

Abstract
Rev-erbα and Rev-erbβ are heme-binding nuclear receptors (NR) that repress the transcription of genes involved in regulating metabolism, inflammation, and the circadian clock. Previous gene expression and co-immunoprecipitation studies led to a model in which heme binding to Rev-erbα recruits nuclear receptor corepressor 1 (NCoR1) into an active repressor complex. However, in contradiction, biochemical and crystallographic studies have shown that heme decreases the affinity of the ligand-binding domain of Rev-erb NRs for NCoR1 peptides. One explanation for this discrepancy is that the ligand-binding domain and NCoR1 peptides used for in vitro studies cannot replicate the key features of the full-length proteins used in cellular studies. However, the combined in vitro and cellular results described here demonstrate that heme does not directly promote interactions between full-length Rev-erbβ (FLRev-erbβ) and an NCoR1 construct encompassing all three NR interaction domains. NCoR1 tightly binds both apo- and heme-replete FLRev-erbβ·DNA complexes; furthermore, heme, at high concentrations, destabilizes the FLRev-erbβ·NCoR1 complex. The interaction between FLRev-erbβ and NCoR1 as well as Rev-erbβ repression at the Bmal1 promoter appear to be modulated by another cellular factor(s), at least one of which is related to the ubiquitin-proteasome pathway. Our studies suggest that heme is involved in regulating the degradation of Rev-erbβ in a manner consistent with its role in circadian rhythm maintenance. Finally, the very slow rate constant (10−6 s−1) of heme dissociation from Rev-erbβ rules out a prior proposal that Rev-erbβ acts as an intracellular heme sensor.
Keywords: DNA-protein interaction, heme, ligand-binding protein, metal ion-protein interaction, nuclear receptor, protein-protein interaction, proteolysis, transcription corepressor, transcription repressor
Introduction
Nuclear receptors (NRs)2 are eukaryotic transcription factors that activate or repress gene expression in response to binding small signaling molecules such as steroid hormones, lipophilic vitamins, and fatty acids (1). Genes regulated by NRs are involved in a myriad of cellular processes ranging from metabolic homeostasis to growth and development. The subjects of this article, Rev-erbα and Rev-erbβ, are NRs that have overlapping functions and tissue expression patterns (2–9) and are under circadian control (10–12). Rev-erb NRs repress the transcription of key genes, e.g. Bmal1, CLOCK, and Rev-erbα itself, involved in regulating the molecular clock (13–15). This is accomplished in part through the formation of a heterodimeric Bmal1-CLOCK complex that activates the transcription of clock-controlled genes including Rev-erbα/β, completing a critical feedback loop required for circadian rhythm maintenance (6, 7). Rev-erbα and Rev-erbβ also regulate the expression of genes involved in gluconeogenesis, lipid homeostasis, and immune responses, ultimately synchronizing these processes to the diurnal cycle (16–21).
The multiple functions of NRs (DNA, ligand, and coregulator binding) are accomplished through their modular structure (22, 23). The N-terminal A/B domain is hypervariable, is involved in ligand-independent transcriptional regulation, and is subject to phosphorylation. This is followed by a conserved DNA-binding domain (DBD, the C domain) containing two zinc-finger motifs that poise the receptor to interact with specific response elements in the promoter of target genes. A flexible hinge region (the D domain) links the DBD to the C-terminal globular ligand-binding domain (LBD or the E domain). The LBD of NRs is generally composed of 12 α-helices arranged in a three-layered bundle that forms a hydrophobic ligand pocket. In the absence of ligand, some NRs, such as the thyroid hormone receptor (THR) and retinoic acid receptor, repress gene expression by interacting with transcription corepressors including nuclear receptor corepressor 1 (NCoR1) and silencing mediator of retinoid and thyroid receptors (SMRT) (24); binding of NR antagonists can also lead to corepressor recruitment (25). Both NCoR1 and SMRT are large (>250 kDa) proteins that bind histone deacetylase complexes and contain multiple NR interaction domains at their C termini (26). Interaction domain motifs are composed of short amphipathic helices with an I/LXXI/VIXXXL/F/Y consensus that associate with NRs by binding to a hydrophobic cleft on the LBD (27). Binding of the specific ligand, i.e. thyroid hormone for the THR, to the LBD causes α-helix 12 of the NR to adopt a compact conformation against the LBD that partially occludes the corepressor cleft and exposes a region that recognizes transcriptional coactivator proteins like the p160 steroid receptor coactivator. These coactivators contain LXXLL NR box motifs that also form helices and bind to the shorter cleft once occupied by corepressor interaction domains (28). Rev-erbα and Rev-erbβ isoforms are unique to the NR superfamily in that they lack α-helix 12, rendering them unable to associate with a coactivator. Thus, these NRs are thought to function exclusively as repressors through binding of NCoR1, although Rev-erbβ has been shown to activate Srebp-1c in skeletal muscle (29, 30).
Identifying the ligand and how binding of the ligand affects various properties of the NR, such as its subcellular localization, stability, oligomeric state, repressor/activator activity, and interactions with DNA and/or a coregulator, are essential for understanding the role of the NR. Many NRs are classed as orphan receptors, meaning that their ligands are unknown. Rev-erbα and Rev-erbβ were first identified as orphan members of the NR family (31, 32). When the Drosophila homolog of Rev-erb, E75, was found to bind heme (33), various studies were performed that ultimately demonstrated that Rev-erbα and Rev-erbβ also bind heme and that this interaction appears to be responsible for recruitment of NCoR1, leading to repression of Rev-erb target genes (16, 34), which include important regulators of glucose and lipid metabolism, immune responses, and the molecular clock (16–21).
Intriguingly, the LBDs of E75, Rev-erbα and Rev-erbβ, contain a heme regulatory motif (HRM), which consists of a Cys-Pro dipeptide generally followed by a hydrophobic residue (35). For Rev-erbβ, the relevant sequence is C374NTGGRMHLVC384PMSK, where Cys-384 (Cys-418 for Rev-erbα) and His-568 serve as heme axial ligands (36). In most HRMs, the Cys residue is followed by a Pro, where the conformational inflexibility of the Pro directs residues C-terminal to the Cys-Pro dipeptide away from the heme face, poising the Cys thiolate for an interaction with heme (37). Cys-384 is involved in a redox-regulated ligand switch in which it forms a disulfide bond with Cys-374, which lowers heme affinity (38). HRMs have also been proposed to control the activity or stability of several regulatory proteins including an iron-responsive regulator in bacteria (39), a kinase (eIF2α kinase or heme-regulated inhibitor (HRI)) (40), a mammalian transcription repressor (Bach1) (41), 5-aminolevulinate synthase (42), and the yeast transcription factor Hap1, which mediates the effects of oxygen on transcription (43). In 5-aminolevulinate synthase, three HRMs facilitate the heme-dependent inhibition of mitochondrial import of the synthase, while in the mammalian iron-responsive protein IRP2, heme binding to the HRM appears to regulate protein degradation (44). In the case of heme oxygenase-2, it was concluded that one heme can bind to each of the two HRMs at the C terminus as well as to the active site, where it undergoes conversion to biliverdin, CO, and iron (45, 46).
The Drosophila Rev-erb homolog, E75, acts as a repressor by heterodimerizing with DHR3, inhibiting its ability to activate the transcription of genes involved in fly development (47). The Fe2+-heme form of E75 preferentially interacts with DHR3, whereas NO binding to form the Fe2+-heme·NO complex destabilizes the heterodimer, thus implicating heme binding, its redox state, and interaction with diatomic gases as regulators of E75 activity (33, 47). Early studies suggested that the heme redox state and NO do not play a role in Rev-erb function, indicating that the regulatory role of heme may differ among the various Rev-erb homologs (34). On the other hand, the results of more recent experiments indicate that NO destabilizes the Rev-erbα/β·NCoR1 interaction akin to the regulatory mechanism of E75 (36), and thus, additional data are required to assess the role of NO in regulating Rev-erb activity.
The results of co-immunoprecipitation (co-IP), chromatin immunoprecipitation (ChIP), and gene expression experiments indicate that heme promotes the interaction of Rev-erbα with NCoR1; thus, Rev-erb NRs were proposed to act as heme sensors (16, 34). These results implied that heme acts as a classical nuclear receptor ligand in which heme binding leads to a conformational change that increases the affinity of the Rev-erb NR for NCoR1, ultimately amplifying gene repression. However, in contrast to the heme-dependent NCoR1 recruitment model, several biochemical studies indicate that heme binding actually destabilizes the Rev-erb·NCoR1 complex. For example, titration of purified Rev-erbα/β LBDs with Fe3+- and Fe2+-heme decreases the affinity of Rev-erb for small peptides encompassing the NCoR1 interaction domains (16, 34, 36). In addition, when the crystal structures of apoRev-erbβ LBD (29), Fe3+-heme-bound Rev-erbβ LBD (36), and Rev-erbα LBD in complex with the NCoR1 interaction domain 1 (ID1) peptide (27) are compared, it is apparent that heme binding induces structural rearrangements that would cause significant steric clash between α-helix 3 of the LBD and a unique anti-parallel β-sheet of ID1. Furthermore, Trp402 and the heme axial ligand His-568 (Rev-erbβ amino acid numbering), which provide hydrophobic interactions in the heme-binding pocket, is unfavorably positioned to accommodate heme in the ID1·Rev-erbα structure. In sum, although studies with full-length proteins in cells provided a model in which heme has a robust stimulatory effect on the interaction between Rev-erbα and NCoR1 (16), biochemical and structural studies (16, 27, 29, 34, 36) indicate that heme binding may destabilize the Rev-erbα/β·NCoR1 complex. However, it should be re-emphasized that these in vitro studies were performed with the isolated LBD and NCoR1 peptides. Because of their generally intractable nature (poor solubility, instability, etc.), there are few studies with full-length NRs and their coregulators. However, concerns have been expressed about the use of domains in studying the biochemistry of NRs (see for example, references 23 and 48). One hypothesis to explain why the in vitro studies cannot reproduce the role of heme in promoting complex formation between Rev-erbα/β and NCoR1 full-length proteins in vivo is that the truncated constructs omit features that are important for heme-dependent regulation.
To test the first hypothesis, we generated an NCoR1 construct encompassing all three NR interaction domains and full-length Rev-erbβ (FLRev-erbβ). Biochemical studies indicate that heme dissociates from Rev-erbβ too slowly to equilibrate with the heme pool and thus would be unable to act, as suggested previously (16, 49), as an intracellular heme sensor. We also demonstrate that heme has only a minimal effect on the binding of NCoR1 to FLRev-erbβ, refuting our original hypothesis that full-length proteins could reproduce the effects of heme observed in the cell. Instead, our co-IP results demonstrate that endogenous NCoR1 in cell extracts interacts with wild-type Rev-erbβ but not with a variant defective in heme binding, indicating that another factor present in extracts mediates the heme-dependent interaction of Rev-erbβ with NCoR1. Potential candidates are proteasome-related proteins or a post-translational modification such as ubiquitination, because we found through mass spectroscopic analyses of the co-IP experiment that an ubiquitin-ligase associates in a heme-dependent manner with Rev-erbβ. In addition, apoRev-erbβ is more stable than the heme-replete protein in cells. Our results also suggest that there are differences in the mechanisms by which heme affects the repressor functions of Rev-erbα and Rev-erbβ. Although heme binding is dispensable for Rev-erbβ repression of the Bmal1 promoter, it modestly enhances Rev-erbα repressor activity.
Experimental Procedures
Plasmids
For the heterologous expression and purification of human FLRev-erbβ in Escherichia coli, two constructs were generated: one contained the sequence from the mammalian gene collection (MGC), pGB1-FLRev-erbβ-MGC, and the other was from the human genome sequence (pGB1-FLRev-erbβ-HGS). The MGC clone differs from the human genome sequence (GenBankTM accession number NM_005126.4) by three missense mutations that result in amino acid substitutions P21H, Q282K, and P288R. The sequences of all plasmids used in this study were verified at the University of Michigan Sequencing Core (Ann Arbor, MI).
To prepare the pGB1-FLRev-erbβ-MGC construct, the Rev-erbβ open reading frame (ORF) lacking the ATG start codon was amplified with PCR using PfuUltra II HS polymerase (Agilent Technologies, Santa Clara, CA), mammalian gene collection clone 5274113 (GenBankTM accession number BC045613.1, coding for human NR1D2) as template, and FLRev-erbβ ligation-independent cloning (LIC) forward and reverse primers (refer to Table 1 for complete primer sequences and plasmid synopses). LIC techniques were utilized to clone the isolated PCR fragment into a modified pMCSG7 vector containing an N-terminal hexahistidine-tagged protein G, B1 domain (GB1) with a C-terminal tobacco etch virus (TEV) proteolytic cleavage site (50, 51). The resulting plasmid encodes a translational fusion between GB1 and FLRev-erbβ with an internal TEV protease cleavage site for downstream removal of GB1 and the His tag. TEV cleavage resulted in three residues from the TEV recognition sequence remaining on the N terminus of FLRev-erbβ with the following sequence, NH2-SNAE-, where Glu (E) is the first residue of Rev-erbβ.
TABLE 1.
Primers, DNA duplexes, and plasmids used in this study
Underlined and italic primer sequences represent LIC 5′-overhangs. Bold sequences indicate short Kozak consensus sequences. Bold and italic sequences indicate Rev-erbβ or THR core hexameric recognition sequences in DNA duplexes.
| Primer, DNA duplex, or plasmid designation | Sequence or synopsis |
|---|---|
| Primers | |
| FLRev-erbβ LIC forward | 5′-TACTTCCAATCCAATGCTGAGGTGAATGCAGGAGGTGT-3′ |
| FLRev-erbβ LIC reverse | 5′-TTATCCACTTCCAATGTTAAGGGTGAACTTTAAAGGCCAAG-3′ |
| KpnI-Kozak-Rev-erbβ forward | 5′-GATCGGTACCATGGAGGTGAATGCAGGAGGTGTGATTGCCTATATCAGTT-3′ |
| XhoI-Rev-erbβ reverse | 5′-GATCCTCGAGTTAAGGGTGAACTTTAAAGGCCAAGAGCTCCTCAGAG-3′ |
| KpnI-Kozak-Rev-erbα forward | 5′-GATCGGTACCATGACGACCCTGGACTCCAACAACAACACAGGTGGCG-3′ |
| NotI-Rev-erbα reverse | 5′-GATCGCGGCCGCTCACTGGGCGTCCACCCGGAAGGACAGCAGCTTCTCG-3′ |
| Rev-erbβ H21P forward | 5′-CCAGCTCAGCCTCAAGCCCGGCCTCTTGTCACAGTGAG-3′ |
| Rev-erbβ H21P reverse | 5′-CTCACTGTGACAAGAGGCCGGGCTTGAGGCTGAGCTGG-3′ |
| Rev-erbβ K282Q forward | 5′-GAGAGCATGCAGCCGCAGAGAGGAGAACGG-3′ |
| Rev-erbβ K282Q reverse | 5′-CCGTTCTCCTCTCTGCGGCTGCATGCTCTC-3′ |
| Rev-erbβ R288P forward | 5′-GAGGAGAACGGATTCCGAAGAACATGGAGC-3′ |
| Rev-erbβ R288P reverse | 5′-GCTCCATGTTCTTCGGAATCCGTTCTCCTC-3′ |
| Rev-erbβ H568F forward | 5′-CGATCTTTAAACAACATGTTCTCTGAGGAGCTCTTGGCC-3′ |
| Rev-erbβ H568F reverse | 5′-GGCCAAGAGCTCCTCAGAGAACATGTTGTTTAAAGATCG-3′ |
| Rev-erbβ C384A forward | 5′-GGAAGAATGCATCTGGTTGCGCCAATGAGTAAGTCTCC-3′ |
| Rev-erbβ C384A reverse | 5′-GGAGACTTACTCATTGGCGCAACCAGATGCATTCTTCC-3′ |
| Rev-erbα H602F forward | 5′-CGGACCCTGAACAACATGTTCTCCGAGAAGCTGCTGTC-3′ |
| Rev-erbα H602F reverse | 5′-GACAGCAGCTTCTCGGAGAACATGTTGTTCAGGGTCCG-3′ |
| NCoR540 LIC forward | 5′-TACTTCCAATCCAATGCATCTGAGGCTGGGAAAGATAAAGGGCCTCCT-3′ |
| NCoR540 LIC reverse | 5′-TTATCCACTTCCAATGCTAGTCATCACTATCCGACAGGGTCTCGTACTG-3′ |
| THRβ1 LIC forward | 5′-TACTTCCAATCCAATGCAATGACTCCCAACAGTATGACAGAAAATGGC-3′ |
| THRβ1 LIC reverse | 5′-TTATCCACTTCCAATGCTAATCCTCGAACACTTCCAAGAACAAAGGGGG-3′ |
| Fluorophore-labeled DNA duplexes | |
| TR/FAM-Rev-DR2 | TR/FAM-5′-CAACTAGGTCACTAGGTCAG-3′ |
| 3′-GTTGATCCAGTGATCCAGTC-5′ | |
| FAM-BMal1-promoter | FAM-5′-CGGAAAGTAGGTTAGTGGTGCGACATTTAGGGAAGGCAGAAAGTAGGTCAGGG-3′ |
| 3′-GCCTTTCATCCAATCACCACGCTGTAAATCCCTTCCGTCTTTCATCCAGTCCC-5′ | |
| FAM-hPL-promoter | FAM-5′-CAGGTGGGGTCAAGCAGGGAGAGAGAA-3′ |
| 3′-GTCCACCCCAGTTCGTCCCTCTCTCTT-5′ | |
| Plasmids | |
| pGB1-FLRev-erbβ-MGC | Rev-erbβ (NR1D2) ORF from the Mammalian Gene Collection cloned into a modified pMCSG7 vector resulting in a translational fusion between the His6-GB1 solubility tag and FLRev-erbβ with an internal TEV protease cleavage site for protein production in E. coli (this study). |
| pGB1-FLRev-erbβ-HGS | pGB1-FLRev-erbβ-MGC carrying H21P, K282Q, and R288P mutations reflecting the Human Genome Sequence; parent vector for H568F and H568F/C384A expression clones (this study). |
| pcDNA3.1(+)-FLRev-erbβ | The Rev-erbβ ORF from pGB1-FLRev-erbβ-HGS was subcloned into pcDNA3.1(+) using a forward primer containing a consensus Kozak sequence for expression in mammalian cells; parent vector for H568F, C384A, and H568F/C384A expression clones (this study). |
| pcDNA3.1(+)-Rev-erbα | The Rev-erbα ORF from mammalian gene collection vector 6190140 was subcloned into pcDNA3.1(+) for overexpression of Rev-erbα in mammalian cells; parent vector for the H602F expression clone (this study). |
| pMBP-NCoR1–540 | DNA coding for the C-terminal 540 amino acids of NCoR1 was cloned into pMCSG9 resulting in a translational fusion between MBP and NCoR1–540 with an internal TEV protease cleavage site (this study). |
| pMBP-THRβ1 | The THRβ1 ORF was cloned into pMCSG9 resulting in a translational fusion between MBP and THRβ1 with an internal TEV protease cleavage site (this study). |
| pCMV-βGal | Mammalian expression vector producing β-galactosidase for measuring transfection efficiency and normalizing luciferase activity (provided by Dr. Daniel Bochar, University of Michigan). |
| pGL3Basic-BMal1 | Luciferase reporter vector driven by the core Bmal1 promoter (encompassing −422 to +108 bp with respect to the transcription start site) from M. musculus (71). |
Site-directed mutagenesis of pGB1-FLRev-erbβ-MGC was carried out to generate an additional clone identical to the human genome sequence, designated pGB1-FLRev-erbβ-HGS, using the QuikChange protocol (Agilent Technologies). PCR was conducted using overlapping primers containing H21P, K282Q, and R288P mutations. Mutant PCR products were digested with DpnI (New England Biolabs, Ipswich, MA) overnight at 37 °C prior to transformation into E. coli Top10 cells (Life Technologies). pGB1-FLRev-erbβ-HGS served as the template for generation of heme axial ligand mutants H568F and H568F/C384A.
To generate a clone of FLRev-erbβ for expression in mammalian cells, the ORF from pGB1-FLRev-erbβ-HGS was amplified with PCR using a forward primer containing a 5′-KpnI restriction site and a consensus Kozak sequence and a reverse primer with a 5′-XhoI restriction site. The PCR fragment was digested with the indicated endonucleases and then ligated into similarly digested pcDNA3.1(+) resulting in pcDNA3.1(+)-FLRev-erbβ. The H568F, C384A, and H568F/C384A mutations were introduced into pcDNA3.1(+)-FLRev-erbβ as described above.
To express Rev-erbα and the corresponding H602F variant, the Rev-erbα ORF from mammalian gene collection clone 6190140 (GenBankTM accession number BC047875.1, coding for human NR1D1) was PCR-amplified using a forward primer consisting of a consensus Kozak sequence and a 5′-KpnI restriction site and a reverse primer with a 5′-NotI restriction site. The PCR fragment was digested with the indicated endonucleases and ligated into similarly digested pcDNA3.1(+) yielding pcDNA3.1(+)-Rev-erbα. The H602F mutation was introduced into this vector as described above.
Expression and purification of soluble forms of NCoR1 containing all three interaction domains and of full-length THRβ1 were accomplished by generating maltose-binding protein (MBP) fusion constructs pMBP-NCoR1–540 and pMBP-THRβ1, respectively. DNA encoding the C-terminal 540 amino acids of NCoR1 was amplified by PCR using ORFeome clone HOC23341 (Genecopoeia, Rockville, MD; GenBankTM accession number BC172437) as template, and NCoR540 LIC forward and reverse primers. The isolated PCR fragment was cloned using LIC methodology into pMCSG9 (52), resulting in a translational fusion between MBP and the C-terminal 540 residues of NCoR1. The same approach was taken when generating an expression clone for THRβ1. The THRβ1 ORF was amplified by PCR using ORFeome clone GC-OG01669 (Genecopoeia; GenBankTM accession number BC106929) as template, THRβ1 LIC as forward primer, and THRβ1 LIC as reverse primer. The resultant product was cloned into pMCSG9 generating pMBP-THRβ1, a translational fusion between MBP and THRβ1 with an internal TEV protease cleavage site for downstream removal of MBP.
Protein Purification
Unless otherwise specified, all materials were reagent grade and were purchased from either Sigma-Aldrich or Thermo Fisher Scientific. FLRev-erbβ MGC, HGS, and heme axial ligand mutants were purified from E. coli BL21(DE3) (Life Technologies) transformed with the appropriate plasmid as described above. The cells were grown with shaking in 2× YT medium (16 g of tryptone, 10 g of yeast extract, and 5 g of NaCl/liter (Fisher Bioreagents)) supplemented with 100 μg ml−1 ampicillin at 37 °C. Once the culture reached an optical density at 600 nm (OD600) of 1.0, the temperature was reduced to 20 °C, and 30–60 min later protein expression was induced with 0.5 mm isopropyl β-d-1-thiogalactopyranoside (IPTG, Gold Biotechnology, St. Louis, MO) and 100 μm zinc acetate. The cells were grown under the same conditions for 16 h prior to harvesting by centrifugation. Cell pellets were suspended in TNG buffer (50 mm Tris, pH 8.0, 300 mm NaCl, and 10% glycerol) with 1 mm 2-mercaptoethanol, 1 mm EDTA, 0.5 mm PMSF, 0.1% Tween 20, 6 mm benzamidine, 2× Complete EDTA-free protease inhibitors (Hoffmann-La Roche), 5 μg ml−1 DNaseI, and 1 mg ml−1 lysozyme. The cell slurry was stirred for 20 min at 4 °C prior to sonication (unless otherwise indicated, all purification steps were at 4 °C). Cell lysates were cleared by centrifugation at 38,000 × g for 1 h, and the soluble extract applied to a 2.5 × 5-cm nickel-nitrilotriacetic acid-agarose (Qiagen, Valencia, CA) column equilibrated in TNG buffer with 1 mm 2-mercaptoethanol. The column was washed with the same buffer for 1 column volume prior to applying a linear gradient from 10 to 250 mm imidazole. Fractions containing partially pure protein verified by SDS-PAGE were pooled and the protein content determined. TEV protease (prepared as described previously (53)) was added to the pool at a mass:mass ratio of 1.5:20 TEV:GB1-FLRev-erbβ and dialyzed against 50 mm Tris, pH 8.0, 150 mm NaCl, 10% glycerol, 5 mm sodium citrate, 5 mm DTT, and 10 μm ZnCl2 at 4 °C for 16 h. The TEV digest reaction was then dialyzed against 50 mm Tris, pH 8.0, 10 mm NaCl, 10% glycerol, and 5 mm DTT at room temperature for 4 h prior to application of the pool onto a 2.5 × 5-cm Q-Sepharose Fast Flow (Sigma) column equilibrated in the dialysate buffer (the remaining purification steps were performed at 4 °C). The column was washed thoroughly (4–5 column volumes) prior to the application of a linear gradient from 10 to 500 mm NaCl. Fractions containing partially pure FLRev-erbβ free of undigested GB1-FLRev-erbβ were identified with SDS-PAGE and pooled, and the NaCl concentration was adjusted to 0.5 m by the slow addition of solid NaCl crystals. The pool was concentrated with 10-kDa molecular weight cutoff (MWCO) Amicon centrifugal filter devices (Millipore, Billerica, MA) and then loaded onto a 1.5 × 90-cm Sephacryl S100 high resolution (Sigma) gel filtration column equilibrated in TNGD buffer (50 mm Tris, pH 8.0, 300 mm NaCl, 10% glycerol, and 5 mm DTT). Fractions containing pure FLRev-erbβ were identified with SDS-PAGE, pooled, concentrated to ∼10 mg ml−1 with 10-kDa MWCO Amicon devices, and snap-frozen in 20–100-μl aliquots in liquid nitrogen prior to storage at −80 °C. Densitometric analysis of SDS-polyacrylamide gels with ImageJ (54) indicated wild-type and variant FLRev-erbβ preparations were >90% pure.
MBP-NCoR1–540 was purified from E. coli BL21(DE3) transformed with pMBP-NCoR1–540. Cells were grown with shaking in Terrific Broth (Fisher Bioreagents) supplemented with 0.4% glycerol and ampicillin (100 μg ml−1) at 37 °C until the culture reached an A600 of 3.0. The temperature was lowered to 18 °C for 1.5 h prior to induction by 0.5 mm isopropyl β-d-1-thiogalactopyranoside, and the cells continued to grow under the same conditions for 3 h before harvesting by centrifugation. Cell pellets were suspended in TNGD buffer and further supplemented with 10 mm EDTA, 1× protease inhibitors, 0.5 mm PMSF, 0.2% Tween 20, 6 mm benzamidine, 20 μg ml−1 DNaseI, and 0.5 mg ml−1 lysozyme. The cell slurry was sonicated and a soluble extract prepared by centrifugation at 100,000 × g for 45 min at 4 °C. The extract was loaded onto a 2.5 × 6-cm amylose column (New England Biolabs) equilibrated in TNGD, and the column was washed with 5 volumes of the same buffer before applying 5 volumes of TNGD containing 20 mm MgCl2 and 10 mm ATP. The ATP wash was necessary to eliminate a contaminant with an approximate molecular mass of 70 kDa (likely DnaK, a common contaminant in preparations of heterologously expressed protein) (55). The column was re-equilibrated in TNGD prior to elution of bound protein in the same buffer containing 20 mm maltose. The presence of partially purified MBP-NCoR1540 was confirmed with SDS-PAGE, and the elution pool was concentrated with 10-kDa MWCO Amicon devices prior to loading onto a 1.5 × 90-cm Sephacryl S300 high-resolution (Sigma) column equilibrated in TNGD. Fractions containing partially pure protein were identified with SDS-PAGE, concentrated as described to ∼20 mg ml−1, and then aliquoted and snap-frozen in liquid nitrogen prior to storage at −80 °C. Densitometric analysis of SDS-polyacrylamide gels indicated that MBP-NCoR1–540 preparations were ∼80% pure. For control experiments utilizing MBP alone, E. coli BL21(DE3) cells transformed with pMCSG9 (encoding an N-terminally hexahistidine-tagged MBP) were grown with shaking in Lennox Broth (Fisher Bioreagents) supplemented with 200 μg ml−1 ampicillin at 37 °C until the culture reached an A600 of 0.7. Expression was induced with 0.5 mm isopropyl β-d-1-thiogalactopyranoside, and the cells were incubated under the same conditions for an additional 16 h prior to harvesting by centrifugation. The pellets were suspended in 25 mm Tris, pH 8.0, 150 mm NaCl, 5% glycerol, and 5 mm DTT (0.5× TNGD buffer) supplemented with 1 mm EDTA and 1× protease inhibitors, sonicated, and a soluble extract prepared as described for MBP-NCoR1–540. The extracts were loaded onto a 2.5 × 6-cm amylose column that had been equilibrated in 0.5× TNGD, and the column was washed with 5 volumes of the same buffer before eluting bound protein with 20 mm maltose in 0.5× TNGD. The presence of highly pure MBP was confirmed by SDS-PAGE, and aliquots were stored at −80 °C.
THRβ1 was purified from E. coli BL21(DE3) transformed with pMBP-THRβ1. The cells were grown, harvested, and lysed as described for GB1-FLRev-erbβ expression, except that the lysis buffer for the THRβ1 cells contained 5 mm DTT instead of 2-mercaptoethanol and 1× protease inhibitors, but it included all other supplements at the aforementioned concentrations. The soluble extract was loaded onto a 2.5 × 10-cm amylose column that had been equilibrated in TNGD and then thoroughly washed prior to eluting the protein with 20 mm maltose. TEV protease was added to the MBP-THRβ1 pool at a ratio of 0.5:20 (w/w), and the digest was dialyzed against 50 mm Tris, pH 8.0, 15 mm NaCl, 10% glycerol, and 5 mm DTT at 4 °C for 16 h. The pool was applied to a 2.5 × 5-cm Q-Sepharose Fast Flow column equilibrated in the dialysate and the column washed with 2 volumes of the same buffer prior to applying a linear gradient from 15 mm to 500 mm NaCl. Fractions containing THRβ1 were confirmed by SDS-PAGE, pooled, and dialyzed against 20 mm Tris, pH 8.0, 5% glycerol, 20 mm MgCl2, and 5 mm DTT for 16 h at 4 °C. The pool was applied to a 1.5 × 1.5-cm heparin-agarose column (Sigma) equilibrated in dialysate buffer and the column washed with 10 volumes of the same buffer before applying 25 volumes of buffer containing 5 mm ATP, which effectively eliminated a ∼70-kDa contaminant, likely DnaK. The protein was eluted from the column in the same buffer (lacking ATP) but with 1.5 m NaCl. The presence of pure THRβ1 (>85%) was confirmed with SDS-PAGE prior to buffer exchanging into 0.5× TNGD by repeated concentration and dilution in 10-kDa MWCO Microcon centrifugal filter devices (Millipore) and storage of the protein at −80 °C after snap freezing the aliquots in liquid nitrogen.
