

Abstract
The Rcs phosphorelay is a two-component signal transduction system that is induced by cell envelope stress. RcsB, the response regulator of this signaling system, is a pleiotropic transcription regulator, which is involved in the control of various stress responses, cell division, motility, and biofilm formation. RcsB regulates transcription either as a homodimer or together with auxiliary regulators, such as RcsA, BglJ, and GadE in Escherichia coli. In this study, we show that RcsB in addition forms heterodimers with MatA (also known as EcpR) and with DctR. Our data suggest that the MatA-dependent transcription regulation is mediated by the MatA-RcsB heterodimer and is independent of RcsB phosphorylation. Furthermore, we analyzed the relevance of amino acid residues of the active quintet of conserved residues, and of surface-exposed residues for activity of RcsB. The data suggest that the activity of the phosphorylation-dependent dimers, such as RcsA-RcsB and RcsB-RcsB, is affected by mutation of residues in the vicinity of the phosphorylation site, suggesting that a phosphorylation-induced structural change modulates their activity. In contrast, the phosphorylation-independent heterodimers BglJ-RcsB and MatA-RcsB are affected by only very few mutations. Heterodimerization of RcsB with various auxiliary regulators and their differential dependence on phosphorylation add an additional level of control to the Rcs system that is operating at the output level.
Keywords: bacterial signal transduction, DNA binding protein, protein phosphorylation, protein-protein interaction, transcription coregulator
Introduction
In bacteria, two-component signaling systems play a central role in modulating the intracellular response to specific extracellular signals (1). In canonical two-component systems, signal sensing by the input domain of the sensor kinase triggers its autophosphorylation at a conserved histidine residue. From there, the phosphoryl group is transferred to a conserved aspartate residue within the receiver domain of the response regulator, whose phosphorylation affects its output function (1). The Rcs phosphorelay is a complex two-component signal transduction system in Enterobacteriaceae that was originally identified as a regulatory system of colanic acid capsule biosynthesis in Escherichia coli (2–4). Its complexity is evident at the levels of signal sensing, signal transfer, and signaling output. Sensing of perturbations of the bacterial cell wall and outer membrane proteins involves the lipoprotein RcsF and the inner membrane protein IgaA that are acting upstream of the inner membrane sensor kinase RcsC and the phosphotransfer protein RcsD (5–7). Intriguingly, signal sensing mediated by RcsF involves a switch in its localization. RcsF is exposed to the cell surface by the outer membrane β-barrel protein assembly machinery, but upon perturbation of outer membrane protein assembly or lipoprotein transport machinery, newly synthesized RcsF is no longer surface-exposed and is then able to trigger Rcs signaling from the periplasm via IgaA (7).
The complexity of the output that is generated by the Rcs phosphorelay via the response regulator RcsB is likewise high and involves additional protein components. RcsB is a transcriptional regulator that acts as a homodimer or by interaction with auxiliary transcriptional regulators, including RcsA, GadE, and BglJ (3, 8, 9). RcsB and its auxiliary partners all belong to the FixJ/NarL family of transcriptional regulators characterized by a conserved C-terminal DNA-binding domain family (3, 10, 11). The interaction of RcsB with the auxiliary partners alters the DNA binding specificity (8, 9, 12) and thus extends the regulatory repertoire of the Rcs system to the control of multiple loci related to motility and biofilm formation, various stress responses, cell surface components, and additional functions (3, 4). As a homodimer, RcsB activates or represses transcription of several loci, including rprA, encoding the small regulatory RNA RprA (3, 13). The heterodimer RcsA-RcsB activates loci coding for exopolysaccharide production and represses transcription of flhDC, encoding the flagella master regulator (14–16). GadE-RcsB activates expression of the gadA gene, encoding a glutamate decarboxylase important in the acid-stress response, whereas phosphorylated RcsB represses expression of gadA as a homodimer by binding to a different site (8). BglJ-RcsB activates expression of >10 loci, including leuO encoding a pleiotropic LysR-type transcription regulator (9, 17, 18).
The flexibility of the Rcs output is increased because only some of the RcsB heterodimers depend on RcsB phosphorylation. Transcriptional activation by RcsB-RcsB homodimers and RcsA-RcsB heterodimers depends on RcsB phosphorylation and thus on induction of the Rcs signaling cascade (3). In contrast, BglJ-RcsB and GadE-RcsB heterodimers are active independent of RcsB phosphorylation (8, 9). Thus, there is a dual control of RcsB activity, by phosphorylation via the Rcs phosphorelay and by interaction with auxiliary partners all belonging to the FixJ/NarL family of transcriptional regulators. To date, heterodimerization of bacterial response regulators in addition to RcsB has only been described for BldM and WhiI in the filamentous bacteria Streptomyces (19).
In this study, we analyzed whether RcsB interacts with additional FixJ/NarL auxiliary partners, and we initiated a characterization of amino acid residues of RcsB relevant for this interaction. A two-hybrid analysis for interaction of RcsB with the 18 members of the FixJ/NarL family present in E. coli K12 allowed us to expand the RcsB regulon by two interaction partners, MatA (also named EcpR) and DctR. MatA activates expression of fimbria synthesis genes, named mat in newborn meningitis- and septicemia-associated E. coli (NMEC)2 and named ecp (for E. coli common pili) in enterohemorrhagic E. coli and other E. coli (20–23). Our data suggest that this activation is mediated by MatA-RcsB heterodimers independently of RcsB phosphorylation. DctR plays a role in protection against organic acids that are metabolic products prevalent at high cell densities under growth in acidic conditions (24–26). However, no target gene of DctR is known. Further, we identified amino acids within the receiver domain of RcsB that are presumably important for interaction with specific auxiliary partners. Our data show that transcriptional activation by RcsB homodimers and RcsA-RcsB heterodimers, those depending on RcsB phosphorylation, is affected by mutation of amino acid residues belonging to the “active quintet” coordinating phosphorylation as well as conserved and further amino acid residues in close proximity to the phosphorylation site. In contrast, the activity of the phosphorylation-independent heterodimers BglJ-RcsB and MatA-RcsB is disturbed by only very few of the specific RcsB mutants that were analyzed, including RcsB-I14A with a mutation mapping in helix α1 and K180A mapping in the DNA-binding domain.
Experimental Procedures
Bacterial Strains, Plasmids, and Media
E. coli K12 strains and their construction are described in Table 1. Strain construction included transductions with phage T4GT7 and phage P1vir (27, 28), integration of reporter constructs at the attachment site attB (29, 30), and generation of chromosomal replacements and deletions by Red-Gam-mediated recombination (31). Mutants were analyzed by allele-specific PCRs, whereas replacements of the matA and rcsA promoters, respectively, were in addition characterized by sequencing. The plasmids and their construction are listed in Table 2, and the sequences of the oligonucleotides are shown in Table 3. Bacterial cultures were grown in LB medium (5 g/liter yeast extract, 10 g/liter tryptone, 5 g/liter NaCl). Antibiotics were added to 50 μg/ml ampicillin and 25 μg/ml kanamycin, respectively, if required.
TABLE 1.
E. coli K12 and UPEC strains
The E. coli K12 strains and the UPEC strain used in this study are listed with their genotype and the source or construction of the strain. For construction of strains by transduction with phage T4GT7 and P1vir, respectively, the number of the source strain is given in parentheses. In the cases of generation of deletions or replacement by Red-Gam-mediated recombination, the fragments were generated by PCR with the indicated oligonucleotide primers and DNA templates (x primer1/primer2 (template plasmid)). Plasmid pCP20 containing the flp gene was used for site-specific deletion of the resistance cassettes (x pCP20). Furthermore, promoter lacZ fusions were integrated at the attachment site attB using helper plasmid pLDR8 (encoding Int) and the religated BamHI fragments of the indicated plasmids (x plasmid number). Constructed and transduced alleles were analyzed by PCR using oligonucleotide primers given in Table 3.
| Strain | Genotype | Source/Construction |
|---|---|---|
| E. coli K12 strains | ||
| BW30270 | MG1655 rph+ | CGSC #7925 |
| S3974 | BW30270 ilvG+ (non-motile) | Ref. 9 |
| T1241 | BW30270 ilvG+ (motile) | BW30270 x T4GT7 (S3974) |
| U89 | BW30270 ilvG+ ΔrcsBcmR | T1241 x T4GT7 (T13) |
| S4197 | BW30270 ilvG+ ΔlacZ (non-motile) | Ref. 9 |
| T21 | S4197 ΔrcsBFRT | Ref. 17 |
| T768 | S4197Δ(rcsDB-rcsC)cmR | S4197/pKD46 x PCR T433/T329 (pKD3) |
| T903 | S4197 ΔrcsBFRT PL-rcsAkanR | Ref. 58 |
| JW1224 | Δ(araD-araB)567 ΔlacZ4787 (::rrnB-3), λ− ΔgalU745kanR rph1 Δ(rhaD-rhaB)568 hsdR514 | CGSC #9110 (59) |
| UPEC E. coli strain | ||
| CFT073 | Uropathogenic E. coli | Ref. 60 |
| E. coli K12 strains with an integration of a promoter lacZ fusions at attB | ||
| S3432 | CSH50 ΔlacZ Δbgl sulA3 lexA71::Tn5 ΔrcsBFRT attB::(SpecR lacIq PsulA+/+ lacZ) | Ref. 9 |
| S3434 | CSH50 ΔlacZ Δbgl sulA3 lexA71::Tn5 ΔrcsBFRTΔ(yjjP-yjjQ-bglJ)FRT attB::(SpecR lacIq PsulA+/+ lacZ) | Ref. 9 |
| S3440 | CSH50 ΔlacZ Δbgl sulA3 lexA71::Tn5 ΔrcsBFRT attB::(SpecR lacIq PsulA408/+ lacZ) | Ref. 9 |
| S3442 | CSH50 ΔlacZ Δbgl sulA3 lexA71::Tn5 ΔrcsBFRTΔ(yjjP-yjjQ-bglJ)FRT attB::(SpecR lacIq PsulA408/+ lacZ) | Ref. 9 |
| T572 | S4197 attB::(SpecR PleuO lacZ) ΔrcsBFRT bglJC | Ref. 17 |
| T2023 | S4197 attB::(SpecR PrprA lacZ) | S4197/pLDR8 x pKES299 |
| T1052 | S4197 attB::(SpecR PrprA lacZ) ΔrcsBFRT | T21/pLDR8 x pKES299 |
| T2041 | S4197 attB::(SpecR PrprA lacZ) ΔgalUkanR | T2023 x P1vir (JW1224) |
| T2037 | S4197 attB::(SpecR Pwza lacZ) | S4197/pLDR8 x pKES260 |
| T2039 | S4197 attB::(SpecR Pwza lacZ) kanRPL-rcsA | T2037 x T4GT7 (T903) |
| T864 | S4197 attB::(SpecR Pwza lacZ) ΔrcsBFRT | T21/pLDR8 x pKES260 |
| T927 | S4197 attB::(SpecR Pwza lacZ) ΔrcsBFRT PL-rcsAFRT | T864 x T4GT7 (T903) x pCP20 |
| T921 | S4197 attB::(SpecR Pwza lacZ) Δ(rcsDB-rcsC)cmR | T864 x T4GT7 (T768) |
| T963 | S4197 attB::(SpecR Pwza lacZ) Δ(rcsDB-rcsC)FRT FRTPL-rcsA | T921 x T4GT7 (T903) x pCP20 |
| T2042 | S4197 attB::(SpecR Pwza lacZ) ΔgalUkanR | T2037 x P1vir (JW1224) |
| T2044 | S4197 attB::(SpecR Pwza lacZ) ΔgalUFRT | T2042 x pCP20 |
| T2045 | S4197 attB::(SpecR Pwza lacZ) ΔgalUFRT kanRPL-rcsA | T2044 x T4GT7 (T903) |
| T1749 | S4197 attB::(SpecR PmatA lacZ) | S4197/pLDR8 x pKEDP49 |
| T1747 | S4197 attB::(SpecR PmatA lacZ) ΔrcsBFRT | T21/pLDR8 x pKEDP49 |
| T1986 | S4197 attB::(SpecR PmatA lacZ)FRT FRTPL-matA | T1749/pKD46 x PCR OA83/OA84 (pKES263) x pCP20 |
| T1987 | S4197 attB::(SpecR PmatA lacZ) ΔrcsBFRT FRTPL-matA | T1747/pKD46 x PCR OA83/OA84 (pKES263) x pCP20 |
TABLE 2.
