

Abstract
A signature event during the cell intrinsic apoptotic pathway is mitochondrial outer membrane permeabilization, leading to formation of the apoptosome, a caspase activation complex. The cellular apoptosis susceptibility protein (CAS) can facilitate apoptosome assembly by stimulating nucleotide exchange on Apaf-1 following binding of cytochrome c. We report here that CAS expression itself is up-regulated during tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis, and knockdown of CAS renders cells resistant to TRAIL. We find that TRAIL induces up-regulation of CAS in a posttranscriptional, caspase-8-dependent manner through degradation of cIAP1, an E3 ligase that targets CAS for ubiquitin-dependent proteasomal degradation. We identified a novel signaling pathway whereby caspase-8 engages a feedforward cascade that leads to CAS up-regulation and amplifies the apoptotic signal. Furthermore, in silico analysis revealed that expression of CAS is up-regulated at both the mRNA and DNA levels in human breast tumors, consistent with its role in promoting cell proliferation. Overexpression of various oncogenes led to CAS up-regulation in non-transformed cells. Intriguingly, oncogene-induced CAS up-regulation also resulted in greater susceptibility to TRAIL-induced cell death, consistent with its proapoptotic function. These findings suggest that CAS plays contrasting roles in proliferation and apoptosis and that overexpression of CAS in tumors could serve as a potential biomarker to guide therapeutic choices.
Keywords: apoptosis, caspase, oncogene, TRAIL, ubiquitylation (ubiquitination), CAS, IAP1
Introduction
Apoptosis in mammalian cells typically proceeds through one of two signaling cascades: the cell-intrinsic or the cell-extrinsic pathway. The cell-intrinsic pathway is initiated by mitochondrial outer membrane permeabilization (MOMP)2 (1). This leads to release of cytochrome c into the cytosol, where it binds to adaptor protein Apaf-1 and triggers assembly of the apoptosome, a heptameric caspase activation complex (2, 3). The apoptosome recruits and activates caspase-9, an initiator caspase that subsequently activates executioner caspase-3/7, which cleave a variety of cellular proteins, leading to cell death (4, 5). In the cell-extrinsic pathway, binding of extracellular ligands leads to activation of caspase-8 at the cell surface (6). Caspase-8 can then directly activate caspase-3 or, additionally, engage the mitochondrial pathway through cleavage of BID, leading to MOMP (7, 8). In so called “type II” cells, BID-mediated MOMP is essential for death receptor-induced apoptosis. On the other hand, direct activation of caspase-3 by active caspase-8 is sufficient for apoptosis in type I cells (9, 10).
MOMP is associated with a loss of mitochondrial function and release of several factors from the mitochondrial intermembrane space that can induce caspase activation as well as caspase-independent cell death. Therefore, MOMP has been postulated to be a “point of no return” for cell death; i.e. following MOMP, cells are committed to death regardless of caspase activation (11). However, although this may be true in some cases, several lines of evidence contradict this claim. For instance, cells lacking Apaf-1 or caspase-9 are highly resistant to various apoptotic stimuli that induce MOMP (12–17). Additionally, pharmacological or genetic inhibition of caspases protects neurons from NGF withdrawal-induced cell death, despite cytochrome c release, and these cells completely recover after NGF restimulation (18, 19). Indeed, cells can survive MOMP, provided executioner caspase activity is inhibited (20, 21).
The ability to survive MOMP has several important physiological consequences. Firstly, it provides a mechanism to protect cells against “accidental” MOMP induced by minor apoptotic insults. This is particularly relevant to the survival of postmitotic cells like cardiomyocytes and neurons, which indeed exhibit a higher threshold of cytosolic cytochrome c needed to induce cell death (22–24). Furthermore, caspase-3 and -9 are involved in several non-apoptotic processes, such as differentiation of various cell types (25–29), development and maintenance of neuronal function (30–32), and proliferation and maturation of immune cells (33, 34). Importantly, caspase-3 activation in these scenarios is not lethal but, rather, leads to changes in cell shape or function, presumably resulting from cleavage of specific substrates. In the context of oncogenesis, tumor cells often evolve mechanisms of inhibiting caspase-3 activation downstream of MOMP, including down-regulation or loss of Apaf-1 (35, 36) or caspase-3 (37) and overexpression of inhibitor of apoptosis (IAP) proteins (38, 39). The ability to survive therapy-induced MOMP by limiting caspase-3 activation can facilitate tumor cell survival and has obvious clinical implications. Intriguingly, when MOMP is limited or incomplete, low levels of caspase-3 activation can directly promote tumorigenesis through genomic instability (40, 41). Finally, it is worth noting that, even in cases where MOMP is sufficient to trigger cell death, caspase-3 activity is essential in preventing an immune response in vivo (42, 43). Collectively, these findings underscore the importance of understanding how caspase-3 activation is regulated post-MOMP.
Regulating apoptosome formation is a critical means through which caspase-3 activity can be fine-tuned following the onset of MOMP. After binding cytochrome c, Apaf-1 undergoes nucleotide exchange. This is a necessary step for apoptosome formation because, in the absence of nucleotide exchange, cytochrome c-bound Apaf-1 forms nonfunctional aggregates (44). The cellular apoptosis susceptibility protein (CAS), functioning together with PHAPI and Hsp70, stimulates apoptosome formation by enhancing nucleotide exchange on Apaf-1 following cytochrome c binding (45). In this study, we investigate the regulation of CAS upon TRAIL-induced apoptosis. Furthermore, we explore the role of CAS in cancer cell growth and apoptosis.
Experimental Procedures
Cell Culture
MCF-10A cells were cultured in DMEM/F12 supplemented with 5% horse serum, EGF (20 ng/ml), hydrocortisone (0.5 μg/ml), cholera toxin (100 ng/ml), insulin (10 μg/ml), and penicillin-streptomycin. 293T and HT-29 cells were cultured in DMEM high-glucose supplemented with 10% FBS, l-glutamine (2 mm), and penicillin-streptomycin. Lentiviral or retroviral constructs were co-transfected with packaging vectors into 293T cells for virus production. Virus containing-medium was passed through a 0.45-μm polyethersulfone filter and supplemented with Polybrene before being used to transduce cells.
