERK, t-ERK, and β-actin proteins in C2C12 cells treated with 5 mm AA for different times (0.5, 1, 3, 6, and 12 h) were analyzed by Western blotting. f, C2C12 cells were pre-treated with the MEK inhibitor PD98059 (20 μm) for 1 h prior to AA (5 mm) treatment. The levels of cyclin D1, p-ERK, t-ERK, and β-actin were determined by Western blotting. g, proliferation of C2C12 cells treated with AA (5 mm) with or without PD98059 (20 μm) was analyzed by FACS. h, cyclin D1 expression was detected in the ERK1/2-knockdown C2C12 cells treated with 5 mm AA or PBS for 12 h. i, cell proliferation was analyzed in ERK1/2-knockdown C2C12 cells treated with 5 mm AA or PBS for 24 h. j, representative sections of AA-treated TA muscles, with and without PD98059 (20 μm) treatment, 2 days after injury immunostained for MyoD (green), and nuclei (DAPI; blue). Right panel shows merged image with DAPI. k, quantification of MyoD+ cells in sections described in j. l, expression of Pax7, cyclin D1, p-ERK, and t-ERK in damaged TA muscles treated with AA in the presence or absence of PD98059 was analyzed by Western blotting. Scale bar, 50 μm. Similar results were obtained in three separate experiments. Data are presented as means ± S.E. (error bars; *, p < 0.05). NS, not significant.
The finding that this function of AA is SCOT-independent and cyclin D1-dependent prompted us to speculate that AA may act as a signaling molecule rather than an energy source in promoting cell proliferation. To test this hypothesis, we sought to determine which signaling pathways were involved in the AA-mediated promotion of cell proliferation through cyclin D1. We examined the p38, PI3K-Akt, and Ras-Raf-MEK1-ERK1/2 signaling pathways in AA-treated cells, all of which have the potential to affect myoblast proliferation. Our results revealed that AA did not change the total levels or phosphorylation status of p38 or Akt proteins (data not shown). However, AA treatment induced a rapid (within 30 min) and sustained (up to 12 h) activation of ERK1/2 in proliferating C2C12 cells (Fig. 5e). As expected, this AA-induced ERK1/2 activation was associated with a comparable increase in the phosphorylation of MEK1 (Fig. 5e), the upstream kinase in this pathway. To further verify the involvement of ERK1/2 in this function of AA, we tested the effects of AA on C2C12 cell proliferation in the presence and absence of the MEK1 inhibitor, PD98059. Inhibition of ERK1/2 kinase activity by PD98059 considerably blocked the AA-induced up-regulation of cyclin D1 (Fig. 5f) and significantly attenuated AA-induced cell proliferation (Fig. 5g). Requirement of ERK1/2 activation for AA function was further supported by showing that AA-induced up-regulation of cyclinD1 (Fig. 5h) and cell proliferation (Fig. 5i) were also remarkably attenuated in the ERK1/2 knockdown C2C12 cells. Taken together, our results reveal that AA stimulates myoblast proliferation by up-regulating cyclin D1 expression through activation of the MEK1-ERK1/2 signaling cascade.
To investigate whether the AA-induced stimulation of muscle regeneration in vivo was also mediated through MEK1-ERK1/2 activation, we injected CTX-injured TA muscles with AA in the presence or absence of PD98059, immunostained the TA muscles with antibodies against MyoD (Fig. 5j), and quantified the number of the MyoD+ cells (Fig. 5k). AA treatment significantly increased the number of MyoD+ cells relative to PBS treatment (Fig. 5k). Consistent with the in vitro results (Fig. 5f), PD98059-induced inhibition of ERK1/2 significantly attenuated AA-induced satellite cell activation and proliferation, as evidenced by marked reductions in the numbers of MyoD+ cells (Fig. 5k). These observations were further corroborated by significant increases in the levels of Pax7, cyclin D1, and phospho-ERK1/2 proteins in damaged muscle treated with AA and the notable reductions in these levels following co-treatment with PD98059 (Fig. 5l). In summary, our in vitro and in vivo results clearly demonstrate that MEK1-ERK1/2 activity is required for the ability of AA to promote muscle cell proliferation and accelerate muscle regeneration in mice.
Ras Is Dispensable for the AA-mediated Promotion of Muscle Cell Proliferation via Activation of MEK1-ERK1/2
Because Ras plays an important role as the upstream kinase for MEK1-ERK1/2 activation, we next determined whether AA-stimulated MEK-ERK1/2 activation depended on Ras/Raf. Intriguingly, our data showed that the levels of total Ras, total Raf, and phosphorylated Raf were not changed in response to AA (Fig. 6, a and b), suggesting that MEK1-ERK1/2 may not be activated by Ras/Raf-mediated signaling in this context. As described in our previous study (69), we functionally blocked Ras activity by expressing the dominant-negative Ras mutant, Ras17N, in C2C12 cells, and we tested whether Ras was required for AA-induced MEK-ERK1/2 activation. Western blot analyses showed that ERK1/2 was still concentration-dependently activated by AA in Ras17N C2C12 cells (Fig. 6, c and d). Consistent with these effects, blockade of the Ras/Raf signaling pathway using pharmacological inhibitors of Ras (FTA) or Raf (GW5074) did not prevent the AA-induced activations of MEK1 and ERK1/2 (Fig. 6e). Moreover, cell proliferation assays showed that AA was still able to concentration-dependently promote cell proliferation in Ras17N C2C12 cells or C2C12 cells treated with FTA or GW5074 (Fig. 6, f and g), further confirming that Ras is dispensable for these actions of AA. Collectively, these findings demonstrate that Ras, the main upstream intracellular regulator of MEK1/ERK1/2, is dispensable for AA-stimulated MEK1-ERK1/2 activation and C2C12 cell proliferation.