Analytical Gel Filtration
50-μl samples of FLRev-erbβ MGC and HGS (0.5–1 mg ml−1) were injected onto a Shodex KW-803 high pressure liquid chromatography (HPLC) column (8 × 300 mm) attached to a Shimadzu HPLC system fitted with a diode array detector. Samples were run at 1 ml min−1 in 50 mm Bis-Tris, pH 7.0, 150 mm NaCl, 5% glycerol, and 0.5 mm TCEP. Prior to injection of FLRev-erbβ, the column was standardized with blue dextran to determine the void volume, Bio-Rad gel filtration standards (thyroglobulin, 670 kDa; γ-globulin, 158 kDa; ovalbumin, 44 kDa; myoglobin, 17 kDa; and vitamin B12, 1.35 kDa), bovine serum albumin (66.4 kDa), apoferritin (443 kDa), and cytochrome c (12.4 kDa). A standard curve was generated by plotting the logarithm of the molecular masses of the standards versus elution volume/void volume (Ve/V0) and fitting the data with a linear line, which yielded an R2 of 0.9905 (data not shown). The molecular masses of FLRev-erbβ MGC and HGS were determined using the equation derived from the standards plot and Ve/V0 of the major eluting species.
Heme and CO Binding Assays
The affinity of Fe3+-heme and Fe2+-heme for FLRev-erbβ MGC, HGS, H568F, and H568F/C384A was assessed with difference UV-visible spectrophotometry. All spectra in this study were acquired using either a Shimadzu model UV-2600 or UV-2501PC double beam spectrophotometer equipped with a TCC temperature controller set to 20 °C. Prior to heme titration, proteins (100–200 μm) were thiol-reduced with 25 mm TCEP at ambient temperature for 1 h in an anaerobic chamber (<2 ppm O2) prior to buffer exchanging to TNG lacking reductant using Micro Bio-Spin 6 gel filtration columns (Bio-Rad). Wild-type FLRev-erbβ (MGC or HGS) was diluted to 0.8 μm in 1 ml of 0.5× TNG (<1% dilution factor), and the sample was sealed with a rubber stopper in a quartz cuvette to maintain oxygen-free conditions during the experiment carried out at the bench. A parallel cuvette containing 1 ml of buffer was used as the reference. Fe3+- and Fe2+-heme titrants were prepared by dissolving solid hemin (5 mg ml−1; Frontier Scientific, Logan, UT) in 0.1 m NaOH with 10% dimethyl sulfoxide (DMSO), filtering the solution, and diluting it to ∼100 μm in 0.5× TNG containing 5% DMSO for Fe3+-heme. Fe2+-heme was prepared by dilution in the same buffer but with 20% DMSO (which is necessary to avoid precipitation of Fe2+-heme) followed by the addition of 10 mm dithionite. Titrant concentrations were determined by dilution in 0.1 m NaOH using ϵ = 58.44 mm−1 cm−1 at 385 nm (56) (prior to the addition of dithionite for Fe2+-heme) and were maintained under anaerobic conditions in stoppered serum vials during the titration. One-μl increments of the stocks were added to both the reference and experimental cuvettes, mixed by inversion, and the spectrum monitored until equilibrium was reached; then the final spectrum was recorded. The absorbance of the Soret band maxima for each titration was plotted as a function of heme concentration, and the data were fit to Equation 1 (all fitting and statistical analysis in this study was performed with SigmaPlot software (Systat Software, Inc., San Jose, CA)), which accounts for the concentration of free ligand in equilibrium titrations and is ideal for tight-binding interactions where Kd ≪ protein concentration; thus the [free ligand] is negligible compared with the [total ligand] (57).
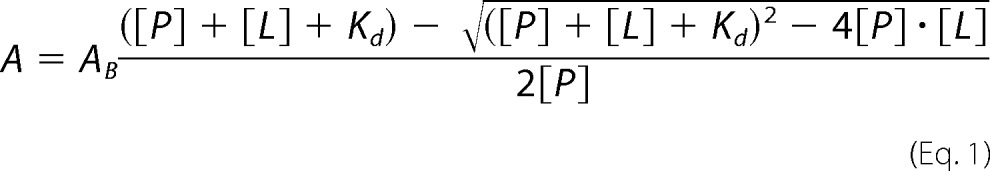 |
In this equation, A is the observed absorbance from the experiment (but can represent any observed signal, i.e. aniosotropy), AB is the difference absorbance of fully bound protein, [P] is the protein concentration, [L] is ligand concentration (heme), and Kd is the dissociation constant.
For titrations of H568F and H568F/C384A, 4 μm protein was used. In addition, a high concentration of DMSO was required to stabilize concentrated (1 mm) Fe2+-heme stocks used in these titrations; thus, titrants were prepared by dissolving hemin at 5 mg ml−1 in 100% DMSO, filtering the solution, and determining the concentration of the stock by serial dilution in DMSO and ϵ = 170 mm−1 cm−1 at 406 nm (58). The stock was diluted to 1 mm in 50% DMSO for Fe3+-heme or further reduced with 10–20 mm dithionite for Fe2+-heme. Two-μl increments of the titrants were added to both the reference and experimental cuvettes, and the spectra were recorded and data analyzed as described for MGC and HGS.
To determine the affinity of CO for FLRev-erbβMGC/HGS, thiol-reduced proteins were diluted under anaerobic conditions to 3 μm in 3 ml total volume 0.5× TNG buffer containing 2 μm Fe3+-heme in a 4-ml capacity quartz cuvette. The cuvette was sealed with a rubber stopper, and dithionite was added to generate the Fe2+-heme complex. One-μl increments of CO-saturated water were titrated into the cuvette using a gas-tight syringe, and the ensuing spectral changes were monitored until equilibrium was achieved. The concentration of CO in the titrant was determined by a second titration against the Fe2+-heme·myoglobin complex using the extinction coefficient for the CO complex of 207 mm−1 cm−1 at 424 nm (59).
Determination of Fe3+-Heme Off-rates with Excess Apomyoglobin
Apomyoglobin was prepared by extracting heme from equine skeletal muscle myoglobin (Sigma) using methyl ethyl ketone as described (60). Briefly, solid myoglobin was dissolved in 10 mm potassium Pi, pH 7.0, and the solution was acidified to ∼pH 3 by the addition of HCl. Heme was removed with three 1:1 methyl ethyl ketone extractions on ice, thereby separating the protein-containing aqueous phase between extractions. Denatured apomyoglobin was refolded by dialysis against 10 mm potassium Pi, pH 7.0, at 4 °C overnight and then concentrated to ∼2 mm (ϵ = 14 mm−1 cm−1 at 280 nm based on the calculated extinction coefficient from the ExPASy server (61) and protein database accession P68082 for equine myoglobin) prior to snap freezing samples in liquid nitrogen and storage at −80 °C. Before an experiment, apomyoglobin was buffer-exchanged to 0.5× TNG by concentration and dilution in 10-kDa MWCO Microcon devices. Heme transfer experiments were set up and performed under anaerobic conditions in cuvettes sealed with rubber septa. 0.7-ml assay volumes were used, which is sufficient to encompass the light beam of the spectrophotometer when a 1.7-ml quartz cuvette is raised by placing a flat Teflon cap in the bottom of the sample cell. Assays were prepared in 0.5× TNG buffer containing 1.5 μm Fe3+-heme and 3 μm thiol-reduced wild-type FLRev-erbβ HGS, H568F, or H568F/C384A (the maximum dilution factor of protein is <5%); an identical experiment with heme alone was performed as a control. The cuvettes were sealed and then brought out of the anaerobic chamber, and the spectrum was recorded at 20 °C. To initiate the heme transfer reaction, apomyoglobin was injected (30 μm final concentration, <2% dilution factor), the sample mixed by inversion (dead time from mixing was ∼20 s), and absorbance changes at 408 nm (the position of the γ-band maximum for holomyoglobin) monitored over time. At the conclusion of the time course, a spectrum was recorded to confirm heme transfer to apomyoglobin. The kinetic data fit best to a double exponential rate equation. All reported rate constants and Δabsorbance % values for these experiments are averages ± S.D. of three independent experiments.
Electron Paramagnetic Resonance (EPR)
As-purified FLRev-erbβMGC was thiol-reduced and buffer exchanged under anaerobic conditions as described above. The sample was concentrated to 97 μm using 10-kDa MWCO Microcon devices, and heme was added from a concentrated stock in TNG buffer (prepared by dissolving heme at 20 mg ml−1 in 0.1 m NaOH, filtering, and then diluting it in TNG buffer, confirming the concentration as described above); this resulted in a final [heme] of 86 μm and [protein] of 93 μm. The sample was transferred to an EPR tube and frozen in liquid nitrogen prior to collecting spectra on an X-band Bruker EMX spectrometer (Bruker Biospin Corp., Billerica, MA) containing an Oxford ITC4 temperature controller, a Hewlett-Packard model 5340 automatic frequency counter, and a Bruker gaussmeter with the following instrument parameters: 10 K, 9.3829 GHz microwave frequency, 0.32 milliwatt power, 6.3 × 105 receiver gain, 8.31 G modulation amplitude, 100 kHz modulation frequency, 163.84 ms time constant, and 8 total scans with 2048 points each.
Fluorescence Anisotropy
To establish a Kd for the interaction of fluorescein-labeled NCoR1 ID1 (F-ID1, Life Technologies; fluorescein-RTHRLITLADHICQIITQDFARN) with FLRev-erbβMGC, a 10–12.5 μm stock of protein in 0.5× TNG buffer with 0.5 mm TCEP was titrated into 1.5 ml of the same buffer containing 10 nm F-ID1 at 25 °C. After the addition of protein, the anisotropy of F-ID1 was measured with a PTI Quantamaster fluorometer (controlled by FeliX software) by vertically polarized excitation at 495 nm (6-nm slit width) and detection of parallel and perpendicular emission intensities at 525 nm (6-nm slit width). Intensity was recorded as the average of a 60-s integration time with 1 reading s−1. Care was taken to assure that no change in intensity occurred during the reading, indicating that equilibrium had been reached. Total fluorescence and anisotropy were calculated using Equation 2,
 |
where A is the anisotropy of F-ID1, Iv and Ih are the vertical and horizontal emission intensities, respectively, and G is an instrument correction factor determined by excitation of the F-ID1 solution with horizontally polarized light and calculating the ratio of the resulting emission intensities Iv/Ih (62). Protein titration led to a concentration-dependent quenching of F-ID1, where the total fluorescence of the system is equal to the denominator of Equation 2. Thus, the raw anisotropy data were corrected for quenching using Equation 3, as described previously (63).
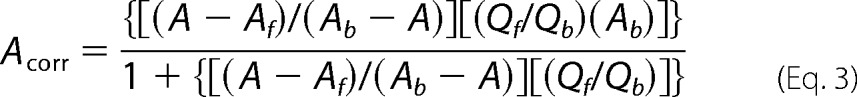 |
Here, Acorr is the corrected anisotropy, A is the measured anisotropy, Af and Ab are the anisotropy of F-ID1 in the free and bound state, respectively, and Qf and Qb are the total fluorescence of free and bound F-ID1, respectively. Ab is estimated from the hyperbolic fit of the raw anisotropy data, whereas Qb is determined from the hyperbolic decay fit of the total fluorescence. The corrected anisotropy data are plotted versus [FLRev-erbβMGC] and fit with Equation 1, holding the [F-ID1] constant at 10 nm. The effect of Fe3+-heme on the F-ID1·FLRev-erbβMGC complex was assessed by titration of heme in 0.5× TNG with 0.5 mm TCEP into 1.5 ml of the same buffer containing 10 nm F-ID1 and 25 nm FLRev-erbβMGC. After the addition of heme the anisotropy of F-ID1 was determined as described above and the data fit with a hyperbolic decay function to estimate IC50.
The DNA binding activities of FLRev-erbβ MGC, HGS, H568F, and H568F/C384A were measured by preparing 0.15-ml assays in 0.5× TNGD containing 1 ng μl−1 poly(deoxyinosinic-deoxycytidylic) acid competitor DNA (poly(dI-dC), Sigma), 10 nm fluorescein amidite (FAM)-labeled Rev-DR2 or FAM-Bmal1 promoter DNA duplexes (see Table 1 for sequence; duplexes were prepared by Integrated DNA Technologies (Coralville, IA) from HPLC-purified single-stranded oligonucleotides and used without further purification), and varying concentrations of the aforementioned proteins. The assay mixtures were incubated at 4 °C for 2 h prior to transferring 0.1 ml of each assay to a black polystyrene nonbinding surface 96-well plate (Corning) and reading the anisotropy of the FAM duplexes with a Tecan Safire microplate reader with the appropriate settings for fluorescein detection. Protein titration had a negligible effect on FAM duplex total fluorescence. Raw anisotropy data were plotted versus [protein] and the data fit with the Hill equation.
To test the effect of Fe2+-heme on the interaction of NCoR1 and FLRev-erbβ, we obtained a Texas Red (TR)-labeled Rev-DR2 duplex, as TR is resistant to dithionite quenching (64) (obtained from Integrated DNA Technologies and prepared as described for FAM duplexes). The Kd for the interaction between MBP-NCoR1–540 and FLRev-erbβHGS·TR-Rev-DR2 was determined by preparing 0.3-ml assay mixtures containing 1 ng μl−1 poly(dI-dC), 20 nm TR-Rev-DR2, 100 nm FLRev-erbβHGS, and increasing concentrations of MBP-NCoR1–540 in 0.5× TNGD. A parallel control experiment was performed with MBP instead of MBP-NCoR1–540. The assays were incubated for 2 h at 4 °C prior to quantifying the anisotropy of TR-Rev-DR2 using the microplate method, 0.2 ml of each sample, and excitation and emission wavelengths of 590 and 630 nm, respectively. The effect of Fe3+-heme and Fe2+-heme on the interaction between MBP-NCoR1–540 and the FLRev-erbβHGS·Rev-DR2 complex was tested using two approaches. Either Fe3+-heme was added at a stoichiometry of 1.5:1 heme:FLRev-erbβ to the titration described above or assays containing a fixed [MBP-NCoR1–540] of 30 nm were mixed with increasing concentrations of Fe3+-heme. In both experiments, Fe3+-heme was converted to Fe2+-heme by the addition of 5 mm dithionite diluted from a freshly prepared stock of 100 mm in 100 mm Tris, pH 8.0, and the anisotropy was recorded within 10 min to avoid heme reoxidation.
Electrophoretic Mobility Shift Assays (EMSA)
20-μl assays were prepared in 0.5× TNGD buffer containing 25 ng μl−1 poly(dI-dC), 1 μm FAM-Rev-DR2 or FAM-Bmal1 promoter, and varying concentrations of wild-type and variant FLRev-erbβ, Fe3+-heme and MBP or MBP-NCoR1–540. Mixtures were incubated on ice for 2 h prior to loading 10 μl of each sample in the wells of a NativePAGE Novex 4–16% gradient polyacrylamide gel (Life Technologies) that had been prerun at 100 V for 1 h at ambient temperature in 50 mm bis-Tris and 50 mm Tricine, pH 6.8, running buffer. Samples were separated under the same conditions prior to imaging the wet gel with an Amersham Biosciences Typhoon Trio+ using the appropriate settings for fluorescein detection.
For experiments with THRβ1, samples were prepared as described above, except they contained 0.25 mg ml−1 BSA, 1 μm FAM-labeled THRβ1 response element from the human placental lactogen gene promoter (65, 66) (FAM-hPL promoter, Table 1; the duplex was prepared as described but required additional purification by gel filtration chromatography to eliminate a high molecular mass contaminant, as described in the analytical gel filtration section above), and varying concentrations of heme or T3 (Sigma) (BSA was omitted from those assays containing heme). T3 was prepared by dissolving the solid to 20 mg ml−1 in 0.1 m NaOH and determining the concentration with ϵ = 4.66 mm−1 cm−1 at 320 nm in 0.1 m NaOH (56). The stock was diluted in 0.5× TNGD containing 1 mg ml−1 BSA prior to dilution in EMSA samples.
Cell Culture and Transfection
HEK293 cells were maintained in Dulbecco's modified Eagle's medium (Life Technologies) with 4.5 g liter−1 glucose, 0.11 g liter−1 pyruvate, 0.584 g liter−1 glutamine, 10% fetal bovine serum, 100 units ml−1 penicillin, and 100 μg ml−1 streptomycin at 37 °C in a humidified incubator maintained at 5.0% CO2 density. Prior to an experiment, confluent cells on 10-cm-diameter plates (60.1 cm2 growth area) were harvested using 1–2 ml of 0.25% trypsin/EDTA dissociation reagent (Life Technologies). Trypsinized cells were brought to a total volume of 10 ml with medium, the cells were pelleted at <1000 × g prior to aspirating the supernatant, and the pellet was suspended in the same volume of Opti-MEM (Life Technologies) with 2.5% FBS and 4.5 g liter−1 glucose (low serum medium). Unless otherwise stated, cells were diluted 5-fold into the same medium (∼5 × 106 cells/10-cm-diameter plate, 10 ml total medium), which resulted in 60–70% confluency 24 h after dilution; this ratio was used to determine the number of cells to seed into plates with different growth areas. Transfections were carried out 24 h after seeding cells using Lipofectamine LTX and Plus reagents (Life Technologies) according to manufacturer's recommendations with a DNA (μg):LTX (μl) ratio of 1:1.9–2.3.
Determination of Cellular Heme Content
The concentration of heme in soluble cell extracts was determined by iron extraction with supersaturated oxalic acid and quantitation of the resulting protoporphyrins with fluorescence spectroscopy as described previously (67) with modifications. Briefly, cell extracts (50–200 μg) prepared with either reporter lysis buffer (Promega, Madison, WI) or Triton X-100 lysis buffer (20 mm Tris, pH 8.0, 150 mm NaCl, 1 mm EDTA, 10% glycerol, 1.5 mm MgCl2, 2 mm TCEP, and 1% Triton X-100) were diluted in supersaturated oxalic acid 1:20 in sealed glass HPLC vials, mixed, and immediately heated at 121 °C for 30 min in an autoclave. A parallel set of samples was left unheated. The samples were cooled and transferred to a black microplate, and the fluorescence emission spectrum of protoporphyrin was recorded from 550–700 nm with excitation at 400 nm in a Tecan Safire microplate reader. Heme concentration (nmol mg protein−1) was determined using a standard curve of hemin prepared in DMSO (the standards were treated the same as those samples containing cell extract) and quantified after subtraction of background emission (662 nm) from protoporphyrin in the unheated samples from the intensity of the heated sample at the same wavelength.
Protein Assays, PAGE, and Western Blotting
Concentrations of purified proteins and mammalian cell extracts were determined with Bradford reagent (68) and a bicinchoninic acid assay (Life Technologies), respectively, using BSA as a standard (except cell extracts prepared in reporter lysis buffer, which were assayed with Bradford reagent). SDS-PAGE was performed with Laemmli buffers (69), 6–12% resolving gels, and 4% stacking gels, and protein bands were visualized with Coomassie Brilliant Blue or transferred to 0.2 μm Immun-Blot PVDF membranes (Bio-Rad) in 25 mm Tris base, 192 mm glycine, and 0.025% SDS at 90 V and 4 °C for 1.5 h. Membranes were blocked in 5% milk prepared in TBST (20 mm Tris, pH 7.4, 200 mm NaCl, 0.1% Tween 20) prior to incubation with primary antibodies diluted (1:250–1:2000) in TBST containing 0.1% sodium azide and 5% BSA or 5% milk overnight at 4 °C. Primary antibodies used in this study included Rev-erbβ QK-6 (mouse), NCoR1 C-20 (goat), and Rev-erbα RS-14 (mouse) from Santa Cruz Biotechnology, actin (Sigma, A2066, rabbit), Hsp90 AC88 (Enzo Life Sciences, mouse), lamin B1 (Abcam, rabbit), MycBP2 (ab86078, Abcam, rabbit), and a polyclonal chicken IgY antibody raised against purified Rev-erbβ LBD residues 247–579 (38) that was excised from SDS-polyacrylamide gels (Aves Labs, Tigard, OR) and affinity-purified with Rev-erbβ LBD coupled to Actigel ALD Superflow resin (Sterogene Bioseparations, Carlsbad, CA). Membranes were washed thoroughly with TBST, incubated with HRP-conjugated secondary antibodies diluted (1:50,000–200,000) in TBST with 5% milk, washed again, and exposed to SuperSignal West Femto enhanced chemiluminescent substrate for HRP (Life Technologies). Immunoreactive bands were detected by exposing the membranes to x-ray film.
Cellular Fractionation
HEK293 cells grown in 10-cm-diameter dishes were transfected with 12.6 μg of pcDNA3.1(+)-FLRev-erbβ wild type, H568F, or H568F/C384A expression vectors. After an additional 24 h incubation, the cells were washed thoroughly with PBS (1.1 mm KH2PO4, 3 mm Na2HPO4-7H2O, and 155 mm NaCl, pH 7.4) prior to fractionation into cytosolic and nuclear compartments as described (70). Briefly, the pelleted cells were suspended in 3-fold the pellet volume of 10 mm HEPES, pH 7.4, 0.33 m sucrose, 1 mm MgCl2, 0.1% Triton X-100, and 1× protease inhibitors and incubated on ice for 15 min; the insoluble debris/nuclei were pelleted by centrifugation at 17,000 × g for 10 min at 4 °C. The soluble fraction comprising the cytosolic components was removed, and the pellet was washed thoroughly with the same buffer prior to suspension in 10 mm HEPES, pH 7.4, 0.45 m NaCl, 1 mm MgCl2, and 1× protease inhibitors. Soluble nuclear components were extracted with frequent agitation and incubation on ice for 30 min prior to centrifuging and pelleting the remaining insoluble material at 17,000 × g for 5 min at 4 °C, with the soluble nuclear proteins remaining in the supernatant.
For experiments determining the effect of intracellular heme on the distribution of endogenous Rev-erbβ, rapidly growing HEK293 cells (∼50% confluent) in low serum medium were incubated with 1 mm succinylacetone (SA) for 42 h prior to harvesting and fractionating as described above, except HEPES was replaced with Tris buffer in the cytoplasmic extraction buffer, and the nuclear pellet was lysed in Triton X-100 lysis buffer. 40–75 μg of each fraction was electroblotted after SDS-PAGE separation as described, and the membranes were probed with Rev-erbβ LBD, Rev-erbβ QK-6, Hsp90, NCoR1 C-20, and lamin B1 antibodies.
Co-immunoprecipitation and Mass Spectrometric Identification of Proteins in SDS Gels
One approach to determining the extent of endogenous NCoR1 co-immunoprecipitation with Rev-erbβ is to make use of recombinant proteins. In those assays, HEK293 cells (>80% confluent) were washed thoroughly with PBS prior to lysing the cells with Triton X-100 lysis buffer containing 1.5× protease inhibitors. For some experiments, the cells were depleted of heme by treatment with 2 mm SA for 24 h prior to harvesting. In 0.5 ml total volume of lysis buffer, 1 mg of soluble extract was mixed with 5 μg of recombinant FLRev-erbβHGS, H568F, or H568F/C384A variants (a parallel set of control experiments contained no recombinant protein) and 0, 5, 10, or 20 μm Fe3+-heme, and the mixtures were incubated on ice for 1 h prior to adding Rev-erbβ LBD antibody to each sample (9.3 μg). A second control experiment contained the same mass of nonspecific IgY antibodies added to assays with recombinant protein. In both cases, control IPs led to no detectable FLRev-erbβHGS or NCoR1 precipitation (data not shown). After an additional hour of incubation on ice, 40 μl of PrecipHen-agarose (50% agarose slurry, Aves Labs) was added, and the samples were incubated overnight at 4 °C with rocking and protected from light. The beads were washed thoroughly with Triton X-100 lysis buffer (except that the Triton concentration was 0.1%, instead of 1.0%) prior to eluting the protein complexes by boiling for 10 min in 2× SDS loading buffer. Complexes were separated by SDS-PAGE and electroblotted as described, and the membranes were probed with Rev-erbβ QK-6, NCoR1 C-20, or MycBP2 antibodies.
A second approach for assessing the role of heme in NCoR1 binding to Rev-erbβ was to overexpress the wild-type receptor or the H568F variant via transient transfection of HEK293FT cells. 40–50% confluent cultures in 10-cm-diameter dishes grown in low serum medium were transfected with 20 μg of pcDNA3.1(+) parent vector as a control, pcDNA3.1(+)-FLRev-erbβ for overexpression of wild-type Rev-erbβ, or the analogous expression vector for the H568F variant. 24 h after transfection, 20 μm MG-132 (Sigma; stock solution is 10 mm in DMSO) was added to the medium, and the cells were incubated for an additional 4 h prior to washing with PBS, lysing in Triton X-100 lysis buffer containing 1.5× protease inhibitors, and preparing IPs as described above for recombinant proteins.
Mass spectrometry was used to identify the proteins that interacted with FLRev-erbβ. The co-IP utilizing recombinant protein was scaled up 2-fold (in terms of recombinant protein and cell extract mass), and the eluted complexes were separated by SDS-PAGE, staining the gel with Coomassie Brilliant Blue. Protein bands of interest were excised from the gel and subjected to in-gel reduction and alkylation (DTT/iodoacetamide) followed by tryptic digestion (at 37 °C for 16 h) and extraction with 5% acetonitrile in 0.1% formic acid. The samples were analyzed by capillary HPLC-electrospray ionization tandem mass spectrometry (HPLC-ESI-MS/MS) on a Thermo Scientific LTQ Orbitrap Velos Pro mass spectrometer. On-line HPLC separation was accomplished with an Eksigent/AB Sciex NanoLC-Ultra 2D HPLC system using a PicoFritTM (New Objective) 75 μm i.d. column packed to 15 cm with C18 adsorbent reverse phase medium (Vydac, 218MS 5 μm, 300 Å). Precursor ions were acquired in the Orbitrap in centroid mode at 60,000 resolution (m/z 400); data-dependent collision-induced dissociation (CID) spectra of the six most intense ions in the precursor scan were acquired at the same time in the linear trap (30% normalized collision energy). Mascot (Matrix Science) was used to search the spectra against the human subset of the NCBInr database (NCBInr 20140522, Homo sapiens (human) (279,814 sequences and 14,178,194,136 residues)) concatenated with a database of common protein contaminants (contaminants 20120713 (247 sequences, 128,130 residues)). Cysteine carbamidomethylation was set as a fixed modification, and methionine oxidation and deamidation of glutamine and asparagine were considered as variable modifications; trypsin was specified as the proteolytic enzyme, with one missed cleavage allowed. A subset search of the identified proteins by X! Tandem, cross-correlation with the Mascot results, and determination of protein and peptide identity probabilities were accomplished by Scaffold (Proteome Software). The thresholds for acceptance of peptide and protein assignments in Scaffold were 95 and 99%, respectively.
Cycloheximide Degradation Assays
For those experiments measuring the half-life of overexpressed Rev-erbβ, HEK293 cells grown in 3.5-cm-diameter dishes (2 ml total volume medium) were transfected with 3 μg of pcDNA3.1(+)-FLRev-erbβ or the expression vectors for H568F/C384A and H568F variants. After 24 h, the medium was replaced, and the cells were incubated for another 24 h prior to adding 20 μg ml−1 cycloheximide (CHX, Santa Cruz Biotechnology; 20 mg ml−1 stocks were prepared in DMSO and used immediately). In some experiments, 20 μm MG-132 was added at the same time as CHX. At the appropriate time points after CHX addition, cells were washed with PBS, lysed in radioimmune precipitation assay buffer (Life Technologies) containing 1× protease inhibitors, and soluble extracts were electroblotted after SDS-PAGE separation as described, probing membranes with either Rev-erbβ QK-6 or actin antibodies.
Repression Assays
HEK293 cells grown in 6-well plates were transfected with 0.1 μg of pCMV-βGal control vector (provided by Dr. Daniel Bochar, University of Michigan), 0.2 μg of pGL3Basic-Bmal1 luciferase reporter (71), varying masses of pcDNA3.1(+)-FLRev-erbβ or pcDNA3.1(+)-Rev-erbα (or the corresponding vectors for expression of heme axial ligand mutants), and parent pcDNA3.1(+) vector to a total mass of 3.5 μg/well. 24 h after transfection, cells were washed with PBS and lysed in reporter lysis buffer with one freeze-thaw cycle, and the soluble extract was assayed for luciferase activity by mixing 20 μl of extract with 100 μl of luciferase substrate (Promega) and detecting luminescence with a Tecan Safire2 microplate reader over a 10-s integration time. The relative luciferase units were normalized to β-galactosidase activity, which was measured by the colorimetric detection of o-nitrophenol from the hydrolysis of o-nitrophenyl-β-d-galactopyranoside (β-galactosidase enzymeassay system, Promega).
Results
Purification of FLRev-erbβ and Its Ability to Bind COand Heme
Two Rev-erbβ (NR1D2) sequences exist in GenBankTM, one corresponding to the mammalian gene collection (72) and the other to the human genome reference sequence. MGC differs from HGS by three missense mutations that result in amino acid substitutions P21H, Q282K, and P288R and may represent single nucleotide polymorphisms of NR1D2. To test whether these amino acid substitutions affect Rev-erbβ ligand binding, both clones were heterologously expressed in E. coli as translational fusions to a His6-GB1 solubility tag with a TEV protease recognition sequence between the fusion partners. GB1-FLRev-erbβ MGC and HGS were purified by nickel-nitrilotriacetic acid-agarose and digested with TEV protease to cleave the GB1 tag, and FLRev-erbβ was isolated from the protease and undigested fusion by anion exchange chromatography. A final preparative gel filtration step resulted in preparations that were >90% pure and free of GB1 (Fig. 1, lanes 1 and 2). After these purification steps, the MGC and HGS forms of FLRev-erbβ contained 2.2 and 2.9% bound heme, respectively, as determined using an oxalic acid fluorescence assay (average of four replicates; see “Experimental Procedures” for details). The native molecular masses of HGS and MGC (with monomeric molecular masses of 64.8 and 64.9 kDa, respectively) were estimated by analytical gel filtration chromatography to be 142 ± 5 and 123 ± 10 kDa, respectively (average of duplicate HPLC runs ± range), indicating that FLRev-erbβ associates into a dimeric quaternary structure (Fig. 2).
FIGURE 1.

