

Abstract
DEAD-box proteins utilize ATP to bind and remodel RNA and RNA-protein complexes. All DEAD-box proteins share a conserved core that consists of two RecA-like domains. The core is flanked by subfamily-specific extensions of idiosyncratic function. The Ded1/DDX3 subfamily of DEAD-box proteins is of particular interest as members function during protein translation, are essential for viability, and are frequently altered in human malignancies. Here, we define the function of the subfamily-specific extensions of the human DEAD-box protein DDX3. We describe the crystal structure of the subfamily-specific core of wild-type DDX3 at 2.2 Å resolution, alone and in the presence of AMP or nonhydrolyzable ATP. These structures illustrate a unique interdomain interaction between the two ATPase domains in which the C-terminal domain clashes with the RNA-binding surface. Destabilizing this interaction accelerates RNA duplex unwinding, suggesting that it is present in solution and inhibitory for catalysis. We use this core fragment of DDX3 to test the function of two recurrent medulloblastoma variants of DDX3 and find that both inactivate the protein in vitro and in vivo. Taken together, these results redefine the structural and functional core of the DDX3 subfamily of DEAD-box proteins.
Keywords: ATPase, conformational change, crystal structure, medulloblastoma, molecular dynamics, RNA-binding protein, RNA helicase, X-ray crystallography
Introduction
DEAD-box proteins are ATP-dependent RNA-binding proteins that remodel RNA structures and RNA-protein complexes, stably clamp RNA, and promote fluidity within RNA granules (1–3). The human DEAD-box protein DDX3 (encoded by DDX3X) and its yeast ortholog Ded1p have been implicated in numerous functions including translation initiation (4–12). Messenger RNA molecules containing especially long or structured 5′ leader sequences are particularly sensitive to DDX3 activity (6, 7, 12, 13). DDX3X is frequently mutated in numerous cancer types (5), such as chronic lymphocytic leukemia (14–16), natural killer/T-cell lymphoma (17), head and neck squamous cell carcinoma (18, 19), and lung cancer (20). DDX3X is also one of the most frequently mutated genes in the highly malignant brain tumor medulloblastoma (21–24). In medulloblastoma, many mutations are predicted to inactivate DDX3, and some have been demonstrated to diminish activities in vitro (17, 25).
DEAD-box proteins are defined by 12 different motifs that function in ATP binding or hydrolysis and RNA binding, or couple ATP and RNA binding (1). Outside of these conserved motifs, each DEAD-box protein subfamily has unique tails that lie N- or C-terminal to the helicase core and contain elements that define the unique properties of that subfamily. For example, DDX21 has a GUCT domain in its C-terminal extension (26, 27), DDX5 has tandem P68HR domains in its C-terminal extension, and DDX43 has a KH1 domain in its N-terminal extension. However, as the tails of each DEAD-box protein subfamily are idiosyncratic, whereas the cores are very similar (28, 29), it is essential to study individual subfamilies of DEAD-box proteins in detail to understand the role of subfamily-specific tails.
DDX3 is a member of the Ded1/DDX3 subfamily, along with the Saccharomyces cerevisiae ortholog DED1, and Vasa/DDX4 (5, 30). The tails of Ded1/DDX3 subfamily members are thought to be largely unstructured and contain diverse motifs with different functions. For example, the N-terminal tail of DDX3 contains a Crm1-dependent nuclear export sequence (31) and an eIF4E-binding motif (10, 11), whereas the C-terminal tail contains conserved sequences of unknown function that are essential for oligomerization (5, 32). The tails of DED1 additionally contain assembly domains that modulate translation and alleviate lethality associated with protein overexpression when deleted (10). The minimal functional core of DEAD-box proteins has been defined as the isolated phenylalanine upstream of the Q-motif through roughly 35 residues beyond Motif VI (29). However, this analysis does not consider subfamily-specific extensions. Similarly, prior structural work has truncated one (25) or both (33) of the Ded1/DDX3 subfamily-specific regions down to the boundary of the helicase core, but it is unknown how active these truncations are when compared with full-length DDX3. Moreover, previous structures of DDX3 are of either inactive constructs (33) or point mutants (25), so the relevance of the crystallized conformations is unclear. Therefore, crucial details of the regions uniquely conserved within the Ded1/DDX3 subfamily and the conformational landscape of active, wild-type DDX3 remain incompletely understood.
Here, we assay the function of the tails of human DDX3 by generating a series of truncations and exploring their activity in vitro and in vivo. We define an active but truncated construct of DDX3 and solve its crystal structure at 2.2 Å resolution in a partially closed state, representing the highest resolution structure of active, wild-type DDX3 to date. We find that this partially closed state is autoinhibited and demonstrate that mutations predicted to destabilize this conformation accelerate RNA duplex unwinding by DDX3. Using molecular dynamics simulations, we show that the ATP-binding loop of DDX3 samples transient interactions with ATP and that the partially closed state is stable in solution. Lastly, we test two recurrent medulloblastoma variants of DDX3 and find that they inactivate duplex unwinding by up to 3 log units. Our work defines a functional truncation of DDX3 that purifies to high yield, elucidates the function of the signature tails found in the Ded1/DDX3 subfamily of DEAD-box proteins, presents high-resolution structural information of active DDX3 of utility for molecular modeling and drug design, and demonstrates the consequences of two medulloblastoma-associated DDX3 variants.
Experimental Procedures
Recombinant Protein Purification
The Ded1/DDX3 subfamily core of DDX3 was expressed using a construct containing Escherichia coli codon-optimized, human DDX3X amino acids 132–607 fused to a His6-MBP (maltose-binding protein) tag and expressed in E. coli BL21 Star by induction with isopropyl-1-thio-β-d-galactopyranoside at 16 °C for 18 h. Cell pellets were lysed by sonication, clarified by centrifugation at ∼30,000 × g, and purified by nickel chromatography including a 1 m NaCl wash to remove bound nucleic acids. The His6-MBP tag was cleaved using tobacco etch virus protease during dialysis into 200 mm NaCl, 10% (v/v) glycerol, 20 mm HEPES, pH 7, and 0.5 mm TCEP.3 The sample was then purified using heparin affinity chromatography, eluted at 400 mm NaCl, 10% glycerol, 20 mm HEPES, pH 7, and 0.5 mm TCEP, and applied to a Superdex 75 gel filtration column equilibrated in 500 mm NaCl, 10% glycerol, 20 mm HEPES, pH 7.5, and 0.5 mm TCEP. Fractions were then concentrated and supplemented with 20% (v/v) glycerol and flash-frozen for kinetics, or used directly for crystallization. Typical yield was ∼10 mg of purified protein per liter. Point mutants were generated by site-directed mutagenesis.
Full-length DDX3X with a His6 tag was cloned into a pET-29a vector and expressed in E. coli BL21 (37 °C). Cells were processed as described previously for the purification of Ded1p (34). Lysates were passed through pre-equilibrated nickel-agarose beads and washed with increasing imidazole concentrations (5–60 mm) (34). DDX3X was eluted in 250 mm imidazole. The His6 tag was cleaved using tobacco etch virus protease in 50 mm Tris-Cl (pH 8.0), 0.5 mm EDTA, 1 mm DTT, and 40% (v/v) glycerol. DDX3X was further purified by adsorption to phosphocellulose resin (P11, Whatman) and elution with NaCl, as described for Ded1p (34). Eluted fractions were analyzed by SDS-PAGE and Western blotting using anti-His antibody to confirm removal of the His6 tag. DDX3X fractions were supplemented to 40% (v/v) glycerol, flash-frozen in liquid nitrogen, and stored at −80 °C.