FIGURE 6.

AA activates the MEK-ERK1/2 pathway and promotes C2C12 cell proliferation independent of Ras/Raf. a, levels of Ras, t-Raf (c-Raf), and p-Raf (p-c-Raf) proteins in C2C12 cells in response to AA (5 mm) treatment were assessed by Western blotting at the indicated times. b, normalized level of Ras and p-Raf. Ras protein was normalized to β-actin, and p-Raf was normalized to total Raf. c, protein levels of Ras, t-Raf/p-Raf, t-MEK/p-MEK, and t-ERK1/2/p-ERK1/2 were determined by Western blot analysis in normal C2C12 and DN-Ras C2C12 cells treated with AA (5 mm) or PBS for 12 h. d, quantified levels of p-MEK and p-ERK relative to t-MEK and t-ERK, respectively, based on blots in c. e, protein levels of Ras, p-Raf, t-Raf, p-MEK, t-MEK, p-ERK, t-ERK, and β-actin in C2C12 cells treated with AA (5 mm) for 12 h in the presence or absence of the inhibitors FTA or GW5074 (GW) were determined by Western blotting. f, cell proliferation was analyzed in C2C12 cells or DN-Ras expressing C2C12 cells treated with 5 mm AA. g, cell proliferation was analyzed in C2C12 cells treated with AA (5 mm) in the presence or absence of FTA or GW5074. h, schematic model of AA function. Similar results were obtained in three separate experiments. Data are presented as means ± S.E. (error bars; *, p < 0.05; ***, p < 0.001).
Discussion
Intermediate metabolites acting as signaling molecules to regulate cellular processes is a newly emerging theme, and researchers are just beginning to reveal how these metabolites communicate with the cell cycle machinery to influence cell growth and fate decision (14, 15, 70). Here, we report a novel function of the small intrinsic metabolite AA in promoting muscle cell proliferation in vitro, improving muscle regeneration in vivo, and ameliorating muscular dystrophy in mdx mice. Most importantly, our study revealed that AA promotes the activation and proliferation of skeletal muscle cells through the MEK-ERK1/2-cyclin D1 signaling pathway, as shown schematically in Fig. 6h. Lithium ions dissociated from acetoacetate lithium are biologically active and inhibit the function of GSK-3α/β and other key kinases (COX, Arrestin-2, CaMK1, PHK, CHK2, NEK6, and IKKγ), which play important roles in regulating cell proliferation (71, 72). Therefore, we treated C2C12 cells with acetoacetate sodium to rule out the possibility that cell proliferation promoted by acetoacetate lithium was due to the activity of lithium effect rather than acetoacetate itself. Consistent with the results from acetoacetate lithium treatment, acetoacetate sodium exerted a similar biological effect on cell proliferation as acetoacetate lithium through activating the MEK/ERK/cyclinD1 signaling pathway (data not shown). We thus conclude the demonstration of an unanticipated function of AA that is distinct from its function as a fuel, and we provide a potential mechanism by which this action of AA couples metabolism with muscle development and regeneration.
AA and 3HB are both prominent ketone bodies that serve as energy-rich compounds to transport energy from the liver to other tissues, particularly the brain and skeletal muscle (41, 73). However, AA differs from 3HB in that it also has unique functions in various biological processes, such as insulin release in vitro, generation of free oxygen radicals, lipid peroxidation, and activation of the ERK1/2 and p38 MAPK signaling pathways (45, 46, 49). In this report, we demonstrate for the first time that AA-mediated promotion of muscle cell proliferation is not a generalized ketone body effect but instead is specific to AA. We hypothesized that these distinctive actions of AA might reflect a regulatory role as a previously unrecognized signaling metabolite, rather than its traditionally recognized role as a fuel. The biochemical pathways of ketolysis have been well defined in mitochondria of ketolytic organs, such as the brain and skeletal muscle tissues, where 3HB is oxidized to AA in a reaction catalyzed by 3HB dehydrogenase. AA, in turn, receives a CoA moiety from succinyl-CoA to generate acetoacetyl-CoA in a reaction catalyzed by SCOT. Finally, acetoacetyl-CoA is converted to acetyl-CoA in a reaction catalyzed by mitochondrial acetoacetyl-CoA thiolase, and the acetyl-CoA is terminally oxidized within the tricarboxylic acid cycle for the production of energy that is used to fulfill energy demands for cell growth and division. Accordingly, if AA functioned as a fuel in the examined process, acetoacetyl-CoA and acetyl-CoA would be predicted to have functions similar to that of AA in terms of ERK1/2 activation and cell proliferation and differentiation. Moreover, SCOT should be essential for AA action. Intriguingly, our experiments revealed that SCOT was not required for this function of AA (Fig. 4a), and mitochondrial biogenesis was unaltered in AA-treated cells (Fig. 4e). In addition, gene set enrichment analysis identified the gene sets involved in muscle cell proliferation as the most significant sets in the cells in response to AA, and in particular, the expression of genes encoding ketolysis enzymes required for energy generation was not significantly altered by AA treatment, indicating a signaling role of AA in regulating expression of genes involved in myogenic cell function. Collectively, these findings support the notion that AA is capable of functioning as a signaling molecule in regulating muscle cell function. Interestingly, recent reports have demonstrated that abnormally accumulated fumarate plays a novel role in the irregular modification of cellular proteins (succination of KEAP1) and Nrf2 (NFE2-related factor 2) signaling in fumarate hydratase-deficient cells and that these functions are independent of its ability to inhibit α-ketoglutarate-dependent dioxygenases (17, 73–75). Thus, our findings and the results of prior studies collectively indicate that AA could regulate cellular activity independent of its energetic role.