Homogeneity of protein preparations determined by SDS-PAGE. Each lane contains 2 μg of purified protein, and the molecular masses (kDa) of the ladder are depicted. Lanes: L, molecular mass ladder; 1, FLRev-erbβMGC; 2, FLRev-erbβHGS; 3, FLRev-erbβ H568F; 4, FLRev-erbβ H568F/C384A; 5, MBP-NCoR1–540; 6, MBP; 7, THRβ1.
FIGURE 2.

Estimation of FLRev-erbβ HGS and MGC molecular mass with analytical gel filtration chromatography. 50 μl of 0.5–1 mg ml−1 FLRev-erbβ HGS (solid line) or MGC (dashed line) was injected onto a Shodex KW-803 HPLC column equilibrated in 50 mm bis-Tris, pH 7.0, 150 mm NaCl, 5% glycerol, and 0.5 mm TCEP. The approximate elution volumes of molecular mass standards are depicted, and the mass of FLRev-erbβ proteins are determined as described under “Experimental Procedures.”
The ability of FLRev-erbβ MGC and HGS to bind heme and CO was assessed by UV-visible and EPR spectroscopy. Because thiol oxidation has been shown to compromise the heme binding activity of the isolated Rev-erbβ LBD (38), we chose to study the thiol-reduced form of FLRev-erbβ. Thus, prior to heme binding analyses, proteins were reduced with an excess of TCEP in an anaerobic chamber, buffer-exchanged to eliminate reductant, and maintained under anaerobic conditions throughout the assay. The UV-visible spectra of Fe3+-heme with a 2-fold excess of FLRev-erbβ MGC or HGS (to ensure the majority of the heme was bound) were identical and exhibited features of a six-coordinate low-spin heme with thiolate ligation (Fig. 3A) (73). The spectra are characterized by a weak shoulder at 650 nm, broad α- and β-bands at 577 and 543 nm, respectively, a Soret (γ band) at 423 nm, and a δ-feature at 358 nm. The EPR spectrum of the Fe3+-heme·FLRev-erbβMGC complex (Fig. 4) exhibits a rhombic feature with g values of 1.88, 2.28, and 2.47, similar to those reported for cystathionine-β-synthase and HRI (Table 2), which bind heme as six-coordinate complexes with His/Cys ligation (74–77). Other minor features in the EPR spectrum are likely due to the heterogeneity of the axial cysteine sulfhydryl protonation state (73). The addition of 1 mm dithionite generates Fe2+-heme complexes (Fig. 3B) with sharper α- and β-bands at 559 and 530 nm, respectively, and a 3 nm red-shifted Soret band at 426 nm, whereas Fe2+-CO adducts (Fig. 3C) have broader α/β-bands (569 and 539 nm) and a 5 nm blue-shifted Soret peak at 421 nm. Further, titration of CO-saturated water into a solution of Fe2+-heme·FLRev-erbβHGS indicates that CO binds with high affinity in a 1:1 complex as described previously for the isolated LBD (Fig. 3D). Similar results were obtained with FLRev-erbMGC (data not shown). Thus, both the UV-visible and EPR spectra of heme/CO-bound MGC/HGS closely resemble those of the equivalent complexes prepared with thiol-reduced Rev-erbβ LBD (38) (Table 2), indicating that the LBD and FLRev-erbβ are competent to bind both redox forms of heme and CO and that additional domains provided by the full-length constructs do not impart unique heme binding characteristics.
FIGURE 3.