X-ray Crystallography
Purified protein was concentrated to ∼5 mg ml−1 and mixed 1:1 with precipitant solution containing from 6 to 12% PEG 3000 and 100 mm sodium citrate, pH 5.0, and crystallization was achieved by hanging drop vapor diffusion at 18 °C within 24 h. Nucleotide-bound crystals were grown identically but supplemented with 10 mm of the nucleotide in solution. Homogeneous pieces of branched crystals were harvested for data collection, which was conducted at Beamline 8.3.1 of the Advanced Light Source. Data were indexed, integrated, and scaled using XDS (35), phased using molecular replacement with PHASER (36) with both domains from Protein Data Bank (PDB) 2I4I (33) as independent search models, and refined and built using PHENIX (37) and Coot (38). High resolution was determined by CC½ ≥ 10% (39). Structures were visualized with PyMOL (40).
RNA Duplex Unwinding Assays
Assays were performed as described (41) with minor modifications. Briefly, duplex RNAs containing a 3′ overhang were formed by radiolabeling a single-stranded RNA, annealing, and using gel purification. The two RNA sequences are 5′-AGCACCGUAAAGACGC-3′ and 5′-GCGUCUUUACGGUGCUUAAAACAAAACAAAACAAAACAAAA-3′. Reactions contained trace duplex RNA and 1 μm protein and were initiated by the addition of 2 mm MgATP.
Yeast Complementation Assays
A yeast strain containing the genomic region surrounding DED1 on a centromeric plasmid was used for all experiments (10). Mutations were made in a HIS3-marked plasmid and exchanged for the wild-type allele by plasmid shuffling using counterselection with 5-fluoroorotic acid. Growth assays show 10-fold dilution from an optical density of 1 and were conducted using rich medium (YPD) plates at the temperature indicated.
Multiple Sequence Alignments
Sequences for Drosophila melanogaster Vasa, Homo sapiens DDX4, Danio rerio PL10, Mus musculus PL10, M. musculus DDX3Y, H. sapiens DDX3Y, M. musculus DDX3X, H. sapiens DDX3X, D. melanogaster belle, S. cerevisiae DED1, and Schizosaccharomyces pombe sum3 were retrieved from the National Center for Biotechnology Information (NCBI) and aligned using MUSCLE (42). Sequence alignments were visualized using Jalview (43). The DEAD-box protein motifs shown in Fig. 6A were generated using WebLogo with input from all human DEAD-box protein sequences aligned with MAFFT (44).
FIGURE 6.
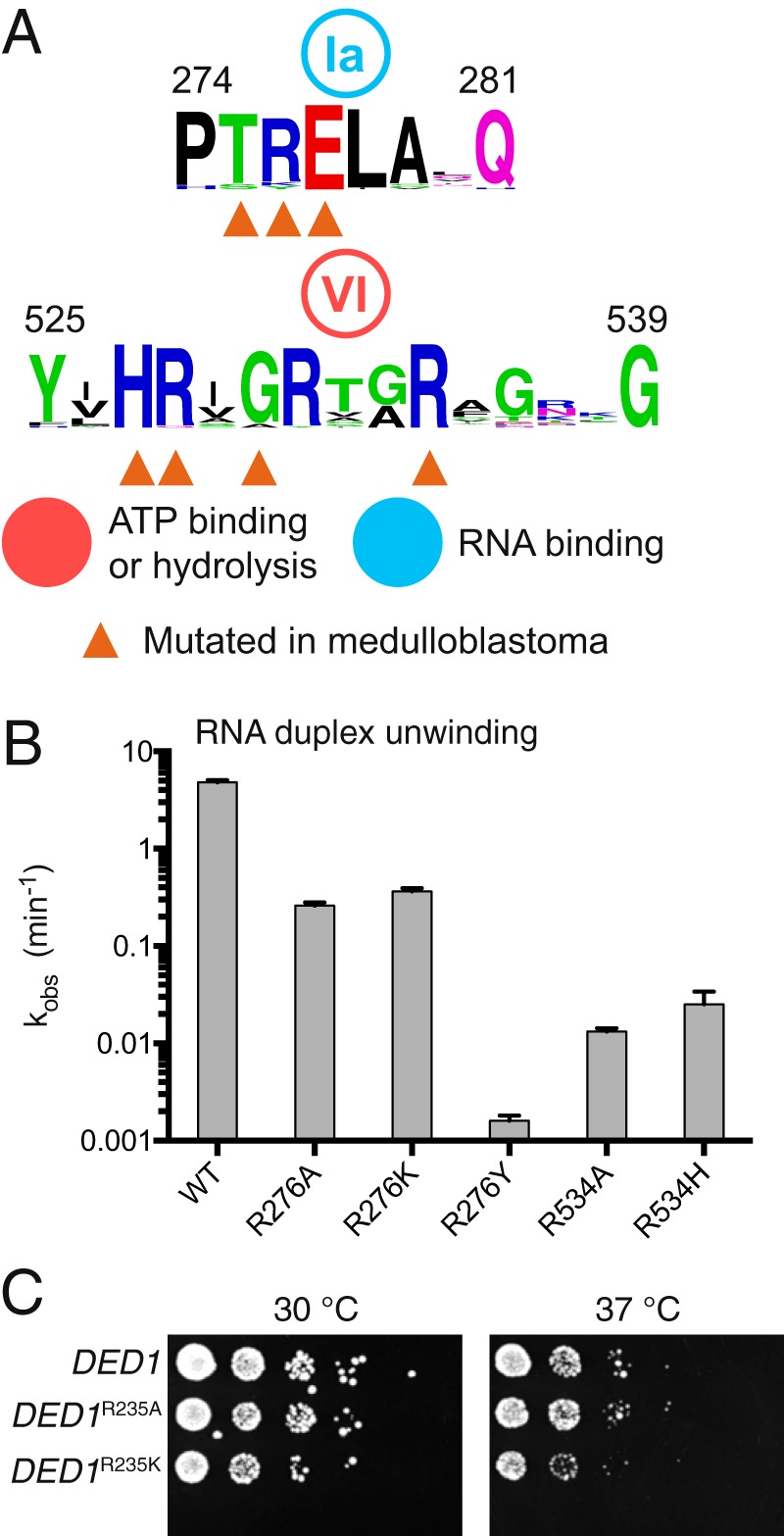
Medulloblastoma-associated variants inactivate DDX3. A, motifs Ia and VI of human DEAD-box proteins with residues altered in the medulloblastoma indicated. B, RNA duplex unwinding conducted with DDX3 132–607 and recurrent medulloblastoma variants indicated. Error bars indicate standard error of the fit parameter. C, yeast with mutations at DED1 Arg-235 to alanine or lysine are tolerated, whereas R235Y, R492A, and R492H were nonviable (not shown). DED1 Arg-235 and Arg-492 correspond to DDX3 Arg-276 and Arg-534, respectively.
Molecular Dynamics Simulations
Molecular dynamics simulations were performed using Gromacs version 4.6.5 (45). The apo structure was used as the starting point for all simulations (PDB 5E7I) with missing loops built using UCSF Chimera version 1.8.1 (46) and Modeller version 9.12 (47). A rhombic dodecahedral, periodic simulation box was used with a buffer of 12 Å between the solute and the boundary. Na+ and Cl− ions were added for charge neutralization followed by vacuum and solvent equilibration using transferable intermolecular potential 3 point (TIP3P) water (48). The production simulation length was 100 ns, and the step size was 2 fs. For modeling, the ATP-bound, Vasa closed-state domains of the AMPPNP structure (PDB 5E7M) were superposed onto the corresponding domains of the Vasa structure using PyMOL. Then, missing loops were added using UCSF Chimera and Modeller. Initial energies were high, likely reflecting a clash due to imperfect alignment, but quickly equilibrated in vacuum. Interatomic distances along trajectories were calculated using VMD version 1.9.1 (49).