A functional role of AA as a signaling molecule has been well documented in a bacterial two-component system. The expression of the atoDAEB operon (whose gene products participate in the catabolism of SCFAs) is highly induced by AA but not by other saturated SCFAs, such as butyrate and valerate (55, 56), indicating that the regulatory role of AA as a signaling metabolite is specific in this bacterial signal-transduction system. Following this line of evidence and considering our finding that AA regulates muscle cell function, it is plausible to hypothesize that the functional role of AA as a signaling metabolite in communicating between metabolism and cellular activity may be evolutionally conserved from bacteria to mammals. This should be of particular interest for studies on when and how the signaling functions of intermediate metabolites evolved in mammals, and how this mechanism acts in concert with their fuel functions to mediate coordinated communication between cellular metabolism and cell growth or division.
In this study, we made the intriguing discovery that Ras/Raf was dispensable for the AA-stimulated activation of the MEK-ERK1/2 MAPK pathway in promoting C2C12 cell proliferation. AA is a small and diffusible molecule (a 4-carbon organic acid) that can freely cross the cell membrane. Given this, it is possible that AA could function as a specific diffusible signaling molecule and intracellularly activate MEK-ERK1/2. We examined this possibility by treating C2C12 cells with various blockers of endocytosis. Interestingly, we found that these inhibitors did not block AA-mediated ERK1/2 activation or cell proliferation (data not shown). Consistent with this finding, we also observed that this function of AA was not inhibited by suramin (data not shown), which is a broad spectrum antagonist of the purinergic P2X and P2Y receptors (and other G-protein-coupled receptors), which can bind and inhibit several growth factors, including IGF, fibroblast growth factor, epidermal growth factor, and platelet-derived growth factor. Together, these results suggest that AA most likely crosses the cell membrane through simple diffusion and then intracellularly activates MEK-ERK1/2. Support for this general hypothesis is provided by a report that the metabolite fumarylacetoacetate, a precursor of AA, activates ERK1/2 in a growth factor receptor-independent manner in human HeLa and Chinese hamster V79 cells (76). Fumarylacetoacetate is converted into fumarate and AA by the enzyme, fumarylacetoacetate hydrolase (EC 3.7.1.2) (76). AA and fumarylacetoacetate are structurally similar, suggesting that they may share a common mechanism for activating ERK1/2 as signaling molecules. A previous study showed that AA can activate the ERK1/2 signaling pathway in primary cultured rat hepatocytes through reactive oxygen species and oxidative stress (45). Our data confirmed that AA plays a role in MEK-ERK1/2 activation in myogenic C2C12 cells, although we found that reactive oxygen species was not required for this activation (data not shown). Thus, although our data indicate that AA activates MEK-ERK1/2 in a Ras-independent manner, future studies are needed to determine the precise molecular mechanism through which AA stimulates MEK-ERK1/2 activation in promoting muscle cell proliferation.
The functional role of AA as a signaling molecule is also indirectly supported by the experimental data from metabolites of leucine and HMB, one of derivatives from leucine. Although AA generally has been attributed from fatty acid by the liver, ketone bodies derived from leucine may also contribute to physiological hyperketonemia after short-term fasting (77, 78). The production of ketone bodies from leucine in muscles was evidenced by the studies that leucine and its 2-oxo analogue, 4-methyl-2-oxopentanoate, increased the release of acetoacetate and 3-hydroxybutyrate from hemidiaphragms isolated from 40-h starved rats (79). It has been reported that both leucine and HMB significantly promoted muscle cell proliferation and differentiation through the MEK/ERK signaling pathway (80–82), and it is conceivable that AA might act as a signaling metabolite participating in leucine- and HMB-mediated functions in promoting muscle cell proliferation and differentiation.
Recently, a large scale yeast study designed to identify in vivo interactions between proteins and small intrinsic metabolites (83) revealed that many proteins bind to small metabolites in interactions that had not been previously recognized. In particular, the authors found that ergosterol bound to many proteins and that its binding specifically enhanced the protein kinase activity of Ypk1 and altered the protein levels of Ssk22; they thus postulated that the functions of some proteins could be modulated by interactions with metabolites that are not their substrates or products (83). Therefore, it is conceivable that small intrinsic metabolites may play a more general role than previously appreciated, globally regulating cellular activities by directly interacting with proteins for controlling their activity and function (84). Early investigations of AA utilization in animals using 14C-labeled AA indicated that most of the AA was recovered as respiratory 14CO2; however, a small amount of AA was incorporated into proteins (85, 86). Intriguingly, while our manuscript was being submitted, T. Nakagawa et al. (87) reported their recent work in which they have provided experimental evidence to show that ketone bodies (3HB and AA) could serve as low glucose signaling by regulating nucleo-cytosolic trafficking of ChREBP through promoting interaction of ChREBP/14-3-3 and inhibiting ChREBP/importin interaction. This is the first demonstration that ketone bodies could physically interact with cellular proteins. Most importantly, it provides direct evidence to support our hypothesis that AA, as a signaling metabolite, regulates cellular activity through interactions with or modifications of cellular proteins. Therefore, future studies should seek to identify AA-modified or -bound proteins, as this will provide important new insights into the actions of AA as a signaling molecule.