Plasmids and their relevant features
Plasmids that were used or constructed are listed with their name, relevant features, and source or construction. For plasmids that were constructed by cloning of PCR fragments, the primers used for amplification of the fragment and the cloning vector are given. The PCR fragments were amplified from plasmids carrying the wild-type gene or from the E. coli K12 genome. Mutagenesis of rcsB was conducted by fusion PCR using oligonucleotide primers (listed in Table 3) that carry the specific mutation and flanking primers T106 and T358; the mutated nucleotides are indicated. Derivatives of plasmids pDP804 and pMS604 carry translational fusions of the indicated genes to lexA(1–87)WT and lexA(1–87)408, respectively.
| Plasmid | Features | Source/Construction |
|---|---|---|
| pCP20 | cI857 λ-PR flp pSC101 repts bla | Ref. 61 |
| pKD46 | araC Para γ-β-exo in pSC101 repts bla | Ref. 31 |
| pLDR8 | cI857 λ-PR int pSC101 repts neo | Ref. 29 |
| pKD3 | FRT cat FRT oriRγ bla | Ref. 31 |
| pKD13 | FRT neo FRT oriRγ bla | Ref. 31 |
| pKES263 | FRT neo FRT PL oriRγ bla in pKD13 | PCR T464/T465 from phage λ |
| pKES148 | MCS lacZ attP aadA p15A neo | Ref. 9 |
| pKES260 | Pwza lacZ attP aadA p15A neo | PCR T460/T461 in pKES148 |
| pKES299 | PrprA lacZ attP aadA p15A neo | PCR T563/T564 in pKES148 |
| pKES268 | PlacUV5 MCS lacZ attP aadA p15A neo | Ref. 18 |
| pKEDP49 | PmatA lacZ attP aadA p15A neo | PCR T908/T909 from CFT073 in pKES268 |
| pDP804 | PlacUV5 lexA(1–87)408-jun p15A bla | Ref. 40 |
| pKEAP28 | PlacUV5 lexA(1–87)408-rcsB in pDP804 | Ref. 9 |
| pKEAP29 | PlacUV5 lexA(1–87)408-bglJ in pDP804 | Ref. 9 |
| pKEDP58 | PlacUV5 lexA(1–87)408-dctR in pDP804 | PCR OA29/OA30 in pDP804 |
| pKEDP59 | PlacUV5 lexA(1–87)408-matA in pDP804 | PCR OA27/OA28 in pDP804 |
| pKEDP60 | PlacUV5 lexA(1–87)408-rcsA in pDP804 | PCR OA25/OA26 in pDP804 |
| pMS604 | PlacUV5 lexA(1–87)WT-fos ColE1 tet | Ref. 40 |
| pKEAP30 | PlacUV5 lexA(1–87)WT-bglJ | Ref. 9 |
| pKEMK17 | PlacUV5 lexA(1–87)WT-rcsB | Ref. 9 |
| pKES192 | PlacUV5 lexA(1–87)WT-rcsA | Ref. 9 |
| pKEAP27 | PlacUV5 lexA(1–87)WT-yjjQ | PCR S691/S692 in pMS604 |
| pKEMK1 | PlacUV5 lexA(1–87)WT-dctR | PCR S956/S957 in pMS604 |
| pKEMK2 | PlacUV5 lexA(1–87)WT-fimZ | PCR S960/S961 in pMS604 |
| pKEMK3 | PlacUV5 lexA(1–87)WT-malT | PCR S964/S965 in pMS604 |
| pKEMK4 | PlacUV5 lexA(1–87)WT-matA | PCR S966/S967 in pMS604 |
| pKEMK5 | PlacUV5 lexA(1–87)WT-narL | PCR S968/S969 in pMS604 |
| pKEMK6 | PlacUV5 lexA(1–87)WT-narP | PCR S970/S971 in pMS604 |
| pKEMK7 | PlacUV5 lexA(1–87)WT-sdiA | PCR S972/S973 in pMS604 |
| pKEMK8 | PlacUV5 lexA(1–87)WT-uhpA | PCR S974/S975 in pMS604 |
| pKEMK9 | PlacUV5 lexA(1–87)WT-uvrY | PCR S976/S977 in pMS604 |
| pKEMK10 | PlacUV5 lexA(1–87)WT-ygeK | PCR S978/S979 in pMS604 |
| pKEMK11 | PlacUV5 lexA(1–87)WT-yhjB | PCR S980/S981 in pMS604 |
| pKEMK12 | PlacUV5 lexA(1–87)WT-yahA | PCR S982/T10 in pMS604 |
| pKEMK15 | PlacUV5 lexA(1–87)WT-evgA | PCR S958/S959 in pMS604 |
| pKEMK16 | PlacUV5 lexA(1–87)WT-gadE | PCR S962/S963 in pMS604 |
| pKES177 | PlacUV5 lexA(1–87)WT-csgD | PCR S885/S886 in pMS604 |
| pBAD24 | araC PBAD pBR bla | Ref. 62 |
| pKEDP51 | araC PBAD matA | PCR T691/T692 in pBAD24 |
| pKEDP57 | araC PBAD dctR | PCR T694/T695 in pBAD24 |
| pKESK22 | lacIq Ptac p15A neo | Ref. 54 |
| pKETS6 | lacIq Ptac rcsB in pKESK22 | Ref. 9 |
| pKETS7 | lacIq Ptac rcsB-D56E (GAT → GAG) | Ref. 9 |
| pKES229 | lacIq Ptac rcsB-D66A (GAT → GCG) | This work |
| pKES230 | lacIq Ptac rcsB-H77A (CAT → GCG) | This work |
| pKES231 | lacIq Ptac rcsB-I14A (ATA → GCA) | This work |
| pKES232 | lacIq Ptac rcsB-M88A (ATG → GCG) | This work |
| pKES234 | lacIq Ptac rcsB-Y64A (TAC → GCC) | This work |
| pKES235 | lacIq Ptac rcsB-D56A (GAT → GCG) | This work |
| pKES271 | lacIq Ptac rcsB-D11A (GAC → GCC) | This work |
| pKES272 | lacIq Ptac rcsB-P60A (CCT → GCT) | This work |
| pKES273 | lacIq Ptac rcsB-G67A (GGC → GCC) | This work |
| pKES274 | lacIq Ptac rcsB-T87A (ACT → GCT) | This work |
| pKES275 | lacIq Ptac rcsB-K109A (AAA → GCA) | This work |
| pKES276 | lacIq Ptac rcsB-K180A (AAA → GCA) | This work |
| pKEDP47 | lacIq Ptac rcsB-D62G (GAT → GGT) | This work |
| pKESL111 | lacIq Ptac rcsB-L95A (CTT → GCT) | This work |
| pKESL112 | lacIq Ptac rcsB-L99A (TTG → GCG) | This work |
| pKESL113 | lacIq Ptac rcsB-D100A (GAT → GCT) | This work |
| pKESL114 | lacIq Ptac rcsB-E104A (GAA → GCA) | This work |
| pKESL115 | lacIq Ptac rcsB-I106A (ATC → GCC) | This work |
| pKESL116 | lacIq Ptac rcsB-L108A (CTG → GCG) | This work |
| pKESL117 | lacIq Ptac rcsB-T114A (ACC → GCC) | This work |
| pKESL118 | lacIq Ptac rcsB-K118A (AAA → GCA) | This work |
| pKESL119 | lacIq Ptac rcsB-S96A (AGT → GCT) | This work |
| pKESL120 | lacIq Ptac rcsB-D115A (GAT → GCT) | This work |
TABLE 3.
Oligonucleotides
The sequences of oligonucleotides used as PCR primers and for mutagenesis are shown, along with a brief description of their use. Bases of the oligonucleotide that match the template are shown in capital letters, whereas other bases are in lowercase letters. Restriction sites are underlined.