UV-visible spectra (A–C) of the complexes of Fe3+-heme, Fe2+-heme, and Fe2+-heme·CO with wild-type and variant FLRev-erbβ and (D) CO titration of the heme-bound protein. A, Fe3+-heme complexes were prepared under anaerobic conditions in 1 ml 0.5× TNG buffer containing 3 μm heme and 6 μm thiol-reduced FLRev-erbβMGC (red trace), FLRev-erbβHGS (green trace), H568F (cyan trace), or H568F/C384A (blue trace). Inset, expanded 500–700 nm region. 3 μm free Fe3+-heme is shown for reference (black trace) and 6 μm as purified FLRev-erbβMGC (dashed black trace). B, Fe2+-heme complexes were prepared by adding 1 mm dithionite anaerobically to the Fe3+-heme complexes described in A. C, Fe2+-heme-CO adducts were generated by purging the headspace of the cuvette containing Fe2+-heme complexes with pure CO for 1 min and mixing by inversion. D, a solution containing 2 μm Fe2+-heme and 3 μm FLRev-erbβHGS was titrated with a calibrated CO-saturated water solution, and the spectra were recorded after reaching equilibrium. Inset, a plot of the change in absorbance at 421 nm from the recorded spectra versus the concentration of CO. The equivalency point of the fit, i.e. ∼1.9 μm, indicates a 1:1 complex of CO with the 2 μm Fe2+-heme·FLRev-erbHGS complex present in the solution. The 426–421 nm blueshift resulting from CO addition is depicted with an arrow.
FIGURE 4.

EPR spectrum of the FLRev-erbβ·Fe3+-heme complex. The FLRev-erbβMGC sample was prepared as described under “Experimental Procedures”; the g values for the major rhombic feature are depicted, although other minor features exist that are likely due to the heterogeneity of the axial Cys-thiolate protonation state.
TABLE 2.
Comparison of UV-visible and EPR spectral features of heme complexes with wild-type and variant FLRev-erbβ, thiol-reduced Rev-erbβ LBD, and other thiolate-ligated hemoproteins
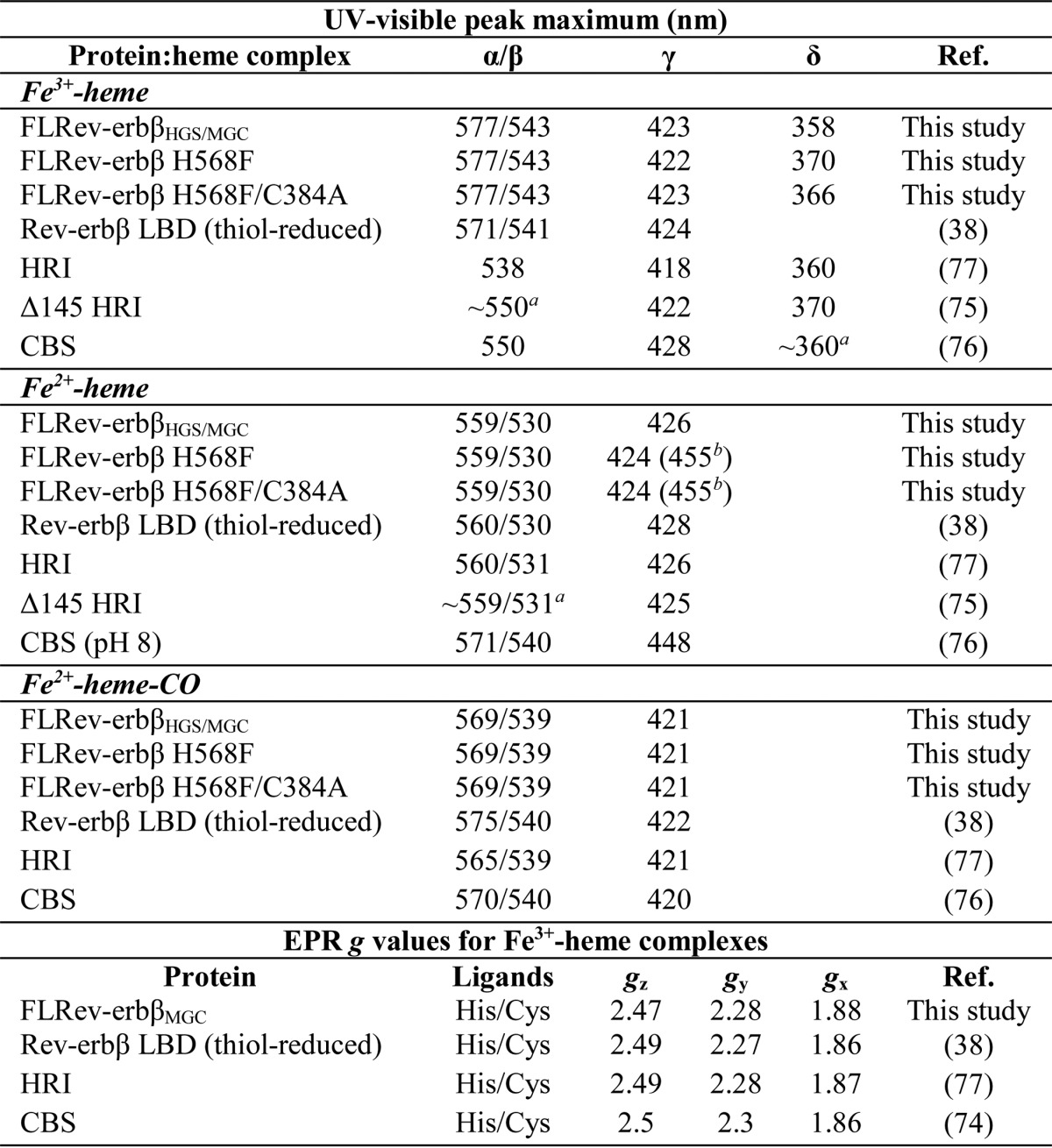
a Estimated from the published spectrum.
b Weak shoulder red-shifted from major γ-band.
The crystal structure of the Rev-erbβ LBD in complex with Fe3+-heme indicates that His-568 and Cys-384 serve as axial ligands coordinating the heme iron atom (36). In an attempt to generate a form of FLRev-erbβ that is deficient in heme binding, the H568F and H568F/C384A variants were purified (Fig. 1, lanes 3 and 4) and their heme binding properties characterized. The UV-visible spectrum of Fe3+-heme in the presence of a 2-fold excess of H568F is distinct from those of the wild-type proteins, most notably by the low relative ratio of the γ-band absorbance (422 nm) to that of the pronounced δ-band at 370 nm (γ422/δ370 = 0.92; wild-type HGS, γ423/δ358 = 2.43 (Fig. 3A)). This spectrum closely resembles that of an HRI truncation mutant that lacks the His axial ligand and binds Fe3+-heme with Cys/H2O ligation (75). The Fe3+-heme·H568F/C384A spectrum is similar to that of H568F, with a Soret band at 423 nm and slightly blue-shifted δ-band at 366 nm, with the γ423/δ366 ratio equal to 1.36. Fe2+-heme complexes of H568F and H568F/C384A resemble those of HGS and MGC with overlapping α/β-bands and slightly blue-shifted Soret bands at 424 nm; a unique feature of the variants is a weak shoulder at 455 nm (Fig. 3B). Similarly, the spectra of the Fe2+-heme·CO complexes with the mutants are nearly identical to those of the wild-type proteins (Fig. 3C).
Having established that wild-type and variant FLRev-erbβ are competent to bind Fe3+/Fe2+-heme, it was imperative to measure the Kd values for the heme·FLRev-erbβ complex to determine whether the full-length proteins could bind heme at the low intracellular concentrations of free/exchangeable heme, which has been estimated to be <0.2 μm in mature erythrocytes (78, 79). Difference UV-visible spectrophotometric titrations of Fe3+-heme into anaerobic solutions of thiol-reduced MGC (Fig. 5A) and HGS establish Kd values of 7 ± 2 and 6 ± 2 nm, respectively, and titration of Fe2+-heme (Fig. 5C for MGC) yields Kd values of 24 ± 4 and 23 ± 5 nm, respectively. All of these values are well below 0.2 μm, suggesting that FLRev-erbβ tightly binds both redox forms of heme in the cell. Furthermore, the identical Kd values shared between HGS and MGC indicate that the minor differences in primary amino sequence between isoforms have no influence on heme binding capacity. The high concentrations of heme required for titration of the H568F and H568F/C384A variants lead to precipitation; thus, only data collected prior to the observation of protein precipitation were included in the fits. Nonetheless, in stark contrast to the wild-type proteins, Fe3+-heme titrations of H568F and H568F/C384A (Fig. 5B) yield Kd values of 23 ± 3 and 14 ± 1 μm, respectively, with similar affinities observed for Fe2+-heme titrations (see Fig. 5D for the double mutant) of 18 ± 2 and 9 ± 1 μm. Considering that the affinity of the mutants for heme is ∼1000-fold lower than wild-type and Kd values are ∼50-fold higher than the estimated concentration of the free heme pool, they will exist predominantly as apoprotein in the cell.
FIGURE 5.

Representative UV-visible difference titrations of Fe3+/Fe2+-heme into thiol-reduced FLRev-erbβ and the H568F/C384A variant under anaerobic conditions. Stock solutions of Fe3+/Fe2+-heme were titrated into 1 ml of 0.5× TNG buffer containing either 0.8 μm thiol-reduced FLRev-erbβMGC (A, Fe3+-heme; C, Fe2+-heme) or 4 μm H568F/C384A (B, Fe3+-heme; D, Fe2+-heme) under anaerobic conditions at 20 °C. A reference cuvette containing only buffer was used to subtract the absorbance of free heme at each concentration. The absorbance of the Soret band maximum for each titration point was plotted as a function of heme concentration and the data fitted with Equation 1 (insets). Because the titration of MGC is stoichiometric, [P] in Equation 1 was left as a variable, as the inflection point of the fit (representative of [P]) was sometimes slightly lower than the known concentration of protein in the cuvette (0.8 μm). Thus, the ratio of [P] determined from the fit to the known [P] is between 0.8 and 1, indicating a 1:1 heme:MGC stoichiometry. For fitting of H568F and H568F/C384A data, [P] = 4 μm. Titrations of HGS and H568F, omitted for brevity, closely resembled those depicted.
The Kd for wild-type proteins were determined by using a quadratic equation (Equation 1), instead of a simple Michaelis-Menten-type binding equation, to fit the data. This is because the Kd values are so far below the protein concentration that the concentration of free heme (the x axis in the simple ligand-binding isotherm) is negligible compared with that of total heme. In some cases, the affinity is so high that even the quadratic treatment does not provide correct Kd values. For example, a recent report from our laboratory showed that the Kd for Fe3+-heme binding to heme oxygenase-2 determined with stoichiometric equilibrium titrations was 70-fold higher (14 nm) than the value (0.2 nm) calculated from heme on- and off-rates (45). Thus, although Equation 1 is useful for fitting data where the Kd ≪ the protein concentration, it may still overestimate the true Kd, suggesting that those low nanomolar values reported herein for FLRev-erbβ may represent an upper limit of Kd.
For the reasons just discussed, we measured the Fe3+-heme off-rates, the controlling factor in distinguishing heme affinity among a variety of heme proteins (49, 75). We mixed FLRev-erbβ·Fe3+-heme complexes with 10-fold excess apomyoglobin and monitored the conversion of apo- to holomyoglobin at 408 nm (Fig. 6 and Table 3). In a control experiment, when we reacted excess apomyoglobin (30 μm) with 1.5 μm free Fe3+-heme (Fig. 6D), complete conversion to holomyoglobin occurred within the mixing dead time of ∼20 s. Mixing excess apomyoglobin with a solution of 1.5 μm Fe3+-heme and 3 μm FLRev-erbβHGS leads to very slow increases in absorbance at 408 nm (Fig. 6A) that fit best to a double exponential equation, yielding off-rate constants of koff-fast = (1.5 ± 0.4) × 10−4 s−1 and koff-slow = (3.9 ± 0.8) × 10−6 s−1. The fast phase makes up 18 ± 3% of the total Δabsorbance, indicating that the second slower phase is representative of the majority of the FLRev-erbβHGS·heme population. Upon completion of the kinetics experiment at 4000 min, the γ-band is centered at 415 nm, approximately halfway between the positions of holomyoglobin (408 nm) and holoHGS (423 nm) (Fig. 6A), revealing incomplete heme transfer. In addition, the total Δabsorbance for both fast and slow phases (∼0.05) is only ∼50% of the expected total (0.10–0.11) based on assays with heme axial ligand variants (discussed below). To ensure that apomyoglobin could still bind heme after the long incubation of the assay, 6 μm Fe3+-heme was added to the cuvette, yielding a spectrum characteristic of holomyoglobin (Fig. 6A, blue spectrum; note the γ-band is sharply centered at 408 nm). However, FLRev-erbβ did not bind any added heme. An identical experiment with the Rev-erbβ LBD alone also exhibits nearly identical biphasic off-rates but with heme transfer proceeding to near completion, suggesting that the FLRev-erbβ undergoes denaturation during the long incubation times of the assay.
FIGURE 6.

UV-visible spectra and time courses of Fe3+-heme transfer from FLRev-erbβ, H568F, and H568F/C384A to excess apomyoglobin. 1.5 μm Fe3+-heme was mixed with 3 μm thiol-reduced FLRev-erbβHGS (A), H568F (B), and H568F/C384A (C) under anaerobic conditions, and the spectrum was recorded (black spectrum). 30 μm apomyoglobin was added, and absorbance changes at 408 nm were monitored over time (insets: 2-s intervals for axial ligand mutants and 60 s for the wild type; raw data are represented as black circles). The kinetic data fit best to a double exponential (red fit, inset) yielding rate constants for the fast and slow phases (see Table 3). At the conclusion of the time course, a spectrum was recorded to confirm the extent of heme transfer to apomyoglobin (red spectrum). Note the incomplete transfer from wild-type FLRev-erbβ to apomyoglobin by the position of the Soret band maximum at 415 nm in the spectrum taken at 4000 min after the addition of apomyoglobin (A). Further, to ensure apomyoglobin was still competent to bind heme after the long time frame of the experiment in A, 6 μm Fe3+-heme was added yielding the blue spectrum characteristic of holomyogobin with a Soret band sharply centered at 408 nm. An identical control experiment performed with 1.5 μm free Fe3+-heme (D) demonstrates heme binding to apomyoglobin goes to completion within the mixing dead time of ∼20 s. A spectrum of 30 μm apomyoglobin is included as a reference (D, blue spectrum).
TABLE 3.
Fe3+-heme dissociation rate constants for FLRev-erbβHGS, H568F, and H568F/C384A
Rate constants are derived from double exponential fits of the raw data (see Fig. 6).
| Protein | koff-fast; ΔAbsorbancea | koff-slow; ΔAbsorbancea |
|---|---|---|
| s−1; % of total | s−1; % of total | |
| FLRev-erbβHGS | (1.5 ± 0.4) × 10−4; 18 ± 3 | (3.9 ± 0.8) × 10−6; 82 ± 3 |
| FLRev-erbβ H568F | (2.1 ± 0.4) × 10−2; 64 ± 9 | (5 ± 1) × 10−3; 36 ± 9 |
| FLRev-erbβ H568F/C384A | (4.3 ± 0.3) × 10−2; 88 ± 1 | (2.4 ± 1.0) × 10−3; 12 ± 1 |
a Rate constants and ΔAbsorbance % values represent the average ± S.D. of three replicate experiments.
An identical experiment performed with H568F demonstrates that heme transfer to apomyoglobin reaches completion within 10 min (Fig. 6B). These data also fit best to a double exponential equation yielding off-rate constants of koff-fast = (2.1 ± 0.4) × 10−2 s−1 and koff-slow = (5 ± 1) × 10−3 s−1, with the first phase representing the majority of the total Δabsorbance at 64 ± 9%. The spectrum acquired at the end of the kinetics experiment indicates complete heme transfer by the sharp γ-band centered at 408 nm (Fig. 6B), essentially identical to that spectrum acquired after mixing free Fe3+-heme with apomyoglobin (Fig. 6D). Complete heme transfer from the H568F/C384A variant also occurred within 10 min, yielding rate constants of koff-fast = (4.3 ± 0.3) × 10−2 s−1 and koff-slow = (2.4 ± 1.0) × 10−3 s−1, with the fast phase comprising the majority (88 ± 1%) of the total Δabsorbance (Fig. 6C). These data demonstrate that koff-slow for HGS is ∼600 times slower than the slowest phase of the heme axial ligand mutants(koff-slow for H568F/C384A), indicating that off-rates are responsible for the vast differences in Kd values observed in equilibrium titrations for wild-type FLRev-erbβ versus H568F and H568F/C384A.
FLRev-erbβ and Its Interactions with DNA and NCoR1
To ensure that FLRev-erbβ MGC, HGS, H568F, and H568F/C384A variants are competent to bind DNA, fluorescence anisotropy was used to measure the affinity of these proteins for two fluorescein-labeled DNA duplexes, which serve as Rev-erb response elements, termed Rev-REs (Table 1 and Fig. 7). FAM-Rev-DR2, is a synthetic construct comprised of two tandem Rev-REs separated by 2 bp, whereas the FAM-Bmal1 promoter encompasses the two Rev-REs found in the core Bmal1 promoter separated by 30 bp (from the 3′-end of the distal Rev-RE to the 5′-start of the proximal hexameric Rev-RE) (13).
FIGURE 7.

Binding of wild-type and variant (H568F and H568F/C384A) FLRev-erbβ to promoter DNA elements. Binding assays containing 10 nm FAM-Rev-DR2 (A) or FAM-BMal1 promoter (B) elements, 1 ng μl−1 poly(dI-dC), and varying concentrations of FLRev-erbβ MGC (closed circles), HGS (open circles), H568F (inverted closed triangles), and H568F/C384A (inverted open triangles) were prepared in 0.5× TNGD, incubated at 4 °C for 2 h, and the anisotropy of the labeled duplexes was determined at ambient temperature. Anisotropy values are plotted (average ± S.D. of three replicate experiments) as a function of protein concentration and the data fit with the Hill equation. C, summary of the K0.5 and Hill coefficients from the fits described in A and B.
Increasing concentrations of recombinant HGS, MGC, H568F, and H568F/C384A correspondingly lead to increased anisotropy of both the FAM-Rev-DR2 and FAM-Bmal1 promoter elements (Fig. 7, A and B). The data fit best to the Hill equation, with K0.5 values for wild-type MGC and HGS proteins between 60 and 70 nm and Hill coefficients ranging from 1.4 to 2.0 (Fig. 7C). In agreement with prior studies, Rev-erb NRs bind cooperatively as a homodimer to the Rev-DR2 construct (80–82). However, our results show that cooperative interactions occur even when the spacing between the Rev-REs is much longer. The affinities of H568F and H568F/C384A for FAM-Rev-DR2 and FAM-Bmal1 promoter were ∼2- and 1.5-fold lower, respectively, than those of the wild-type proteins, suggesting that mutations in the LBD may affect DNA binding. However, these minor changes in affinity could be due to variable levels of Zn2+ loading of the two 4-Cys zinc-fingers in the DBDs of the recombinant proteins. In any case, wild-type and heme axial ligand mutants bind Rev-REs with high affinity, a result supported by an EMSA demonstrating that both H568F and H568F/C384A variants bind FAM-Rev-DR2 and FAM-Bmal1 promoter elements with similar capacity to wild-type FLRev-erbβMGC (Fig. 8, compare odd numbered lanes).
FIGURE 8.

EMSA experiments show that wild-type FLRev-erbβ and H568F and H568F/C384A variants bind DNA and MBP-NCoR1–540 with similar affinity. Binding assays were prepared in 0.5× TNGD buffer containing 25 ng μl−1 poly(dI-dC), 1 μm FAM-Rev-DR2 (A), or FAM-Bmal1 promoter (B) and the indicated concentrations of wild-type FLRev-erbβMGC, or variant FLRev-erbβ, and MBP-NCoR1–540. Assays were incubated on ice for 2 h prior to separation by native gradient gel electrophoresis and imaging of the protein-DNA complexes.
The interaction of FLRev-erbβMGC with a fluorescein-labeled peptide corresponding to NCoR1 ID1 (F-ID1) was assessed with fluorescence anisotropy. Titration of heme-free FLRev-erbβMGC into a solution containing 10 nm F-ID1 led to a concentration-dependent increase in anisotropy that was accompanied by quenching of peptide fluorescence (Fig. 9A). The anisotropy data were corrected for the quenching effect and fit well to Equation 1 yielding a Kd of 15 ± 11 nm (average ± S.D. of three independent titrations), thus indicating that recombinant FLRev-erbβ is competent to bind NCoR1 with high affinity in the absence of heme. In fact, FLRev-erbβ exhibits a higher affinity for this NCoR1 peptide than that of the isolated allophycocyanin-labeled apoRev-erbα LBD for a europium chelate-labeled NCoR1 ID1 peptide (Kd of 135 nm) determined with fluorescence energy transfer (27). Thus, the above experiments clearly demonstrate that FLRev-erbβ binds heme, CO, DNA, and an NCoR1 peptide with a high affinity similar to the isolated domains.
FIGURE 9.