Our discovery that AA functions in accelerating muscle regeneration and improving muscular dystrophy in mdx mice may have significant clinical implications, providing a rationale for the potential use of AA to treat muscular dystrophy and other diseases associated with muscle wasting in humans. Perhaps more importantly, our findings suggest that small intrinsic metabolites can play general roles during development, hinting that they may have therapeutic potential for treating other human diseases. The future identification of additional intrinsic metabolites involved in various biological processes should help us decipher how these metabolites mediate the regulatory interplay between metabolism and the cellular events associated with development and diseases.
Author Contributions
D. H. Z. conceived and coordinated the study and wrote the paper. X. T. Z. designed, performed, and analyzed the experiments shown in Figs. 2, 3, and 5. J. M. and L. L. designed, performed, and analyzed the experiments shown in Figs. 1 and 4–6. W. H. H., R. Z., C. Y. L., X. X. M., J. C., and Y. Z. provided technical assistance and contributed to the preparation of the figures. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Drs. Ye-Guang Chen, Xiang Gao, Jian Zhang, and Duanqing Pei for their useful discussions and critical reading of the manuscript.
This work was supported by National Basic Research Program of China Grants 2011CBA01104, 2015CB943103, and 2014CB964703 and National Natural Science Foundation of China Grants 31471289 and 31030041. The authors declare that they have no conflicts of interest with the contents of this article.
- 2-HG
- 2-hydroxyglutarate
- AA
- acetoacetate
- 3HB
- 3-hydroxybutyrate
- ChREBP
- carbohydrate-response element-binding protein
- HMB
- β-hydroxy-β-methylbutyrate
- AMPK
- AMP-activated protein kinase
- EDL
- extensor digitorum longus
- SCFA
- short-chain fatty acid
- FTA
- farnesyl-thioacetic acid
- CTX
- cardiotoxin
- TA
- tibialis anterior
- SCOT
- succinyl-CoA:3-oxo-acid CoA-transferase
- TFAM
- mitochondrial transcription factor A
- Cyto-c
- cytochrome c.
References
- 1. Mohanty S., Singhal R., Sood S., Dhawan B., Das B. K., and Kapil A. (2005) Comparative in vitro activity of β-lactam/β-lactamase inhibitor combinations against Gram negative bacteria. Indian J. Med. Res. 122, 425–428 [PubMed] [Google Scholar]
- 2. Wang Y. X., and Rudnicki M. A. (2012) Satellite cells, the engines of muscle repair. Nat. Rev. Mol. Cell Biol. 13, 127–133 [DOI] [PubMed] [Google Scholar]
- 3. Braun T., and Gautel M. (2011) Transcriptional mechanisms regulating skeletal muscle differentiation, growth and homeostasis. Nat. Rev. Mol. Cell Biol. 12, 349–361 [DOI] [PubMed] [Google Scholar]
- 4. Wagers A. J., and Conboy I. M. (2005) Cellular and molecular signatures of muscle regeneration: current concepts and controversies in adult myogenesis. Cell 122, 659–667 [DOI] [PubMed] [Google Scholar]
- 5. Kuang S., Gillespie M. A., and Rudnicki M. A. (2008) Niche regulation of muscle satellite cell self-renewal and differentiation. Cell Stem Cell 2, 22–31 [DOI] [PubMed] [Google Scholar]
- 6. Kamrava M., Simpkins F., Alejandro E., Michener C., Meltzer E., and Kohn E. C. (2005) Lysophosphatidic acid and endothelin-induced proliferation of ovarian cancer cell lines is mitigated by neutralization of granulin-epithelin precursor (GEP), a prosurvival factor for ovarian cancer. Oncogene 24, 7084–7093 [DOI] [PubMed] [Google Scholar]
- 7. Usas A., Mačiulaitis J., Mačiulaitis R., Jakubonienė N., Milašius A., and Huard J. (2011) Skeletal muscle-derived stem cells: implications for cell-mediated therapies. Medicina 47, 469–479 [PubMed] [Google Scholar]
- 8. Bhagavati S. (2008) Stem cell based therapy for skeletal muscle diseases. Curr. Stem Cell Res. Ther. 3, 219–228 [DOI] [PubMed] [Google Scholar]
- 9. Endo T. (2007) Stem cells and plasticity of skeletal muscle cell differentiation: potential application to cell therapy for degenerative muscular diseases. Regen. Med. 2, 243–256 [DOI] [PubMed] [Google Scholar]
- 10. Shi X., and Garry D. J. (2006) Muscle stem cells in development, regeneration, and disease. Genes Dev. 20, 1692–1708 [DOI] [PubMed] [Google Scholar]
- 11. Zeng L., Akasaki Y., Sato K., Ouchi N., Izumiya Y., and Walsh K. (2010) Insulin-like 6 is induced by muscle injury and functions as a regenerative factor. J. Biol. Chem. 285, 36060–36069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cantini M., Massimino M. L., Rapizzi E., Rossini K., Catani C., Dalla Libera L., and Carraro U. (1995) Human satellite cell proliferation in vitro is regulated by autocrine secretion of IL-6 stimulated by a soluble factor(s) released by activated monocytes. Biochem. Biophys. Res. Commun. 216, 49–53 [DOI] [PubMed] [Google Scholar]