| Name | Nucleotide sequence | Use |
|---|---|---|
| T106 | cagggatcctctagattaGTCTTTATCTGCCGGACTTAAGGTCAC | rcsB cloning |
| T358 | gaccgaattcTTGCTGTAGCAAGGTAGCCTATTACATG | rcsB cloning |
| T329 | gagaacattgcggtaacacgcttttaccgctaccttaaccacactGTGTAGGCTGGAGCTGCTTCG | rcsDB-rcsC deletion |
| T433 | ggtaagagtctggaatttcacactgtaccctttatactgccctatCATATGAATATCCTCCTTAGTTCCTATTCC | rcsDB-rcsC deletion |
| T460 | agcagtcgacCTCACATTATCCCTGAATTAAAAGTGG | Pwza cloning |
| T461 | agcgtctagattaCATCATTGTTTATTTATCACTTTGGCAG | Pwza cloning |
| T464 | agcagtcgacCTCTCACCTACCAAACAATGCCC | PL cloning |
| T465 | agcaggatccTCATGGTGGTCAGTGCGTCC | PL cloning |
| T466 | aatacctacgaacatcttccaggatactcctgcagcgaaatattGTGTAGGCTGGAGCTGCTTCG | Replacement of PrcsA by PL |
| T467 | cataccctcactcaatgcgtaacgataattccccttacctgaaTCATGGTGGTCAGTGCGTCC | Replacement of PrcsA by PL |
| T563 | agcagtcgacAATTGATATTTGCTTGCTCTTCCCC | PrprA cloning |
| T564 | agcgtctagaCCGTGAGCTAATAGTAGGCATACGG | PrprA cloning |
| T691 | agcagaattcAATTACAGGTTTGGAAAGTAGTGACATG | matA cloning |
| T692 | agcgTCTAGATTACTGAACCAACTTATATATTTTTGAGTACAGC | matA cloning |
| T694 | agcagaattcGTCCGCACCAGGAGTCGG | dctR cloning |
| T695 | agcgTCTAGATCACACCAGATAATCAATATGCTGATG | dctR cloning |
| T908 | agcagtcgacGCCATCGTTCCTGTGACAACTG | PmatACFT073 cloning |
| T909 | agcgtctagaTTGCCATGTCACTACTTTCCAAACC | PmatACFT073 cloning |
| OA25 | agcactcgagTCAACGATTATTATGGATTTATGTAGTTACAC | LexA (1–87)408 rcsA cloning |
| OA26 | agcgagatctTTAGCGCATGTTGACAAAAATACC | LexA (1–87)408 rcsA cloning |
| OA27 | agcactcgagACATGGCAAAGTGATTACAGTAGGGAC | LexA (1–87)408 matA cloning |
| OA28 | agcgagatctTTACTGAACCAACTTATATATTTTTGAGTACAGCTT | LexA (1–87)408 matA cloning |
| OA29 | agcactcgagTTTCTTATAATTACCAGGGATACGATGTTC | LexA (1–87)408 dctR cloning |
| OA30 | agcgagatctTCACACCAGATAATCAATATGCTGATG | LexA (1–87)408 dctR cloning |
| OA83 | tcttcaatgacagctcatcatagttttatattctatcccttaGTGTAGGCTGGAGCTGCTTCG | Replacement of PmatA by PL |
| OA84 | actgtaatcactttgccatgtcactactttccaaacctgtaaTCATGGTGGTCAGTGCGTCC | Replacement of PmatA by PL |
| S691 | ttctgcagTTGCCAGGATGCTGCAAAA | LexA (1–87)WT yjjQ cloning |
| S692 | ttctcgagACTCTCAATACCGATACTACTCATGACG | LexA (1–87)WT yjjQ cloning |
| S885 | ccatctgcagTTTAATGAAGTCCATAGTATTCATGGTCATAC | LexA (1–87)WT csgD cloning |
| S886 | ccatctcgagTTATCGCCTGAGGTTATCGTTTGC | LexA (1–87)WT csgD cloning |
| S956 | ccatctgcagTTTCTTATAATTACCAGGGATACGATGTTC | LexA (1–87)WT dctR cloning |
| S957 | ccatctcgagTCACACCAGATAATCAATATGCTGATG | LexA (1–87)WT dctR cloning |
| S958 | ccatggatcctgcagAACGCAATAATTATTGATGACCATCCT | LexA (1–87)WT evgA cloning |
| S959 | ccatggatcctcgagTTAGCCGATTTTGTTACGTTGTGC | LexA (1–87)WT evgA cloning |
| S960 | ccatctgcagAAACCAACGTCGGTGATCATTATG | LexA (1–87)WT fimZ cloning |
| S961 | ccatctcgagTTATATTAATTCGTATAATTTGGCGTAGTCGAT | LexA (1–87)WT fimZ cloning |
| S962 | ccatggatcctgcagATTTTTCTCATGACGAAAGATTCTTTTCTT | LexA (1–87)WT gadE cloning |
| S963 | ccatggatcctcgagCTAAAAATAAGATGTGATACCCAGGGTGAC | LexA (1–87)WT gadE cloning |
| S964 | ccatctgcagCTGATTCCGTCAAAACTAAGTCGTCC | LexA (1–87)WT malT cloning |
| S965 | ccatctcgagTTACACGCCGTACCCCATCAT | LexA (1–87)WT malT cloning |
| S966 | ccatctgcagACATGGCAAAGTGATTACAGTAGGGAC | LexA (1–87)WT matA cloning |
| S967 | ccatctcgagTTACTGAACCAACTTATATATTTTTGAGTACAGCTT | LexA (1–87)WT matA cloning |
| S968 | ccatctgcagAGTAATCAGGAACCGGCTACTATCCTG | LexA (1–87)WT narL cloning |
| S969 | ccatctcgagTCAGAAAATGCGCTCCTGATG | LexA (1–87)WT narL cloning |
| S970 | ccatctgcagCCTGAAGCAACACCTTTTCAGGT | LexA (1–87)WT narP cloning |
| S971 | ccatctcgagTTATTGTGCCCCGCGTTGTT | LexA (1–87)WT narP cloning |
| S972 | ccatctgcagCAGGATAAGGATTTTTTCAGCTGG | LexA (1–87)WT sdiA cloning |
| S973 | ccatctcgagTCAAATTAAGCCAGTAGCGGCC | LexA (1–87)WT sdiA cloning |
| S974 | ccatctgcagATCACCGTTGCCCTTATAGACGAT | LexA (1–87)WT uhpA cloning |
| S975 | ccatctcgagTCACCAGCCATCAAACATGCG | LexA (1–87)WT uhpA cloning |
| S976 | ccatctgcagATCAACGTTCTACTTGTTGATGACCAC | LexA (1–87)WT uvrY cloning |
| S977 | ccatctcgagTCACTGACTTGATAATGTCTCCGCAT | LexA (1–87)WT uvrY cloning |
| S978 | ccatctgcagATGGGGGCCGAACTCGTAAA | LexA (1–87)WT ygeK cloning |
| S979 | ccatctcgagTTATATAGTGCAAACACCCATACGTAAAGC | LexA (1–87)WT ygeK cloning |
| S980 | ccatctgcagCAAATAGTCATGTTTGACAGGCAGTCA | LexA (1–87)WT yhjB cloning |
| S981 | ccatctcgagTCAGGAGGAGATATTTAACATCATTGC | LexA (1–87)WT yhjB cloning |
| S982 | ccatctgcagAATTCATGTGATTTTCGTGTTTTTCTG | LexA (1–87)WT yahA cloning |
| T10 | ccatagatctTCAACCACCTGCTTTCATTACCC | LexA (1–87)WT yahA cloning |
RNA Isolation and Microarray Analysis
For identification of target genes that are regulated by MatA/EcpR or DctR, E. coli strain BW30270 was transformed with plasmids pKEDP30 (encoding MatA), pKEDP31 (encoding DctR), and pKESK22 (control plasmid), respectively. Overnight cultures of these transformants were used to inoculate a 15-ml culture to an A600 of 0.05 in LB medium supplemented with kanamycin. The cultures were grown in a shaker at 37 °C until A600 = 0.3. Then isopropyl β-d-1-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mm, and the cultures were harvested after 30 min of further incubation using RNAprotect bacteria reagent (Qiagen, Hilden, Germany). Total RNA was isolated using the RNeasy minikit system, including an on-column DNase I treatment according to the manufacturer's instructions (Qiagen, Germany). For each strain background, three biological replicates of the RNA preparations were used for microarray analysis, which was performed by the Cologne Center for Genomics using Affymetrix GeneChip E. coli Genome 2.0 microarrays, as described (17). Differential expression levels were calculated as -fold change relative to the empty vector control.
β-Galactosidase Assay
For expression analyses of promoter lacZ fusions, β-galactosidase assays were carried out as described (28). Briefly, exponential cultures were inoculated from a fresh overnight culture to an A600 of 0.05 in LB medium, which contained appropriate antibiotics in the case of transformants. Where indicated, IPTG (1 mm final concentration) was added to the overnight and the exponential culture for induction. Bacteria were grown at 37 °C and harvested on ice at an A600 of 0.5. The β-galactosidase assays were repeated from at least three independent biological replicates.
Motility Assays
For motility assays, 3 μl of a fresh overnight culture grown in LB medium, which in the case of transformants was supplemented with 25 μg/ml kanamycin and 1 mm IPTG, was pipetted to the center of freshly poured LB soft agar plates (0.2% agar). In the case of transformants, the plates were supplemented with 25 μg/ml kanamycin and 0.2 mm IPTG. The motility assay plates were incubated for 5 h at 37 °C and then scanned using an Epson Perfection V700 Photo transparency scanner.
Structure Prediction of the RcsB Receiver Domain
The RcsB protein consists of 216 amino acids with the N-terminal receiver domain comprising residues 1–124 and the C-terminal DNA-binding domain comprising residues 144–209 (3). The structure of the DNA-binding domain was solved for RcsB from Erwinia amylovora (PDB code 1P4W) (32), which is 92% identical to E. coli RcsB. The structure of the RcsB receiver domain has not been solved. Therefore, we used structural modeling by Phyre2 (33), I-TASSER (34), and HHPred (35), using the E. coli K12 RcsB amino acid sequence from residue 1 to 125 as a query. For Phyre2, the highest scoring model was based on the crystal structure of NarL from Mycobacterium tuberculosis (PDB code 3EUL) (36) and a 26% protein identity. The model is shown under “Results.” For I-Tasser, the model was based on the crystal structure of NarL from E. coli (PDB code 1A04) (37, 38), and for HHPred, the model was based on a response regulator from Aurantimonas sp. S185-9A1 (PDB code 3CZ5). Structures of receiver domains exhibit a conserved (αβ)5 fold (39). Accordingly, the models of the RcsB receiver domain are very similar in the well ordered (αβ)5 fold but show variations in the loops that connect the α helices and β sheets.
Results
RcsB Interacts with MatA (EcpR) and DctR
The response regulator RcsB is known to interact with auxiliary regulators, such as RcsA, BglJ, and GadE, which likewise carry a FixJ/NarL-type DNA-binding domain (3, 8, 9). In the E. coli K12 genome, in total, 18 proteins with a FixJ/NarL-type DNA-binding domain are encoded. Here we investigated whether RcsB interacts with any other member of these FixJ/NarL-type proteins. To this end, we performed interaction studies using the bacterial LexA-based one-hybrid and two-hybrid system (40). The one-hybrid reporter for examining homodimer formation consists of the native sulA promoter fused to lacZ (Fig. 1A). Only homodimers of proteins fused to the wild-type DNA-binding domain (amino acid residues 1–87) of the LexA repressor (LexA(1–87)WT) are able to bind to the lexA operator and repress PsulA lacZ expression. For analyzing heterodimer formation, the sulA promoter carries a hybrid lexA operator (408/+) with a mutation in one half-site (Fig. 1B) (40). Only heterodimers, in which one partner is fused to LexA(1–87)WT and the other partner is fused to the 408 mutant DNA-binding domain (LexA(1–87)408), are able to bind the hybrid operator and repress PsulA lacZ expression (40).
FIGURE 1.