Reagents, Antibodies, and Plasmids
SuperKiller TRAIL (catalog no. ALX-201-115-3010) and Z-VAD-fmk (catalog no. ALX-260-020) were from Enzo Life Sciences. Caspase-8 inhibitor (IETD-fmk, catalog no. 550380) and caspase-3 inhibitor (DEVD-fmk, catalog no. 550378) were from BD Biosciences. MG132 was from EMD Millipore (catalog no. 474790). Bafilomycin A1 was from Sigma. For Western blot analysis, the following antibodies were used: anti-CAS (Bethyl, catalog no. A300-473A), anti-caspase-3 (Cell Signaling Technology, catalog no. 9662), anti-caspase-8 (Cell Signaling Technology, catalog no. 9746), anti-cIAP1 (Cell Signaling Technology, catalog nos. 7065 and 4952), anti-XIAP (Cell Signaling Technology, catalog no. 2045), anti-CYLD (Cell Signaling Technology, catalog no. 8462), anti β-Actin (Sigma, catalog nos. A1978 and 5316), anti α-actinin (Santa Cruz Biotechnology, catalog no. sc-17829), anti-ubiquitin (Millipore, catalog no. 05-944), anti-GFP (Roche, catalog no. 11814460001), and anti-HA clone 16B12 (Covance, catalog no. MMS-101P). The SMAC-cherry reporter construct was generated by fusing the N-terminal 171 bp of human SMAC cDNA to the N terminus of mCherry on the pBabe-puro vector. Bcl-xL was subcloned into the pQCXIP vector using the EcoRI and BamHI sites. N-terminally tagged GFP-His-CAS and GFP-CAS were generated by subcloning the respective fragments into the pQCXIP-GFP vector. cIAP1 cDNA was purchased from Addgene (WT, catalog no. 8311; H588A, catalog no. 8334) and subcloned into the pQCXIP-HA vector (N-terminally tagged) at the XhoI and EcoRI sites or the pET28A vector at the Nhe1 and Xho1 sites. pcDNA-Myc-His-Ubiquitin, pBabe-Erbb2, MSCV-mCherry-Myc, pBabe-PI3KH1047R, and pBabe-KRASG12V were provided by Filippo Giancotti (Memorial Sloan Kettering Cancer Center).
Co-immunoprecipitation
For co-immunoprecipitation assays, 293T cells grown in 10-cm plates were transfected with the indicated plasmids. 24 h later, cells were lysed in-plate with IP buffer (10 mm Hepes (pH 7.5), 150 mm NaCl, 0.5% Nonidet P-40, 1 mm EDTA, 1 mm DTT, and 10% Glycerol) supplemented with protease inhibitors. Lysates were cleared by centrifugation at 14,000 rpm for 10 min at 4 °C and incubated with HA-agarose beads (Sigma, catalog no. A2095) overnight at 4 °C. Beads were then washed five times in IP buffer and eluted by boiling in 1× SDS sample buffer.
In Vivo Ubiquitination Assays
For assays in MCF10A cells, cells stably expressing GFP-His-CAS were lysed in 100 μl of radio-immunoprecipitation assay buffer supplemented with 25 μm MG132, 10 mm N-ethylmaleimide (Sigma), and protease inhibitors. Ni-NTA-agarose beads (Qiagen) were then used to pull down His-tagged protein as described previously (46). For assays in 293T cells, cells were transfected with Myc-His-Ubiquitin and the indicated plasmids in 6-cm plates. 24 h later, cells were treated with 25 μm MG132 for 4 h. After washing with PBS, a small fraction of the cell pellet was saved for input and lysed in radio-immunoprecipitation assay buffer. The rest of the cell pellet was resuspended in lysis buffer (6 m guanidine, 0.1 m NaH2PO4, 10 mm Tris (pH 8.0), 10 mm β-mercaptoethanol) freshly supplemented with 15 mm imidazole, 10 μm MG132, 10 mm N-ethylmaleimide, and protease inhibitors and sonicated to reduce viscosity. Lysates were then incubated with prewashed Ni-NTA-agarose beads for 3 h at room temperature. Beads were then washed once with buffer A (lysis buffer + 0.2% Triton), once with buffer B (8 m urea, 0.1 m NaH2PO4, 10 mm Tris (pH 8.0), 10 mm β-mercaptoethanol and 0.2% Triton), twice with buffer C (8 m urea, 0.1 m NaH2PO4, 10 mm Tris (pH 6.3), 10 mm β-mercaptoethanol and 0.2% Triton), and twice with buffer D (8 m urea, 0.1 m NaH2PO4, 10 mm Tris (pH 6.3), 10 mm β-mercaptoethanol and 0.1% Triton). All wash buffers were freshly supplemented with 15 mm imidazole. Finally, bound proteins were eluted by boiling in radio-immunoprecipitation assay buffer containing 1× SDS loading buffer and 200 mm imidazole for 10 min. Samples were then subjected to Western blot analysis with the indicated antibodies.
In Vitro Ubiquitination Assay
His-tagged cIAP1 proteins were cloned into the pET28A vector, expressed in BL21-DE3 cells (Novagen), and purified from bacterial lysates using Ni-NTA-agarose beads. GFP-CAS was expressed in 293T cells and purified using GFP-Trap beads (ChromoTek). Briefly, cells were lysed in IP buffer and incubated with GFP-Trap beads for 1 h, followed by sequentially washing three times in IP buffer and twice in ubiquitination reaction buffer (50 mm Tris (pH 7.5), 5 mm MgCl2, and 2 mm DTT). Beads were then resuspended in reaction buffer containing 75 nm E1, 600 nm E2, 5 mm ATP, and 10 μg/μl ubiquitin in a final volume of 20 μl. Where indicated, 800 ng of WT- or H588A-cIAP1 were included, and reactions were incubated at 30 °C for 2 h with gentle agitation. Following the reaction, beads were washed four times in denaturing buffer (8 m urea and 1% SDS in PBS) to remove nonspecific binding. Following a final wash in IP buffer, bound proteins were eluted by boiling in 1× SDS sample buffer and subjected to immunoblotting with the indicated antibodies.
Knockdown by siRNA and shRNA
siGenome SMARTpool siRNAs (CAS, cIAP1, and XIAP), control siRNA (D-001210-05-20), and individual siRNA against CYLD (M-004413-02-0005) were purchased from Dharmacon. Cells were seeded in 6-well plates at a density of 80,000 cells/well (MCF10A) or 200,000 cells/well (HT-29) and, 24 h later, transfected with 25–30 nm CAS siRNA using Oligofectamine (Invitrogen) or Dharmafect 1 (Dharmacon), respectively. Cells were routinely assayed 64–72 h post-transfection. For cell growth assays, a higher concentration of CAS siRNA (75 nm) was used. Knockdown of cIAP1, XIAP, and CYLD was achieved with 100 nm siRNA. The caspase-8 shRNA sequences were as follows: sh1, GACATGAACCTGCTGGATATT; sh2, GCCTTGATGTTATTCCAGAGA. Lentiviruses harboring the knockdown sequences were used to infect MCF10A cells. Cells when then selected with puromycin for at least 3 days before testing for knockdown.
Cell Viability and Growth Assays
Cell viability was measured using the CellTiter-Glo luminescent cell viability assay kit (Promega). Briefly, cells were plated in triplicate at a density of 1500–3000 (MCF10A) or 6000 (HT-29) cells/well and allowed to adhere for 24–48 h. Following treatment with TRAIL, the percentage of viable cells was quantified relative to untreated cells according to the directions of the manufacturer. All cell viability data are values from at least three independent experiments. To measure cell growth, following transfection with siRNA, cells were split into 12-well plates at a density of 10,000 cells/well. At 24-h intervals, cells were fixed in 10% formalin for 10 min. Following washing with PBS, cells were stained with 0.1% crystal violet solution for 20 min, washed three times with water, and allowed to air-dry. The stain was extracted by incubating in a 10% acetic acid solution for 20 min with shaking. The relative number of cells was then quantified by measuring the absorbance of the extracted stain at 590 nm.