Results
The Ded1/DDX3 Subfamily Contains Conserved Regions outside the Helicase Core
To define the minimal active construct of human DDX3X, we aligned Ded1/DDX3 family members from diverse species. In addition to the RecA-like core domains, five regions of sequence conservation unique to this family are apparent (Fig. 1A). The N-terminal conserved sequences correspond to a Crm1-dependent nuclear export sequence (31)and an eIF4E-binding site (10, 11), and the C terminus contains an RDYR motif and an invariant WW dipeptide motif (Fig. 1) (5). In addition, there are regions adjacent to the helicase core conserved between DDX3, Ded1, and Vasa/DDX4 (Fig. 1A) (5, 25). The N-terminal extension (NTE; residues 132–168; Fig. 1B) is predicted by PSIPRED (50) to form a short α-helix from residues 145 to 151, whereas the C-terminal extension (CTE; residues 582–607; Fig. 1B) has no predicted structure but is highly positively charged (pI ∼12). Recent structural and biochemical work demonstrated that DDX3 constructs containing the NTE are competent for ATP hydrolysis (25), but it is unknown how this activity compares with full-length human DDX3.
FIGURE 1.

Conserved regions specific to the Ded1/DDX3 subfamily of DEAD-box proteins. A, a linear diagram of the features of the Ded1/DDX3 family, including the core helicase DEAD and HELICc RecA-like domains, the Crm1-dependent nuclear export sequence (NES), and the eIF4E-binding site. Conserved regions specific to the Ded1/DDX3 subfamily are indicated in gray; GINF, RDYR, and WW refer to amino acid motifs. The NTE and CTE are indicated. B, sequence alignments showing the N- and C-terminal extensions found in the Ded1/DDX3 family and indicating construct boundaries used in this study and two previous crystal structures. Numbers correspond to human DDX3X.
The CTE Is Essential for RNA Duplex Unwinding and Affects Yeast Growth
To compare the activity of full-length DDX3 with truncated variants, we expressed and purified full-length DDX3 and truncations of the NTE, CTE, or both and then measured RNA duplex unwinding activities. Truncation of 131 residues from the N terminus and 55 residues from the C terminus yields a functional core of DDX3 that robustly unwinds RNA duplexes, although this construct has a roughly 5-fold lower functional affinity for RNA when compared with the full-length protein (Fig. 2A). Both full-length and truncated protein show sigmoidal functional binding isotherms, suggesting that DDX3 functions as an oligomer, as yeast DED1 (32).
FIGURE 2.

Subfamily-specific extensions to the DEAD-box core are essential for DDX3 function. A, RNA duplex unwinding rates at the indicated concentrations for four truncations of DDX3. Error bars indicate S.D. B, yeast growth assays for strains containing truncated copies of the essential gene DED1 demonstrate a requirement for the region conserved in the Ded1/DDX3 family. DED1 residues 16, 30, 85, 543, 561, and 604 correspond to DDX3 residues 23, 47, 122, 592, 607, and 661, respectively. Strains containing truncations of both tails were nonviable (data not shown).
Removal of the CTE containing the RDYR motif severely diminishes duplex unwinding, independent of the presence of the NTE (Fig. 2A). It is possible that removal of this positively charged region in the CTE negatively impacts RNA binding, as suggested by weaker binding to heparin resin (data not shown) and by the RNA binding defect caused by deletion of the entire C-terminal tail of Ded1p up to the helicase core (29). Alternatively, or in addition, this region might be critical for oligomerization (32).
To assess the biological function of the truncated proteins, we tested the ability of truncated versions of DED1 to support yeast growth. We generated truncations of DED1 in a plasmid and shuffled these into a yeast strain containing the sole copy of DED1 on a plasmid under control of its endogenous promoter (10). Under normal growth conditions, DED1 truncations lacking both the nuclear export sequence and the eIF4E-binding site complement yeast growth fully. Truncation up to the boundary of the NTE is viable but confers a slow growth phenotype (Fig. 2B). It is intriguing that this strain fully complements DED1 because Ded1p is thought to be primarily nuclear localized when the nuclear export sequence is deleted (51). We were unable to generate strains lacking the ATP-binding loop (ABL; DED1130–604; data not shown). The DED130–604 and DED185–604 strains are cold-sensitive, indicating a special requirement for the N-terminal tail at cold temperatures. Similarly, truncation up to the boundary of the CTE is tolerated, but deletion of the CTE results in slowed growth (Fig. 2B), whereas further truncation into the CTE exhibits cold sensitivity (10) or very weak complementation (29). These data demonstrate that the activity of the Ded1/DDX3 subfamily of DEAD-box proteins is surprisingly resilient to truncation of the subfamily-specific tails, but only up to conserved regions adjacent to the helicase core. Furthermore, inclusion of the CTE is essential for activity in vitro and in vivo.
The 2.2 Å Crystal Structure of Wild-type DDX3 132–607
To better understand the role of the NTE and CTE, we solved the crystal structure of AMP-bound wild-type DDX3 132–607 to ∼2.2 Å resolution (Table 1). The DEAD-domain is oriented uniquely with respect to the HELICc domain when compared with two other structures of DDX3 (25, 33) (Fig. 3A). Interestingly, the orientation of the present structure and DDX3 135–582 (ΔCTE; PDB 4PXA) is more similar than DDX3 168–582 (ΔNTE and ΔCTE; PDB 2I4I), suggesting that inclusion of the NTE biases crystallization toward this conformation. However, the interdomain orientation between the present structure and 4PXA is different, likely because PDB 4PXA contains a D354V mutation, which is located at the interdomain interface in both structures. Comparison of the present crystal structure with the structure of Vasa bound to RNA shows that the crystallized conformation of DDX3 is refractory to RNA binding, as the HELICc domain overlaps with the bound RNA in the Vasa structure (Fig. 3B; PDB 2DB3) (52). The CTE is predicted to be disordered by PSIPRED, and we observe no density past residue 584 in any of our structures, despite the obvious requirement for this region for protein function in vitro and in vivo (Fig. 2). Therefore, DDX3 preferentially crystallizes in a partially closed conformation with interdomain interactions between the DEAD and HELICc domains that are refractory to catalysis.
TABLE 1.
Data collection and refinement statistics
Statistics for the highest-resolution shell are shown in parentheses. RMS, root mean square.
| DDX3 132–607 AMPPNP (5E7M) | DDX3 132–607 AMP (5E7J) | DDX3 132–607 apo (5E7I) | |
|---|---|---|---|
| Wavelength (Å) | 1.115869 | 1.115869 | 1.115869 |
| Resolution range | 47.29–2.304 (2.386–2.304) | 46.18–2.229 (2.309–2.229) | 84.33–2.223 (2.303–2.223) |
| Space group | P 21 21 21 | P 21 21 21 | P 1 21 1 |
| Unit cell | 53.93 100.28 107.26 90 90 90 | 51.35 89.91 107.65 90 90 90 | 92.71 105.38 94.61 90 114.544 90 |
| Total reflections | 198,184 (19,421) | 96,467 (5247) | 311,845 (18,681) |
| Unique reflections | 26,335 (1930) | 24,246 (1660) | 79,732 (6607) |
| Multiplicity | 7.5 (7.6) | 4.0 (2.8) | 3.9 (2.8) |
| Completeness (%) | 0.97 (0.98) | 0.96 (0.76) | 0.98 (0.82) |
| Mean I/σ(I) | 10.80 (0.12) | 16.59 (2.42) | 8.24 (0.95) |
| Wilson B-factor | 75.58 | 31.24 | 33 |
| Rmerge | 0.1274 (15.21) | 0.06914 (0.5794) | 0.1617 (1.306) |
| Rmeas | 0.1372 (16.32) | 0.07978 (0.7047) | 0.1873 (1.593) |
| CC½ | 0.999 (0.111) | 0.998 (0.931) | 0.991 (0.362) |
| CC* | 1 (0.447) | 1 (0.982) | 0.998 (0.729) |
| Reflections used in refinement | 25,653 (1928) | 24,004 (1644) | 79,682 (6593) |
| Reflections used for Rfree | 1281 (95) | 1200 (81) | 3985 (332) |
| Rwork | 0.2279 (0.5683) | 0.2328 (0.4521) | 0.2270 (0.3966) |
| Rfree | 0.2856 (0.5753) | 0.2649 (0.4761) | 0.2599 (0.4120) |
| CCwork | 0.953 (0.377) | 0.940 (0.556) | 0.950 (0.446) |
| CCfree | 0.942 (0.283) | 0.950 (0.563) | 0.940 (0.477) |
| Number of non-hydrogen atoms | 3390 | 3446 | 10,060 |
| Macromolecules | 3354 | 3446 | 10,060 |
| Ligands | 31 | 23 | 0 |
| Protein residues | 424 | 433 | 1272 |
| RMS (bonds) | 0.01 | 0.003 | 0.004 |
| RMS (angles) | 1.43 | 0.82 | 0.84 |
| Ramachandran favored (%) | 93 | 98 | 98 |
| Ramachandran allowed (%) | 6.5 | 2.1 | 2.1 |
| Ramachandran outliers (%) | 0.48 | 0 | 0.24 |
| Rotamer outliers (%) | 2.7 | 0.27 | 1 |
| Clashscore | 16.42 | 4.22 | 5.37 |
| Average B-factor | 100.35 | 37.4 | 46.72 |
| Macromolecules | 100.18 | 37.4 | 46.72 |
| Ligands | 118.05 | 42.53 | |
| Solvent | 102.92 |
FIGURE 3.