Binding affinity of F-ID1 peptide for FLRev-erbβ measured by fluorescence anisotropy and the effect of Fe3+-heme on the peptide-protein interaction. A, titration of 10 nm F-ID1 with FLRev-erbβMGC in 0.5× TNG buffer containing 0.5 mm TCEP at 25 °C. A [FLRev-erbβMGC]-dependent quenching of F-ID1 fluorescence (open triangles) was accounted for using Equation 3. Thus, the corrected change in anisotropy of the peptide is plotted as a function of [FLRev-erbβMGC], and the data are fit with Equation 1, holding the [F-ID1] term constant at 10 nm (open circles). The uncorrected anisotropy data fit with a square hyperbola is shown for clarity (closed circles). The data given in A are from a representative experiment; averaging three replicate titrations yielded Kd = 15 ± 11 nm (error is S.D.). B, titration of 10 nm F-ID1 and 25 nm FLRev-erbβMGC with Fe3+-heme performed under the same conditions as the experiment described in A. Raw anisotropy values are plotted and the data fit with a hyperbolic decay function. The data are from a representative titration with duplicate titrations yielding an average IC50 of 124 nm.
Binding Studies Using Purified Components Showing That Heme Does Not Promote the Interaction of FLRev-erbβ with NCoR1
Because heme titration has been shown to lead to dissociation of the complex between NCoR1 peptides and Rev-erbα/β LBDs, we performed a similar experiment with the FLRev-erbβMGC·F-ID1 complex. As observed with the LBD, the addition of Fe3+-heme destabilizes the complex between ID1 and the full-length NR with an IC50 of 124 nm (average IC50 of two independent titrations) (Fig. 9B). We were unable to test the effect of Fe2+-heme in this system, as F-ID1 fluorescence was paradoxically quenched under anaerobic conditions. Nonetheless, we can unambiguously conclude that additional domains provided by FLRev-erbβ do not impart Fe3+-heme-dependent stimulation of NCoR1 peptide binding. Thus, we developed a new hypothesis that, because the two N-terminal NCoR1 interaction domains are critical for an interaction with Rev-erbα in the cell (83), the disconnect between in vivo and in vitro results originates from the use of a peptide encompassing only one of three NR interaction domains found in NCoR1.
To mimic the interactions of Rev-erbβ with full-length NCoR1, we generated a translational fusion between MBP and the C-terminal 540 residues of NCoR1 encompassing all three interaction domains (MBP-NCoR1–540). The construct was heterologously expressed in E. coli and purified to ∼80% homogeneity by successive chromatography on amylose affinity resin and Sephacryl S300 gel filtration medium (Fig. 1, lane 5). Using EMSAs with native gradient polyacrylamide gels, we resolved the ternary complexes among the FAM-Bmal1 promoter (or the FAM-Rev-DR2 elements), FLRev-erbβ, and MBP-NCoR1–540, ultimately imaging the protein-DNA complexes using the fluorescein tags. The interaction of FLRev-erbβMGC with FAM-Bmal1 promoter reveals at least three protein-DNA complexes (Fig. 10A, lanes 1–4), likely representing a mixed population of MGC monomers and dimers binding to the two distant Rev-REs or a cooperative complex where Rev-erbβ binding bends the DNA to allow an interaction between the two sites. The addition of MBP-NCoR1–540 causes a supershift in the MGC·FAM-Bmal1 promoter complex, resolving at least four distinct corepressor complexes (Fig. 10A, lanes 5–8). MBP alone does not affect migration of the MGC-promoter complex (Fig. 10A, lane 9), indicating that the supershift is the result of a specific interaction between the C-terminal 540 residues of NCoR1 and FLRev-erbβ. Because the purified receptors are predominantly heme-free, these results indicate that heme is not required for the interaction between NCoR1 and FLRev-erbβ.
FIGURE 10.

Effect of heme on NCoR1 binding to the FLRev-erbβ·DNA complex. EMSA was utilized to observe the interaction between FLRev-erbβ and FAM-labeled DNA promoter elements. Binding of MBP-NCoR1–540 leads to a concentration-dependent supershift of the Rev-erbβ·DNA complex, which is marginally affected by the addition of Fe3+-heme. A, binding assays were prepared in 0.5× TNGD buffer containing 25 ng μl−1 poly(dI-dC), 1 μm FAM-Bmal1 promoter, and the indicated concentrations of FLRev-erbβMGC, Fe3+-heme, MBP, or MBP-NCoR1–540. Assays were incubated on ice for 2 h prior to separation by native gradient gel electrophoresis. Protein-DNA complexes were imaged using the appropriate excitation and emission wavelengths for fluorescein. B, same as the experiment described for A except FLRev-erbβHGS was added (lane orientation is the same for all panels; only protein-DNA complexes are shown for B–D). C, same as described for A except binding assays contained 1 μm FAM-Rev-DR2 instead of FAM-Bmal1 promoter. D, same as described for C except with FLRev-erbβHGS.
To further address the effect of heme on the interaction between Rev-erbβ and NCoR1, we added Fe3+-heme at a 1:1 stoichiometry (with respect to the concentration of FLRev-erbβ) to the assay mixture and found no effect on this interaction at different concentrations of corepressor (Fig. 10A, compare lanes 6 and 8 (heme-free samples) with lanes 10 and 12 (same samples with 1:1 heme)). An identical experiment with FLRev-erbβHGS yielded the same results (Fig. 10B). High levels of Fe3+-heme (10:1, heme:FLRev-erbβ ratio) decreased the overall intensity of the protein-DNA complexes, suggesting that heme at high concentrations may inhibit DNA binding (Fig. 10A, lanes 11 and 13). An alternative interpretation is that heme induces DNA degradation, perhaps by catalyzing the generation of reactive oxygen species by Fenton chemistry. Similar experiments show that the H568F and H568F/C384A variants bind MBP-NCoR1–540 to the extent of the wild type with identical supershifting patterns on both the FAM-Bmal1 promoter and FAM-Rev-DR2 elements (Fig. 8, compare even numbered lanes).
FLRev-erbβ MGC and HGS binding to FAM-Rev-DR2 reveals an intense band likely representing the homodimer (Fig 10, C and D, lanes 1–4). MBP-NCoR1–540 supershifts the protein-DNA complex (Fig 10, C and D, lanes 5–8) and a 1:1 heme:FLRev-erbβ ratio has no effect on the Rev-erbβ-NCoR1 interaction at two concentrations of corepressor (Fig 10, C and D, lanes 10 and 12), although we do observe a low intensity supershifted band at elevated heme concentrations (10:1 heme:FLRev-erbβ ratio) and an intermediate concentration of MBP-NCoR1–540 (Fig 10, C and D, lane 11). However, it is unclear if this supershift is due to heme promoting the interaction between Rev-erbβ and NCoR1 or simply to an effect of heme on the electrophoretic mobility of the heme-bound complex. If heme stimulated the interaction we would expect this band to be at least the same intensity as the complex in Fig 10, C and D, lane 10, with 1:1 heme, supporting the notion that heme impacts electrophoretic mobility. This phenomenon is restricted to the Rev-DR2 element, as we did not see pronounced heme-dependent supershifting of bands in the experiments with the FAM-Bmal1 promoter.
Because of these apparent minor effects of Fe3+-heme on Rev-erbβ and MBP-NCoR1–540 binding, it was important to quantitatively assess how much heme is bound to FLRev-erbβ under the EMSA conditions and to what extent MBP-NCoR1–540 affects heme binding. The concentrations of the proteins and DNA described for the assay mixtures in Fig. 10D, lanes 4 (FLRev-erbβHGS·Rev-DR2 complex) and 8 (FLRev-erbβHGS·Rev-DR2 complex bound by MBP-NCoR1–540), were scaled up 3-fold and included 3 μm Fe3+-heme, i.e. retaining a 1:1 heme:FLRev-erbβ ratio. Parallel assays were performed without heme, and the UV-visible spectra were recorded for all of the samples. The spectra of heme-free samples were subtracted from those of the heme-containing samples to yield the difference spectra shown in Fig. 11A. Then, 5 mm dithionite was added to the samples to generate the Fe2+-heme complexes, and the UV-visible spectra were recorded after reaching equilibrium (Fig. 11B). For both the Fe3+-heme and Fe2+-heme complexes, the Soret absorbance of samples containing the corepressor is slightly (∼10%) lower than that of the sample lacking MBP-NCoR1–540 (also observed in an independent experiment). If heme displaces the ID1 corepressor peptide, excess corepressor should reciprocally displace heme. To assess the extent of heme loading in each sample, extinction coefficients were calculated for Fe3+-heme and Fe2+-heme complexes with FLRev-erbβHGS/MGC based on those spectra acquired in Fig. 3, A and B, with excess protein-heme. For Fe3+-heme, we took into account the minor absorbance at 423 nm from residual heme in the as purified protein (Fig. 3A, dashed black trace), which yielded ϵ = 90 mm−1 cm−1 at 423 nm. This value is in good agreement with the estimated extinction coefficient for the Fe3+-heme complex with thiol-reduced Rev-erbβ LBD of ϵ = 79 mm−1 cm−1 at 424 nm (based on the spectrum described in Ref. 38). For the Fe2+-heme complexes, ϵ = 127 mm−1 cm−1 and 130 mm−1 cm−1 at 426 nm for FLRev-erbβMGC and FLRev-erbβHGS, respectively. Based on these extinction coefficients and those spectra shown in Fig. 11A, the concentration of Fe3+-heme bound to FLRev-erbβHGS in complex with Rev-DR2 and MBP-NCoR1–540 is 2.6 μm, indicating 87% heme loading based on the total [FLRev-erbβHGS] of 3 μm in the assay; the sample lacking corepressor contains 2.8 μm bound heme or 93% holoprotein. Reduction to Fe2+-heme uniformly decreased the levels of heme loading (Fig. 11B); the sample containing MBP-NCoR1–540 is loaded with 2.2 μm heme or 73% holoprotein, whereas the sample without corepressor is loaded with 2.4 μm heme, i.e. 80% holoprotein. In either case, these data demonstrate that FLRev-erbβ binds both redox forms of heme under the EMSA conditions and that corepressor binding has only a minor effect on the level of heme loading.
FIGURE 11.
Rev-DR2·FLRev-erbβHGS binds Fe3+/Fe2+-heme in a ternary complex with MBP-NCoR1–540. To test whether heme and NCoR1 can bind simultaneously to the FLRev-erbβHGS·Rev-DR2 complex, the assay mixtures described in Fig. 10D, lane 4 (FLRev-erbβHGS·Rev-DR2 complex, black spectra) and lane 8 (FLRev-erbβHGS·Rev-DR2 complex fully bound by MBP-NCoR1–540, red spectra), were scaled-up 3-fold and included 3 μm Fe3+-heme, i.e. a 1:1 ratio of heme to FLRev-erbβHGS. Unlabeled Rev-DR2 duplex was used to avoid overlap of the fluorescein spectrum with that of heme. A parallel set of assays was prepared without heme. Samples were incubated on ice for at least 2 h prior to recording the UV-visible spectrum. The spectra of heme-free samples were subtracted from heme-containing samples, generating the difference spectra shown in A. 5 mm dithionite was added to the samples in A under anaerobic conditions, the cuvettes were sealed, and the UV-visible spectrum was recorded until no change was observed. Difference spectra of Fe2+-heme complexes are depicted in B.
Thus, our results support a model in which heme, Rev-erbβ, and NCoR1 form a ternary complex in which stoichiometric heme does not affect corepressor binding. As a positive control and to ensure that the EMSA technique is sensitive enough to detect ligand-dependent changes in NCoR1 binding to a NR, we tested the effect of T3 on the interaction of MBP-NCoR1–540 with purified THRβ1. Previous EMSA experiments have shown that T3 binding to THRβ1 decreases the affinity of the receptor for NCoR1 and DNA (84). Full-length THRβ1 was heterologously expressed in E. coli as a fusion with MBP, and the protein was purified with amylose affinity resin. After TEV digestion and successive chromatography on anion exchange and heparin-agarose, tagless THRβ1 preparations were >85% pure (Fig. 1, lane 7). For DNA we use a FAM-labeled duplex corresponding to a region of the human placental lactogen gene promoter that is composed of two tandem THRβ1 response elements separated by 4 base pairs (FAM-hPL promoter; see Table 1) (65, 66). THRβ1 interacts with the FAM-hPL promoter yielding two distinct complexes likely representing the receptor binding as a monomer or homodimer (Fig. 12A, lanes 1–4). MBP-NCoR1–540, but not MBP alone, supershifts the protein-DNA complexes (Fig. 12A, lanes 5–9), and inclusion of 1:1 T3:THRβ1 decreases the intensity of the corepressor complex by 65% (determined with densitometry) with a concomitant increase in the intensity of the apparent monomer-DNA complex (Fig. 12A, lane 10). Higher concentrations of T3 did not lead to increased dissociation of MBP-NCoR1–540, indicating that the system had reached equilibrium at stoichiometric concentrations of T3. These data demonstrate that our EMSA with purified components could distinguish ligand-dependent changes in corepressor affinity.
FIGURE 12.

EMSA reveals T3-induced dissociation of MBP-NCoR1–540 from the THRβ1·FAM-hPL promoter complex, whereas Fe3+-heme causes dissociation of THRβ1 from DNA. A, binding assays were prepared in 0.5× TNGD buffer containing 0.25 mg ml−1 BSA, 25 ng μl−1 poly(dI-dC), 1 μm FAM-hPL promoter, and the indicated concentrations of THRβ1, MBP, MBP-NCoR1–540, and T3. B, same experimental setup as described for A except containing no BSA or T3 and with the indicated concentrations of Fe3+-heme. Assays were incubated on ice for 2 h prior to separation by native gradient gel electrophoresis and imaging of the protein-DNA complexes.
To further assess whether heme has a specific effect on the interactions between FLRev-erbβ and NCoR1, we performed heme titrations with the THRβ1 system, which is responsive to T3 but not to heme. Intriguingly, as seen with FLRev-erbβ, high levels of Fe3+-heme cause dissociation of THRβ1 from the FAM-hPL promoter without effecting corepressor binding at two concentrations of MBP-NCoR1–540 (Fig. 12B, compare lanes 2 and 5 (heme-free samples) with lanes 4 and 7 (the same samples with a 10:1 ratio of heme:THRβ1). On the other hand, there was little effect of a 1:1 ratio of heme:THRβ1 on band intensities (Fig. 12B, lanes 3 and 6). This result suggests that, at high concentrations, free heme may inhibit the binding of NRs to DNA, perhaps because of some Fenton chemistry-related reactions.
To obtain a more quantitative assessment of the interaction between MBP-NCoR1–540 and DNA-bound FLRev-erbβHGS and the effect of Fe3+/Fe2+-heme on the complex, we developed a fluorescence anisotropy assay. We obtained a Texas Red (TR)-labeled Rev-DR2 element because, unlike other fluorescence probes, TR is resistant to dithionite quenching (64), which allowed us to determine the effect of Fe2+-heme on the system. When increasing concentrations of MBP-NCoR1–540 were mixed with 20 nm TR-Rev-DR2 and 100 nm HGS, we observed increases in anisotropy that could be ascribed to MBP-NCoR1–540 binding to the binary complex between FLRev-erbβHGS and TR-Rev-DR2. The effect was saturable with corepressor concentration, and the addition of MBP alone yielded no change in anisotropy (Fig. 13A). A titration of 20 nm TR-Rev-DR2 with FLRev-erbβHGS yielded a K0.5 of 83 ± 1 nm and a Hill coefficient of 1.6 ± 0.4, demonstrating that a mixture of 20 nm TR-Rev-DR2 and 100 nm HGS would contain 11.6 nm protein-DNA complex. Thus, we fit the MBP-NCoR1–540 titration data with Equation 1 holding the [P] term (in this case, [P] = binary complex of TR-Rev-DR2 and HGS) constant at 11.6 nm, yielding a Kd of 35 ± 4 nm. We caution that MBP-NCoR1–540 preparations were only ∼80% pure, thus the concentrations displayed in Fig. 13 are best estimates based on a determination with the Bradford assay. Nonetheless, the measured Kd is similar to that for the F-ID1 peptide (15 ± 11 nm), verifying this value and suggesting that studies using the ID1 peptide accurately reflect interactions with the full-length protein. Inclusion of Fe3+/Fe2+-heme at a 1.5:1 heme:HGS ratio did not affect the Kd (Fig. 13A). Another approach to assay the effect of heme is to hold the concentration of MBP-NCoR1–540 constant at 30 nm (∼Kd) and increase the [Fe3+/Fe2+-heme] (Fig. 13B). Both redox forms of heme cause a decrease in anisotropy at high concentrations, with no effect at concentrations close to that of FLRev-erbβ (100 nm). Conversion to Fe2+-heme by the addition of dithionite leads to <10% quenching of TR-Rev-DR2 fluorescence, likely accounting for the uniform decrease in anisotropy of these samples (Fig. 13B). Fitting the data with a four-parameter logistics curve generates IC50 values for Fe3+/Fe2+-heme of 1.0 ± 0.2 and 1.0 ± 0.1 μm, respectively. Comparing these data with the anisotropy values of free 20 nm TR-Rev-DR2, 20 nm TR-Rev-DR2 plus 100 nm FLRev-erbβHGS, and the same mixture but with saturating levels of MBP-NCoR1–540 (400 nm) shows that heme titration decreases the anisotropy to the level of free DNA, suggesting that HGS dissociates from TR-Rev-DR2 at high heme concentrations (Fig. 13C). These data agree with our EMSA results in which high levels of Fe3+-heme decrease the intensity of bands correlating to protein-DNA complexes.
FIGURE 13.

Fe3+/Fe2+-heme has a negligible effect on the affinity of MBP-NCoR1–540 for FLRev-erbβHGS bound to TR-Rev-DR2 determined with fluorescence anisotropy. A, assay mixtures containing 1 ng μl−1 poly(dI-dC), 20 nm TR-Rev-DR2, 100 nm FLRev-erbβHGS, and increasing concentrations of MBP (open triangles) or MBP-NCoR1–540 in the absence (filled circles) or presence of 150 nm Fe3+-heme (open circles) in 0.5× TNGD were incubated for 2 h at 4 °C prior to quantifying the anisotropy of TR-Rev-DR2 using a microplate reader. Fe2+-heme complexes were generated by the addition of 5 mm dithionite and the anisotropy read within 10 min to avoid oxidation (solid inverted triangles). Data represent the average of three independent experiments ± S.D. and are fit with Equation 1, holding the concentration of the TR-Rev-DR2·FLRev-erbβHGS complex constant at 11.6 nm (see “Results” for details). B, assay mixtures were prepared as described in A, except the concentration of MBP-NCoR1–540 was held constant at 30 nm (∼Kd) with increasing concentrations of Fe3+-heme (closed circles) or Fe2+-heme (open circles). Dithionite addition led to a <10% quenching of total fluorescence, which likely accounts for the uniform decrease in anisotropy (compare total fluorescence data in the absence (closed inverted triangles) and presence (open inverted triangles) of dithionite), which likely accounts for the uniform decrease in anisotropy. Raw anisotropy values are plotted and represent the average of triplicate experiments ± S.D. The data are fit with a four-parameter logistics curve to determine the IC50. C, heme-free samples of 20 nm TR-Rev-DR2 (Free DR2), 20 nm TR-Rev-DR2, and 100 nm FLRev-erbβHGS (DR2:Rβ), and the same mixture but with saturating levels of 400 nm MBP-NCoR1–540 (DR2:Rβ:NCoR1) ± 5 mm dithionite are shown for reference. The data represent the average of three experiments ± S.D.
In summary, the data presented in this section clearly demonstrate that heme and NCoR1 can bind simultaneously to Rev-erbβ. Furthermore, at stoichiometric levels, the binding of heme has no impact on the affinity of the receptor for NCoR1; only high [heme] causes the breakdown of the NCoR1·Rev-erbβ·DNA complex.
An Unknown Factor Present in Cell Extracts Mediates the Heme-dependent Interaction of Full-length NCoR1 with FLRev-erbβ
To extend our results to a system with full-length NCoR1 and to assess whether there is a missing cellular component that might induce the heme-dependent regulation of this interaction, we tested the effect of heme binding on the ability of FLRev-erbβ to interact with endogenous NCoR1 from HEK293 cell extracts in a co-IP experiment. Initially we were unable to detect co-precipitation of NCoR1 with endogenous Rev-erbβ. However, the levels of the endogenous receptor in HEK293 cells are low, i.e. <5 ng/100 μg of cell extract or <0.005% (based on comparing the relative intensities of bands for endogenous Rev-erbβ and known masses of recombinant full-length protein; data not shown). The addition of recombinant FLRev-erbβHGS to heme-depleted HEK293 extracts at a 100-fold excess to endogenous protein co-precipitated appreciable levels of NCoR1; however, this was unaffected by heme (Fig. 14A), suggesting that the interaction with NCoR1 in this assay is dependent on the concentration of Rev-erbβ, requiring high levels of the receptor to perhaps outcompete other NCoR1-binding proteins. Although FLRev-erbβHGS interacts with NCoR1 in heme-depleted extracts, the H568F and H568F/C384A variants paradoxically failed to interact with the corepressor (Fig. 14B). This result is especially puzzling considering that both recombinant H568F and H568F/C384A interacted with MBP-NCoR1–540 to the same extent as wild-type protein in our EMSA (Fig. 8). Additionally, it suggests that the low levels of heme present in SA-depleted extracts were sufficient to bind excess apoFLRev-erbβHGS and impart the heme-dependent NCoR1 interaction.
FIGURE 14.