Homo- and heterodimer formation by RcsB, BglJ, RcsA, MatA, and DctR. The LexA two-hybrid system exploits repression of the sulA promoter by dimeric LexA (40). Expression is repressed in cases when a fusion of a protein (×) to the LexA-DNA-binding domain forms homodimers (A). For analysis of heterodimerization, a sulA promoter variant carrying a hybrid lexA 408/+ operator is used, which is repressed by heterodimers of proteins X and Y that are fused to the LexA(1–87)WT and LexA(1–87)408 DNA-binding domains, respectively (B). The LexA fusion proteins were expressed from compatible plasmids under the control of the IPTG-inducible lacUV5 promoter. C, heterodimer formation by RcsB, BglJ, RcsA, MatA, and DctR. The -fold repression of the lexAop408/+ sulA promoter lacZ fusion, as a measure of heterodimerization, is calculated as the ratio of expression values (given in smaller type) directed by the PsulA lacZ reporter when bacteria are grown without and with induction, respectively, of LexA fusion protein expression. Strain S3440 (ΔrcsB) or S3442 (ΔrcsB Δ(yjjP-yjjQ-bglJ) were co-transformed with plasmids encoding for LexA(1–87)-X and LexA(1–87)408-Y fusions, respectively. The following plasmids were used: pKEMK17 (LexA(1–87)-RcsB), pKEAP30 (LexA(1–87)-BglJ), pKES192 (LexA(1–87)-RcsA), pKEMK4 (LexA(1–87)-MatA), and pKEMK1 (LexA(1–87)-DctR) as well as pKEAP28 (LexA(1–87)408-RcsB), pKEAP29 (LexA(1–87)408-BglJ), and pKEDP59 (LexA(1–87)408-MatA). The cultures were grown to A600 of 0.5 in LB medium supplemented with ampicillin and tetracyclin. IPTG was added where indicated. Values for RcsB, BglJ, and RcsA homo- and heterodimer analysis are taken from Ref. 9. D, homodimer formation of RcsB, BglJ, RcsA, MatA, and DctR. The -fold repression, as a measure for dimerization, was calculated as the ratio of the β-galactosidase activities determined of cultures grown without and with induction of the LexA fusion proteins. Strain S3434 (ΔrcsB Δ(yjjP-yjjQ-bglJ)) was transformed with plasmids pKEMK17 (LexA(1–87)-RcsB), pKEAP30 (LexA(1–87)-BglJ), and pKES192 (LexA(1–87)-RcsA), respectively. Strain S3432 (ΔrcsB) was transformed with plasmid pKEMK4 (LexA(1–87)-MatA) and pKEMK1 (LexA(1–87)-DctR). Cultures were grown in LB tetracycline medium to A600 of 0.5 without and with 1 mm IPTG.
The two-hybrid assays for heterodimer formation of RcsB with any of the additional 17 other FixJ/NarL-type proteins encoded in E. coli K12 were conducted in the lexA ΔrcsB strain S3440 or S3442 carrying the sulA 408/+ hybrid promoter fused to lacZ (Fig. 1 and Table 4). The reporter strain was co-transformed with plasmid pKEAP28 harboring RcsB fused to the LexA(1–87)408 DNA-binding domain, together with a plasmid harboring one of the other FixJ/NarL-type proteins fused to the wild-type LexA(1–87)WT DNA-binding domain (Fig. 1B). The co-induction of LexA(1–87)408-RcsB with LexA(1–87)WT-MatA as well as the co-induction of LexA(1–87)408-RcsB with LexA(1–87)WT-DctR resulted in a 9- and 12-fold repression, respectively, suggesting that RcsB forms heterodimers with both MatA and DctR (Fig. 1C). This repression was similar to previous results obtained for BglJ-RcsB and RcsA-RcsB heterodimerization (shown for comparison in Fig. 1C) (9). Beyond MatA, DctR, RcsA, and BglJ, heterodimerization between RcsB and any of the other FixJ/NarL type proteins was weaker or could not be observed at all (Table 4) (see below).
TABLE 4.
Homo- and heterodimerization of RcsB and other FixJ/NarL-type proteins encoded by the E. coli K12 genome
Homodimerization of fusions of FixJ/NarL-type proteins to the LexA(1–87) wild-type DNA-binding domain was analyzed in strain S3432, whereas homodimerization of LexA(1–87)WT-YjjQ was analyzed in strain S3434 and other strains. Heterodimerization of RcsB fused to LexA(1–87)408 was analyzed in strain S3440 co-transformed with a plasmid expressing a fusion of the indicated FixJ/NarL-type protein to LexA(1–87) wild type. The -fold repression of the sulA promoter-lacZ reporter is a measure of dimerization and is calculated as the quotient of the β-galactosidase activity obtained without induction (−IPTG) and with induction of expression of the fusion proteins (+IPTG).
| Fusion to LexA(1–87)WT | Homodimerization |
Heterodimerization with LexA(1–87)408-RcsB |
||||
|---|---|---|---|---|---|---|
| β-Galactosidase activity |
-Fold repression | β-Galactosidase activity |
-Fold repression | |||
| −IPTG | +IPTG | −IPTG | +IPTG | |||
| EvgAa | 672 | 93 | 7.2 | 2108 | 278 | 7.6 |
| FimZ | 1874 | 122 | 15.4 | 2080 | 612 | 3.4 |
| NarL | 2626 | 150 | 17.5 | 1987 | 575 | 3.5 |
| NarP | 2256 | 120 | 18.8 | 2037 | 702 | 2.9 |
| UhpA | 2684 | 109 | 24.6 | 2171 | 409 | 5.3 |
| UvrY | 2860 | 116 | 24.7 | 2255 | 591 | 3.8 |
| YgeK | 3164 | 1057 | 3.0 | 1838 | 1781 | 1.0 |
| CsgD | 3145 | 167 | 18.8 | 2084 | 1113 | 1.9 |
| GadE | 3141 | 2273 | 1.4 | 1787 | 564 | 3.2 |
| SdiA | 1037 | 124 | 8.4 | 2302 | 1407 | 1.6 |
| YhjB | 3159 | 107 | 29.5 | 2225 | 1085 | 2.0 |
| YjjQ | 2695 | 152 | 17.7b | 2180 | 595 | 3.7 |
| MalT | 2182 | 112 | 19.5 | 2142 | 1557 | 1.4 |
| YahA/PdeLa | 414 | 101 | 4.1 | 2126 | 732 | 2.9 |
a For these proteins, a repression could be observed even without induction by IPTG.
b Values taken from Ref. 9.
Because RcsB interacts with RcsA, BglJ, MatA, and DctR, these proteins may form heterodimers in other combinations as well. To analyze such a potential mutual interaction between BglJ, RcsA, MatA, and DctR, all other pairs of combinations were tested by the two-hybrid assay (Fig. 1C). However, the co-expression of none of these protein pairs resulted in repression of the sulA 408/+ hybrid promoter lacZ reporter (Fig. 1C). These results suggest that neither BglJ nor RcsA nor MatA nor DctR forms a heterodimer with any other of these four proteins.
RcsB forms homodimers, whereas for the known auxiliary regulators BglJ and RcsA no or only weak homodimer formation was detected by the one-hybrid assay (9). Here we tested homodimer formation of MatA, DctR (Fig. 1D), and the other FixJ/NarL-type proteins (Table 4). Homodimer formation was analyzed in the ΔrcsB reporter strain S3432 or S3434, carrying the wild-type sulA promoter lacZ fusion (Fig. 1B). Neither the induction of LexA(1–87)WT-MatA nor the induction of LexA(1–87)WT-DctR resulted in repression of PsulA lacZ expression, suggesting that neither MatA nor DctR forms homodimers (Fig. 1D). The other FixJ/NarL type proteins tested, except GadE, all formed homodimers (Table 4). For GadE, we could detect neither homodimer nor heterodimer formation, at least under standard growth conditions used in this study. In the case of GadE, it is possible that its interaction with RcsB might be acid stress-dependent. For EvgA, the data indicate that it forms homodimers, as expected, and heterodimers with RcsB (Table 4). However, EvgA plays a pleiotropic role in activating genes related to acid resistance, osmotic adaptation, and drug resistance (41), and we observed a slow culture growth upon induction of LexA(1–87)WT-EvgA fusions. Therefore, the implication of a possible interaction of RcsB with EvgA was not analyzed further. Taken together, the one-hybrid and two-hybrid analyses suggest that RcsB interacts with RcsA, BglJ, MatA, and DctR, respectively, and that these auxiliary partners do not form homodimers. The data for the remaining FixJ/NarL-type proteins (except for EvgA, GadE, and YgeK) suggest that these form homodimers but no or only weak heterodimers with RcsB, indicating that those proteins that form heterodimers with RcsB do not form homodimers, and vice versa.
Activation of the mat Promoter by MatA (EcpR) Depends on RcsB
Our two-hybrid results suggest that the FixJ/NarL-type proteins MatA (EcpR) and DctR form heterodimers with RcsB, whereas they do not form homodimers. However, no target loci of DctR (25) are known, whereas MatA (EcpR) activates the mat (ecp) operon in NMEC, enterohemorrhagic E. coli, and other E. coli but not in E. coli K12 (20, 22, 23). To analyze the relevance of the MatA-RcsB and DctR-RcsB protein interaction in gene regulation, we first needed to identify target genes that may serve as reporters for DctR and MatA, respectively. For this, we performed a microarray analysis and compared the transcriptome of E. coli K12 strain BW30270 harboring plasmids encoding MatA (pKEDP30) and DctR (pKEDP31), respectively, with the same strain harboring vector pKESK22, as control. However, this microarray analysis did not reveal any specific target locus, neither of MatA nor of DctR in E. coli K12. Therefore, a reporter system for analysis of the relevance of the DctR-RcsB protein interaction remains to be established. For MatA, it is known that activation of the H-NS-repressed mat operon in NMEC strain IHE3034 and other E. coli strains depends on the specific sequence of the mat promoter and regulatory region (23). The sequence of the mat regulatory region is divergent between the E. coli lineages B2, D, and E as compared with the E. coli A and B1 lineages, whereas the nucleotide sequence of the matABCDEF coding region is highly conserved among all E. coli strains (23). To establish a suitable reporter system, we constructed a lacZ fusion of the matA promoter and regulatory region of UPEC strain CFT073 encompassing positions −552 to +68 relative to the T1 transcription start site, as described before (23). The nucleotide sequence of the matA promoter and regulatory region is almost identical in UPEC strain CFT073 and NMEC strain IHE3034, which both belong to the B2 E. coli lineage (23).