- 13. Riuzzi F., Sorci G., Sagheddu R., and Donato R. (2012) HMGB1-RAGE regulates muscle satellite cell homeostasis through p38-MAPK- and myogenin-dependent repression of Pax7 transcription. J. Cell Sci. 125, 1440–1454 [DOI] [PubMed] [Google Scholar]
- 14. Buchakjian M. R., and Kornbluth S. (2010) The engine driving the ship: metabolic steering of cell proliferation and death. Nat. Rev. Mol. Cell Biol. 11, 715–727 [DOI] [PubMed] [Google Scholar]
- 15. Ward P. S., and Thompson C. B. (2012) Signaling in control of cell growth and metabolism. Cold Spring Harb. Perspect. Biol. 4, a006783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pollard P. J., and Ratcliffe P. J. (2009) Cancer. Puzzling patterns of predisposition. Science 324, 192–194 [DOI] [PubMed] [Google Scholar]
- 17. Bardella C., El-Bahrawy M., Frizzell N., Adam J., Ternette N., Hatipoglu E., Howarth K., O'Flaherty L., Roberts I., Turner G., Taylor J., Giaslakiotis K., Macaulay V. M., Harris A. L., Chandra A., et al. (2011) Aberrant succination of proteins in fumarate hydratase-deficient mice and HLRCC patients is a robust biomarker of mutation status. J. Pathol. 225, 4–11 [DOI] [PubMed] [Google Scholar]
- 18. Sasaki M., Knobbe C. B., Munger J. C., Lind E. F., Brenner D., Brüstle A., Harris I. S., Holmes R., Wakeham A., Haight J., You-Ten A., Li W. Y., Schalm S., Su S. M., Virtanen C., Reifenberger G., et al. (2012) IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature 488, 656–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang M., Soga T., Pollard P. J., and Adam J. (2012) The emerging role of fumarate as an oncometabolite. Front. Oncol. 2, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dang L., White D. W., Gross S., Bennett B. D., Bittinger M. A., Driggers E. M., Fantin V. R., Jang H. G., Jin S., Keenan M. C., Marks K. M., Prins R. M., Ward P. S., Yen K. E., Liau L. M., et al. (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhao S., Lin Y., Xu W., Jiang W., Zha Z., Wang P., Yu W., Li Z., Gong L., Peng Y., Ding J., Lei Q., Guan K. L., and Xiong Y. (2009) Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1α. Science 324, 261–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gross S., Cairns R. A., Minden M. D., Driggers E. M., Bittinger M. A., Jang H. G., Sasaki M., Jin S., Schenkein D. P., Su S. M., Dang L., Fantin V. R., and Mak T. W. (2010) Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J. Exp. Med. 207, 339–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ward P. S., Patel J., Wise D. R., Abdel-Wahab O., Bennett B. D., Coller H. A., Cross J. R., Fantin V. R., Hedvat C. V., Perl A. E., Rabinowitz J. D., Carroll M., Su S. M., Sharp K. A., Levine R. L., and Thompson C. B. (2010) The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17, 225–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Epstein A. C., Gleadle J. M., McNeill L. A., Hewitson K. S., O'Rourke J., Mole D. R., Mukherji M., Metzen E., Wilson M. I., Dhanda A., Tian Y. M., Masson N., Hamilton D. L., Jaakkola P., Barstead R., et al. (2001) C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107, 43–54 [DOI] [PubMed] [Google Scholar]
- 25. Yu F., White S. B., Zhao Q., and Lee F. S. (2001) HIF-1α binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc. Natl. Acad. Sci. U.S.A. 98, 9630–9635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Isaacs J. S., Jung Y. J., Mole D. R., Lee S., Torres-Cabala C., Chung Y. L., Merino M., Trepel J., Zbar B., Toro J., Ratcliffe P. J., Linehan W. M., and Neckers L. (2005) HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 8, 143–153 [DOI] [PubMed] [Google Scholar]
- 27. Selak M. A., Armour S. M., MacKenzie E. D., Boulahbel H., Watson D. G., Mansfield K. D., Pan Y., Simon M. C., Thompson C. B., and Gottlieb E. (2005) Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 7, 77–85 [DOI] [PubMed] [Google Scholar]
- 28. Baysal B. E., Ferrell R. E., Willett-Brozick J. E., Lawrence E. C., Myssiorek D., Bosch A., van der Mey A., Taschner P. E., Rubinstein W. S., Myers E. N., Richard C. W. 3rd, Cornelisse C. J., Devilee P., and Devlin B. (2000) Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 287, 848–851 [DOI] [PubMed] [Google Scholar]
- 29. Astuti D., Douglas F., Lennard T. W., Aligianis I. A., Woodward E. R., Evans D. G., Eng C., Latif F., and Maher E. R. (2001) Germline SDHD mutation in familial phaeochromocytoma. Lancet 357, 1181–1182 [DOI] [PubMed] [Google Scholar]