Co-immunoprecipitation reveals heme-dependent interaction of Rev-erbβ with endogenous NCoR1 and MycBP2 from HEK293 cell extracts. A, co-IP of NCoR1 with FLRev-erbβHGS from soluble extracts depleted of heme. HEK293 cells were treated with 2 mm SA for 24 h and lysed, and a soluble extract was prepared. The heme content of those extracts was determined with an oxalic acid fluorescence assay, indicating control extracts contained 0.103 ± 0.005 nmol heme (mg protein)−1, whereas SA treatment lowered the heme content by 35% to 0.0669 ± 0.0003 nmol (mg protein)−1 (values represent the average of triplicate experiments ± S.D.). 1 mg of soluble extract was mixed with increasing concentrations of heme (denoted by a black triangle above the corresponding lanes; concentrations are 5, 10, and 20 μm) and 5 μg of recombinant FLRev-erbβHGS. Rev-erbβ LBD antibody was added to each sample, and the antibody-protein complexes were immunoprecipitated (IP), separated by SDS-PAGE, and electroblotted. Membranes were probed with NCoR1 C-20 (top panel) and Rev-erbβ QK-6 (bottom panel) antibodies. The input lane is 100 μg of cell extract (1/10th of the mass used in the IP) without recombinant protein added to demonstrate endogenous levels of NCoR1 and Rev-erbβ; the latter is undetectable under the exposure time necessary to observe the high levels of recombinant protein. B, a similar experiment as described for A, except the IPs did not contain heme, and recombinant FLRev-erbβHGS, H568F, and H568F/C384A were added to extracts. A control IP containing no recombinant protein was added to demonstrate the specificity of the NCoR1 interaction with the recombinant proteins (Cont.). C, HEK293FT cells were transiently transfected with pcDNA3.1(+) parent vector as a control, pcDNA3.1(+)-FLRev-erbβ for overexpression of wild-type Rev-erbβ (wt), or the analogous expression vector for the H568F variant. Rev-erbβ LBD antibody was added to the cell extracts, and the level of NCoR1 co-purifying with Rev-erbβ was assessed with immunoblotting. Input is 50 μg of extract (1/20th of the mass used in the IP) and Lane R = 50 ng of recombinant FLRev-erbβHGS. D, Coomassie-stained SDS-PAG demonstrating high molecular weight bands associated with wild-type FLRev-erbβMGC versus the H568F/C384A mutant. The asterisk indicates the location of the gel that was excised from the wild-type and H568F/C384A IP lanes, and those proteins within the gel slabs were identified with mass spectrometry. 5 μg each of pure FLRev-erbβMGC (wt) and H568F/C384A are shown for reference (denoted by black bars above the appropriate lanes). E, proteins with high total spectrum counts from the MS analysis are listed for both the wild-type and H568F/C384A samples. F, a similar IP as described in B, except the membrane was probed with a MycBP2 antibody showing a modest heme dependence to the MycBP2/Rev-erbβ interaction. The absence of MycBP2 in the control lane (Cont.) lacking recombinant protein indicates that the interaction of recombinant Rev-erbβ with the E3 ligase is specific.
To ensure that the use of recombinant proteins in the aforementioned IPs did not lead to artifactual data, we carried out a similar IP but overexpressed wild-type and H568F Rev-erbβ in HEK293FT cells. In addition, the proteasome inhibitor MG-132 was added to cells prior to lysis to increase Rev-erbβ levels (later we describe that MG-132 rescues Rev-erbβ from degradation, implicating the proteasome in Rev-erbβ turnover). In accord with the results using recombinant H568F, wild-type Rev-erbβ immunoprecipitated higher levels of NCoR1 than did the H568F variant (Fig. 14C). Of note, when comparing the relative intensities of the bands for overexpressed wild-type and H568F Rev-erbβ in extracts versus 50 ng of recombinant protein, we can estimate that the levels of Rev-erbβ in these cells is also >100-fold higher than the endogenous protein. In any case, these data disagree with our binding studies carried out with purified components, suggesting that another cellular factor in HEK293 extracts mediates the apparent heme-dependent NCoR1 interaction with Rev-erbβ under these conditions, including high receptor concentrations and high detergent (from the cell lysis buffer) levels.
In an attempt to identify the component that mediates the heme-dependent recruitment of NCoR1 to Rev-erbβ, the co-IP experiment described in Fig. 14B was scaled up 2-fold, and the precipitated complexes were separated by SDS-PAGE (Fig. 14D). At least one high molecular mass band of >250 kDa appeared to bind uniquely to the wild-type protein versus H568F/C384A. The portion of the gel encompassing this region was excised from both the wild-type and H568F/C384A lanes, and the proteins in both gel slabs were identified by mass spectrometry. As expected, NCoR1 was enriched in the gel slab from the wild-type co-IP over that of H568F/C384A, validating the assay (Fig. 14E). Interestingly, we also identified MycBP2 associated with both wild-type and H568F/C384A proteins (Fig. 14, D and E). A Western blot confirmed that 2.6 ± 1.0-fold (average of three replicate experiments ± S.D.) more MycBP2 co-precipitated with wild-type Rev-erbβ versus H568F/C384A (Fig. 14F), indicating that the interaction is modestly heme-dependent but not to the same degree as NCoR1. MycBP2 is an E3 ubiquitin-ligase that has been identified previously as interacting with and facilitating the degradation of Rev-erbα; however, in that study (94) it was shown that heme had no impact on the apparent rate of Rev-erbα degradation. Nonetheless, we chose to investigate whether heme had any influence on regulating Rev-erbβ degradation.
Heme Binding Inversely Controls Rev-erbβ Protein Levels, Likely through a Proteasome-dependent Pathway
Rev-erbα is a critical regulator of adipogenesis where its degradation is important for potentiating differentiation. Given that heme leads to a decrease in the steady-state level of Rev-erbα (85, 86), we tested whether heme binding affects the stability of Rev-erbβ. Indeed, steady-state levels of Rev-erbβ H568F are 5.7-fold higher than wild-type protein in transiently transfected HEK293 cells (Fig. 15, A and C). Likewise, the analogous H602F variant of Rev-erbα is at a 1.9-fold higher level than the wild type, indicating that heme promotes degradation of both Rev-erb isoforms; however this effect appears to be more pronounced for Rev-erbβ (Fig. 15, B and C). Similarly, the half-life of the H568F and H568F/C384A Rev-erbβ variants in CHX-treated cells increased ∼2-fold versus the wild type (Fig. 15, D and E). The addition of MG-132, a proteasome inhibitor, completely reversed the effects of CHX on the wild-type protein (Fig. 15, F and G), demonstrating that the proteasome is responsible for degradation of Rev-erbβ, as suggested for Rev-erbα (85). Thus, heme binding inversely controls the concentration of Rev-erbβ in the cell, likely by targeting the protein for proteasomal degradation. It remains unclear what aspect of this process is regulated by heme (perhaps the rate of ubiquitination, likely through MycBP2, or some other facet of the degradation pathway). In any case, it is tempting to speculate that whatever the degradation process regulated by heme, it may also play a role in mediating the interaction between Rev-erbβ and NCoR1 as observed in our co-IP.
FIGURE 15.

Western blotting analysis indicates that heme induces Rev-erbβ protein degradation through a proteasome-dependent pathway. A, a Western blot demonstrating steady-state levels of Rev-erbβ in HEK293 cells transiently transfected with pcDNA3.1(+) parent vector (Cont., control), or the corresponding expression vectors for wild-type Rev-erbβ (wt) or the H568F variant (H568F). Cells (60–70% confluent in 35-mm-diameter plates) were transfected with 4 μg of vector and incubated for 24 h, and a soluble extract was prepared in radioimmune precipitation assay buffer. 30 μg of each extract was electroblotted and probed with Rev-erbβ QK-6 antibody (top panel) or actin as a loading control (bottom panel). B, same experiment as described for A, except cells were transfected with expression vectors for wild-type Rev-erbα (wt) or the H602F variant (H602F). Membranes were probed with Rev-erbα RS-14 antibody (top panel) or actin as a loading control (bottom panel). C, the intensities of the bands in A and B were quantified by densitometry, and the mean intensities ± S.D. of three experiments were plotted. For the Rev-erbα analysis, the low level intensity of the band in the control lane was subtracted from the intensities of the bands for wild-type and H602F Rev-erbα from the same experiment. D, HEK293 cells transiently transfected with pcDNA3.1(+)-FLRev-erbβ (wild-type) or the analogous expression vector for H568F (top experiment) or the H568F/C384A variant (bottom experiment) were treated with 20 μg ml−1 CHX. Cells were lysed and soluble extracts prepared at the indicated time points post-CHX addition. 50 μg of each extract was electroblotted and probed with Rev-erbβ QK-6 or an actin antibody as a loading control. Lane R, 25 ng of FLRev-erbβHGS. E, the intensities of the bands in D were quantified by densitometry, and the mean intensities ± S.D. of three experiments were plotted. F, a replicate CHX half-life experiment as described in D for cells overexpressing wild-type Rev-erbβ. Lane R, 5 ng of FLRev-erbβHGS. G, the same cells as described in F were treated with either 20 μg ml−1 CHX or 20 μg ml−1 CHX plus 20 μm MG-132 (denoted by black bars above the appropriate lanes; three biological replicates of each set are depicted). Cells were incubated for an additional 4 h prior to preparing extracts and blotting with Rev-erbβ QK-6, indicating that the proteasome degrades Rev-erbβ (i.e. a direct comparison can be made between time points from F and G, as these experiments were carried out on the same day).
Heme Has a Minor Effect on Rev-erbβ Subcellular Localization
Given that heme binding regulates Rev-erbβ protein levels in the cell, we were curious as to whether the accumulation of apoRev-erbβ was specific to the cytosol or nucleus. Further, although Rev-erbα/β have been shown to be predominantly nuclear proteins (87), the effect of heme on Rev-erbβ subcellular localization has not been tested. We transiently transfected HEK293 cells with expression vectors for the wild-type, H568F, and H568F/C384A forms of Rev-erbβ, fractionated the cells into cytosolic and nuclear compartments, and determined the relative amounts of Rev-erbβ in each fraction (Fig. 16A). Extracts from cells that had not been transfected with a Rev-erbβ expression vector yielded undetectable levels of endogenous protein under similar Western blotting conditions and x-ray film exposure times (data not shown). The purity of the fractions was determined by blotting for compartment-specific proteins including Hsp90 for the cytosol and lamin B1 and NCoR1 for the nucleus (Fig. 16B). Wild-type and heme axial ligand mutants were similarly distributed throughout the cytosol and nucleus, suggesting that heme binding does not regulate subcellular localization.
FIGURE 16.

Effects of heme on the subcellular localization and steady-state levels of Rev-erbβ. A, HEK293 cells transiently transfected with pcDNA3.1(+)-FLRev-erbβ, pcDNA3.1(+)-FLRev-erbβ-H568F, or pcDNA3.1(+)-FLRev-erbβ-H568F/C384A were fractionated into cytosolic (Cyto) and nuclear (Nuc) compartments, and the fractions were immunoblotted with Rev-erbβ LBD antibody; 25 ng of recombinant FLRev-erbβHGS, used as a positive control (lane R). The cytosolic and nuclear fractions are indicated by black bars above the corresponding lanes, and duplicate biological replicates (Rep.) are shown for each fraction. B, the purity of cytosolic and nuclear fractions assessed by probing the membranes described in A with antibodies against lamin B1 (nuclear envelope), NCoR1 (nucleus), and Hsp90 (cytosol). A representative blot is shown for each protein with duplicate biological replicates. C, the effect of altering intracellular heme on the distribution and steady-state level of endogenous Rev-erbβ. HEK293 cells were incubated with 1 mm SA for 42 h or left untreated as a control (indicated by black bars above the appropriate lanes). The blot is arranged as described in A, with duplicate biological replicates shown for each fraction; 5 ng of FLRev-erbβMGC as a positive control (lane R). Blots assessing the purity of the fractions are also depicted.
We also examined the effect of heme on the steady-state levels and cellular localization of endogenous Rev-erbβ in HEK293 cells. SA treatment for 42 h, which lowers heme levels from 0.095 ± 0.008 nmol mg protein−1 for control cells to 0.044 ± 0.001 nmol mg protein−1 (the average of three experiments ± S.D.) (Fig. 16C), leads to a 2.3-fold increase in cytosolic Rev-erbβ with little effect on protein levels in the nucleus (based on densitometry analysis of the bands in Fig 16C). These results are consistent with the increase in steady-state levels observed with the overexpressed H568F variant (Fig 15A). Further, lowering the intracellular heme did not have a pronounced effect on the overall distribution of Rev-erbβ between cellular compartments, in agreement with those studies in transiently transfected cells overexpressing wild-type Rev-erbβ and heme axial ligand variants (Fig. 16A).
Heme Does Not Affect Repressor Activity of Rev-erbβ, but It Enhances That of Rev-erbα
The conflicting data on the role of heme binding in recruitment of NCoR1 to Rev-erbβ in our co-IP and binding studies prompted us to test whether heme regulates the repressor activity of Rev-erbβ in the cell. Further, gene expression studies indicate that heme markedly stimulates the interaction of Rev-erbα with NCoR1 (16, 34). Thus, we tested the ability of wild-type Rev-erbβ and heme axial ligand mutants H568F, C384A, and H568F/C384A to repress the transcription of a luciferase reporter construct driven by the core Bmal1 promoter from Mus musculus encompassing both Rev-REs and representing −422 to +108 bp with respect to the transcription start site (pGL3Basic-Bmal1 (71)). It should be noted that the region of the Bmal1 promoter encompassing the proximal and distal Rev-REs is identical in mouse, rat, and human (13) and that both Rev-erbα/β isoforms are known to bind to the Bmal1 promoter and repress transcription in a complex with NCoR1 (6). After transient transfection with pGL3Basic-Bmal1, pCMV-βGal (as a transfection efficiency control) and wild-type or mutant Rev-erbβ expression vector, HEK293 cells were incubated for 24 h, lysed, and assayed for repressor activity expressed as fold change compared with the reporter control (Fig. 17A). Transfection of 0.5 μg of wild-type Rev-erbβ vector repressed luciferase expression to 0.33 ± 0.04-fold of the control, and increasing the amount of Rev-erbβ vector to 2.5 μg enhanced repression to 0.072 ± 0.007-fold relative to the control, consistent with higher levels of Rev-erbβ in those cells (Fig. 17B, compare lanes 1–3). The observed repression is strictly dependent on overexpressed Rev-erbβ because undetectable levels of endogenous protein are visible in the control lane of the immunoblot. Transfection of 0.5 μg of H568F, C384A, and H568F/C384A vectors repressed luciferase expression to 0.41 ± 0.05-, 0.36 ± 0.01-, and 0.42 ± 0.06-fold, respectively, indicating that the mutations have a consistent but minor inhibitory effect on the repressor activity of Rev-erbβ. Further, protein levels of axial ligand variants are similar to the wild type (Fig. 17B, compare lane 2 with lanes 4–6), indicating that repression of the Bmal1 promoter depends on the concentration of Rev-erbβ and not on the ability of the receptor to bind heme.
FIGURE 17.