The PmatACFT073 lacZ reporter for analysis of regulation by RcsB and MatA was integrated into the chromosome of the E. coli K12 ΔlacZ strain S4197 (rcsB+) and an isogenic ΔrcsB strain (yielding strains T1749 and T1747, respectively). In addition, isogenic PL-matA and PL-matA ΔrcsB strains were constructed (yielding strains T1986 and T1987). In the two latter strains, the native mat promoter in the K12 genome was replaced by the phage λ PL promoter, for constitutive expression of matA. To analyze regulation by MatA and RcsB, these reporter strains were transformed either with the control plasmid pKESK22 or plasmid pKETS6 carrying rcsB under control of the IPTG-inducible tac promoter (Fig. 2A). The matACFT073 promoter lacZ fusion directed basal expression levels of 167 units in the rcsB+ strain T1749 that was transformed with the control plasmid (Fig. 2A). In this strain (T1749), RcsB is expressed by the native chromosomal rcsB gene, whereas the chromosomal matA gene is not expressed, because its K12-specific promoter is silenced by H-NS. In the isogenic ΔrcsB strain (T1747), the expression level was 135 units, and thus it was slightly lower than in the rcsB+ strain (Fig. 2A). Complementation of the ΔrcsB strain (T1747) with plasmid-encoded rcsB using plasmid pKETS6 caused a moderate increase of expression to 319 units (Fig. 2A). In the presence of MatA encoded by allele PL-matA, the expression level of the PmatACFT073 lacZ reporter increased to 1367 units in the rcsB+ strain T1986 (Fig. 2A). However, in the PL-matA ΔrcsB strain (T1987), the activity of the Pmat lacZ reporter was very low (89 units) (Fig. 2A). Complementation of this PL-matA ΔrcsB strain with rcsB using plasmid pKETS6 restored expression of Pmat lacZ to 2468 units (Fig. 2A). The data are in accordance with previous results showing that MatA (EcpR) activates the mat operon and that RcsB is required for mat operon expression (22, 23). Taken together, the results confirm that both proteins, MatA and RcsB, are required for activation of the matACFT073 promoter, and they indicate that a MatA-RcsB heterodimer activates the mat promoter.
FIGURE 2.

MatA-RcsB activates the matACFT073 promoter and inhibits motility. A, β-galactosidase expression levels directed by the matACFT073 promoter lacZ reporter were determined in rcsB+ strain T1749, ΔrcsB strain T1747, rcsB+ PL-matA strain T1986, and PL-matA ΔrcsB strain T1987. These strains were either transformed with control plasmid pKESK22 (pCtrl) or plasmids pKETS6 (pRcsB), pKET7 (pD56E), and pKES235 (pD56A), respectively. Cultures for β-galactosidase assays were grown to A600 of 0.5 in LB medium, supplemented with 1 mm IPTG and 25 mg/ml kanamycin. B, motility was determined of wild-type strain T1241 and ΔrcsB strain U89 and of transformants of these strains ectopically expressing MatA under the control of Ptac using plasmid pKEDP30 (pMatA). Overnight cultures were grown in LB medium, which was supplemented with 1 mm IPTG and 25 mg/ml kanamycin for growth of the transformants. Three μl of each culture was spotted on the center of a soft agar plate (0.2% agar), supplemented with 0.2 mm IPTG and 25 mg/ml kanamycin in the case of transformants, and the plates were incubated at 37 °C for 5 h. The plates were scanned, and the motility radii that are indicated by arrows were measured in mm. Error bars represent S.D. The images of the plates have been scaled to 25% of the original size.
MatA-RcsB Activates the mat Promoter Independently of Phosphorylation and Inhibits Motility
The activity of RcsA-RcsB and RcsB homodimers is phosphorylation-dependent, whereas BglJ-RcsB and GadE-RcsB are active independent of RcsB phosphorylation (3, 4, 8, 9, 18). Therefore, we assessed whether the activity of MatA-RcsB depends on RcsB phosphorylation. To this end, the Pmat lacZ reporter strain T1987 (ΔrcsB PL-matA) was transformed with plasmids encoding RcsB mutants RcsB-D56E (pKETS7), mimicking phosphorylated RcsB, and RcsB-D56A (pKES235), mimicking non-phosphorylated RcsB (42). Upon complementation of this reporter strain (T1987) with rcsB-D56E and rcsB-D56A, expression levels increased to 3207 and 2177 units, respectively (Fig. 2A). The moderate difference in activation of Pmat by the RcsB-D56 mutants and by wild-type RcsB (2468 units) suggests that transcriptional activation of the matACFT073 promoter by MatA-RcsB heterodimers is not (or is only weakly) dependent on RcsB phosphorylation. These results are in accordance with expression studies in NMEC strain IHE3034, where deletion of the Rcs phosphorelay genes rcsC and rcsD did not affect mat expression (22).
Recently, it was shown that ectopic expression of matA impairs the swimming behavior of E. coli NMEC strain IHE3034 and K12 strain MG1655, presumably by repression of the flhDC operon encoding the master regulator of flagella synthesis, FlhD4C2 (43). Here we tested whether this repression of motility by ectopically expressed MatA likewise depends on RcsB. To this end, 3 μl of an overnight culture of motile E. coli K12 wild-type strain (T1241) was spotted to the center of a soft agar plate (0.2% agar), and the motility radius was measured after growth of 5 h at 37 °C. When the motile K12 wild-type strain T1241 was transformed with a plasmid containing matA under control of the tac promoter (pKEDP30), its motility on soft agar plates (supplemented with 0.2 mm IPTG for matA induction and kanamycin for plasmid selection) was completely abolished (Fig. 2B). The motility of an isogenic ΔrcsB strain (U89) was similar to that of the wild-type strain T1241 (16 mm), and this ΔrcsB strain remained motile upon additional expression of matA (18 mm) (Fig. 2B). These data demonstrate that inhibition of motility by MatA requires RcsB, indicating that MatA-RcsB heterodimers repress motility.
Characterization of Amino Acid Residues of RcsB That Affect Its Activity as Homo- and Heterodimer
Our results suggest that MatA-RcsB heterodimers are active independent of RcsB phosphorylation, as shown previously for BglJ-RcsB and GadE-RcsB (8, 9, 18). In contrast, the activity of RcsA-RcsB heterodimers and RcsB homodimers is phosphorylation-dependent (3, 4). Phosphorylation of response regulators at the conserved aspartic acid residue is considered to induce a structural change that stabilizes the active form (39, 44). This presumptive structural change is apparently not relevant for the RcsB-heterodimers that are independent of RcsB phosphorylation, indicating that their active form is more stable and/or that they interact differently. Receiver domains of two-component response regulators typically exhibit a (βα)5 topology with five parallel β sheets in the center surrounded by two α helices on the one and three on the other side (39). To identify amino acids of the receiver domain of RcsB that are important for its activity as homodimer and heterodimer, respectively, we performed an alanine mutagenesis of amino acid residues specified below and tested the activity of these RcsB mutants using suitable promoter lacZ reporter fusions. For measuring MatA-RcsB activity, the PmatACFT073 PL-matA ΔrcsB ΔlacZ strain T1987 was used, as described above. For BglJ-RcsB, we used a PleuO bglJC ΔrcsB ΔlacZ reporter strain (T572), in which bglJ is expressed constitutively (17). For analyzing the RcsB homodimer activity, we constructed a PrprA lacZ fusion, as described (13), and for analyzing RcsA-RcsB activity, we constructed a fusion of the RcsA-RcsB-activated Pwza to lacZ (45).
First, we analyzed the suitability of the PrprA lacZ fusion as reporter for analyzing mutant RcsB homodimers. The reporter for RcsB homodimer activity, PrprA lacZ, was integrated into the chromosome of ΔlacZ strain S4197, resulting in strain T2023 (Fig. 3). In this rcsB+ strain T2023, the PrprA lacZ was poorly expressed, as expected (7 units; Fig. 3). Rcs signaling is known to be induced in ΔgalU strains, which cannot produce UDP-d-glucose (46). Accordingly, PrprA lacZ expression increased to 39 units upon deletion of galU (strain T2041) (Fig. 3). In the isogenic ΔrcsB strain (T1052) PrprA lacZ expression was very low (2 units; Fig. 3), whereas expression increased to 52 units upon complementation with plasmidic rcsB using pKETS6 and induction of rcsB expression by IPTG (Fig. 3). Upon complementation with RcsB mutant D56E, mimicking the phosphorylated form, the expression of PrprA lacZ increased to 100 units, whereas complementation with the RcsB-D56A mutant plasmid resulted in lower activation of the PrprA promoter (35 units) (Fig. 3). Thus, complementation with plasmidic rcsB overcomes the requirement for induction of the Rcs signaling cascade, whereas mutation of the RcsB phosphorylation site still affects the activation of PrprA by RcsB homodimers. Accordingly, the PrprA lacZ ΔrcsB strain is suitable for analysis of the activity of RcsB alanine mutants.
FIGURE 3.

Validation of a PrprA-lacZ fusion as a reporter system of RcsB activity. Expression levels directed by the chromosomal PrprA lacZ fusion were determined in rcsB+ strain T2023, ΔgalU strain T2041, and ΔrcsB strain T1052. The strains were either untransformed or complemented with rcsB, encoded by plasmid pKETS6 (pRcsB). RcsB derivatives D56E (pD56E) and D56A (pD56A) were expressed from plasmids pKETS7 and pKES235, respectively. Cultures for β-galactosidase assays were grown in LB medium to an A600 of 0.5, which was supplemented with 1 mm IPTG and 25 μg/ml kanamycin in the case of the transformants. Error bars represent S.D.
Second, for analyzing the activity of RcsA-RcsB heterodimers, a chromosomal Pwza lacZ fusion was used as reporter. The wza promoter is derived from the RcsA-RcsB regulated capsular exopolysaccharide (EPS) biosynthesis gene cluster wza-wca (45). Expression analyses of this Pwza reporter revealed that strain PL-rcsA ΔrcsBCD ΔlacZ (T963) is suitable. Briefly, the Pwza lacZ reporter was not expressed (1 unit) in the rcsB+ strain T2037 (Fig. 4). Induction of Rcs signaling by deletion of galU (T2042) induced Pwza lacZ expression slightly, to 5 units (Fig. 4). Complementation of the isogenic ΔrcsB strain (T864) with plasmidic rcsB did not confer an increase in expression (1 unit), whereas complementation of the isogenic ΔrcsBCD strain T921 induced the reporter slightly (5 units), suggesting that the ΔrcsBCD background in which RcsB cannot be dephosphorylated by the RcsCD phosphorelay might be more suitable. Further, the data suggest that induction of Rcs signaling or plasmidic rcsB expression is not sufficient for activation of the rcsA gene, which itself is H-NS-repressed and positively autoregulated by RcsA-RcsB (3, 47). Therefore, the rcsA promoter was replaced by the phage λ PL promoter, causing constitutive expression of rcsA (allele PL-rcsA). This resulted in activation of Pwza lacZ to 217 units in the rcsB+ PL-rcsA strain T2039 (Fig. 4). Additional deletion of galU (strain T2045) caused a further increase of expression to 287 units (Fig. 4). For the RcsB mutant characterization, complementation of Pwza lacZ PL-rcsA ΔrcsBCD (T963) with plasmidic rcsB was tested. In the PL-rcsA ΔrcsBCD strain (T963), the Pwza promoter was inactive (1 unit), as expected, whereas complementation with plasmidic rcsB (pKETS6) activated the Pwza promoter (223 units) (Fig. 4). Complementation with RcsB mutant D56E resulted in a further increase of Pwza activity to 323 units, whereas complementation with RcsB-D56A caused only a weak activation of Pwza (20 units), confirming that activation of Pwza by RcsA-RcsB in the reporter strain T963 remains RcsB phosphorylation-dependent (Fig. 4).