Caspase Assay
To quantify caspase activity, cell lysates were collected after treatment, and 20 μg of total protein was incubated with 15 μm fluorogenic caspase-3 rhodamine-DEVD substrate (AnaSpec, catalog no. 60304) in a final volume of 20 μl. Samples were then loaded immediately on 384-well plates, and fluorescence was measured at 30 °C at an interval of 2 min for 3 h using a SpectraFluor Plus spectrometry reader (Tecan) with an excitation wavelength of 485 nm and an emission wavelength of 535 nm. Average relative fluorescence units ± S.E. of three independent experiments were plotted.
Quantitative RT-PCR
Total RNA was extracted using the Aurum Total RNA mini kit (Bio-Rad), and reverse transcription was performed using the iScript cDNA synthesis kit (Bio-Rad). Quantitative RT-PCR was performed with iQ SYBR Green Supermix (Bio-Rad) using a CFX Connect real-time system (Bio-Rad). The relative level of mRNA was calculated by the comparative Ct method using GAPDH as a control. The primers for CAS were TGCCTCGTTTTGTTACAGCC (forward) and GGTCTCTCACAAACTGAAGCC (reverse). The primers for GAPDH were TGCACCACCAACTGCTTAGC (forward) and GGCATGGACTGTGGTCATGAG (reverse).
Genomic Analysis
The Cancer Genome Atlas dataset used has been described previously (47). All data were retrieved and analyzed using the cBioPortal for Cancer Genomics (48, 49).
Statistical Analysis
Unless noted otherwise, results were analyzed by Student's t test performed using GraphPad Prism 6 software. Significance was defined as p < 0.05.
Results
CAS Promotes TRAIL-induced Apoptosis
We and others have shown previously that CAS plays a significant role in apoptosis induced by select stimuli (45, 50). To specifically examine the role of CAS in TRAIL-induced apoptosis, we used siRNA to silence CAS expression in MCF10A and HT-29 cells. Relative to control siRNA, we observed that knockdown of CAS significantly inhibited TRAIL induced caspase-3/7 activation in both cell types (Fig. 1, a and b). Consistently, overall cell viability following TRAIL-treatment was greater in CAS-siRNA transfected cells (Fig. 1, c and d). These results clearly indicate that CAS facilitates TRAIL-induced cell death.
FIGURE 1.

Knockdown of CAS inhibits TRAIL-induced apoptosis. a and c, MCF10A cells transfected with non-target (NT) or CAS siRNA were treated with TRAIL (15 ng/ml for 4 h) for measurement of caspase-3/7 activity (a) or for 8 h at the indicated concentration for measurement of cell viability (c). b and d, HT-29 cells transfected with NT or CAS siRNA were treated with TRAIL (12.5 ng/ml for 6 h) for measurement of caspase-3/7 activity (b) or for 12 h at the indicated concentration for measurement of cell viability (d). Representative phase-contrast images displaying cell morphology and Western blots confirming knockdown of CAS are shown. All values are mean ± S.E. from at least three independent experiments. *, p < 0.05. RFU, relative fluorescence units.
TRAIL Induces Robust Up-regulation of CAS through Activation of Caspase-8 and Independent of Mitochondrial Outer Membrane Permeabilization
In the course of our experiments, we observed that TRAIL induces a robust increase in CAS protein levels within a few hours of treatment (Fig. 2a). Because CAS clearly plays a functional role in TRAIL-induced cell death, we sought to explore the mechanism of TRAIL-induced CAS up-regulation. Apoptotic signaling by TRAIL proceeds through activation of the initiator caspase-8, followed by executioner caspase-3/7. We hypothesized that activation of one or more caspases plays a critical role in up-regulating CAS levels upon TRAIL treatment. In support of this hypothesis, we found that addition of a pan-caspase inhibitor (Z-VAD-fmk) completely blocked TRAIL-induced CAS up-regulation (Fig. 2b). Furthermore, specific inhibition (Fig. 2b) or shRNA-mediated knockdown of caspase-8 (Fig. 2c) inhibited an increase in CAS levels upon TRAIL treatment. Intriguingly, co-treatment with a caspase-3-specific inhibitor failed to inhibit CAS up-regulation (Fig. 2b). All three inhibitors completely abolished downstream activation of caspase-3, confirming their respective activity (Fig. 2b). These results demonstrate that TRAIL-induced CAS up-regulation is caspase-8-dependent but caspase-3-independent.
FIGURE 2.

TRAIL induces robust up-regulation of CAS in a caspase-8-dependent manner and independently of MOMP. a, MCF10A cells were treated with 30 ng/ml TRAIL and harvested for Western blot analysis at the indicated time points. b, MCF10A cells were pretreated for 30 min with 20 μm of a pan-caspase inhibitor (Z-VAD-fmk), a caspase-3 inhibitor (C3i, DEVD-fmk), or a caspase-8 inhibitor (C8i, IETD-fmk), followed by 40 ng/ml TRAIL for 3 h, and subsequently harvested for Western blot analysis. Right panel, quantification of CAS levels (mean ± S.D.) relative to untreated cells from three independent experiments. c, MCF10A cells stably expressing control shRNA or one of two different shRNAs against caspase-8 were treated with 30 ng/ml TRAIL for 3 h, and expression of the indicated proteins was analyzed by Western blot. The asterisk indicates a nonspecific band. d, WT and Bcl-xL-overexpressing MCF10A cells stably expressing SMAC-cherry were treated with 40 ng/ml TRAIL for 2.5 h, and imaged for the onset of MOMP. e, WT and Bcl-xL-overexpressing MCF10A cells were treated with 40 ng/ml TRAIL for 3 h and harvested for Western blot analysis with the indicated antibodies.
Upon activation, caspase-8 can, in turn, activate caspase-3 by two distinct mechanisms: direct cleavage of caspase-3 or cleavage of the BH3-only protein BID, leading to MOMP and cytochrome c release. To determine whether MOMP is required for up-regulation of CAS, we generated an MCF10A cell line stably overexpressing Bcl-xL. Bcl-xL is an anti-apoptotic Bcl-2 family protein that antagonizes mitochondrial permeabilization by Bax/Bak (51). To monitor MOMP, we used a fluorescent SMAC-cherry reporter construct. mCherry fluorescence was localized to mitochondria in untreated cells but released into the cytosol following TRAIL stimulation, indicating the onset of MOMP. Bcl-xL-overexpressing MCF10A cells were resistant to TRAIL-induced MOMP (Fig. 2d). Consistently, TRAIL-induced caspase-3 activation was also blocked by Bcl-xL overexpression (Fig. 2e), demonstrating that MCF10A are type II cells. Importantly, overexpression of Bcl-xL failed to inhibit CAS up-regulation (Fig. 2e), indicating that TRAIL induces CAS up-regulation upstream of MOMP.