- 30. Hao H. X., Khalimonchuk O., Schraders M., Dephoure N., Bayley J. P., Kunst H., Devilee P., Cremers C. W., Schiffman J. D., Bentz B. G., Gygi S. P., Winge D. R., Kremer H., and Rutter J. (2009) SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science 325, 1139–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kaelin W. G., Jr. (2009) SDH5 mutations and familial paraganglioma: somewhere Warburg is smiling. Cancer Cell 16, 180–182 [DOI] [PubMed] [Google Scholar]
- 32. Bayley J. P., Kunst H. P., Cascon A., Sampietro M. L., Gaal J., Korpershoek E., Hinojar-Gutierrez A., Timmers H. J., Hoefsloot L. H., Hermsen M. A., Suárez C., Hussain A. K., Vriends A. H., Hes F. J., Jansen J. C., et al. (2010) SDHAF2 mutations in familial and sporadic paraganglioma and phaeochromocytoma. Lancet Oncol. 11, 366–372 [DOI] [PubMed] [Google Scholar]
- 33. Xu W., Yang H., Liu Y., Yang Y., Wang P., Kim S. H., Ito S., Yang C., Xiao M. T., Liu L. X., Jiang W. Q., Liu J., Zhang J. Y., Wang B., Frye S., et al. (2011) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Oermann E. K., Wu J., Guan K. L., and Xiong Y. (2012) Alterations of metabolic genes and metabolites in cancer. Semin. Cell Dev. Biol. 23, 370–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. King A., Selak M. A., and Gottlieb E. (2006) Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene 25, 4675–4682 [DOI] [PubMed] [Google Scholar]
- 36. Xiao M., Yang H., Xu W., Ma S., Lin H., Zhu H., Liu L., Liu Y., Yang C., Xu Y., Zhao S., Ye D., Xiong Y., and Guan K. L. (2012) Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 26, 1326–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Menendez J. A., and Lupu R. (2007) Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 7, 763–777 [DOI] [PubMed] [Google Scholar]
- 38. Bauer D. E., Hatzivassiliou G., Zhao F., Andreadis C., and Thompson C. B. (2005) ATP citrate lyase is an important component of cell growth and transformation. Oncogene 24, 6314–6322 [DOI] [PubMed] [Google Scholar]
- 39. Minetti G. C., Feige J. N., Rosenstiel A., Bombard F., Meier V., Werner A., Bassilana F., Sailer A. W., Kahle P., Lambert C., Glass D. J., and Fornaro M. (2011) Gαi2 signaling promotes skeletal muscle hypertrophy, myoblast differentiation, and muscle regeneration. Sci. Signal. 4, ra80. [DOI] [PubMed] [Google Scholar]
- 40. Lu C., Ward P. S., Kapoor G. S., Rohle D., Turcan S., Abdel-Wahab O., Edwards C. R., Khanin R., Figueroa M. E., Melnick A., Wellen K. E., O'Rourke D. M., Berger S. L., Chan T. A., Levine R. L., et al. (2012) IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Robinson A. M., and Williamson D. H. (1980) Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol. Rev. 60, 143–187 [DOI] [PubMed] [Google Scholar]
- 42. Macdonald M. J., Hasan N. M., and Longacre M. J. (2008) Studies with leucine, β-hydroxybutyrate and ATP citrate lyase-deficient beta cells support the acetoacetate pathway of insulin secretion. Biochim. Biophys. Acta 1780, 966–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schönfeld P., Wieckowski M. R., Lebiedzińska M., and Wojtczak L. (2010) Mitochondrial fatty acid oxidation and oxidative stress: lack of reverse electron transfer-associated production of reactive oxygen species. Biochim. Biophys. Acta 1797, 929–938 [DOI] [PubMed] [Google Scholar]
- 44. Samartsev V. N., and Kozhina O. V. (2010) Acetoacetate as regulator of palmitic acid-induced uncoupling involving liver mitochondrial ADP/ATP antiporter and aspartate/glutamate antiporter. Biochemistry 75, 598–605 [DOI] [PubMed] [Google Scholar]
- 45. Abdelmegeed M. A., Kim S. K., Woodcroft K. J., and Novak R. F. (2004) Acetoacetate activation of extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase in primary cultured rat hepatocytes: role of oxidative stress. J. Pharmacol. Exp. Ther. 310, 728–736 [DOI] [PubMed] [Google Scholar]
- 46. Jain S. K., and McVie R. (1999) Hyperketonemia can increase lipid peroxidation and lower glutathione levels in human erythrocytes in vitro and in type 1 diabetic patients. Diabetes 48, 1850–1855 [DOI] [PubMed] [Google Scholar]
- 47. Finn P. F., and Dice J. F. (2005) Ketone bodies stimulate chaperone-mediated autophagy. J. Biol. Chem. 280, 25864–25870 [DOI] [PubMed] [Google Scholar]
- 48. Uehara Y., Engel T., Li Z., Goepfert C., Rust S., Zhou X., Langer C., Schachtrup C., Wiekowski J., Lorkowski S., Assmann G., and von Eckardstein A. (2002) Polyunsaturated fatty acids and acetoacetate downregulate the expression of the ATP-binding cassette transporter A1. Diabetes 51, 2922–2928 [DOI] [PubMed] [Google Scholar]