Heme binding enhances the repressor activity of Rev-erbα but has a negligible effect on Rev-erbβ repression of the Bmal1 promoter. A, HEK293 cells were transfected with 0.1 μg of β-galactosidase control vector, 0.2 μg of pGL3Basic-Bmal1 luciferase reporter, and varying masses of pcDNA3.1(+)-FLRev-erbβ wild-type (wt) or variant expression vectors (indicated below the graph). 24 h after transfection, luciferase activity was measured as described under “Experimental Procedures.” Data are normalized to β-galactosidase activity and are represented as -fold change in luciferase activity based on the reporter control. Three biological replicates were performed with mean ± S.D. shown. B, Western blotting analysis of 50 μg of soluble extracts from a representative replicate experiment described in A, probed with Rev-erbβ QK-6 antibody (top panel), or actin (bottom panel) as a loading control. 5 ng of recombinant FLRev-erbβHGS as a positive control (lane R). A nonspecific reactive band (NS) and a potential Rev-erbβ degradation product (Deg) are indicated with arrows. C, a similar experiment as described in A, except cells were co-transfected with the β-galactosidase control vector, Bmal1 luciferase reporter, and varying masses of pcDNA3.1(+)-Rev-erbα (wt) or the corresponding expression vector for the H602F variant (indicated below graph). Bars represent the average ± S.D. of three biological replicates.
In the repression assays (Fig. 17B), we did not observe the higher steady-state protein levels of the heme axial ligand variants relative to the wild type described in Fig. 15A. Perhaps this is because of a much higher expression level of Rev-erbβ due to the increased amount of Rev-erbβ expression vector used in the assay described in Fig. 15A. Furthermore, it is likely that there is a subtle balance between the rate of Rev-erbβ degradation and the amount of the NR such that the stabilization is observed only in cells expressing high levels of Rev-erbβ or depleted of heme for long periods of time. Differences in cell lysis reagents between studies may also affect the amount of Rev-erbβ extraction. In any case, the nearly equivalent levels of wild-type Rev-erbβ and heme-binding variants in the repression assays underscore the conclusion that it is the concentration of Rev-erbβ in cells that dictates repression of the reporter and not its ability to bind heme.
A similar experiment was performed with cells co-transfected with 0.5 μg of expression vectors for wild-type or H602F Rev-erbα (Fig. 17C). The wild-type Rev-erbα vector represses luciferase expression to 0.53 ± 0.07-fold of the control versus the H602F mutant with a value of 0.82 ± 0.15-fold. Similarly, transfection of 2.5 μg wild-type Rev-erbα vector enhances repression to 0.19 ± 0.02-fold of the control, whereas the same mass of H602F vector leads to 0.40 ± 0.03-fold repression. These data agree with previously published results for similar reporter-based assays with Rev-erbα and the H602F variant (16, 34) and suggest that heme enhances Rev-erbα repressor activity to a greater degree than Rev-erbβ. The results also indicate that heme binding is only partially responsible for Rev-erbα repressor activity. It may be that Rev-erbα/β competition with the transcriptional activator, human retinoic acid receptor-related orphan receptor, at Rev-REs in the Bmal1 promoter is predominantly responsible for the observed repression. Perhaps promoter elements with closely spaced Rev-REs, e.g. those resembling the canonical Rev-DR2, are subject to higher degrees of NCoR1 contribution to repression. Another interpretation is that the reporter system is insensitive to regulation by endogenous NCoR1 due to overexpression of the promoter element and Rev-erb NRs. In any case, although heme binding is dispensable for Rev-erbβ repressor activity, it modestly stimulates that of Rev-erbα.
Discussion
Determination of the Properties of FLRev-erbβ and Demonstration That All Its Domains Are Intact
To test our hypothesis related to the discrepancy between cellular and in vitro studies, we developed a method to obtain the full-length receptor and showed that it binds all of its ligands, e.g. heme, CO, DNA, and NCoR1 (both a peptide and a construct containing all three NR-binding domains), with high affinity. In doing so, our work represents the first report of a homogeneous preparation of an active full-length Rev-erb NR and legitimizes the use of the construct to test the effect of heme on its functional properties.
FLRev-erbβ purifies as a homodimer, in accord with studies of the LBD (apoRev-erbβ and the Rev-erbα LBD·ID1 complex) (27, 29) and DBD (Rev-erbα DBD bound to the Rev-DR2 promoter element) (80, 81). FLRev-erbβ binds with high affinity (K0.5 values <100 nm) and cooperatively to DNA Rev-REs, yielding Hill coefficients between 1.6 and 2.0 for interactions with the TR/FAM-Rev-DR2 and FAM-Bmal1 promoter. These results are similar to those of THRβ DBD binding as a homodimer to an inverted palindrome (with a K0.5 of 130 nm) (88) and of the isolated Rev-erbβ DBD binding to Rev-DR2 with an apparent K0.5 of 270 nm (82).
FLRev-erbβ also tightly binds heme, with Kd values below 30 nm for the complex between FLRev-erbβMGC/HGS and either ferric or ferrous heme, values that are similar to the low nm Kd value for the complex between heme and thiol-reduced Rev-erbβ LBD (38). Furthermore, the UV-visible spectra of the heme·FLRev-erbβ complexes are nearly identical to those of heme·LBD (38) (see Table 2). Thus, the LBD in the context of the full-length protein retains its heme coordination environment and its high affinity for heme. Furthermore, because these Kd values are below the 0.1–0.2 μm concentration of the intracellular exchangeable heme pool (78, 79), the receptors would be expected to exist predominantly within the cell in the heme-bound state. On the other hand, the H568F and H568F/C384A variants, with 1000-fold lower heme affinity than the wild-type protein, would exist as apoproteins within the cell.
In accord with the results of equilibrium titrations, heme dissociates at least 600-fold faster from the H568F and H568F/C384A variants relative to wild-type protein. The heme dissociation rates correlate with the role of heme in a particular protein, from relatively fast (10−2-10−3 s−1) off-rate constants for heme sensors and transporters such as HRI and BSA, which must bind and release heme quickly as metabolic conditions (such as heme availability) change to 10−4-10−7 s−1 for hemoproteins that interact with diatomic gases (75, 89). Thus, the slow Fe3+-heme dissociation rate constant for FLRev-erbβ (0.3 day−1 at 20 °C) is more akin to those for globins than those for heme sensors, indicating that Rev-erbβ would be ineffective in its proposed (16) role as a heme sensor. The instability of wild-type FLRev-erbβ during the long off-rate measurements prevented us from performing them at physiological temperatures; nonetheless, increasing the temperature from 20 to 37 °C increased the rate of heme dissociation from myoglobin by ∼3-fold (60). Assuming a similar Arrhenius relationship for Rev-erbβ, the rate constant (10−5 s−1) for heme dissociation remains well within the range of proteins like globins that cannot readily exchange heme.
Related to the potential role of Rev-erbβ in diatomic gas binding, both Rev-erb isoforms bind NO and CO, and our laboratory has shown avid CO binding (Kd = 6–60 nm) (Ref. 38 and herein). Although CO is reported to have little effect on Rev-erbβ repressor activity (36), one study reports that NO negligibly affects Rev-erbα repression (34), whereas another indicates that NO relieves Rev-erbα/β repression of target genes by 2–4 fold (36). This effect of repression could occur by disrupting interactions with corepressors (like NCoR1) or proteins that regulate steady-state levels of Rev-erbβ. Further studies are needed to characterize the interactions of NO and CO with Rev-erb and their potential effects on activity.
The FLRev-erbβ construct tightly binds an NCoR1 ID1 peptide (Kd = 15 nm) with similar affinity to that of the isolated Rev-erbα LBD (27). Thus, in sum, our experiments validate the idea that the functions of the LBD and the DBD are all intact in our FLRev-erbβ construct. This is important, because few full-length NRs have been expressed in an active state. Furthermore, although the two subunits of the dimer (either full-length or DBDs) exhibit cooperative DNA binding, each of the domains of FLRev-erbβ apparently act as independent modular units.
An Unknown Factor Present in Cell Extracts Mediates the Effects of Heme on the Interaction of Rev-erbβ and NCoR1
Although both the ID1 and ID3 motifs of NCoR1 are important for interacting with Rev-erbα (83), the peptide encompassing only ID1 appears to accurately mimic interactions between FLRev-erbβ and the MBP-NCoR1–540 construct, which comprises all three NR interaction domains. Furthermore, gel shift and fluorescence anisotropy assays show that the apo form of FLRev-erbβ, as well as variants that poorly bind heme, interacts with MBP-NCoR1–540 and promoter DNA (Rev-DR2 or the core Bmal1 promoter). Also, the addition of Fe2+/Fe3+-heme only marginally affects the affinity of FLRev-erbβ for NCoR1.
On the other hand, unlike wild-type Rev-erbβ, the H568F and H568F/C384A variants (whether added as purified recombinant protein or expressed in the cell) failed to interact with NCoR1 in extracts, reminiscent of a previous co-IP study with the Rev-erbα H602F variant (16). Furthermore, we could only co-immunoprecipitate NCoR1 when Rev-erbβ was in >100-fold excess of endogenous levels. It is likely that this dose effect results from the competition of Rev-erbβ with other NCoR1-binding proteins under the conditions of this assay.
These combined results strongly suggest that an additional cellular factor present in HEK293 extracts is required in order to observe the effect of heme on the interaction between NCoR1 and Rev-erbβ, at least in the context of the co-IP assay. Our MS/bioinformatics studies suggest that one factor is the proteasomal E3 ligase MycBP2. For example, binding of heme to Rev-erbβ may enhance its ubiquitination by MycBP2. Such interactions may also be directly or indirectly affected by a post-translational modification. For example, glycogen synthase kinase 3β phosphorylates two conserved serine residues in the A/B region of Rev-erbα, stabilizing the protein; however the effect of heme on the interaction of these proteins has not been explored (90). Acetylation is another potential post-translational modification regulated by heme binding. The histone acetyltransferase Tip60 acetylates a conserved Rev-erbβ lysine within a consensus RXKK sequence, abrogating repression of the apoC-III gene. Repressor activity is restored by HDAC1, which interacts with Rev-erbβ and deacetylates the regulatory lysine (91). In light of our results, it is imperative to determine whether heme affects the interaction of Rev-erbβ with Tip60 or HDAC1.
Another possibility is that heme regulates the interaction of Rev-erbβ with a protein in HEK293 extracts that regulates NCoR1 recruitment. One possibility is ZNHIT-1, a zinc-finger HIT domain protein known to interact with the Rev-erbβ LBD through a FXXLL motif similar to the LXXLL motif found in NR coactivators. ZNHIT-1 binds Rev-erbβ on the apoC-III promoter and inhibits its repressor activity; whether ZNHIT-1 accomplishes this through competitive binding with NCoR1 has not been tested (92). Further, because heme, ZNHIT-1, and NCoR1 bind to the LBD and have differential effects on repression, it will be critical to ascertain whether heme binding affects the interaction of Rev-erbβ or NCoR1 with ZNHIT-1.
Cellular Studies of the Role of Heme in Regulating Rev-erbβ Repressor Activity and Turnover
The Rev-erbα H602F variant, which is deficient in heme binding, is unable to repress transcription or to interact with NCoR1 at the Bmal1 promoter (16, 34). In part, these results laid the groundwork for the heme-dependent NCoR1 recruitment model described in the introduction to this work. We replicated those results, demonstrating that heme binding enhances Rev-erbα repressor activity by ∼2-fold over basal repression by the apoprotein. However, surprisingly, although the Rev-erbβ H568F and H568F/C384A variants are highly deficient in binding heme, they still interact with NCoR1 and DNA (Fig. 8) and retain repressor activity in promoter fusion studies (Fig. 17), providing consistency between in vitro and cell-based experiments in indicating that heme has different effects on the α- versus β-isoforms of Rev-erb. Again, the co-IP study demonstrating that H568F failed to bind NCoR1 provides the only conflicting data that we observed against a model in which heme binding is dispensable for Rev-erbβ repressor activity and an interaction with NCoR1.
Heme-dependent regulation of the proteolysis/stability of Rev-erb NRs would be important in at least two physiological processes: adipogenesis and the circadian cycle. Although peroxisome proliferator-activated receptor γ induces the high levels of Rev-erbα necessary for the early stages of preadipocyte differentiation (93), repression of peroxisome proliferator-activated receptor γ2 and proteasomal degradation of Rev-erbα are required for later differentiation stages (85). Heme levels increase throughout adipogenesis at a rate closely matching that of Rev-erbα degradation. Similarly, decreasing cellular heme levels with 3-amino-1,2,4-triazole increases the steady-state levels of Rev-erbα in adipocytes (86). However, heme did not affect degradation of Rev-erbα in cells treated with the translation inhibitor cycloheximide (94).
The role of heme in controlling the proteolysis of Rev-erbβ is consistent with prior results on other proteins that function in the negative limb of the circadian clock (94–96), where Rev-erbα and Period (Per) have been shown to undergo ubiquitination and proteasomal degradation. In this process, Bmal1-CLOCK heterodimers activate the transcription of clock-controlled genes, including Rev-erbα/β, Per, and Cry. The binding of heme to the HRM of Period 2 (Per2) enhances its proteasomal degradation in immortalized cells (97). Heme also has been shown to dampen Per2 rhythm in mouse suprachiasmatic nucleus explants (98). Per forms a heterodimeric complex with Cry that inhibits the transcriptional activator function of Bmal1-CLOCK, whereas Rev-erb NRs feed back, repressing the transcription of Bmal1 and CLOCK, which also has been shown to bind heme (99), although this binding is claimed to be nonspecific (100). Significantly, the CLOCK homolog in the forebrain, NPAS2, is also a hemoprotein that binds CO, which ultimately disrupts NPAS2 DNA binding (101), although heme binds to the CO-sensing PAS-A domain with bis-His ligation and not via an HRM (102). Thus, heme binding to clock proteins appears to represent an important mechanism in circadian rhythm maintenance.
The circadian clock interconnects various aspects of heme and cellular metabolism. The expression of key heme proteins involved in electron transport and xenobiotic metabolism (P450s) is entrained with the circadian rhythm (103). Furthermore, heme biosynthesis itself is controlled by the molecular clock through the regulation of expression of the rate-determining enzyme aminolevulinic acid synthase 1 (ALAS1). The Bmal1-NPAS heterodimer and PGC-1α (a coactivator of nuclear receptors and regulator of metabolism), which are both under circadian control, activate expression of ALAS1 and Rev-erbα, with Rev-erbα completing the feedback loop by binding heme as a ligand, recruiting NCoR1, and repressing expression of Bmal1 and PGC-1α (34, 104–107). The interconnecting regulatory loops of heme biosynthesis, the molecular clock, and metabolism are exemplified in a recent study demonstrating that a diet high in iron increases the amplitude of circadian PGC-1α expression, which raises intracellular heme levels. In turn, elevated heme leads to NCoR1 recruitment to Rev-erbα and repression of key enzymes involved in gluconeogenesis, ultimately lowering serum glucose concentrations. Thus, dietary signals like iron control metabolism through the regulation of core clock protein expression and heme biosynthesis (108).
Conclusion
Many of the key studies supporting a role for heme in recruiting NCoR1 to Rev-erb NRs are based on experiments with the α-isoform (16, 21, 34, 86, 109), which is more extensively studied, likely because of its discovery prior to Rev-erbβ and its unique context in the genome being transcribed on the opposite strand of the gene coding for THRα on chromosome 17 (2, 9), whereas Rev-erbβ is encoded on chromosome 3. Because the DBD and LBD of the two receptors share a high level of homology and nearly identical heme binding properties (9, 36), it has been assumed that the mechanism of heme regulation would be the same for both isoforms. Further, several reports demonstrate that Rev-erbβ and Rev-erbα share a significant overlap in the genes they repress, especially those involved in lipid homeostasis and the molecular clock, namely Bmal1 (6, 7, 110). This redundancy likely provides protection against clock and metabolic dysfunction, as knockdown or deletion of one Rev-erb isoform has only modest effects on these processes, whereas loss of both leads to marked dyslipidemia and loss of rhythmic clock gene expression (6, 7). In this study we have shown that Rev-erbβ behaves like Rev-erbα in terms of heme-dependent NCoR1 binding in co-IPs, Nonetheless, heme appears to have a more pronounced effect on stabilizing Rev-erbβ H568F versus Rev-erbα H602F, but it also enhances Rev-erbα repressor activity to a greater degree than the β-isoform (although this effect is modest). Thus, there seems to be subtle differences in the regulatory role of heme in terms of Rev-erb turnover and repressor activity. Two recent reports demonstrate that Rev-erbα and -β bind at or near the same Rev-REs in the promoters of target genes (6, 7), provoking the question, do the isoforms form heterodimers that cooperatively repress transcription? Perhaps, the effects of heme and NCoR1 act on a Rev-erbα/β heterodimer. Intriguingly, both Rev-erbα and Rev-erbβ repress the transcription of Rev-erbα; however, neither receptor binds to the Rev-erbβ promoter (6). Thus, heme-induced degradation may act as a post-translational mechanism to control the protein levels of Rev-erbβ, maintaining its ability to rhythmically repress the transcription of genes in metabolic pathways, thereby synchronizing metabolism with the molecular clock.
Author Contributions
E. L. C., N. G., and S. W. R. designed the study, and E. L. C. and N. G. performed the experiments. E. L. C. and S. W. R. composed the manuscript, and all authors reviewed and approved its content.
Acknowledgments
We thank Yanil Rodriguez-Breton, Holly Cooperrider, and Janet Rhee for assistance with some of the experiments, and Dr. Angela Fleischhacker for helpful discussions. We thank the University of Michigan Proteomics Core for data interpretation and discussion and the Institutional Mass Spectrometry Laboratory, University of Texas Health Science Center at San Antonio, for performing LC-MS/MS analysis.
This work was supported by National Institutes of Health Grants 5R01-HL-102662 (to S. W. R.) and 1F32HL114150 (to E. L. C.) and by Grant T32ES007062 from the NIEHS (to E. L. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- NR
- nuclear receptor
- bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- CHX
- cycloheximide
- IP
- immunoprecipitate or immunoprecipitation
- DBD
- DNA-binding domain
- DMSO
- dimethyl sulfoxide
- F-ID1
- fluorescein-labeled NCoR1 ID1 peptide
- FAM
- fluorescein amidite
- GB1
- protein G, B1 domain
- HGS
- Rev-erbβ sequence corresponding to the human genome sequence
- hPL
- human placental lactogen
- HPLC
- high pressure liquid chromatography
- HRI
- heme-regulated inhibitor
- HRM
- heme regulatory motif
- LIC
- ligation-independent cloning
- LBD
- ligand-binding domain
- MBP
- maltose-binding protein
- MGC
- Rev-erbβ sequence corresponding to the mammalian gene collection
- MWCO
- molecular weight cutoff
- NCoR1
- nuclear receptor corepressor 1
- Rev-RE
- Rev-erb response element
- SA
- succinylacetone
- T3
- triiodothyronine
- TCEP
- tris(2-carboxyethyl)phosphine
- TEV
- tobacco etch virus
- THR
- thyroid hormone receptor
- TR
- Texas Red
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine.
References
- 1. Aranda A., and Pascual A. (2001) Nuclear hormone receptors and gene expression. Physiol. Rev. 81, 1269–1304 [DOI] [PubMed] [Google Scholar]
- 2. Lazar M. A., Hodin R. A., Darling D. S., and Chin W. W. (1989) A novel member of the thyroid/steroid hormone receptor family is encoded by the opposite strand of the rat c-erbAα transcriptional unit. Mol. Cell. Biol. 9, 1128–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dumas B., Harding H. P., Choi H. S., Lehmann K. A., Chung M., Lazar M. A., and Moore D. D. (1994) A new orphan member of the nuclear hormone receptor superfamily closely related to Rev-Erb. Mol. Endocrinol. 8, 996–1005 [DOI] [PubMed] [Google Scholar]
- 4. Enmark E., Kainu T., Pelto-Huikko M., and Gustafsson J. A. (1994) Identification of a novel member of the nuclear receptor superfamily which is closely related to Rev-ErbA. Biochem. Biophys. Res. Commun. 204, 49–56 [DOI] [PubMed] [Google Scholar]
- 5. Peña-de-Ortiz S., and Jamieson G. A. Jr. (1997) Molecular cloning and brain localization of HZF-2α, a new member of the Rev-erb subfamily of orphan nuclear receptors. J. Neurobiol. 32, 341–358 [DOI] [PubMed] [Google Scholar]
- 6. Bugge A., Feng D., Everett L. J., Briggs E. R., Mullican S. E., Wang F., Jager J., and Lazar M. A. (2012) Rev-erbα and Rev-erbβ coordinately protect the circadian clock and normal metabolic function. Genes Dev. 26, 657–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cho H., Zhao X., Hatori M., Yu R. T., Barish G. D., Lam M. T., Chong L. W., DiTacchio L., Atkins A. R., Glass C. K., Liddle C., Auwerx J., Downes M., Panda S., and Evans R. M. (2012) Regulation of circadian behaviour and metabolism by REV-ERB-α and REV-ERB-β. Nature 485, 123–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Retnakaran R., Flock G., and Giguère V. (1994) Identification of RVR, a novel orphan nuclear receptor that acts as a negative transcriptional regulator. Mol. Endocrinol. 8, 1234–1244 [DOI] [PubMed] [Google Scholar]
- 9. Bonnelye E., Vanacker J. M., Desbiens X., Begue A., Stehelin D., and Laudet V. (1994) Rev-erbβ, a new member of the nuclear receptor superfamily, is expressed in the nervous system during chicken development. Cell Growth Differ. 5, 1357–1365 [PubMed] [Google Scholar]
- 10. Zvonic S., Ptitsyn A. A., Conrad S. A., Scott L. K., Floyd Z. E., Kilroy G., Wu X., Goh B. C., Mynatt R. L., and Gimble J. M. (2006) Characterization of peripheral circadian clocks in adipose tissues. Diabetes 55, 962–970 [DOI] [PubMed] [Google Scholar]
- 11. Torra I. P., Tsibulsky V., Delaunay F., Saladin R., Laudet V., Fruchart J. C., Kosykh V., and Staels B. (2000) Circadian and glucocorticoid regulation of Rev-erbα expression in liver. Endocrinology 141, 3799–3806 [DOI] [PubMed] [Google Scholar]
- 12. Balsalobre A., Damiola F., and Schibler U. (1998) A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell 93, 929–937 [DOI] [PubMed] [Google Scholar]
- 13. Preitner N., Damiola F., Lopez-Molina L., Zakany J., Duboule D., Albrecht U., and Schibler U. (2002) The orphan nuclear receptor REV-ERBα controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell 110, 251–260 [DOI] [PubMed] [Google Scholar]