Proteasomal Degradation of CAS Is Inhibited during TRAIL-induced Apoptosis
We next asked whether TRAIL induces an increase in transcription of CAS. Quantitative RT-PCR experiments revealed that CAS mRNA levels are not increased upon TRAIL treatment (Fig. 3a). Additionally, TRAIL treatment led to a comparable up-regulation of a GFP-CAS fusion protein expressed under the control of a naïve retroviral promoter (Fig. 3b), indicating that TRAIL-induced CAS up-regulation is posttranscriptional in nature.
FIGURE 3.
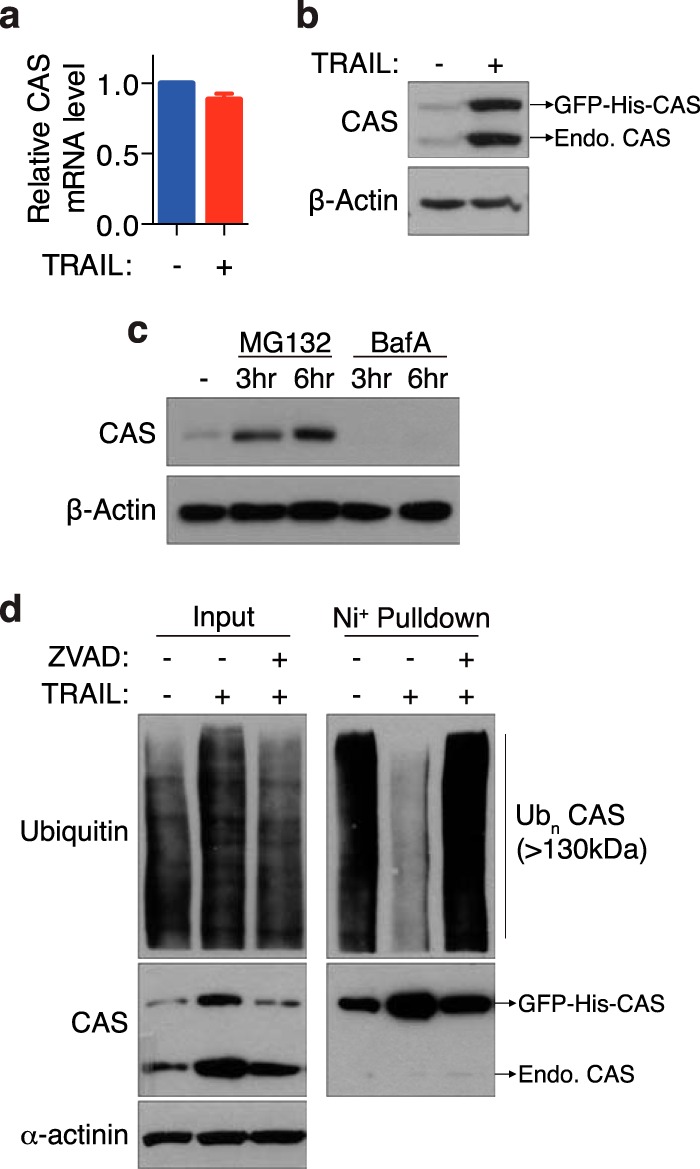
TRAIL-induced CAS up-regulation is posttranscriptional and occurs through inhibition of its proteasomal degradation. a, quantitative RT-PCR was used to detect expression of CAS mRNA in MCF10A cells treated with 30 ng/ml TRAIL for 1 h. GAPDH was used as an internal control. Values are mean ± S.E. from three independent experiments. b, Western blot of MCF10A cells stably expressing a GFP-His-CAS fusion protein following stimulation with TRAIL (30 ng/ml, 3 h). c, MCF10A cells were treated with the proteasome inhibitor MG132 (75 μm) or the lysosomal inhibitor Bafilomycin A1 (BafA, 20 nm) for the indicated time and then harvested for Western blot analysis. d, lysates from MCF10A cells stably expressing GFP-His-CAS were subjected to pulldown with nickel beads under denaturing conditions and subsequently probed with the indicated antibodies. Where indicated, cells were treated with 10 μm Z-VAD-fmk for 30 min prior to addition of TRAIL (30 ng/ml, 4 h). Endo, endogenous; Ubn CAS, polyubiquitinated CAS.
Interestingly, we observed that inhibition of proteasome activity by MG132 was sufficient to elevate basal levels of CAS, whereas inhibition of lysosome activity by Bafilomycin A1 failed to do so (Fig. 3c), suggesting that CAS is prone to ubiquitination-mediated proteasomal turnover. We therefore tested whether CAS ubiquitination is decreased in response to TRAIL. Indeed, TRAIL stimulation significantly reduced the accumulation of ubiquitinated CAS but not global ubiquitinated proteins (Fig. 3d). Furthermore, consistent with the essential role of caspase activation in promoting CAS up-regulation, addition of the pan-caspase inhibitor Z-VAD-fmk abolished the decrease in CAS ubiquitination upon TRAIL treatment.
Degradation of cIAP1 Mediates CAS Accumulation during TRAIL-induced Apoptosis
On the basis of these data, we reasoned that TRAIL treatment could lead to decreased CAS ubiquitination through two possible mechanisms: inhibition of an E3 ubiquitin ligase that targets CAS for degradation or activation of a deubiquitinase. To further dissect this signaling pathway, we sought to identify the proteins involved in this process. We undertook a candidate-based approach, focusing on proteins that met two criteria: those documented to possess E3 ligase or deubiquitinase catalytic activity and those known to be regulated by caspase-8 and/or TRAIL signaling. Intriguingly, a literature search revealed that cIAP1, a potent E3 ligase and anti-apoptotic protein, is degraded during TRAIL-induced apoptosis (52). Consistently, we observed that TRAIL treatment leads to a significant decrease in cIAP1 levels, which is rescued by addition of a caspase-8 inhibitor (Fig. 4a), fitting our model. To test whether cIAP1 controls the stability of CAS, we transfected siRNA targeting cIAP1 into MCF10A cells. Notably, silencing cIAP1 expression led to a modest increase in the steady-state levels of CAS relative to control siRNA (Fig. 4b). Meanwhile, silencing XIAP (another E3 ligase degraded upon TRAIL treatment (52)) or CYLD (a deubiquitinase known to be targeted by active caspase-8 (53)) had no effect on basal CAS expression or TRAIL-induced CAS up-regulation, respectively (data not shown). Furthermore, co-immunoprecipitation in HEK293T cells revealed that cIAP1 and CAS interact in vivo (Fig. 4c). We observed that transfection of a high dose of WT but not E3-dead cIAP1 in 293T cells promoted CAS degradation in a dose-dependent manner (Fig. 4d). Furthermore, WT-cIAP1 overexpression significantly increased polyubiquitination of CAS in vivo, whereas E3-dead cIAP1 had no such effect (Fig. 4e). To confirm that cIAP1 is a direct E3 ligase for CAS, we used purified proteins to perform an in vitro assay and found that WT, but not E3-dead, cIAP1 can catalyze in vitro polyubiquitination of CAS (Fig. 4f). Collectively, these results demonstrate that cIAP1 directly ubiquitinates and targets CAS for proteasomal degradation. TRAIL-induced caspase-8 activation leads to a marked decrease in cIAP1 levels, thereby mitigating CAS ubiquitination and allowing for its accumulation (Fig. 4g).