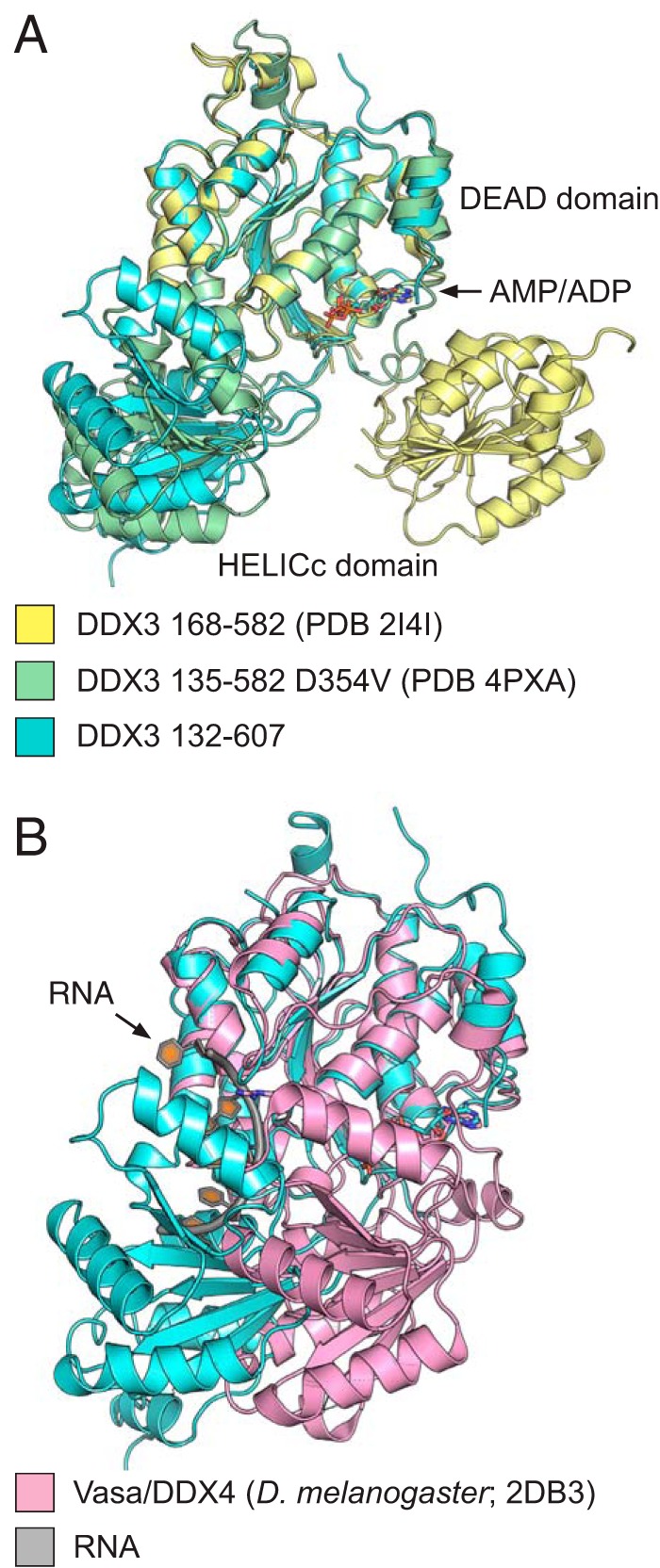
The 2.2 Å crystal structure of the conserved core of wild-type DDX3. A, the structure of DDX3 132–607 bound to AMP (blue) is shown along with the structure of DDX3 135–582 D354V (green; PDB 4PXA) and 168–582 (yellow; PDB 2I4I). Structures are aligned by the DEAD domain, highlighting the rotation of the C-terminal HELICc domain between the three structures. B, the partially closed state of DDX3 (blue) clashes with the RNA-binding site based on a comparison with the DEAD-box protein Vasa bound to RNA (Vasa: pink; RNA: gray).
We observe a short α-helix from residues 146–151 (the 150′s helix) in the NTE as predicted by PSIPRED and seen in the DDX3 135–582 structure (25). The NTE additionally contains the ABL, which is disordered in our structure and 4PXA (25) but forms a short α-helix in the structure of Vasa bound to RNA (52). Therefore, we crystallized DDX3 132–607 in the presence of no ligand, ADP, or AMPPNP to see whether the conformation of the ABL was altered (Table 1). In all cases, the ABL was disordered and difficult to model robustly. We elected to not build residues 155–165 of the NTE in any of these structures as repeated model building and refinement indicated insufficient density in this region to specify a unique structure. Thus, the ABL is dynamic in the presence of adenosine phosphates, and may fold into an α-helix cooperatively with interdomain closure and RNA binding, as seen in the Vasa structure bound to ssRNA (52).
Mutation of an Interdomain Interacting Residue Accelerates Duplex Unwinding
If the crystallized conformation of DDX3 is present in solution, then it should inhibit duplex unwinding by DDX3 because it is refractory to RNA binding (Fig. 3). The interdomain interface buries ∼560 Å2, suggesting that it may be stable in solution. Therefore, we hypothesized that mutation of the interdomain interface should accelerate duplex unwinding. Four residues make apparent interdomain contacts: Asp-354 and Glu-388 form a salt bridge with His-527 (separated by 4.0 and 2.7 Å, respectively), and Asp-506 hydrogen-bonds to the backbone of Arg-276 and caps an α-helix (Fig. 4A; 2.8 Å distance). We targeted residues Asp-354 and Glu-388 for mutation because Asp-506 and His-527 are members of conserved motifs Va and VI, respectively (1). Mutation of residue Asp-354 to either alanine or tryptophan (to sterically block formation of the partially closed state) results in decreased activity (Fig. 4B). Although Asp-354 is not part of a pan-DEAD-box motif, it is conserved within the Ded1/DDX3 subfamily, including Vasa. Interestingly, the structure of DDX3 with a D354V mutation shows a different closed state (25), supporting the role of this residue in interdomain interactions. In contrast, mutation of Glu-388 to alanine, arginine, or tryptophan accelerates duplex unwinding by a factor of two (Fig. 4B). In the closed, RNA-bound structure of D. melanogaster Vasa, residue Glu-438 (DDX3 Glu-388) is solvent-exposed and distal to the RNA-binding site, making it unlikely that these mutations increase activity by altered RNA or interdomain interactions. As a comparison, we tested mutation of the conserved GINF motif in the NTE (Fig. 1A) and found that it decreases duplex unwinding by a factor of two, supporting the conclusion that the ABL is necessary for catalysis (Fig. 4B) (25). In sum, mutations predicted to destabilize the crystallized interdomain interface accelerate duplex unwinding by DDX3, consistent with the presence of the partially closed structure in solution and its inhibitory nature.