- 14. Crumbley C., and Burris T. P. (2011) Direct regulation of CLOCK expression by REV-ERB. PLoS One 6, e17290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Adelmant G., Bègue A., Stéhelin D., and Laudet V. (1996) A functional Rev-erbα responsive element located in the human Rev-erbα promoter mediates a repressing activity. Proc. Natl. Acad. Sci. U.S.A. 93, 3553–3558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yin L., Wu N., Curtin J. C., Qatanani M., Szwergold N. R., Reid R. A., Waitt G. M., Parks D. J., Pearce K. H., Wisely G. B., and Lazar M. A. (2007) Rev-erbα, a heme sensor that coordinates metabolic and circadian pathways. Science 318, 1786–1789 [DOI] [PubMed] [Google Scholar]
- 17. Raspé E., Duez H., Mansén A., Fontaine C., Fiévet C., Fruchart J. C., Vennström B., and Staels B. (2002) Identification of Rev-erbα as a physiological repressor of apoC-III gene transcription. J. Lipid Res. 43, 2172–2179 [DOI] [PubMed] [Google Scholar]
- 18. Coste H., and Rodríguez J. C. (2002) Orphan nuclear hormone receptor Rev-erbα regulates the human apolipoprotein CIII promoter. J. Biol. Chem. 277, 27120–27129 [DOI] [PubMed] [Google Scholar]
- 19. Ramakrishnan S. N., Lau P., Burke L. J., and Muscat G. E. (2005) Rev-erbβ regulates the expression of genes involved in lipid absorption in skeletal muscle cells: evidence for cross-talk between orphan nuclear receptors and myokines. J. Biol. Chem. 280, 8651–8659 [DOI] [PubMed] [Google Scholar]
- 20. Gibbs J. E., Blaikley J., Beesley S., Matthews L., Simpson K. D., Boyce S. H., Farrow S. N., Else K. J., Singh D., Ray D. W., and Loudon A. S. (2012) The nuclear receptor REV-ERBα mediates circadian regulation of innate immunity through selective regulation of inflammatory cytokines. Proc. Natl. Acad. Sci. U.S.A. 109, 582–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chandra V., Mahajan S., Saini A., Dkhar H. K., Nanduri R., Raj E. B., Kumar A., and Gupta P. (2013) Human IL10 gene repression by Rev-erbα ameliorates Mycobacterium tuberculosis clearance. J. Biol. Chem. 288, 10692–10702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Carter E. L., and Ragsdale S. W. (2014) Modulation of nuclear receptor function by cellular redox poise. J. Inorg. Biochem. 133, 92–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jin L., and Li Y. (2010) Structural and functional insights into nuclear receptor signaling. Adv. Drug Deliv. Rev. 62, 1218–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cohen R. N., Brzostek S., Kim B., Chorev M., Wondisford F. E., and Hollenberg A. N. (2001) The specificity of interactions between nuclear hormone receptors and corepressors is mediated by distinct amino acid sequences within the interacting domains. Mol. Endocrinol. 15, 1049–1061 [DOI] [PubMed] [Google Scholar]
- 25. Burris T. P., Solt L. A., Wang Y., Crumbley C., Banerjee S., Griffett K., Lundasen T., Hughes T., and Kojetin D. J. (2013) Nuclear receptors and their selective pharmacologic modulators. Pharmacol. Rev. 65, 710–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mottis A., Mouchiroud L., and Auwerx J. (2013) Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes Dev. 27, 819–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Phelan C. A., Gampe R. T. Jr., Lambert M. H., Parks D. J., Montana V., Bynum J., Broderick T. M., Hu X., Williams S. P., Nolte R. T., and Lazar M. A. (2010) Structure of Rev-erbα bound to N-CoR reveals a unique mechanism of nuclear receptor-co-repressor interaction. Nat. Struct. Mol. Biol. 17, 808–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Watson P. J., Fairall L., and Schwabe J. W. (2012) Nuclear hormone receptor co-repressors: structure and function. Mol. Cell. Endocrinol. 348, 440–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Woo E. J., Jeong D. G., Lim M. Y., Kim S. J., Kim K. J., Yoon S. M., Park B. C., and Ryu S. E. (2007) Structural insight into the constitutive repression function of the nuclear receptor Rev-erbβ. J. Mol. Biol. 373, 735–744 [DOI] [PubMed] [Google Scholar]
- 30. Ramakrishnan S. N., Lau P., Crowther L. M., Cleasby M. E., Millard S., Leong G. M., Cooney G. J., and Muscat G. E. (2009) Rev-erbβ regulates the Srebp-1c promoter and mRNA expression in skeletal muscle cells. Biochem. Biophys. Res. Commun. 388, 654–659 [DOI] [PubMed] [Google Scholar]
- 31. Miyajima N., Horiuchi R., Shibuya Y., Fukushige S., Matsubara K., Toyoshima K., and Yamamoto T. (1989) Two erbA homologs encoding proteins with different T3 binding capacities are transcribed from opposite DNA strands of the same genetic locus. Cell 57, 31–39 [DOI] [PubMed] [Google Scholar]
- 32. Miyajima N., Kadowaki Y., Fukushige S., Shimizu S., Semba K., Yamanashi Y., Matsubara K., Toyoshima K., and Yamamoto T. (1988) Identification of two novel members of erbA superfamily by molecular cloning: the gene products of the two are highly related to each other. Nucleic Acids Res. 16, 11057–11074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reinking J., Lam M. M., Pardee K., Sampson H. M., Liu S., Yang P., Williams S., White W., Lajoie G., Edwards A., and Krause H. M. (2005) The Drosophila nuclear receptor E75 contains heme and is gas responsive. Cell 122, 195–207 [DOI] [PubMed] [Google Scholar]
- 34. Raghuram S., Stayrook K. R., Huang P., Rogers P. M., Nosie A. K., McClure D. B., Burris L. L., Khorasanizadeh S., Burris T. P., and Rastinejad F. (2007) Identification of heme as the ligand for the orphan nuclear receptors REV-ERBα and REV-ERBβ. Nat. Struct. Mol. Biol. 14, 1207–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang L., and Guarente L. (1995) Heme binds to a short sequence that serves a regulatory function in diverse proteins. EMBO J. 14, 313–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pardee K. I., Xu X., Reinking J., Schuetz A., Dong A., Liu S., Zhang R., Tiefenbach J., Lajoie G., Plotnikov A. N., Botchkarev A., Krause H. M., and Edwards A. (2009) The structural basis of gas-responsive transcription by the human nuclear hormone receptor REV-ERBβ. PLoS Biol. 7, e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kühl T., Wiβbrock A., Goradia N., Sahoo N., Galler K., Neugebauer U., Popp J., Heinemann S. H., Ohlenschläger O., and Imhof D. (2013) Analysis of Fe(III) heme binding to cysteine-containing heme-regulatory motifs in proteins. ACS Chem. Biol. 8, 1785–1793 [DOI] [PubMed] [Google Scholar]
- 38. Gupta N., and Ragsdale S. W. (2011) Thiol-disulfide redox dependence of heme binding and heme ligand switching in nuclear hormone receptor Rev-erbβ. J. Biol. Chem. 286, 4392–4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Qi Z., Hamza I., and O'Brian M. R. (1999) Heme is an effector molecule for iron-dependent degradation of the bacterial iron response regulator (Irr) protein. Proc. Natl. Acad. Sci. U.S.A. 96, 13056–13061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Igarashi J., Murase M., Iizuka A., Pichierri F., Martinkova M., and Shimizu T. (2008) Elucidation of the heme binding site of heme-regulated eukaryotic initiation factor 2α kinase and the role of the regulatory motif in heme sensing by spectroscopic and catalytic studies of mutant proteins. J. Biol. Chem. 283, 18782–18791 [DOI] [PubMed] [Google Scholar]
- 41. Zenke-Kawasaki Y., Dohi Y., Katoh Y., Ikura T., Ikura M., Asahara T., Tokunaga F., Iwai K., and Igarashi K. (2007) Heme induces ubiquitination and degradation of the transcription factor Bach1. Mol. Cell. Biol. 27, 6962–6971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Munakata H., Sun J. Y., Yoshida K., Nakatani T., Honda E., Hayakawa S., Furuyama K., and Hayashi N. (2004) Role of the heme regulatory motif in the heme-mediated inhibition of mitochondrial import of 5-aminolevulinate synthase. J. Biochem. 136, 233–238 [DOI] [PubMed] [Google Scholar]
- 43. Hon T., Hach A., Lee H. C., Cheng T., and Zhang L. (2000) Functional analysis of heme regulatory elements of the transcriptional activator Hap1. Biochem. Biophys. Res. Commun. 273, 584–591 [DOI] [PubMed] [Google Scholar]
- 44. Ishikawa H., Kato M., Hori H., Ishimori K., Kirisako T., Tokunaga F., and Iwai K. (2005) Involvement of heme regulatory motif in heme-mediated ubiquitination and degradation of IRP2. Mol. Cell 19, 171–181 [DOI] [PubMed] [Google Scholar]
- 45. Fleischhacker A. S., Sharma A., Choi M., Spencer A. M., Bagai I., Hoffman B. M., and Ragsdale S. W. (2015) The C-terminal heme regulatory motifs of heme oxygenase-2 are redox-regulated heme binding sites. Biochemistry 54, 2709–2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bagai I., Sarangi R., Fleischhacker A. S., Sharma A., Hoffman B. M., Zuiderweg E. R., and Ragsdale S. W. (2015) Spectroscopic studies reveal that the heme regulatory motifs of heme oxygenase-2 are dynamically disordered and exhibit redox-dependent interaction with heme. Biochemistry 54, 2693–2708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cáceres L., Necakov A. S., Schwartz C., Kimber S., Roberts I. J., and Krause H. M. (2011) Nitric oxide coordinates metabolism, growth, and development via the nuclear receptor E75. Genes Dev. 25, 1476–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bain D. L., Heneghan A. F., Connaghan-Jones K. D., and Miura M. T. (2007) Nuclear receptor structure: Implications for function. Annu. Rev. Physiol. 69, 201–220 [DOI] [PubMed] [Google Scholar]
- 49. Owens C. P., Du J., Dawson J. H., and Goulding C. W. (2012) Characterization of heme ligation properties of Rv0203, a secreted heme-binding protein involved in Mycobacterium tuberculosis heme uptake. Biochemistry 51, 1518–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stols L., Gu M., Dieckman L., Raffen R., Collart F. R., and Donnelly M. I. (2002) A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr. Purif. 25, 8–15 [DOI] [PubMed] [Google Scholar]
- 51. Zhou P., and Wagner G. (2010) Overcoming the solubility limit with solubility-enhancement tags: successful applications in biomolecular NMR studies. J. Biomol. NMR 46, 23–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Eschenfeldt W. H., Stols L., Millard C. S., Joachimiak A., and Donnelly M. I. (2009) A family of LIC vectors for high-throughput cloning and purification of proteins. Methods Mol. Biol. 498, 105–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Spencer A. L., Bagai I., Becker D. F., Zuiderweg E. R., and Ragsdale S. W. (2014) Protein/protein interactions in the mammalian heme degradation pathway: heme oxygenase-2, cytochrome P450 reductase, and biliverdin reductase. J. Biol. Chem. 289, 29836–29858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schneider C. A., Rasband W. S., and Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rial D. V., and Ceccarelli E. A. (2002) Removal of DnaK contamination during fusion protein purifications. Protein Expr. Purif. 25, 503–507 [DOI] [PubMed] [Google Scholar]
- 56. Dawson R. M., Elliott D. C., Elliott W. H., and Jones K. M. (eds) (1969) Data for Biochemical Research, 2nd Ed., Oxford University Press, Oxford [Google Scholar]
- 57. Copeland R. A. (2002) Protein-ligand binding equilibria, in Enzymes, pp 76–108, John Wiley & Sons, Inc., New York [Google Scholar]
- 58. Rish K. R., Swartzlander R., Sadikot T. N., Berridge M. V., and Smith A. (2007) Interaction of heme and heme-hemopexin with an extracellular oxidant system used to measure cell growth-associated plasma membrane electron transport. Biochim. Biophys. Acta 1767, 1107–1117 [DOI] [PubMed] [Google Scholar]
- 59. Antonini E., and Brunori M. (1971) Hemoglobin and Myoglobin in Their Reactions with Ligands, North-Holland Publishing Co., Amsterdam [Google Scholar]
- 60. Hargrove M. S., Singleton E. W., Quillin M. L., Ortiz L. A., Phillips G. N. Jr., Olson J. S., and Mathews A. J. (1994) His64(E7) → Tyr apomyoglobin as a reagent for measuring rates of hemin dissociation. J. Biol. Chem. 269, 4207–4214 [DOI] [PubMed] [Google Scholar]
- 61. Gasteiger E., Gattiker A., Hoogland C., Ivanyi I., Appel R. D., and Bairoch A. (2003) ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 31, 3784–3788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Suzuki H., Obara M., Kubo K., and Kanazawa T. (1989) Changes in the steady-state fluorescence anisotropy of N-iodoacetyl-N′-(5-sulfo-1-naphthyl)ethylenediamine attached to the specific thiol of sarcoplasmic reticulum Ca2+-ATPase throughout the catalytic cycle. J. Biol. Chem. 264, 920–927 [PubMed] [Google Scholar]
- 63. Mitkova A. V., Khopde S. M., and Biswas S. B. (2003) Mechanism and stoichiometry of interaction of DnaG primase with DnaB helicase of Escherichia coli in RNA primer synthesis. J. Biol. Chem. 278, 52253–52261 [DOI] [PubMed] [Google Scholar]
- 64. Bergen J. M., Kwon E. J., Shen T. W., and Pun S. H. (2008) Application of an environmentally sensitive fluorophore for rapid analysis of the binding and internalization efficiency of gene carriers. Bioconjug. Chem. 19, 377–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Voz M. L., Peers B., Belayew A., and Martial J. A. (1991) Characterization of an unusual thyroid response unit in the promoter of the human placental lactogen gene. J. Biol. Chem. 266, 13397–13404 [PubMed] [Google Scholar]
- 66. Wu Y., Xu B., and Koenig R. J. (2001) Thyroid hormone response element sequence and the recruitment of retinoid X receptors for thyroid hormone responsiveness. J. Biol. Chem. 276, 3929–3936 [DOI] [PubMed] [Google Scholar]
- 67. Morrison G. R. (1965) Fluorometric microdetermination of heme protein. Anal. Chem. 37, 1124–1126 [DOI] [PubMed] [Google Scholar]
- 68. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 69. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 70. Hogan K. A., Ravindran A., Podolsky M. A., and Glick A. B. (2013) The TGFβ1 pathway is required for NFκB-dependent gene expression in mouse keratinocytes. Cytokine 64, 652–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sato T. K., Panda S., Miraglia L. J., Reyes T. M., Rudic R. D., McNamara P., Naik K. A., FitzGerald G. A., Kay S. A., and Hogenesch J. B. (2004) A functional genomics strategy reveals Rora as a component of the mammalian circadian clock. Neuron 43, 527–537 [DOI] [PubMed] [Google Scholar]
- 72. Mammalian Gene Collection Program Team (2002) Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc. Natl. Acad. Sci. U.S.A. 99, 16899–16903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Marvin K. A., Reinking J. L., Lee A. J., Pardee K., Krause H. M., and Burstyn J. N. (2009) Nuclear receptors Homo sapiens Rev-erbβ and Drosophila melanogaster E75 are thiolate-ligated heme proteins which undergo redox-mediated ligand switching and bind CO and NO. Biochemistry 48, 7056–7071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ojha S., Hwang J., Kabil O., Penner-Hahn J. E., and Banerjee R. (2000) Characterization of the heme in human cystathionine β-synthase by X-ray absorption and electron paramagnetic resonance spectroscopies. Biochemistry 39, 10542–10547 [DOI] [PubMed] [Google Scholar]
- 75. Miksanova M., Igarashi J., Minami M., Sagami I., Yamauchi S., Kurokawa H., and Shimizu T. (2006) Characterization of heme-regulated eIF2α kinase: Roles of the N-terminal domain in the oligomeric state, heme binding, catalysis, and inhibition. Biochemistry 45, 9894–9905 [DOI] [PubMed] [Google Scholar]
- 76. Omura T., Sadano H., Hasegawa T., Yoshida Y., and Kominami S. (1984) Hemoprotein H-450 identified as a form of cytochrome P-450 having an endogenous ligand at the 6th coordination position of the heme. J. Biochem. (Tokyo) 96, 1491–1500 [DOI] [PubMed] [Google Scholar]
- 77. Igarashi J., Sato A., Kitagawa T., Yoshimura T., Yamauchi S., Sagami I., and Shimizu T. (2004) Activation of heme-regulated eukaryotic initiation factor 2α kinase by nitric oxide is induced by the formation of a five-coordinate NO-heme complex: Optical absorption, electron spin resonance, and resonance Raman spectral studies. J. Biol. Chem. 279, 15752–15762 [DOI] [PubMed] [Google Scholar]
- 78. Liu S. C., Zhai S., and Palek J. (1988) Detection of hemin release during hemoglobin S denaturation. Blood 71, 1755–1758 [PubMed] [Google Scholar]
- 79. Garrick M. D., Scott D., Kulju D., Romano M. A., Dolan K. G., and Garrick L. M. (1999) Evidence for and consequences of chronic heme deficiency in Belgrade rat reticulocytes. Biochim. Biophys. Acta 1449, 125–136 [DOI] [PubMed] [Google Scholar]
- 80. Zhao Q., Khorasanizadeh S., Miyoshi Y., Lazar M. A., and Rastinejad F. (1998) Structural elements of an orphan nuclear receptor-DNA complex. Mol. Cell 1, 849–861 [DOI] [PubMed] [Google Scholar]
- 81. Harding H. P., and Lazar M. A. (1995) The monomer-binding orphan receptor Rev-Erb represses transcription as a dimer on a novel direct repeat. Mol. Cell. Biol. 15, 4791–4802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Terenzi H., Cassia R. O., and Zakin M. M. (1996) Expression, purification, and functional analysis of the DNA binding domain of the nuclear receptor Rev-Erbβ. Protein Expr. Purif. 8, 313–318 [DOI] [PubMed] [Google Scholar]
- 83. Kim J. Y., Son Y. L., Kim J. S., and Lee Y. C. (2010) Molecular determinants required for selective interactions between the thyroid hormone receptor homodimer and the nuclear receptor corepressor N-CoR. J. Mol. Biol. 396, 747–760 [DOI] [PubMed] [Google Scholar]
- 84. Cohen R. N., Wondisford F. E., and Hollenberg A. N. (1998) Two separate NCoR (nuclear receptor corepressor) interaction domains mediate corepressor action on thyroid hormone response elements. Mol. Endocrinol. 12, 1567–1581 [DOI] [PubMed] [Google Scholar]
- 85. Wang J., and Lazar M. A. (2008) Bifunctional role of Rev-erbα in adipocyte differentiation. Mol. Cell. Biol. 28, 2213–2220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kumar N., Solt L. A., Wang Y., Rogers P. M., Bhattacharyya G., Kamenecka T. M., Stayrook K. R., Crumbley C., Floyd Z. E., Gimble J. M., Griffin P. R., and Burris T. P. (2010) Regulation of adipogenesis by natural and synthetic REV-ERB ligands. Endocrinology 151, 3015–3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Chopin-Delannoy S., Thénot S., Delaunay F., Buisine E., Begue A., Duterque-Coquillaud M., and Laudet V. (2003) A specific and unusual nuclear localization signal in the DNA binding domain of the Rev-erb orphan receptors. J. Mol. Endocrinol. 30, 197–211 [DOI] [PubMed] [Google Scholar]
- 88. Chen Y., and Young M. A. (2010) Structure of a thyroid hormone receptor DNA-binding domain homodimer bound to an inverted palindrome DNA response element. Mol. Endocrinol. 24, 1650–1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hargrove M. S., Barrick D., and Olson J. S. (1996) The association rate constant for heme binding to globin is independent of protein structure. Biochemistry 35, 11293–11299 [DOI] [PubMed] [Google Scholar]
- 90. Yin L., Wang J., Klein P. S., and Lazar M. A. (2006) Nuclear receptor Rev-erbα is a critical lithium-sensitive component of the circadian clock. Science 311, 1002–1005 [DOI] [PubMed] [Google Scholar]
- 91. Wang J., Liu N., Liu Z., Li Y., Song C., Yuan H., Li Y. Y., Zhao X., and Lu H. (2008) The orphan nuclear receptor Rev-erbβ recruits Tip60 and HDAC1 to regulate apolipoprotein CIII promoter. Biochim. Biophys. Acta 1783, 224–236 [DOI] [PubMed] [Google Scholar]
- 92. Wang J., Li Y., Zhang M., Liu Z., Wu C., Yuan H., Li Y. Y., Zhao X., and Lu H. (2007) A zinc finger HIT domain-containing protein, ZNHIT-1, interacts with orphan nuclear hormone receptor Rev-erbβ and removes Rev-erbβ-induced inhibition of apoCIII transcription. FEBS J. 274, 5370–5381 [DOI] [PubMed] [Google Scholar]
- 93. Fontaine C., Dubois G., Duguay Y., Helledie T., Vu-Dac N., Gervois P., Soncin F., Mandrup S., Fruchart J. C., Fruchart-Najib J., and Staels B. (2003) The orphan nuclear receptor Rev-Erbα is a peroxisome proliferator-activated receptor (PPAR) γ target gene and promotes PPARγ-induced adipocyte differentiation. J. Biol. Chem. 278, 37672–37680 [DOI] [PubMed] [Google Scholar]
- 94. Yin L., Joshi S., Wu N., Tong X., and Lazar M. A. (2010) E3 ligases Arf-bp1 and Pam mediate lithium-stimulated degradation of the circadian heme receptor Rev-erbα. Proc. Natl. Acad. Sci. U.S.A. 107, 11614–11619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Virshup D. M., Eide E. J., Forger D. B., Gallego M., and Harnish E. V. (2007) Reversible protein phosphorylation regulates circadian rhythms. Cold Spring Harb. Symp. Quant. Biol. 72, 413–420 [DOI] [PubMed] [Google Scholar]
- 96. Reppert S. M., and Weaver D. R. (2002) Coordination of circadian timing in mammals. Nature 418, 935–941 [DOI] [PubMed] [Google Scholar]
- 97. Yang J., Kim K. D., Lucas A., Drahos K. E., Santos C. S., Mury S. P., Capelluto D. G., and Finkielstein C. V. (2008) A novel heme-regulatory motif mediates heme-dependent degradation of the circadian factor Period 2. Mol. Cell. Biol. 28, 4697–4711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Guenthner C. J., Bickar D., and Harrington M. E. (2009) Heme reversibly damps PERIOD2 rhythms in mouse suprachiasmatic nucleus explants. Neuroscience 164, 832–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lukat-Rodgers G. S., Correia C., Botuyan M. V., Mer G., and Rodgers K. R. (2010) Heme-based sensing by the mammalian circadian protein, CLOCK. Inorg. Chem. 49, 6349–6365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Airola M. V., Du J., Dawson J. H., and Crane B. R. (2010) Heme binding to the mammalian circadian clock protein period 2 is non-specific. Biochemistry 49, 4327–4338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Dioum E. M., Rutter J., Tuckerman J. R., Gonzalez G., Gilles-Gonzalez M. A., and McKnight S. L. (2002) NPAS2: A gas-responsive transcription factor. Science 298, 2385–2387 [DOI] [PubMed] [Google Scholar]
- 102. Uchida T., Sagami I., Shimizu T., Ishimori K., and Kitagawa T. (2012) Effects of the bHLH domain on axial coordination of heme in the PAS-A domain of neuronal PAS domain protein 2 (NPAS2): conversion from His119/Cys170 coordination to His119/His171 coordination. J. Inorg. Biochem. 108, 188–195 [DOI] [PubMed] [Google Scholar]
- 103. Panda S., Antoch M. P., Miller B. H., Su A. I., Schook A. B., Straume M., Schultz P. G., Kay S. A., Takahashi J. S., and Hogenesch J. B. (2002) Coordinated transcription of key pathways in the mouse by the circadian clock. Cell 109, 307–320 [DOI] [PubMed] [Google Scholar]
- 104. Kaasik K., and Lee C. C. (2004) Reciprocal regulation of haem biosynthesis and the circadian clock in mammals. Nature 430, 467–471 [DOI] [PubMed] [Google Scholar]
- 105. Handschin C., Lin J., Rhee J., Peyer A. K., Chin S., Wu P. H., Meyer U. A., and Spiegelman B. M. (2005) Nutritional regulation of hepatic heme biosynthesis and porphyria through PGC-1α. Cell 122, 505–515 [DOI] [PubMed] [Google Scholar]
- 106. Wu N., Yin L., Hanniman E. A., Joshi S., and Lazar M. A. (2009) Negative feedback maintenance of heme homeostasis by its receptor, Rev-erbα. Genes Dev. 23, 2201–2209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Liu C., Li S., Liu T., Borjigin J., and Lin J. D. (2007) Transcriptional coactivator PGC-1α integrates the mammalian clock and energy metabolism. Nature 447, 477–481 [DOI] [PubMed] [Google Scholar]
- 108. Simcox J. A., Mitchell T. C., Gao Y., Just S. F., Cooksey R., Cox J., Ajioka R., Jones D., Lee S. H., King D., Huang J., and McClain D. A. (2015) Dietary iron controls circadian hepatic glucose metabolism through heme synthesis. Diabetes 64, 1108–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Estall J. L., Ruas J. L., Choi C. S., Laznik D., Badman M., Maratos-Flier E., Shulman G. I., and Spiegelman B. M. (2009) PGC-1α negatively regulates hepatic FGF21 expression by modulating the heme/Rev-Erbα axis. Proc. Natl. Acad. Sci. U.S.A. 106, 22510–22515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Liu A. C., Tran H. G., Zhang E. E., Priest A. A., Welsh D. K., and Kay S. A. (2008) Redundant function of REV-ERBα and β and non-essential role for Bmal1 cycling in transcriptional regulation of intracellular circadian rhythms. PLoS Genet. 4, e1000023. [DOI] [PMC free article] [PubMed] [Google Scholar]