FIGURE 4.

cIAP1 mediates the turnover of CAS and is degraded upon TRAIL treatment. a, MCF10A cells were treated with TRAIL (30 ng/ml, 4 h) and harvested for Western blot analysis. Where indicated, cells were pretreated with a caspase-8 inhibitor (C8i, 20 μm) for 30 min prior to addition of TRAIL. b, MCF10A cells were transfected with NT siRNA or siRNA against cIAP1 and harvested for Western blot analysis 72 h later. Right panel, quantification of CAS levels (mean ± S.D.) relative to NT siRNA cells from three independent experiments. c, lysates from 293T cells expressing GFP-CAS and empty vector or HA-cIAP1 were subjected to immunoprecipitation with HA-agarose beads. Bound proteins were then probed by Western blotting with the indicated antibodies. d, 293T cells were transfected with GFP-CAS and WT or E3-dead (H588A) cIAP1 and subjected to Western blot analysis 24 h later. The total amount of DNA transfected was balanced using empty vector DNA. Right panel, quantification of CAS levels (mean ± S.D.) relative to control cells from three independent experiments. e, HEK293T cells transfected with the indicated plasmids were treated with 25 μm MG132 for 4 h to allow for accumulation of ubiquitinated proteins. Whole cell lysates were then subjected to a nickel bead pulldown under denaturing conditions, and bound proteins were probed with the indicated antibodies. Note that, in d and e, expression of E3-dead cIAP1 is significantly higher than WT, presumably because of the lack of autoubiquitination-induced degradation. f, in vitro ubiquitination of GFP-CAS purified from 293T cells was performed using recombinant cIAP1, as described under “Experimental Procedures.” Note that WT-cIAP1 shows a smearing pattern because of autoubiquitination activity. IB, immunoblot. g, model illustrating the regulation of CAS. Upon stimulation by TRAIL, cIAP1 is degraded in a caspase-8-dependent manner, allowing for accumulation of CAS, which then feeds forward into the apoptotic pathway by stimulating apoptosome formation. Cyt C, cytochrome c.
Aberrant Expression of CAS in Human Tumors
The proapoptotic function of CAS implies that it is a potential tumor suppressor, and it would therefore be expected to be deleted or undergo loss-of-function mutations in cancer. Intriguingly however, multiple studies have reported that CAS is actually amplified in various human tumors (54–57), implicating CAS as a potential oncogene. Notably, studies have pointed toward an essential role for CAS in cell division (58, 59), and CAS expression has been documented to be higher in rapidly proliferating tissue like testis and fetal liver (50). We therefore reasoned that elevation of CAS levels in tumor cells increases the proliferative capacity of these cells. Consistently, we observed that thorough depletion of CAS using higher doses of siRNA leads to a clear inhibition of cell growth (Fig. 5a).
FIGURE 5.

CAS expression is elevated in human breast tumors, and this is recapitulated by overexpression of various oncogenes in untransformed cells. a, MCF10A cells were transfected with NT or CAS siRNA and seeded at equal density on day 0. At the indicated time points, cells were stained with crystal violet, and the relative number of cells was quantified. A representative image of stained cells and a Western blot confirming knockdown of CAS are shown. Values are mean ± S.E. from two independent experiments. *, p < 0.05. b, frequency of alterations in CAS in The Cancer Genome Atlas breast carcinoma dataset (from the cBioPortal). c, lysates from MCF10A cells stably expressing the indicated oncogenes were subjected to Western blot analysis to assay the expression of CAS protein. d, quantitative RT-PCR was used to measure the expression of CAS mRNA in the indicated MCF10A cell lines. Values are mean ± S.E. from three independent experiments. *, p < 0.05. e, oncoprint (from the cBioPortal) showing the co-occurrence of CAS mRNA up-regulation with alterations in select oncogenes (Fisher's exact test; CAS-Myc, p < 0.001; CAS-Erbb2/PI3K/KRAS, not significant). Only altered cases are displayed.
We sought to understand the cause and consequences of aberrant expression of CAS specifically in breast cancer. Analysis of the breast carcinoma dataset from The Cancer Genome Atlas project revealed alterations in CAS in about 16% of tumors, with mRNA up-regulation being the most frequent (Fig. 5b). Given the role of CAS in cell proliferation, we hypothesized that oncogenic signaling pathways could drive the enhanced expression of CAS in tumors. To test this idea, we introduced various oncogenes, including Erbb2, Myc, PI3KH1047R, and KRASG12V, into untransformed MCF10A cells. This led to a clear increase in expression of CAS at the protein level, driven by an increase in the transcription of CAS mRNA (Fig. 5, c and d). These results suggest that multiple oncogenic signaling pathways are capable of driving CAS mRNA expression, likely to support cell growth. We then examined the co-occurrence of CAS mRNA up-regulation with alterations in the same set of oncogenes in The Cancer Genome Atlas breast cancer dataset (Fig. 5e). Notably, we observed a significant co-occurrence of increased CAS mRNA expression with alterations in Myc (p < 0.001) but not with the other oncogenes tested.
Increased CAS Expression Contributes to the Heightened Apoptotic Sensitivity of Oncogene-overexpressing Cells
To understand the functional significance of increased basal CAS expression, we used c-Myc-overexpressing MCF10A cells (MCF10A-Myc), given the high prevalence of Myc amplification in breast cancer and the tendency toward its co-occurrence with CAS up-regulation. Because CAS can facilitate caspase-3 activation by driving apoptosome formation, we hypothesized that MCF10A-Myc cells (with higher basal CAS expression) would be more sensitive to TRAIL than their wild-type counterparts. Indeed, in response to TRAIL, we observed increased caspase-3 activation and lower cell survival in MCF10A-Myc cells (Fig. 6, a and b). Importantly, knockdown of CAS by siRNA decreased caspase-3 activation and improved cell viability in MCF10A-Myc cells (Fig. 6, c and d). Collectively, these results indicate that, although elevated basal levels of CAS promote breast cancer proliferation, they also lead to an increased sensitivity to TRAIL.
FIGURE 6.

Elevated CAS expression contributes to increased sensitivity to TRAIL in Myc-overexpressing cells. a, WT and Myc- MCF10A cells were treated with 5 ng/ml TRAIL for the indicated time and then harvested for Western blot analysis to detect caspase-3 activation. b, WT and Myc- MCF10A cells were treated with the indicated concentration of TRAIL for 8 h and assayed for cell viability. c and d, MCF10A-Myc cells transfected with NT or CAS siRNA were treated with TRAIL (1.25 ng/ml for 4 h) for measurement of caspase-3/7 activity (c) or for 8 h with the indicated concentrations for measurement of cell viability (d). Representative phase-contrast images displaying cell morphology and Western blots confirming knockdown of CAS are shown. All values are mean ± S.E. from three independent experiments. *, p < 0.05.