FIGURE 4.

Establishing Pwza-lacZ as reporter system of RcsA-RcsB activity. The expression levels of the chromosomally encoded Pwza lacZ fusion were determined in rcsB+ strain T2037, ΔgalU strain T2042, ΔrcsB strain T864, ΔrcsBCD strain T921, PL-rcsA strain T2039, PL-rcsA ΔgalU strain T2045, and PL-rcsA ΔrcsBCD strain T963. Expression levels were determined of non-transformed strains or of strains complemented with plasmidic rcsB, expressed from plasmid pKETS6 (pRcsB), pKET7 (pD56E), and pKES235 (pD56A), respectively. Cultures for β-galactosidase assays were grown to A600 of 0.5 in LB medium, which was supplemented with 1 mm IPTG and 25 μg/ml kanamycin in the case of transformants. Error bars represent S.D.
For identification of amino acid residues of RcsB that are important for RcsB activity, we mutated (i) amino acids residues of the active quintet that is coordinating phosphorylation; (ii) additional highly conserved amino acid residues adjacent to the active quintet; (iii) presumably surface-exposed residues, including residues of the α1 helix, which has been identified as the dimerization surface in NarL-type response regulators, and the α4-β5-α5 surface, which is the dimerization interface of PhoB-type response regulators; and (iv) residues in the helix-turn-helix DNA-binding domain (39, 44, 48). Because the structure of the RcsB receiver domain has not been solved, sequence alignments and comparison of structural models of the RcsB receiver domain (see “Experimental Procedures”) with conserved features of response regulators (39) were used to choose amino acid residues for alanine scanning mutagenesis. The models of the RcsB receiver domain suggest that the active quintet comprises amino acid residue Asp-56 (phosphorylation site) (3) as well as Asp-10, Asp-11, Thr-87, and Lys-109 (Fig. 5A). The three aspartate residues of the presumptive active quintet (Asp-10, Asp-11, and Asp-56) coordinate the metal ion that is essential for the phosphoryl group chemistry and hence for receiver domain function (39). Of the active quintet, alanine substitutions of residues Asp-11, Asp-56, Thr-87, and Lys-109 were analyzed. In addition, conserved amino acid residues Pro-60 and Gly-67 as well as residue Met-88 were exchanged to alanine (Fig. 5A). At the Met-88 position, a small amino acid (alanine or glycine) is conserved (39). Furthermore, the structural model (Fig. 5B) was used to choose presumably surface-exposed residues mapping in α4-β5-α5 (Leu-95, Ser-96, Leu-99, Asp-100, Glu-104, Ile-106, Leu-108, Thr-114, Asp-115, and Lys-118) as well as six additional amino acid residues (Ile-14, Asp-62, Tyr-64, Asp-66, Arg-76, and His-77), including Ile-14 mapping in the α1 helix.
FIGURE 5.

Structure model of the receiver and helix-turn-helix domains of RcsB. The structure model of the RcsB receiver domain predicted by the Phyre2 server (57) on the basis of the crystal structure (PDB code 3EUL) of M. tuberculosis NarL (36). Colored amino acids were replaced by alanine. Blue, active site residues Asp-11, Thr-87, and Lys-109 and highly conserved residues Pro-60, Gly-67, and Met-88. Green, the phosphorylation site Asp-56. Pink, presumably surface-exposed residues. For structure presentation, we used the PyMOL Molecular Graphics System, version 1.7.4 (Schrödinger, LLC). A and B show the same side and top views of the RcsB receiver domain. In A, residues of the active quintet and conserved residues of RcsB are labeled, whereas in B, other surface-exposed residues that were mutated are labeled. C, structure of the RcsB helix-turn-helix domain of E. amylovora (PDB code 1P4W) (32), which is 92% identical to E. coli RcsB. α helices 8 and 9, which bind to the DNA, are shown in blue. Residue Lys-180, which was mutated, is labeled K180.
For RcsB mutant analyses the reporter strains (T1052 for RcsB-RcsB, T963 for RcsA-RcsB, T572 for BglJ-RcsB, and T1987 for MatA-RcsB) were transformed with plasmids coding for wild-type RcsB (pKETS6) as well as the RcsB mutants (Fig. 6). To allow data comparison, the expression levels directed by the respective lacZ reporter fusions were normalized to those obtained in the presence of the active RcsB derivative D56E, which were defined as 100% (Fig. 6). A brief summary of the data is as follows. First, mutation of residues of the active quintet, including Asp-56, Asp-11, Thr-87, and Lys-109 had the highest impact on the phosphorylation-dependent RcsA-RcsB heterodimer and on RcsB-RcsB homodimers. Among these mutants, only K109A had a strong impact on activation by BglJ-RcsB and MatA-RcsB (Fig. 6). Second, mutation G67A strongly impaired RcsB-RcsB, RcsA-RcsB, and BglJ-RcsB, but not MatA-RcsB, whereas P60A impaired only the phosphorylation-dependent RcsB-RcsB and RcsA-RcsB (Fig. 6). In contrast, M88A resulted in high activity of all RcsB homo-/heterodimers (Fig. 6). Third, of the mutations of presumptive surface-exposed residues of α4-β5-α5, those that are located close to the phosphorylation site (including L95A, I106A, L108A, and D115A) reduced the activity of RcsB and RcsA-RcsB but had little to no impact on BglJ-RcsB and MatA-RcsB (Fig. 6). Fourth, of the six additional presumptive surface-exposed amino acid residues, only mutation of Ile-14 impaired the activity of all dimers, whereas mutation of Asp-62, Tyr-64, Asp-66, Arg-76, and His-77 affected RcsB-RcsB, RcsA-RcsB, and BglJ-RcsB and had no effect on MatA-RcsB (Fig. 6). Fifth, mutation of Lys-180 in the helix-turn-helix DNA-binding domain impaired all dimers (Fig. 6).
FIGURE 6.

Activation by RcsB mutants as RcsB-RcsB homodimer and as RcsA-RcsB, BglJ-RcsB, and MatA-RcsB heterodimers. The activity of RcsB mutants with exchanges of residues of the active quintet and conserved residues, surface-exposed residues, and a mutation mapping in the DNA-binding domain was analyzed using reporters for RcsB-RcsB (A, strain T1052), RcsA-RcsB (B, strain T963), BglJ-RcsB (C, strain T572), and MatA-RcsB (D, strain T1987). The strains were transformed with empty plasmid pKESK22, as control, and plasmids expressing wild-type and mutant RcsB under control of the IPTG-inducible tac promoter. The pKETS6-derived RcsB plasmids are listed in Table 2. β-Galactosidase expression levels were normalized to the values obtained for RcsB-D56E, which was defined as 100%. Values of bars marked with 1 are from Ref. 17. Cultures for β-galactosidase assays were grown to A600 of 0.5 in LB medium supplemented with 1 mm IPTG and 25 mg/ml kanamycin. Error bars represent S.D.
Taken together, RcsB homodimers and RcsA-RcsB heterodimers, both depending on RcsB phosphorylation, are impaired by mutations of residues Asp-11, Asp-56, Pro-60, Gly-67, Thr-87, Lys-109 (active quintet and highly conserved) as well as the surface-exposed Ile-14, Asp-62, Tyr-64, Asp-66, Arg-76, His-77, Leu-95, Ile-106, Leu-108, and Asp-115. For the activity of BglJ-RcsB dimers, amino acids Ile-14, Tyr-64, Gly-67, Lys-109, and Lys-180 have a crucial role and Asp-62 and Arg-76 are important. For the activity of MatA-RcsB, mutations of amino acids Ile-14, Lys-109, and Lys-180 are relevant. These data suggest that a structural change that is presumably elicited by phosphorylation and considered to stabilize the active form controls RcsB-RcsB and RcsA-RcsB activity and that the heterodimers of BglJ-RcsB and MatA-RcsB are intrinsically active.
Discussion
The interaction of the transcriptional response regulator RcsB with auxiliary proteins modulates the DNA binding specificity, adds a further level of output control to the Rcs system, and expands the regulatory role of RcsB in E. coli. RcsB functions as homodimer and in interaction with the auxiliary response regulator-like proteins RcsA and BglJ of the FixJ/NarL-family as well as with GadE (3, 8, 9). Our two-hybrid data suggest that RcsB in addition forms heterodimers with MatA (also known as EcpR) and DctR, which likewise belong to the FixJ/NarL family of transcriptional regulators (Fig. 7). Further data suggest that the activation of the matA promoter and repression of motility (23, 43) is mediated by the MatA-RcsB heterodimer independently of RcsB phosphorylation, whereas no target gene of DctR-RcsB could be identified. An alanine scanning mutagenesis of the RcsB receiver domain revealed that the majority of mutations in the vicinity of the phosphorylation site Asp-56 impair the activity of the phosphorylation-dependent RcsB-RcsB homodimer and RcsA-RcsB heterodimer but have little effect on the BglJ-RcsB and MatA-RcsB heterodimers, which are phosphorylation-independent. These residues are presumably affected by the structural change that is elicited by phosphorylation. Mutation of very few amino acid residues within the receiver domain, including Ile-14 and Lys-109, impaired or reduced the activity of all of the RcsB heterodimers that we tested. Further, mutation of residues Asp-62, Gly-67, Tyr-64, and Arg-76 significantly impaired the activity of BglJ-RcsB, RcsA-RcsB, and RcsB-RcsB but had little effect on MatA-RcsB. These data suggest that the stability and/or interaction surface of RcsB with its auxiliary partner proteins varies.
FIGURE 7.