FIGURE 4.
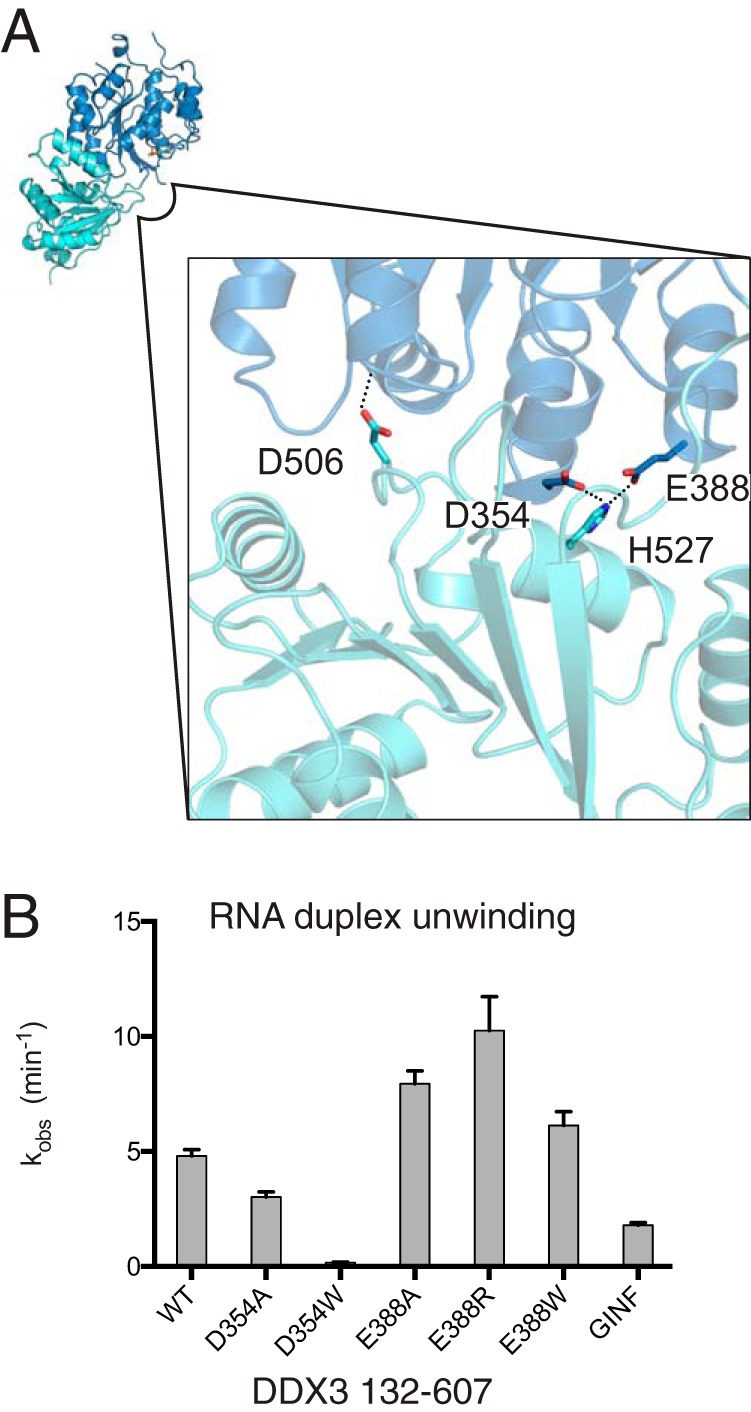
Mutations that destabilize the partially closed state accelerate duplex unwinding. A, contacts between the DEAD and HELICc domains oriented on the view as in Fig. 3 (upper left). Asp-354 and Glu-388 are separated from His-527 by 4.0 and 2.7 Å, respectively, and Asp-506 is separated from the backbone of Arg-276 by 2.8 Å. B, RNA duplex unwinding conducted at 1 μm protein with DDX3 132–607 and mutations indicated. GINF is a mutation of the GINF motif to AAAA (residues 157–160). Error bars indicate standard error of the fit parameter.
The Partially Closed Structure Is Stable over 100 ns of Molecular Dynamics Simulation
As the crystal structure was solved at pH 5 and crystals can trap transient structures, we tested the stability of the partially closed structure and the interactions of the ABL with ATP at neutral pH by performing 100-ns molecular dynamics simulations of apo DDX3 132–607 apo and ATP-bound DDX3 132–607. The ATP-bound state was modeled off the closed form of Vasa bound to RNA to attempt to induce structure formation in the ABL, but instead quickly equilibrated toward an unobserved, alternative closed state, which was not pursued further. In contrast, the partially closed interdomain interface remained stable for the full 100-ns simulation when started from the apo crystal structure (PDB 5E7I) (Fig. 5A). Similarly, in both the partially closed and the ATP-bound states, the 150′s helix remained stably docked to the side of the DEAD domain (Fig. 5B). In contrast, the ABL is dynamic. In the ATP-bound simulation, the ABL makes transient interactions with the adenine moiety of ATP (Fig. 5C) involving interactions between Lys-162 and ATP and Phe-160 cation-π stacking with Lys-162 (Fig. 5D). In this state, Lys-162 forms a bipartite hydrogen bond with ∼3 Å separation from both a ribose oxygen and the adenine N3 of ATP and is separated from the Phe-160 aromatic ring by ∼4 Å. These simulations show that the partially closed state of DDX3 (Fig. 3) is stable in silico and suggest that the ABL becomes structured in a cooperative manner with RNA binding.
FIGURE 5.

Molecular dynamics simulations suggest a stable interdomain interface and transient ABL-ATP interactions. A, the partially closed structure is stable over a 100-ns trajectory. Separation between the side chains Glu-388 (Cδ) and His-527 (Cγ) is shown for apo DDX3 132–607 (black) and a closed-state model based off the Vasa structure (cyan; PDB 2DB3; see “Experimental Procedures”). RMSD, root mean square deviation. B, the 150′s α-helix forms stable interactions with the DEAD domain (colors as in A). Distance is measured between Phe-151 (Cγ) and Leu-197 (Cγ). C, the ABL makes transient, stable interactions with the adenine group of ATP. Black: distance between the Cγ of Phe-160 and the H2 of ATP; cyan: distance between the Nζ of Lys-162 and the N3 of ATP. In A–C, distances are smoothed over a sliding window of 20 frames. D, structural model of the ABL interacting with ATP at 50 ns of simulation.
Medulloblastoma Variants of DDX3 Inactivate the Protein
DDX3 is among the most frequently mutated genes in the highly malignant brain tumor medulloblastoma (21–24). Most variants are predicted to inactivate the catalytic activity of DDX3, and some have been shown to decrease the ATPase activity (25). The truncated construct of DDX3 containing the CTE and NTE is highly active and easy to purify, facilitating analysis of disease-associated variants of DDX3. We therefore selected three recurrent variants found in the DEAD-box motifs Ia and VI, R276K, R276Y, and R534H (Fig. 6A) (21–23), and additionally tested alanine substitutions and made these mutations in the DDX3 132–607 construct. The corresponding residue of Arg-276 in Vasa is Arg-328, which binds to the RNA backbone, and Arg-534 is Vasa Arg-582, which interacts with the γ-phosphate of ATP. All mutant proteins purified to high yield (>5 mg l−1) and decreased the rate of duplex unwinding, from 1-fold to 1000-fold (Fig. 6B). In concert with the in vitro results, mutations R276A and R276K support yeast growth (Fig. 6C), whereas the other three mutations could not complement DED1 (data not shown). Similarly, mutation of the residue corresponding to Arg-534 in S. cerevisiae PRP28 causes dominant negative lethality in vivo (53). In sum, medulloblastoma variants of DDX3 at Arg-276 or Arg-534 are inactivating, further confirming that full-length, inactive DDX3 is selected for by this tumor (21–23, 25), as well as demonstrating the utility of DDX3 132–607 in determining the functional consequences of disease-associated variants of DDX3.