Discussion
Multiple studies have revealed that cells can survive incomplete MOMP provided that downstream caspase-3 activity is limited. Therefore, the decision to live or die following MOMP is determined, at least in part, by the extent of caspase-3 activation. However, our understanding of how caspase-3 activity is regulated following MOMP remains incomplete. We have shown previously that CAS can drive caspase activity by stimulating apoptosome formation (45). Here we demonstrate that, during TRAIL-induced apoptosis, CAS levels are up-regulated rapidly and that knockdown of CAS inhibits TRAIL-induced caspase-3 activity and cell death. On the basis of these results, we propose that up-regulation of CAS amplifies the apoptotic signal, increasing the commitment to cell death following MOMP.
Mechanistically, the up-regulation of CAS in response to TRAIL is caspase-8-dependent. Active caspase-8 induces degradation of cIAP1, an E3 ligase that ubiquitinates and promotes the degradation of CAS. How caspase-8 targets cIAP1 for degradation remains an unanswered question. The most straightforward explanation is a direct cleavage of cIAP1 by caspase-8. Alternatively, caspase-8 could target a binding partner that stabilizes cIAP1 or potentially regulate the autoubiquitination activity of cIAP1. Our studies also reveal that caspase-8 induces up-regulation of CAS independent of stimulating MOMP. Therefore, increased levels of CAS can feed forward into the apoptotic cascade following the onset of MOMP.
Our mechanistic studies focused exclusively on TRAIL-induced apoptosis. However, it is worth noting that we have reported previously that other extrinsic apoptotic stimuli, such as TNF-α and IFN-γ, can also induce CAS up-regulation (45) and that, as with TRAIL, knockdown of CAS inhibits apoptosis induced by these stimuli. Although these stimuli target distinct receptors, leading to accumulation of different adaptor proteins, they all induce caspase-8 activation. Therefore, it is likely that the caspase 8-cIAP1 pathway we described here for TRAIL mediates up-regulation of CAS induced by these stimuli as well. Intriguingly, UV radiation is also capable of up-regulating CAS (45). UV radiation-induced DNA damage leads to activation of the cell-intrinsic apoptotic pathway, and this is thought to occur independently of caspase-8 activity. How UV radiation up-regulates CAS remains an open question.
Contrary to its prototumor-suppressive role in facilitating apoptosis, CAS is frequently amplified in human tumors. Notably, CAS levels are higher in proliferating fibroblasts and decrease upon growth arrest, suggesting its potential role in cell proliferation (50). Furthermore, homozygous deletion of CAS in mice leads to embryonic lethality (59), and mutations in the yeast homologue (CSE1) are lethal as well (60). The precise molecular function of CAS in cell proliferation remains to be identified. Interestingly, we found that overexpression of multiple oncogenes led to increased expression of CAS mRNA and protein. This indicates the existence of a regulatory network closely linking cell growth and CAS levels that can be co-opted during oncogenic transformation.
Our studies show that increased basal expression of CAS in oncogene-overexpressing cells contributes to heightened sensitivity to TRAIL. This raises the intriguing possibility that the increased levels of CAS seen in several human tumors could represent an “Achilles' heel.” Tumor cells up-regulate CAS to support their enhanced proliferation, but this, in turn, could make them more vulnerable to certain apoptotic stimuli. In this regard, it is worth noting that not all apoptotic stimuli induce CAS up-regulation and that those that do not (e.g. staurosporine) are unaffected by knockdown of CAS (45). Therefore, profiling the expression of CAS in tumor tissue could potentially be used to guide decisions regarding therapeutic choices.
In conclusion, our studies uncovered a novel signaling pathway whereby caspase-8 amplifies the TRAIL-induced apoptotic signal by mediating up-regulation of CAS. Furthermore, we highlighted the increased transcription of CAS in response to oncogene overexpression and the consequences of this up-regulation. The regulation of CAS at both the transcriptional and posttranslational levels is reflective of its multiple roles in proliferation and apoptosis. It is likely that such intricate regulation is key in maintaining the proper function of this dynamic protein in these two contrasting cellular processes.
Author Contributions
P. M. and X. J. designed the study, interpreted the results, and wrote the manuscript. P. M. conducted the experiments.
Acknowledgments
We thank Dr. Filippo Giancotti (Memorial Sloan Kettering Cancer Center) for key constructs and all members of the Jiang laboratory for critical discussions.
This work was supported by William H. and Alice Goodwin and the Commonwealth Foundation for Cancer Research of the Experimental Therapeutics Center of Memorial Sloan Kettering Cancer Center; a Cycle for Survival grant; National Institutes of Health Cancer Center Core Grant P30 CA008748; and National Institutes of Health Grants R01GM113013 and R01CA166413 (to X. J.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- MOMP
- mitochondrial outer membrane permeabilization
- IAP
- inhibitor of apoptosis
- CAS
- cellular apoptosis susceptibility protein
- Z-VAD-fmk
- benzyloxycarbonyl-VAD-fluoromethyl ketone
- IP
- immunoprecipitation
- Ni-NTA
- nickel-nitrilotriacetic acid
- NT
- non-target
- TRAIL
- TNF-related apoptosis inducing ligand.