- 49. Jain S. K., Kannan K., Lim G., Matthews-Greer J., McVie R., and Bocchini J. A. Jr. (2003) Elevated blood interleukin-6 levels in hyperketonemic type 1 diabetic patients and secretion by acetoacetate-treated cultured U937 monocytes. Diabetes Care 26, 2139–2143 [DOI] [PubMed] [Google Scholar]
- 50. Noh H. S., Hah Y. S., Nilufar R., Han J., Bong J. H., Kang S. S., Cho G. J., and Choi W. S. (2006) Acetoacetate protects neuronal cells from oxidative glutamate toxicity. J. Neurosci. Res. 83, 702–709 [DOI] [PubMed] [Google Scholar]
- 51. Hoffman W. H., Cheng C., Passmore G. G., Carroll J. E., and Hess D. (2002) Acetoacetate increases expression of intercellular adhesion molecule-1 (ICAM-1) in human brain microvascular endothelial cells. Neurosci. Lett. 334, 71–74 [DOI] [PubMed] [Google Scholar]
- 52. Russell R. R. 3rd, Cline G. W., Guthrie P. H., Goodwin G. W., Shulman G. I., and Taegtmeyer H. (1997) Regulation of exogenous and endogenous glucose metabolism by insulin and acetoacetate in the isolated working rat heart. A three tracer study of glycolysis, glycogen metabolism, and glucose oxidation. J. Clin. Invest. 100, 2892–2899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pan J. W., Bebin E. M., Chu W. J., and Hetherington H. P. (1999) Ketosis and epilepsy: 31P spectroscopic imaging at 4.1 T. Epilepsia 40, 703–707 [DOI] [PubMed] [Google Scholar]
- 54. Nordli D. R. Jr., Kuroda M. M., Carroll J., Koenigsberger D. Y., Hirsch L. J., Bruner H. J., Seidel W. T., and De Vivo D. C. (2001) Experience with the ketogenic diet in infants. Pediatrics 108, 129–133 [DOI] [PubMed] [Google Scholar]
- 55. Pauli G., and Overath P. (1972) ato Operon: a highly inducible system for acetoacetate and butyrate degradation in Escherichia coli. Eur. J. Biochem. / FEBS 29, 553–562 [DOI] [PubMed] [Google Scholar]
- 56. Kyriakidis D. A., and Tiligada E. (2009) Signal transduction and adaptive regulation through bacterial two-component systems: the Escherichia coli AtoSC paradigm. Amino Acids 37, 443–458 [DOI] [PubMed] [Google Scholar]
- 57. Rosenblatt J. D., Lunt A. I., Parry D. J., and Partridge T. A. (1995) Culturing satellite cells from living single muscle fiber explants. In vitro Cell. Dev. Biol. Anim. 31, 773–779 [DOI] [PubMed] [Google Scholar]
- 58. Williamson D. H., Bates M. W., Page M. A., and Krebs H. A. (1971) Activities of enzymes involved in acetoacetate utilization in adult mammalian tissues. Biochem. J. 121, 41–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chen Y., Ye J., Cao L., Zhang Y., Xia W., and Zhu D. (2010) Myostatin regulates glucose metabolism via the AMP-activated protein kinase pathway in skeletal muscle cells. Int. J. Biochem. Cell Biol. 42, 2072–2081 [DOI] [PubMed] [Google Scholar]
- 60. Seale P., Sabourin L. A., Girgis-Gabardo A., Mansouri A., Gruss P., and Rudnicki M. A. (2000) Pax7 is required for the specification of myogenic satellite cells. Cell 102, 777–786 [DOI] [PubMed] [Google Scholar]
- 61. Creuzet S., Lescaudron L., Li Z., and Fontaine-Pérus J. (1998) MyoD, myogenin, and desmin-nls-lacZ transgene emphasize the distinct patterns of satellite cell activation in growth and regeneration. Exp. Cell Res. 243, 241–253 [DOI] [PubMed] [Google Scholar]
- 62. Zammit P. S. (2008) All muscle satellite cells are equal, but are some more equal than others? J. Cell Sci. 121, 2975–2982 [DOI] [PubMed] [Google Scholar]
- 63. Bogdanovich S., Krag T. O., Barton E. R., Morris L. D., Whittemore L. A., Ahima R. S., and Khurana T. S. (2002) Functional improvement of dystrophic muscle by myostatin blockade. Nature 420, 418–421 [DOI] [PubMed] [Google Scholar]
- 64. Minetti G. C., Colussi C., Adami R., Serra C., Mozzetta C., Parente V., Fortuni S., Straino S., Sampaolesi M., Di Padova M., Illi B., Gallinari P., Steinkühler C., Capogrossi M. C., Sartorelli V., et al. (2006) Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat. Med. 12, 1147–1150 [DOI] [PubMed] [Google Scholar]
- 65. Orii K. E., Fukao T., Song X. Q., Mitchell G. A., and Kondo N. (2008) Liver-specific silencing of the human gene encoding succinyl-CoA:3-ketoacid CoA transferase. Tohoku J. Exp. Med. 215, 227–236 [DOI] [PubMed] [Google Scholar]
- 66. Fenselau A., and Wallis K. (1974) Substrate specificity and mechanism of action of acetoacetate coenzyme A transferase from rat heart. Biochemistry 13, 3884–3888 [DOI] [PubMed] [Google Scholar]
- 67. Fenselau A., and Wallis K. (1974) Ketone body usage by mammals. Acetoacetate substrate inhibition of CoA transferase from various rat tissues. Life Sci. 15, 811–818 [DOI] [PubMed] [Google Scholar]
- 68. Garedew A., Andreassi C., and Moncada S. (2012) Mitochondrial dynamics, biogenesis, and function are coordinated with the cell cycle by APC/C CDH1. Cell Metab. 15, 466–479 [DOI] [PubMed] [Google Scholar]