Discussion
DEAD-box proteins consist of two RecA-like domains that comprise the “helicase” core surrounded by variable regions that are unique to individual subfamilies (1, 29). Here, we studied the role of the N- and C-terminal extensions that are essential for function in the Ded1/DDX3 subfamily of DEAD-box proteins (5). We found that removal of the N-terminal 131 and C-terminal 55 residues yields an active construct of DDX3, but further truncation is deleterious (Fig. 2). We then solved the highest resolution crystal structure of an active construct of DDX3 to date (Fig. 3), illustrating a unique autoinhibited state of the protein (Fig. 4) and providing an excellent starting structure for molecular dynamics simulations (Fig. 5). We find that two untested variants found in the highly malignant brain tumor medulloblastoma inactivate the protein in vitro and are lethal in yeast (Fig. 6). In sum, our data characterize the essential, conserved core of the Ded1/DDX3 subfamily, which will prove useful when interpreting variants found in human malignancies.
DDX3 132–607 crystallized with a unique interdomain interface not seen in previous crystal structures (Fig. 4A). The interface buries ∼560 Å2 of surface area and overlaps with the RNA-binding surface, suggesting that it is inhibitory to RNA duplex unwinding (Fig. 3B). Indeed, introduction of point mutations predicted to destabilize the interdomain interface accelerated the rate of duplex unwinding, despite being distal to the RNA, ATP, or closed-state interdomain interface (Fig. 4). In support of the presence of this state in solution at neutral pH, it was stable during our molecular dynamics simulation (Fig. 5). Therefore, DDX3 contains a cryptic second binding site for the HELICc domain on the DEAD-domain, which is present in solution and is inhibitory for function. S. cerevisiae Prp5p was also crystallized in an inhibitory conformation, and destabilization of this “twisted” state accelerated catalysis (54). It is possible that specific proteins may bind and stabilize inhibitory conformations of DEAD-box proteins to negatively regulate catalysis, as opposed to the many MIF4G domains that bind and activate catalysis (55–59).
Other DEAD-box subfamilies have different subfamily-specific NTE and CTEs. For example, the structure of RNA-bound DDX19 (PDB 3GOH) (60) contains an NTE helix similar to DDX3 and Vasa. However, this region forms part of a β-sheet in the ADP-bound structure of DDX19 (PDB 3EWS) (60). In the crystal structure of S. cerevisiae Prp5p, an NTE helix stabilizes the twisted conformation by interacting with the DEAD and HELICc domains (54). Therefore, structural plasticity at the N-terminal boundary of the DEAD domain may be a feature common to many DEAD-box proteins. Perhaps the most extreme example characterized to date of a CTE is in the mitochondrial DEAD-box protein Mss116p, where the CTE forms an entire domain essential for RNA binding and is a fundamental piece of the helicase core (61, 62), unlike most DEAD-box proteins. Thus, to understand the function of an individual DEAD-box subfamily, it is essential to characterize sequences conserved within the subfamily and beyond the DEAD-box helicase core.
Interestingly, the Ded1/DDX3 subfamily of DEAD-box proteins multimerizes both in vitro and in vivo (32). Oligomerization depends on the C-terminal tail of Ded1, and truncation of the C-terminal 69 residues blocks multimerization and hinders duplex unwinding (32). Our data show that removal of this region strongly suppresses duplex unwinding in vitro, but only if the conserved RDYR motif is deleted (Figs. 1A and 2A). Future experiments will test the role of the RDYR motif and C-terminal sequences in oligomerization of the Ded1/DDX3 subfamily.
Human DDX3X is altered in numerous malignancies, and different disorders have unique mutation spectrums indicative of their distinct requirements for DDX3 function. For example, nearly all variants found in patients afflicted by the malignant brain tumor medulloblastoma are nonsynonymous single nucleotide variants yielding point mutants predicted to inactivate the protein, yet there are no premature stop codons, frameshifts, or splice variants (21–24). Thus, full-length but inactive protein is selected by this tumor type. In contrast, in blood cancers such as natural-killer/T-cell lymphoma (17), Burkitt lymphoma (63), or chronic lymphocytic leukemia (15, 16), DDX3X variants include nonsynonymous single nucleotide variants but also many premature stop codons, frameshifts, and splice variants. The elucidation of the minimal conserved functional core and the new, high-resolution structures of DDX3 presented here is of broad utility for molecular modeling and when predicting the function of truncating variants of DDX3 present in patient samples.
Author Contributions
S. N. F. and J. A. D. conceptualized the study; S. N. F., D. S., and K. J. C. designed the methodology; S. N. F., D. S., and K. J. C. investigated the study; S. N. F. and J. A. D. wrote the original draft; S. N. F., J. A. D., and E. J. performed funding acquisition; and J. A. D. and E. J. supervised the study. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Angie Hilliker and the lab of Jasper Rine for help with the yeast experiments and for sharing yeast strains, Kaihong Zhou and Alison Smith for early protein characterization, Yoon-Jae Cho for advice regarding the medulloblastoma variants, and Yun Bai for critical reading of the manuscript. Structural data were collected at Beamline 8.3.1 of the Advanced Light Source at the Lawrence Berkeley National Laboratory, which acknowledges support from the University of California Office of the President, Multicampus Research Programs and Initiatives Grant MR-15-328599, and the Program for Breakthrough Biomedical Research, which is partially funded by the Sandler Foundation. Molecular dynamics starting models were preprocessed with the UCSF Chimera package, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS National Institutes of Health Grant P41-GM103311).
This work was supported by National Institutes of Health NIGMS Grants GM0067700 and GM099720 (to E. J.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (codes 5E7I, 5E7J, and 5E7M) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- TCEP
- tris(2-carboxyethyl)phosphine
- AMPPNP
- 5′-adenylyl-β,γ-imidodiphosphate
- CTE
- C-terminal extension
- NTE
- N-terminal extension
- ABL
- ATP-binding loop
- CC
- correlation coefficient.