References
- 1. Jiang X., and Wang X. (2004) Cytochrome C-mediated apoptosis. Annu. Rev. Biochem. 73, 87–106 [DOI] [PubMed] [Google Scholar]
- 2. Zou H., Henzel W. J., Liu X., Lutschg A., and Wang X. (1997) Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 90, 405–413 [DOI] [PubMed] [Google Scholar]
- 3. Zou H., Li Y., Liu X., and Wang X. (1999) An APAF-1·cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol. Chem. 274, 11549–11556 [DOI] [PubMed] [Google Scholar]
- 4. Srinivasula S. M., Ahmad M., Fernandes-Alnemri T., and Alnemri E. S. (1998) Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol. Cell 1, 949–957 [DOI] [PubMed] [Google Scholar]
- 5. Jiang X., and Wang X. (2000) Cytochrome c promotes caspase-9 activation by inducing nucleotide binding to Apaf-1. J. Biol. Chem. 275, 31199–31203 [DOI] [PubMed] [Google Scholar]
- 6. Ashkenazi A., and Dixit V. M. (1998) Death receptors: signaling and modulation. Science 281, 1305–1308 [DOI] [PubMed] [Google Scholar]
- 7. Li H., Zhu H., Xu C.-J., and Yuan J. (1998) Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94, 491–501 [DOI] [PubMed] [Google Scholar]
- 8. Luo X., Budihardjo I., Zou H., Slaughter C., and Wang X. (1998) Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94, 481–490 [DOI] [PubMed] [Google Scholar]
- 9. Yin X.-M., Wang K., Gross A., Zhao Y., Zinkel S., Klocke B., Roth K. A., and Korsmeyer S. J. (1999) Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 400, 886–891 [DOI] [PubMed] [Google Scholar]
- 10. Jost P. J., Grabow S., Gray D., McKenzie M. D., Nachbur U., Huang D. C., Bouillet P., Thomas H. E., Borner C., Silke J., Strasser A., and Kaufmann T. (2009) XIAP discriminates between type I and type II FAS-induced apoptosis. Nature 460, 1035–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Green D. R., and Kroemer G. (2004) The pathophysiology of mitochondrial cell death. Science 305, 626–629 [DOI] [PubMed] [Google Scholar]
- 12. Soengas M. S., Alarcón R. M., Yoshida H., Giaccia A. J., Hakem R., Mak T. W., and Lowe S. W. (1999) Apaf-1 and caspase-9 in p53-dependent apoptosis and tumor inhibition. Science 284, 156–159 [DOI] [PubMed] [Google Scholar]
- 13. Yoshida H., Kong Y.-Y., Yoshida R., Elia A. J., Hakem A., Hakem R., Penninger J. M., and Mak T. W. (1998) Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell 94, 739–750 [DOI] [PubMed] [Google Scholar]
- 14. Kuida K., Haydar T. F., Kuan C.-Y., Gu Y., Taya C., Karasuyama H., Su M. S., Rakic P., and Flavell R. A. (1998) Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell 94, 325–337 [DOI] [PubMed] [Google Scholar]
- 15. Hakem R., Hakem A., Duncan G. S., Henderson J. T., Woo M., Soengas M. S., Elia A., de la Pompa J. L., Kagi D., Khoo W., Potter J., Yoshida R., Kaufman S. A., Lowe S. W., Penninger J. M., and Mak T. W. (1998) Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell 94, 339–352 [DOI] [PubMed] [Google Scholar]
- 16. Cecconi F., Alvarez-Bolado G., Meyer B. I., Roth K. A., and Gruss P. (1998) Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell 94, 727–737 [DOI] [PubMed] [Google Scholar]
- 17. Jia L., Srinivasula S. M., Liu F.-T., Newland A. C., Fernandes-Alnemri T., Alnemri E. S., and Kelsey S. M. (2001) Apaf-1 protein deficiency confers resistance to cytochrome c-dependent apoptosis in human leukemic cells. Blood 98, 414–421 [DOI] [PubMed] [Google Scholar]
- 18. Martinou I., Desagher S., Eskes R., Antonsson B., André E., Fakan S., and Martinou J.-C. (1999) The release of cytochrome c from mitochondria during apoptosis of NGF-deprived sympathetic neurons is a reversible event. J. Cell Biol. 144, 883–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Deshmukh M., Kuida K., and Johnson E. M. (2000) Caspase inhibition extends the commitment to neuronal death beyond cytochrome c release to the point of mitochondrial depolarization. J. Cell Biol. 150, 131–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tait S. W., Parsons M. J., Llambi F., Bouchier-Hayes L., Connell S., Muñoz-Pinedo C., and Green D. R. (2010) Resistance to caspase-independent cell death requires persistence of intact mitochondria. Dev. Cell 18, 802–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Colell A., Ricci J.-E., Tait S., Milasta S., Maurer U., Bouchier-Hayes L., Fitzgerald P., Guio-Carrion A., Waterhouse N. J., Li C. W., Mari B., Barbry P., Newmeyer D. D., Beere H. M., and Green D. R. (2007) GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell 129, 983–997 [DOI] [PubMed] [Google Scholar]
- 22. Neame S. J., Rubin L. L., and Philpott K. L. (1998) Blocking cytochrome c activity within intact neurons inhibits apoptosis. J. Cell Biol. 142, 1583–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Deshmukh M., and Johnson E. M. Jr. (1998) Evidence of a novel event during neuronal death: development of competence-to-die in response to cytoplasmic cytochrome c. Neuron 21, 695–705 [DOI] [PubMed] [Google Scholar]
- 24. Potts M. B., Vaughn A. E., McDonough H., Patterson C., and Deshmukh M. (2005) Reduced Apaf-1 levels in cardiomyocytes engage strict regulation of apoptosis by endogenous XIAP. J. Cell Biol. 171, 925–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ishizaki Y., Jacobson M. D., and Raff M. C. (1998) A role for caspases in lens fiber differentiation. J. Cell Biol. 140, 153–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Weil M., Raff M. C., and Braga V. M. (1999) Caspase activation in the terminal differentiation of human epidermal keratinocytes. Curr. Biol. 9, 361–365 [DOI] [PubMed] [Google Scholar]
- 27. Fernando P., Kelly J. F., Balazsi K., Slack R. S., and Megeney L. A. (2002) Caspase 3 activity is required for skeletal muscle differentiation. Proc. Natl. Acad. Sci. U.S.A. 99, 11025–11030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fernando P., Brunette S., and Megeney L. A. (2005) Neural stem cell differentiation is dependent upon endogenous caspase 3 activity. FASEB J. 19, 1671–1673 [DOI] [PubMed] [Google Scholar]
- 29. Murray T. V., McMahon J. M., Howley B. A., Stanley A., Ritter T., Mohr A., Zwacka R., and Fearnhead H. O. (2008) A non-apoptotic role for caspase-9 in muscle differentiation. J. Cell Sci. 121, 3786–3793 [DOI] [PubMed] [Google Scholar]
- 30. Campbell D. S., and Holt C. E. (2003) Apoptotic pathway and MAPKs differentially regulate chemotropic responses of retinal growth cones. Neuron 37, 939–952 [DOI] [PubMed] [Google Scholar]
- 31. Huesmann G. R., and Clayton D. F. (2006) Dynamic role of postsynaptic caspase-3 and BIRC4 in zebra finch song-response habituation. Neuron 52, 1061–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li Z., Jo J., Jia J.-M., Lo S.-C., Whitcomb D. J., Jiao S., Cho K., and Sheng M. (2010) Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell 141, 859–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Santambrogio L., Potolicchio I., Fessler S. P., Wong S.-H., Raposo G., and Strominger J. L. (2005) Involvement of caspase-cleaved and intact adaptor protein 1 complex in endosomal remodeling in maturing dendritic cells. Nat. Immunol. 6, 1020–1028 [DOI] [PubMed] [Google Scholar]
- 34. Woo M., Hakem R., Furlonger C., Hakem A., Duncan G. S., Sasaki T., Bouchard D., Lu L., Wu G. E., Paige C. J., and Mak T. W. (2003) Caspase-3 regulates cell cycle in B cells: a consequence of substrate specificity. Nat. Immunol. 4, 1016–1022 [DOI] [PubMed] [Google Scholar]