- 69. Yang W., Chen Y., Zhang Y., Wang X., Yang N., and Zhu D. (2006) Extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase pathway is involved in myostatin-regulated differentiation repression. Cancer Res. 66, 1320–1326 [DOI] [PubMed] [Google Scholar]
- 70. Wellen K. E., and Thompson C. B. (2012) A two-way street: reciprocal regulation of metabolism and signalling. Nat. Rev. Mol. Cell Biol. 13, 270–276 [DOI] [PubMed] [Google Scholar]
- 71. Bain J., Plater L., Elliott M., Shpiro N., Hastie C. J., McLauchlan H., Klevernic I., Arthur J. S., Alessi D. R., and Cohen P. (2007) The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408, 297–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Freland L., and Beaulieu J. M. (2012) Inhibition of GSK3 by lithium, from single molecules to signaling networks. Front. Mol. Neurosci. 5, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Laffel L. (1999) Ketone bodies: a review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab. Res. Rev. 15, 412–426 [DOI] [PubMed] [Google Scholar]
- 74. Adam J., Hatipoglu E., O'Flaherty L., Ternette N., Sahgal N., Lockstone H., Baban D., Nye E., Stamp G. W., Wolhuter K., Stevens M., Fischer R., Carmeliet P., Maxwell P. H., Pugh C. W., et al. (2011) Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 20, 524–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ooi A., Wong J. C., Petillo D., Roossien D., Perrier-Trudova V., Whitten D., Min B. W., Tan M. H., Zhang Z., Yang X. J., Zhou M., Gardie B., Molinié V., Richard S., Tan P. H., et al. (2011) An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell 20, 511–523 [DOI] [PubMed] [Google Scholar]
- 76. Jorquera R., and Tanguay R. M. (2001) Fumarylacetoacetate, the metabolite accumulating in hereditary tyrosinemia, activates the ERK pathway and induces mitotic abnormalities and genomic instability. Hum. Mol. Genet. 10, 1741–1752 [DOI] [PubMed] [Google Scholar]
- 77. Rous S., and Favarger P. (1973) The role of acetoacetate in the transfer of acetyl units outside the mitochondria in liver and adipose tissue of rats or mice. FEBS Lett. 37, 231–234 [DOI] [PubMed] [Google Scholar]
- 78. Rous S., Bas S., and Sengupta S. (1980) Contribution of leucine in the fatty acid synthesis and ketogenesis in mice adipose tissue. Int. J. Biochem. 11, 337–340 [DOI] [PubMed] [Google Scholar]
- 79. Palmer T. N., Gossain S., and Sugden M. C. (1993) Partial oxidation of leucine in skeletal muscle. Biochem. Mol. Biol. Int. 29, 255–262 [PubMed] [Google Scholar]
- 80. Kornasio R., Riederer I., Butler-Browne G., Mouly V., Uni Z., and Halevy O. (2009) β-Hydroxy-β-methylbutyrate (HMB) stimulates myogenic cell proliferation, differentiation and survival via the MAPK/ERK and PI3K/Akt pathways. Biochim. Biophys. Acta 1793, 755–763 [DOI] [PubMed] [Google Scholar]
- 81. Averous J., Gabillard J. C., Seiliez I., and Dardevet D. (2012) Leucine limitation regulates myf5 and myoD expression and inhibits myoblast differentiation. Exp. Cell Res. 318, 217–227 [DOI] [PubMed] [Google Scholar]
- 82. Areta J. L., Hawley J. A., Ye J. M., Chan M. H., and Coffey V. G. (2014) Increasing leucine concentration stimulates mechanistic target of rapamycin signaling and cell growth in C2C12 skeletal muscle cells. Nutr. Res. 34, 1000–1007 [DOI] [PubMed] [Google Scholar]
- 83. Li X., Gianoulis T. A., Yip K. Y., Gerstein M., and Snyder M. (2010) Extensive in vivo metabolite-protein interactions revealed by large-scale systematic analyses. Cell 143, 639–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Li X., and Snyder M. (2011) Metabolites as global regulators: a new view of protein regulation: systematic investigation of metabolite-protein interactions may help bridge the gap between genome-wide association studies and small molecule screening studies. BioEssays 33, 485–489 [DOI] [PubMed] [Google Scholar]
- 85. Balasse E. O., and Havel R. J. (1971) Evidence for an effect of inulin on the peripheral utilization of ketone bodies in dogs. J. Clin. Invest. 50, 801–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Cremer J. E. (1971) Incorporation of label from d-β-hydroxy(14C)butyrate and (3–14C)acetoacetate into amino acids in rat brain in vivo. Biochem. J. 122, 135–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Nakagawa T., Ge Q., Pawlosky R., Wynn R. M., Veech R. L., and Uyeda K. (2013) Metabolite regulation of nucleo-cytosolic trafficking of carbohydrate response element-binding protein (ChREBP): role of ketone bodies. J. Biol. Chem. 288, 28358–28367 [DOI] [PMC free article] [PubMed] [Google Scholar]