References
- 1. Linder P., and Jankowsky E. (2011) From unwinding to clamping: the DEAD box RNA helicase family. Nat. Rev. Mol. Cell Biol. 12, 505–516 [DOI] [PubMed] [Google Scholar]
- 2. Hubstenberger A., Noble S. L., Cameron C., and Evans T. C. (2013) Translation repressors, an RNA helicase, and developmental cues control RNP phase transitions during early development. Dev. Cell 27, 161–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Elbaum-Garfinkle S., Kim Y., Szczepaniak K., Chen C. C., Eckmann C. R., Myong S., and Brangwynne C. P. (2015) The disordered P granule protein LAF-1 drives phase separation into droplets with tunable viscosity and dynamics. Proc. Natl. Acad. Sci. U.S.A. 112, 7189–7194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chuang R. Y., Weaver P. L., Liu Z., and Chang T. H. (1997) Requirement of the DEAD-Box protein Ded1p for messenger RNA translation. Science 275, 1468–1471 [DOI] [PubMed] [Google Scholar]
- 5. Sharma D., and Jankowsky E. (2014) The Ded1/DDX3 subfamily of DEAD-box RNA helicases. Crit. Rev. Biochem. Mol. Biol. 49, 343–360 [DOI] [PubMed] [Google Scholar]
- 6. Sen N. D., Zhou F., Ingolia N. T., and Hinnebusch A. G. (2015) Genome-wide analysis of translational efficiency reveals distinct but overlapping functions of yeast DEAD-box RNA helicases Ded1 and eIF4A. Genome Res. 25, 1196–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lai M. C., Chang W. C., Shieh S. Y., and Tarn W. Y. (2010) DDX3 regulates cell growth through translational control of cyclin E1. Mol. Cell. Biol. 30, 5444–5453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee C. S., Dias A. P., Jedrychowski M., Patel A. H., Hsu J. L., and Reed R. (2008) Human DDX3 functions in translation and interacts with the translation initiation factor eIF3. Nucleic Acids Res. 36, 4708–4718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. de la Cruz J., Iost I., Kressler D., and Linder P. (1997) The p20 and Ded1 proteins have antagonistic roles in eIF4E-dependent translation in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 94, 5201–5206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hilliker A., Gao Z., Jankowsky E., and Parker R. (2011) The DEAD-box protein Ded1 modulates translation by the formation and resolution of an eIF4F-mRNA complex. Mol. Cell 43, 962–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shih J. W., Wang W. T., Tsai T. Y., Kuo C. Y., Li H. K., and Wu Lee Y. H. (2012) Critical roles of RNA helicase DDX3 and its interactions with eIF4E/PABP1 in stress granule assembly and stress response. Biochem. J. 441, 119–129 [DOI] [PubMed] [Google Scholar]
- 12. Lai M. C., Sun H. S., Wang S. W., and Tarn W. Y. (2015) DDX3 functions in antiviral innate immunity through translational control of PACT. FEBS J. 10.1111/febs.13553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Soto-Rifo R., Rubilar P. S., Limousin T., de Breyne S., Décimo D., and Ohlmann T. (2012) DEAD-box protein DDX3 associates with eIF4F to promote translation of selected mRNAs. EMBO J. 31, 3745–3756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Landau D. A., Tausch E., Taylor-Weiner A. N., Stewart C., Reiter J. G., Bahlo J., Kluth S., Bozic I., Lawrence M., Böttcher S., Carter S. L., Cibulskis K., Mertens D., Sougnez C. L., Rosenberg M., Hess J. M., Edelmann J., Kless S., Kneba M., Ritgen M., Fink A., Fischer K., Gabriel S., Lander E. S., Nowak M. A., Döhner H., Hallek M., Neuberg D., Getz G., Stilgenbauer S., and Wu C. J. (2015) Mutations driving CLL and their evolution in progression and relapse. Nature 526, 525–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ojha J., Secreto C. R., Rabe K. G., Van Dyke D. L., Kortum K. M., Slager S. L., Shanafelt T. D., Fonseca R., Kay N. E., and Braggio E. (2015) Identification of recurrent truncated DDX3X mutations in chronic lymphocytic leukaemia. Br. J. Haematol. 169, 445–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang L., Lawrence M. S., Wan Y., Stojanov P., Sougnez C., Stevenson K., Werner L., Sivachenko A., DeLuca D. S., Zhang L., Zhang W., Vartanov A. R., Fernandes S. M., Goldstein N. R., Folco E. G., Cibulskis K., Tesar B., Sievers Q. L., Shefler E., Gabriel S., Hacohen N., Reed R., Meyerson M., Golub T. R., Lander E. S., Neuberg D., Brown J. R., Getz G., and Wu C. J. (2011) SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N. Engl. J. Med. 365, 2497–2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jiang L., Gu Z. H., Yan Z. X., Zhao X., Xie Y. Y., Zhang Z. G., Pan C. M., Hu Y., Cai C. P., Dong Y., Huang J. Y., Wang L., Shen Y., Meng G., Zhou J. F., Hu J. D., Wang J. F., Liu Y. H., Yang L. H., Zhang F., Wang J. M., Wang Z., Peng Z. G., Chen F. Y., Sun Z. M., Ding H., Shi J. M., Hou J., Yan J. S., Shi J. Y., Xu L., Li Y., Lu J., Zheng Z., Xue W., Zhao W. L., Chen Z., and Chen S. J. (2015) Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nat. Genet. 47, 1061–1066 [DOI] [PubMed] [Google Scholar]
- 18. Seiwert T. Y., Zuo Z., Keck M. K., Khattri A., Pedamallu C. S., Stricker T., Brown C., Pugh T. J., Stojanov P., Cho J., Lawrence M. S., Getz G., Brägelmann J., DeBoer R., Weichselbaum R. R., Langerman A., Portugal L., Blair E., Stenson K., Lingen M. W., Cohen E. E., Vokes E. E., White K. P., and Hammerman P. S. (2015) Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin. Cancer Res. 21, 632–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stransky N., Egloff A. M., Tward A. D., Kostic A. D., Cibulskis K., Sivachenko A., Kryukov G. V., Lawrence M. S., Sougnez C., McKenna A., Shefler E., Ramos A. H., Stojanov P., Carter S. L., Voet D., Cortés M. L., Auclair D., Berger M. F., Saksena G., Guiducci C., Onofrio R. C., Parkin M., Romkes M., Weissfeld J. L., Seethala R. R., Wang L., Rangel-Escareño C., Fernandez-Lopez J. C., Hidalgo-Miranda A., Melendez-Zajgla J., Winckler W., Ardlie K., Gabriel S. B., Meyerson M., Lander E. S., Getz G., Golub T. R., Garraway L. A., and Grandis J. R. (2011) The mutational landscape of head and neck squamous cell carcinoma. Science 333, 1157–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bol G. M., Vesuna F., Xie M., Zeng J., Aziz K., Gandhi N., Levine A., Irving A., Korz D., Tantravedi S., Heerma van Voss M. R., Gabrielson K., Bordt E. A., Polster B. M., Cope L., van der Groep P., Kondaskar A., Rudek M. A., Hosmane R. S., van der Wall E., van Diest P. J., Tran P. T., and Raman V. (2015) Targeting DDX3 with a small molecule inhibitor for lung cancer therapy. EMBO Mol. Med. 7, 648–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jones D. T., Jäger N., Kool M., Zichner T., Hutter B., Sultan M., Cho Y. J., Pugh T. J., Hovestadt V., Stütz A. M., Rausch T., Warnatz H. J., Ryzhova M., Bender S., Sturm D., Pleier S., Cin H., Pfaff E., Sieber L., Wittmann A., Remke M., Witt H., Hutter S., Tzaridis T., Weischenfeldt J., Raeder B., Avci M., Amstislavskiy V., Zapatka M., Weber U. D., Wang Q., Lasitschka B., Bartholomae C. C., Schmidt M., von Kalle C., Ast V., Lawerenz C., Eils J., Kabbe R., Benes V., van Sluis P., Koster J., Volckmann R., Shih D., Betts M. J., Russell R. B., Coco S., Tonini G. P., Schüller U., Hans V., Graf N., Kim Y. J., Monoranu C., Roggendorf W., Unterberg A., Herold-Mende C., Milde T., Kulozik A. E., von Deimling A., Witt O., Maass E., Rössler J., Ebinger M., Schuhmann M. U., Frühwald M. C., Hasselblatt M., Jabado N., Rutkowski S., von Bueren A. O., Williamson D., Clifford S. C., McCabe M. G., Collins V. P., Wolf S., Wiemann S., Lehrach H., Brors B., Scheurlen W., Felsberg