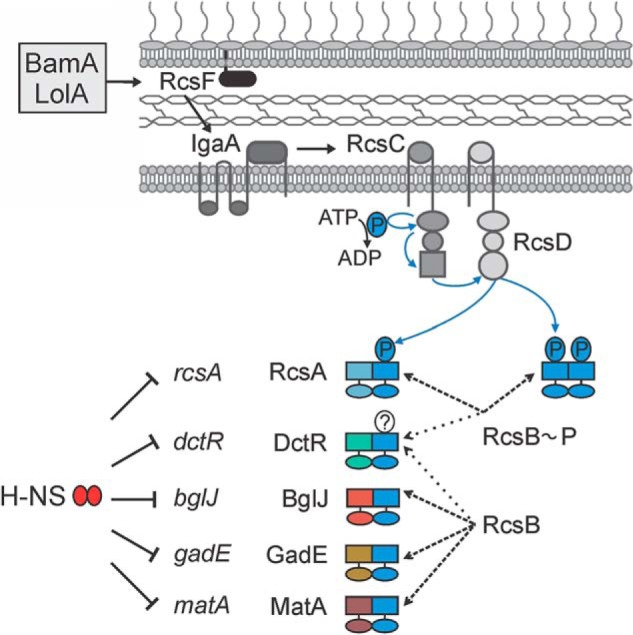
Model of the complex Rcs phosphorelay. The lipoprotein RcsF monitors the lipoprotein transport by LolA and the outer membrane protein assembly by BamA. Upon perturbations, RcsF activates the sensor kinase RcsC via IgaA (7). Upon induction of the Rcs phosphorelay and autophosphorylation of RcsC, the phosphate is transferred via the receiver domain of RcsC to the histidine transfer domain of RcsD and from there to the receiver domain of RcsB. Regulation of target genes by RcsB homodimers and by RcsA-RcsB heterodimers is dependent on RcsB phosphorylation. RcsB also forms heterodimers with DctR, BglJ, GadE, and MatA. Transcriptional activation by heterodimers of RcsB with BglJ, GadE, and MatA is RcsB phosphorylation-independent. For DctR-RcsB, it is not known whether the activity is phosphorylation-dependent. The genes rcsA, dctR, bglJ, gadE, and matA, which are encoding the auxiliary regulators of RcsB, are all repressed by the nucleoid-associated global repressor H-NS, and activation of their expression provides an additional level of control of the Rcs output.
The auxiliary interaction partners of RcsB, which include RcsA, BglJ, MatA, DctR, and GadE, all belong to the family of FixJ/NarL-type helix-turn-helix DNA-binding proteins. These auxiliary proteins do not form or only weakly form homodimers, and they do not interact among each other, as analyzed by a two-hybrid assay. Other members of the FixJ/NarL family present in E. coli K12 form homodimers but no heterodimers with RcsB. The interaction of RcsB with the auxiliary proteins alters the DNA binding specificity because the DNA-binding motifs of RcsB homodimers and heterodimers with RcsA, BglJ, and MatA are similar in one half-site, which is presumably RcsB-bound, whereas the DNA sequence of the other half-site varies (9, 12, 18, 23, 49). Note that mutation of RcsB residue Lys-180 in the helix-turn-helix motif renders RcsB and its heterodimers inactive and that acetylation of this residue inhibits RcsB DNA binding (50, 51).
Heterodimerization of response regulators in bacteria is rare. Currently, it has been described for BldM and WhiI in the filamentous bacteria Streptomyces (19). BldM and WhiI are atypical and orphan response regulators, which are presumably not phosphorylated by cognate sensor kinases. Rather, their expression is controlled by alternative σ factors. Notably, BldM and WhiI likewise belong to the FixJ/NarL family of transcriptional regulators. The BldM homodimer and BldM-WhiI heterodimers play key roles in the morphological differentiation of Streptomyces. BldM homodimers activate transcription of group I genes required for early stages of development, whereas BldM-WhiI heterodimers regulate expression of group II genes involved in the late stages of development. Correspondingly, the BldM consensus DNA-binding site is palindromic, whereas the BldM-WhiI consensus DNA-binding site is asymmetric with one half-site matching the BldM consensus sequence (19). This example demonstrates that heterodimerization of a response regulator is a means to control a developmental program (19).
RcsB and its auxiliary proteins all carry a FixJ/NarL-type helix-turn-helix DNA-binding domain. For other response regulators of this family, such as the homodimeric NarL, VraR, and DesR, it was shown that the DNA-binding domain is buried or inhibited by the non-phosphorylated form of the receiver domain (48, 52, 53). The phosphorylation of the receiver domain induces an interdomain structural change that releases the DNA-binding domain, allowing its dimerization and/or DNA binding. Our RcsB mutant analysis indicates that the activities of the phosphorylation site-dependent RcsB-RcsB and RcsA-RcsB may be regulated by such a structural change because mutations of residues in the vicinity of the phosphorylation site impaired their activity. In addition, Ile-14 (located in the α1 helix) impaired the activity, whereas the mutation of additional presumably surface-exposed residues in the α4β5α5 surface had no effect. In contrast, for the activity of BglJ-RcsB and MatA-RcsB, only a few residues were found to be important; these are Ile-14 (located in the α1 helix) and Lys-109 next to the active center for both heterodimers and further residues (Asp-62, Gly-67, Tyr-64, and Arg-76) for BglJ-RcsB. These data indicate that the presumptive phosphorylation-induced structural change of the receiver domain and an interdomain interaction are irrelevant for heterodimers formed by RcsB with BglJ and MatA, respectively. Furthermore, the data indicate that RcsB and its auxiliary proteins interact via a surface that includes helix α1, similar to NarL and related proteins (48, 53), because the mutation of RcsB Ile-14 to alanine renders all dimers inactive.
How is the interaction of RcsB with the auxiliary proteins regulated? One of the main elements in this control is the availability of the interaction partners. Expression of all of the corresponding genes is repressed by the nucleoid-associated and global repressor protein H-NS (22, 47, 54, 55) (Fig. 7). Still, little is known about the induction of these genes. The rcsA gene is autoregulated by RcsA-RcsB (12, 56), but our data indicate that induction of Rcs signaling is not sufficient to induce expression of the RcsA-RcsB target cps/wza, suggesting that the cellular RcsA concentration is still low, at least at 37 °C. Further, the RcsA protein is a substrate of the ATP-dependent Lon protease, which adds a further level of control on RcsA (14). The bglJ gene is the second gene of the H-NS repressed yjjQ-bglJ operon that can be activated by the pleiotropic transcription regulator LeuO (54). However, the leuO gene is also repressed by H-NS at standard laboratory growth conditions. Although the leuO gene can be activated by BglJ-RcsB and the mutual regulation of leuO and bglJ resembles a double-positive feedback regulation, inducing conditions are not known (17). The dctR gene is encoded in the acid stress island and presumably activated by YdeO, a regulator of genes involved in the cellular response to acid resistance. This is in agreement with the role of DctR in protection against metabolic end products under acidic conditions (26). The matA gene is the first gene of the mat (ecp) operon that encodes a conserved fimbrial adhesin in E. coli. Expression of this locus is induced at low temperature, acidic pH, and high acetate concentration in some lineages of E. coli, including B2 strains, whereas in other strains, mat is not expressed due to variations in the promoter region (23). Taken together, it seems that the activity of the RcsB heterodimers is largely controlled by the regulation of the auxiliary partners. Further, it is an open question whether the auxiliary partners do compete for RcsB under certain conditions.
Author Contributions
D. P. conducted most of the experiments, analyzed the results, and wrote the paper with K. S. M. F. conducted experiments on homodimer and heterodimer formation and analyzed the results. L. G. conducted experiments on RcsB mutants. K. S. conceived the project and wrote most of the paper.
Acknowledgments
We thank the Cologne Center of Genomics for microarray analyses and thank Prof. Ulrich Baumann (Institute for Biochemistry, University of Cologne) for discussions.
This work was supported by Deutsche Forschungsgemeinschaft Grant SCHN 371/10-2. The authors declare that they have no conflicts of interest with the contents of this article.
- NMEC
- meningitis- and septicemia-associated E. coli
- UPEC
- uropathogenic E. coli
- IPTG
- isopropyl β-d-1-thiogalactopyranoside
- PDB
- Protein Data Bank.
References
- 1. Stock A. M., Robinson V. L., and Goudreau P. N. (2000) Two-component signal transduction. Annu. Rev. Biochem. 69, 183–215 [DOI] [PubMed] [Google Scholar]
- 2. Gottesman S., Trisler P., and Torres-Cabassa A. (1985) Regulation of capsular polysaccharide synthesis in Escherichia coli K-12: characterization of three regulatory genes. J. Bacteriol. 162, 1111–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Majdalani N., and Gottesman S. (2005) The Rcs phosphorelay: a complex signal transduction system. Annu. Rev. Microbiol. 59, 379–405 [DOI] [PubMed] [Google Scholar]
- 4. Clarke D. J. (2010) The Rcs phosphorelay: more than just a two-component pathway. Future Microbiol. 5, 1173–1184 [DOI] [PubMed] [Google Scholar]
- 5. Laubacher M. E., and Ades S. E. (2008) The Rcs phosphorelay is a cell envelope stress response activated by peptidoglycan stress and contributes to intrinsic antibiotic resistance. J. Bacteriol. 190, 2065–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Evans K. L., Kannan S., Li G., de Pedro M. A., and Young K. D. (2013) Eliminating a set of four penicillin binding proteins triggers the Rcs phosphorelay and Cpx stress responses in Escherichia coli. J. Bacteriol. 195, 4415–4424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cho S. H., Szewczyk J., Pesavento C., Zietek M., Banzhaf M., Roszczenko P., Asmar A., Laloux G., Hov A. K., Leverrier P., Van der Henst C., Vertommen D., Typas A., and Collet J. F. (2014) Detecting envelope stress by monitoring β-barrel assembly. Cell 159, 1652–1664 [DOI] [PubMed] [Google Scholar]
- 8. Castanié-Cornet M. P., Cam K., Bastiat B., Cros A., Bordes P., and Gutierrez C. (2010) Acid stress response in Escherichia coli: mechanism of regulation of gadA transcription by RcsB and GadE. Nucleic Acids Res. 38, 3546–3554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Venkatesh G. R., Kembou Koungni F. C., Paukner A., Stratmann T., Blissenbach B., and Schnetz K. (2010) BglJ-RcsB heterodimers relieve repression of the Escherichia coli bgl operon by H-NS. J. Bacteriol. 192, 6456–6464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Henikoff S., Wallace J. C., and Brown J. P. (1990) Finding protein similarities with nucleotide sequence databases. Methods Enzymol. 183, 111–132 [DOI] [PubMed] [Google Scholar]
- 11. Gao R., Mack T. R., and Stock A. M. (2007) Bacterial response regulators: versatile regulatory strategies from common domains. Trends Biochem. Sci. 32, 225–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wehland M., and Bernhard F. (2000) The RcsAB box: characterization of a new operator essential for the regulation of exopolysaccharide biosynthesis in enteric bacteria. J. Biol. Chem. 275, 7013–7020 [DOI] [PubMed] [Google Scholar]
- 13. Majdalani N., Hernandez D., and Gottesman S. (2002) Regulation and mode of action of the second small RNA activator of RpoS translation, RprA. Mol. Microbiol. 46, 813–826 [DOI] [PubMed] [Google Scholar]
- 14. Stout V., Torres-Cabassa A., Maurizi M. R., Gutnick D., and Gottesman S. (1991) RcsA, an unstable positive regulator of capsular polysaccharide synthesis. J. Bacteriol. 173, 1738–1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ferrières L., Aslam S. N., Cooper R. M., and Clarke D. J. (2007) The yjbEFGH locus in Escherichia coli K-12 is an operon encoding proteins involved in exopolysaccharide production. Microbiology 153, 1070–1080 [DOI] [PubMed] [Google Scholar]