- 35. Soengas M. S., Capodieci P., Polsky D., Mora J., Esteller M., Opitz-Araya X., McCombie R., Herman J. G., Gerald W. L., Lazebnik Y. A., Cordón-Cardó C., and Lowe S. W. (2001) Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature 409, 207–211 [DOI] [PubMed] [Google Scholar]
- 36. Wolf B. B., Schuler M., Li W., Eggers-Sedlet B., Lee W., Tailor P., Fitzgerald P., Mills G. B., and Green D. R. (2001) Defective cytochrome c-dependent caspase activation in ovarian cancer cell lines due to diminished or absent apoptotic protease activating factor-1 activity. J. Biol. Chem. 276, 34244–34251 [DOI] [PubMed] [Google Scholar]
- 37. Devarajan E., Sahin A. A., Chen J. S., Krishnamurthy R. R., Aggarwal N., Brun A. M., Sapino A., Zhang F., Sharma D., Yang X. H., Tora A. D., and Mehta K. (2002) Down-regulation of caspase 3 in breast cancer: a possible mechanism for chemoresistance. Oncogene 21, 8843–8851 [DOI] [PubMed] [Google Scholar]
- 38. Tamm I., Kornblau S. M., Segall H., Krajewski S., Welsh K., Kitada S., Scudiero D. A., Tudor G., Qui Y. H., Monks A., Andreeff M., and Reed J. C. (2000) expression and prognostic significance of IAP-family genes in human cancers and myeloid leukemias. Clin. Cancer Res. 6, 1796–1803 [PubMed] [Google Scholar]
- 39. Ferreira C. G., van der Valk P., Span S. W., Ludwig I., Smit E. F., Kruyt F. A., Pinedo H. M., van Tinteren H., and Giaccone G. (2001) Expression of X-linked inhibitor of apoptosis as a novel prognostic marker in radically resected non-small cell lung cancer patients. Clin. Cancer Res. 7, 2468–2474 [PubMed] [Google Scholar]
- 40. Ichim G., Lopez J., Ahmed S. U., Muthalagu N., Giampazolias E., Delgado M. E., Haller M., Riley J. S., Mason S. M., Athineos D., Parsons M. J., van de Kooij B., Bouchier-Hayes L., Chalmers A. J., Rooswinkel R. W., Oberst A., Blyth K., Rehm M., Murphy D. J., and Tait S. W. (2015) Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol. Cell 57, 860–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu X., He Y., Li F., Huang Q., Kato T. A., Hall R. P., and Li C.-Y. (2015) Caspase-3 promotes genetic instability and carcinogenesis. Mol. Cell 58, 284–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rongvaux A., Jackson R., Harman C. C., Li T., West A. P., de Zoete M. R., Wu Y., Yordy B., Lakhani S. A., Kuan C.-Y., Taniguchi T., Shadel G. S., Chen Z. J., Iwasaki A., and Flavell R. A. (2014) Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 159, 1563–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. White M. J., McArthur K., Metcalf D., Lane R. M., Cambier J. C., Herold M. J., van Delft M. F., Bedoui S., Lessene G., Ritchie M. E., Huang D. C., and Kile B. T. (2014) Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 159, 1549–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim H.-E., Du F., Fang M., and Wang X. (2005) Formation of apoptosome is initiated by cytochrome c-induced dATP hydrolysis and subsequent nucleotide exchange on Apaf-1. Proc. Natl. Acad. Sci. U.S.A. 102, 17545–17550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kim H.-E., Jiang X., Du F., and Wang X. (2008) PHAPI, CAS, and Hsp70 promote apoptosome formation by preventing Apaf-1 aggregation and enhancing nucleotide exchange on Apaf-1. Mol. Cell 30, 239–247 [DOI] [PubMed] [Google Scholar]
- 46. Wang X., Trotman L. C., Koppie T., Alimonti A., Chen Z., Gao Z., Wang J., Erdjument-Bromage H., Tempst P., Cordon-Cardo C., Pandolfi P. P., and Jiang X. (2007) NEDD4–1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell 128, 129–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. The Cancer Genome Atlas Network (2012) Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cerami E., Gao J., Dogrusoz U., Gross B. E., Sumer S. O., Aksoy B. A., Jacobsen A., Byrne C. J., Heuer M. L., Larsson E., Antipin Y., Reva B., Goldberg A. P., Sander C., and Schultz N. (2012) The cBio Cancer Genomics Portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discovery 2, 401–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gao J., Aksoy B. A., Dogrusoz U., Dresdner G., Gross B., Sumer S. O., Sun Y., Jacobsen A., Sinha R., Larsson E., Cerami E., Sander C., and Schultz N. (2013) Integrative Analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pp.pl1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brinkmann U., Brinkmann E., Gallo M., and Pastan I. (1995) Cloning and characterization of a cellular apoptosis susceptibility gene, the human homologue to the yeast chromosome segregation gene CSE1. Proc. Natl. Acad. Sci. U.S.A. 92, 10427–10431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Martinou J.-C., and Youle R. J. (2011) Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev. Cell 21, 92–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Guicciardi M. E., Mott J. L., Bronk S. F., Kurita S., Fingas C. D., and Gores G. J. (2011) Cellular inhibitor of apoptosis 1 (cIAP-1) degradation by caspase 8 during TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis. Exp. Cell Res. 317, 107–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. O'Donnell M. A., Perez-Jimenez E., Oberst A., Ng A., Massoumi R., Xavier R., Green D. R., and Ting A. T. (2011) Caspase 8 inhibits programmed necrosis by processing CYLD. Nat. Cell Biol. 13, 1437–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Böni R., Wellmann A., Man Y.-G., Hofbauer G., and Brinkmann U. (1999) Expression of the proliferation and apoptosis-associated CAS protein in benign and malignant cutaneous melanocytic lesions. Am. J. Dermatopathol. 21, 125–128 [DOI] [PubMed] [Google Scholar]
- 55. Wellmann A., Flemming P., Behrens P., Wuppermann K., Lang H., Oldhafer K., Pastan I., and Brinkmann U. (2001) High expression of the proliferation and apoptosis associated CSE1L/CAS gene in hepatitis and liver neoplasms: correlation with tumor progression. Int. J. Mol. Med. 7, 489–494 [DOI] [PubMed] [Google Scholar]
- 56. Bar-Shira A., Pinthus J. H., Rozovsky U., Goldstein M., Sellers W. R., Yaron Y., Eshhar Z., and Orr-Urtreger A. (2002) Multiple genes in human 20q13 chromosomal region are involved in an advanced prostate cancer xenograft. Cancer Res. 62, 6803–6807 [PubMed] [Google Scholar]
- 57. Brustmann H. (2004) Expression of cellular apoptosis susceptibility protein in serous ovarian carcinoma: a clinicopathologic and immunohistochemical study. Gynecol. Oncol. 92, 268–276 [DOI] [PubMed] [Google Scholar]
- 58. Ogryzko V. V., Brinkmann E., Howard B. H., Pastan I., and Brinkmann U. (1997) Antisense inhibition of CAS, the human homologue of the yeast chromosome segregation gene CSE1, interferes with mitosis in HeLa cells. Biochemistry 36, 9493–9500 [DOI] [PubMed] [Google Scholar]
- 59. Bera T. K., Bera J., Brinkmann U., Tessarollo L., and Pastan I. (2001) Cse1l is essential for early embryonic growth and development. Mol. Cell. Biol. 21, 7020–7024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Xiao Z., McGrew J. T., Schroeder A. J., and Fitzgerald-Hayes M. (1993) CSE1 and CSE2, two new genes required for accurate mitotic chromosome segregation in Saccharomyces cerevisiae. Mol. Cell. Biol. 13, 4691–4702 [DOI] [PMC free article] [PubMed] [Google Scholar]