- 16. Francez-Charlot A., Laugel B., Van Gemert A., Dubarry N., Wiorowski F., Castanié-Cornet M. P., Gutierrez C., and Cam K. (2003) RcsCDB His-Asp phosphorelay system negatively regulates the flhDC operon in Escherichia coli. Mol. Microbiol. 49, 823–832 [DOI] [PubMed] [Google Scholar]
- 17. Stratmann T., Pul Ü., Wurm R., Wagner R., and Schnetz K. (2012) RcsB-BglJ activates the Escherichia coli leuO gene, encoding an H-NS antagonist and pleiotropic regulator of virulence determinants. Mol. Microbiol. 83, 1109–1123 [DOI] [PubMed] [Google Scholar]
- 18. Salscheider S. L., Jahn A., and Schnetz K. (2014) Transcriptional regulation by BglJ–RcsB, a pleiotropic heteromeric activator in Escherichia coli. Nucleic Acids Res. 42, 2999–3008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Al-Bassam M. M., Bibb M. J., Bush M. J., Chandra G., and Buttner M. J. (2014) Response regulator heterodimer formation controls a key stage in Streptomyces development. PLoS Genet. 10, e1004554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Martínez-Santos V. I., Medrano-López A., Saldaña Z., Girón J. A., and Puente J. L. (2012) Transcriptional regulation of the ecp operon by EcpR, IHF, and H-NS in attaching and effacing Escherichia coli. J. Bacteriol. 194, 5020–5033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lehti T. A., Bauchart P., Heikkinen J., Hacker J., Korhonen T. K., Dobrindt U., and Westerlund-Wikström B. (2010) Mat fimbriae promote biofilm formation by meningitis-associated Escherichia coli. Microbiology 156, 2408–2417 [DOI] [PubMed] [Google Scholar]
- 22. Lehti T. A., Heikkinen J., Korhonen T. K., and Westerlund-Wikstrom B. (2012) The response regulator RcsB activates expression of Mat fimbriae in meningitic Escherichia coli. J. Bacteriol. 194, 3475–3485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lehti T. A., Bauchart P., Kukkonen M., Dobrindt U., Korhonen T. K., and Westerlund-Wikström B. (2013) Phylogenetic group-associated differences in regulation of the common colonization factor Mat fimbria in Escherichia coli. Mol. Microbiol. 87, 1200–1222 [DOI] [PubMed] [Google Scholar]
- 24. Masuda N., and Church G. M. (2003) Regulatory network of acid resistance genes in Escherichia coli. Mol. Microbiol. 48, 699–712 [DOI] [PubMed] [Google Scholar]
- 25. Mates A. K., Sayed A. K., and Foster J. W. (2007) Products of the Escherichia coli acid fitness island attenuate metabolite stress at extremely low pH and mediate a cell density-dependent acid resistance. J. Bacteriol. 189, 2759–2768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yamanaka Y., Oshima T., Ishihama A., and Yamamoto K. (2014) Characterization of the YdeO regulon in Escherichia coli. PLoS One 9, e111962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wilson G. G., Young K. Y., Edlin G. J., and Konigsberg W. (1979) High-frequency generalised transduction by bacteriophage T4. Nature 280, 80–82 [DOI] [PubMed] [Google Scholar]
- 28. Miller J. H. (1972) in Experiments in Molecular Genetics, pp. 352–355, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 29. Diederich L., Rasmussen L. J., and Messer W. (1992) New cloning vectors for integration in the λ attachment site attB of the Escherichia coli chromosome. Plasmid 28, 14–24 [DOI] [PubMed] [Google Scholar]
- 30. Dole S., Kühn S., and Schnetz K. (2002) Post-transcriptional enhancement of Escherichia coli bgl operon silencing by limitation of BglG-mediated antitermination at low transcription rates. Mol. Microbiol. 43, 217–226 [DOI] [PubMed] [Google Scholar]
- 31. Datsenko K. A., and Wanner B. L. (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pristovsek P., Sengupta K., Löhr F., Schäfer B., von Trebra M. W., Rüterjans H., and Bernhard F. (2003) Structural analysis of the DNA-binding domain of the Erwinia amylovora RcsB protein and its interaction with the RcsAB box. J. Biol. Chem. 278, 17752–17759 [DOI] [PubMed] [Google Scholar]
- 33. Kelley L. A., Mezulis S., Yates C. M., Wass M. N., and Sternberg M. J. (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang J., Yan R., Roy A., Xu D., Poisson J., and Zhang Y. (2015) The I-TASSER Suite: protein structure and function prediction. Nat. Methods 12, 7–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Söding J., Biegert A., and Lupas A. N. (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–W248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schnell R., Agren D., and Schneider G. (2008) 1.9 Å structure of the signal receiver domain of the putative response regulator NarL from Mycobacterium tuberculosis. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 64, 1096–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baikalov I., Schröder I., Kaczor-Grzeskowiak M., Cascio D., Gunsalus R. P., and Dickerson R. E. (1998) NarL dimerization? suggestive evidence from a new crystal form. Biochemistry 37, 3665–3676 [DOI] [PubMed] [Google Scholar]
- 38. Baikalov I., Schröder I., Kaczor-Grzeskowiak M., Grzeskowiak K., Gunsalus R. P., and Dickerson R. E. (1996) Structure of the Escherichia coli response regulator NarL. Biochemistry 35, 11053–11061 [DOI] [PubMed] [Google Scholar]
- 39. Bourret R. B. (2010) Receiver domain structure and function in response regulator proteins. Curr. Opin. Microbiol. 13, 142–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dmitrova M., Younès-Cauet G., Oertel-Buchheit P., Porte D., Schnarr M., and Granger-Schnarr M. (1998) A new LexA-based genetic system for monitoring and analyzing protein heterodimerization in Escherichia coli. Mol. Gen. Genet. 257, 205–212 [DOI] [PubMed] [Google Scholar]
- 41. Nishino K., Inazumi Y., and Yamaguchi A. (2003) Global analysis of genes regulated by EvgA of the two-component regulatory system in Escherichia coli. J. Bacteriol. 185, 2667–2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Scharf B. E. (2010) Summary of useful methods for two-component system research. Curr. Opin. Microbiol. 13, 246–252 [DOI] [PubMed] [Google Scholar]
- 43. Lehti T. A., Bauchart P., Dobrindt U., Korhonen T. K., and Westerlund-Wikström B. (2012) The fimbriae activator MatA switches off motility in Escherichia coli by repression of the flagellar master operon flhDC. Microbiology 158, 1444–1455 [DOI] [PubMed] [Google Scholar]
- 44. Gao R., and Stock A. M. (2010) Molecular strategies for phosphorylation-mediated regulation of response regulator activity. Curr. Opin. Microbiol. 13, 160–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stevenson G., Andrianopoulos K., Hobbs M., and Reeves P. R. (1996) Organization of the Escherichia coli K-12 gene cluster responsible for production of the extracellular polysaccharide colanic acid. J. Bacteriol. 178, 4885–4893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Girgis H. S., Liu Y., Ryu W. S., and Tavazoie S. (2007) A comprehensive genetic characterization of bacterial motility. PLoS Genet. 3, 1644–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sledjeski D., and Gottesman S. (1995) A small RNA acts as an antisilencer of the H-NS-silenced rcsA gene of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 92, 2003–2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Trajtenberg F., Albanesi D., Ruétalo N., Botti H., Mechaly A. E., Nieves M., Aguilar P. S., Cybulski L., Larrieux N., de Mendoza D., and Buschiazzo A. (2014) Allosteric activation of bacterial response regulators: the role of the cognate histidine kinase beyond phosphorylation. mBio 5, e02105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sturny R., Cam K., Gutierrez C., and Conter A. (2003) NhaR and RcsB independently regulate the osmCp1 promoter of Escherichia coli at overlapping regulatory sites. J. Bacteriol. 185, 4298–4304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hu L. I., Chi B. K., Kuhn M. L., Filippova E. V., Walker-Peddakotla A. J., Bäsell K., Becher D., Anderson W. F., Antelmann H., and Wolfe A. J. (2013) Acetylation of the response regulator RcsB controls transcription from a small RNA promoter. J. Bacteriol. 195, 4174–4186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Thao S., Chen C.-S., Zhu H., and Escalante-Semerena J. C. (2010) Nϵ-Lysine acetylation of a bacterial transcription factor inhibits its DNA-binding activity. PLoS One 5, e15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Leonard P. G., Golemi-Kotra D., and Stock A. M. (2013) Phosphorylation-dependent conformational changes and domain rearrangements in Staphylococcus aureus VraR activation. Proc. Natl. Acad. Sci. U.S.A. 110, 8525–8530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang J. H., Xiao G., Gunsalus R. P., and Hubbell W. L. (2003) Phosphorylation triggers domain separation in the DNA binding response regulator narL. Biochemistry 42, 2552–2559 [DOI] [PubMed] [Google Scholar]
- 54. Stratmann T., Madhusudan S., and Schnetz K. (2008) Regulation of the yjjQ-bglJ operon, encoding LuxR-type transcription factors, and the divergent yjjP gene by H-NS and LeuO. J. Bacteriol. 190, 926–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sayed A. K., and Foster J. W. (2009) A 750 bp sensory integration region directs global control of the Escherichia coli GadE acid resistance regulator. Mol. Microbiol. 71, 1435–1450 [DOI] [PubMed] [Google Scholar]
- 56. Ebel W., and Trempy J. E. (1999) Escherichia coli RcsA, a positive activator of colanic acid capsular polysaccharide synthesis, functions to activate its own expression. J. Bacteriol. 181, 577–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kelley L. A., and Sternberg M. J. (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4, 363–371 [DOI] [PubMed] [Google Scholar]
- 58. Wiebe H., Gürlebeck D., Gross J., Dreck K., Pannen D., Ewers C., Wieler L. H., and Schnetz K. (2015) YjjQ represses transcription of flhDC and additional loci in Escherichia coli. J. Bacteriol. 197, 2713–2720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K. A., Tomita M., Wanner B. L., and Mori H. (2006) Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Welch R. A., Burland V., Plunkett G. 3rd, Redford P., Roesch P., Rasko D., Buckles E. L., Liou S. R., Boutin A., Hackett J., Stroud D., Mayhew G. F., Rose D. J., Zhou S., Schwartz D. C., Perna N. T., Mobley H. L., Donnenberg M. S., and Blattner F. R. (2002) Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 99, 17020–17024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cherepanov P. P., and Wackernagel W. (1995) Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158, 9–14 [DOI] [PubMed] [Google Scholar]
- 62. Guzman L. M., Belin D., Carson M. J., and Beckwith J. (1995) Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177, 4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]