J., Reifenberger G., Northcott P. A., Taylor M. D., Meyerson M., Pomeroy S. L., Yaspo M. L., Korbel J. O., Korshunov A., Eils R., Pfister S. M., and Lichter P. (2012) Dissecting the genomic complexity underlying medulloblastoma. Nature 488, 100–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kool M., Jones D. T., Jäger N., Northcott P. A., Pugh T. J., Hovestadt V., Piro R. M., Esparza L. A., Markant S. L., Remke M., Milde T., Bourdeaut F., Ryzhova M., Sturm D., Pfaff E., Stark S., Hutter S., Seker-Cin H., Johann P., Bender S., Schmidt C., Rausch T., Shih D., Reimand J., Sieber L., Wittmann A., Linke L., Witt H., Weber U. D., Zapatka M., König R., Beroukhim R., Bergthold G., van Sluis P., Volckmann R., Koster J., Versteeg R., Schmidt S., Wolf S., Lawerenz C., Bartholomae C. C., von Kalle C., Unterberg A., Herold-Mende C., Hofer S., Kulozik A. E., von Deimling A., Scheurlen W., Felsberg J., Reifenberger G., Hasselblatt M., Crawford J. R., Grant G. A., Jabado N., Perry A., Cowdrey C., Croul S., Zadeh G., Korbel J. O., Doz F., Delattre O., Bader G. D., McCabe M. G., Collins V. P., Kieran M. W., Cho Y. J., Pomeroy S. L., Witt O., Brors B., Taylor M. D., Schüller U., Korshunov A., Eils R., Wechsler-Reya R. J., Lichter P., Pfister S. M., and ICGC PedBrain Tumor Project (2014) Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell 25, 393–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pugh T. J., Weeraratne S. D., Archer T. C., Pomeranz Krummel D. A., Auclair D., Bochicchio J., Carneiro M. O., Carter S. L., Cibulskis K., Erlich R. L., Greulich H., Lawrence M. S., Lennon N. J., McKenna A., Meldrim J., Ramos A. H., Ross M. G., Russ C., Shefler E., Sivachenko A., Sogoloff B., Stojanov P., Tamayo P., Mesirov J. P., Amani V., Teider N., Sengupta S., Francois J. P., Northcott P. A., Taylor M. D., Yu F., Crabtree G. R., Kautzman A. G., Gabriel S. B., Getz G., Jäger N., Jones D. T., Lichter P., Pfister S. M., Roberts T. M., Meyerson M., Pomeroy S. L., and Cho Y. J. (2012) Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 488, 106–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Robinson G., Parker M., Kranenburg T. A., Lu C., Chen X., Ding L., Phoenix T. N., Hedlund E., Wei L., Zhu X., Chalhoub N., Baker S. J., Huether R., Kriwacki R., Curley N., Thiruvenkatam R., Wang J., Wu G., Rusch M., Hong X., Becksfort J., Gupta P., Ma J., Easton J., Vadodaria B., Onar-Thomas A., Lin T., Li S., Pounds S., Paugh S., Zhao D., Kawauchi D., Roussel M. F., Finkelstein D., Ellison D. W., Lau C. C., Bouffet E., Hassall T., Gururangan S., Cohn R., Fulton R. S., Fulton L. L., Dooling D. J., Ochoa K., Gajjar A., Mardis E. R., Wilson R. K., Downing J. R., Zhang J., and Gilbertson R. J. (2012) Novel mutations target distinct subgroups of medulloblastoma. Nature 488, 43–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Epling L. B., Grace C. R., Lowe B. R., Partridge J. F., and Enemark E. J. (2015) Cancer-associated mutants of RNA helicase DDX3X are defective in RNA-stimulated ATP hydrolysis. J. Mol. Biol. 427, 1779–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Calo E., Flynn R. A., Martin L., Spitale R. C., Chang H. Y., and Wysocka J. (2015) RNA helicase DDX21 coordinates transcription and ribosomal RNA processing. Nature 518, 249–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ohnishi S., Pääkkönen K., Koshiba S., Tochio N., Sato M., Kobayashi N., Harada T., Watanabe S., Muto Y., Güntert P., Tanaka A., Kigawa T., and Yokoyama S. (2009) Solution structure of the GUCT domain from human RNA helicase II/Guβ reveals the RRM fold, but implausible RNA interactions. Proteins 74, 133–144 [DOI] [PubMed] [Google Scholar]
- 28. Schütz P., Karlberg T., van den Berg S., Collins R., Lehtiö L., Högbom M., Holmberg-Schiavone L., Tempel W., Park H. W., Hammarström M., Moche M., Thorsell A. G., and Schüler H. (2010) Comparative structural analysis of human DEAD-box RNA helicases. PLoS ONE 5, e12791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Banroques J., Cordin O., Doère M., Linder P., and Tanner N. K. (2011) Analyses of the functional regions of DEAD-box RNA “helicases” with deletion and chimera constructs tested in vivo and in vitro. J. Mol. Biol. 413, 451–472 [DOI] [PubMed] [Google Scholar]
- 30. Tarn W. Y., and Chang T. H. (2009) The current understanding of Ded1p/DDX3 homologs from yeast to human. RNA Biol. 6, 17–20 [DOI] [PubMed] [Google Scholar]
- 31. Yedavalli V. S., Neuveut C., Chi Y. H., Kleiman L., and Jeang K. T. (2004) Requirement of DDX3 DEAD box RNA helicase for HIV-1 Rev-RRE export function. Cell 119, 381–392 [DOI] [PubMed] [Google Scholar]
- 32. Putnam A. A., Gao Z., Liu F., Jia H., Yang Q., and Jankowsky E. (2015) Division of labor in an oligomer of the DEAD-Box RNA helicase Ded1p. Mol. Cell 59, 541–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Högbom M., Collins R., van den Berg S., Jenvert R. M., Karlberg T., Kotenyova T., Flores A., Karlsson Hedestam G. B., and Schiavone L. H. (2007) Crystal structure of conserved domains 1 and 2 of the human DEAD-box helicase DDX3X in complex with the mononucleotide AMP. J. Mol. Biol. 372, 150–159 [DOI] [PubMed] [Google Scholar]
- 34. Fairman M. E., Maroney P. A., Wang W., Bowers H. A., Gollnick P., Nilsen T. W., and Jankowsky E. (2004) Protein displacement by DExH/D “RNA helicases” without duplex unwinding. Science 304, 730–734 [DOI] [PubMed] [Google Scholar]
- 35. Kabsch W. (2010) Xds. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., and Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Karplus P. A., and Diederichs K. (2012) Linking crystallographic model and data quality. Science 336, 1030–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. DeLano W. L. (2010) The PyMOL Molecular Graphics System, version 1.3r1, Schrödinger, LLC, New York [Google Scholar]
- 41. Jankowsky E., and Putnam A. (2010) Duplex unwinding with DEAD-box proteins. Methods Mol. Biol. 587, 245–264 [DOI] [PubMed] [Google Scholar]
- 42. Edgar R. C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Waterhouse A. M., Procter J. B., Martin D. M., Clamp M., and Barton G. J. (2009) Jalview Version 2: a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Katoh K., and Standley D. M. (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pronk S., Páll S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M. R., Smith J. C., Kasson P. M., van der Spoel D., Hess B., and Lindahl E. (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 47. Fiser A., Do R. K., and Sali A. (2000) Modeling of loops in protein structures. Protein Sci. 9, 1753–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., and Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926 [Google Scholar]
- 49. Humphrey W., Dalke A., and Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–28 [DOI] [PubMed] [Google Scholar]
- 50. Buchan D. W., Minneci F., Nugent T. C., Bryson K., and Jones D. T. (2013) Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 41, W349–W357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Senissar M., Le Saux A., Belgareh-Touzé N., Adam C., Banroques J., and Tanner N. K. (2014) The DEAD-box helicase Ded1 from yeast is an mRNP cap-associated protein that shuttles between the cytoplasm and nucleus. Nucleic Acids Res. 42, 10005–10022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sengoku T., Nureki O., Nakamura A., Kobayashi S., and Yokoyama S. (2006) Structural basis for RNA unwinding by the DEAD-box protein Drosophila Vasa. Cell 125, 287–300 [DOI] [PubMed] [Google Scholar]
- 53. Jacewicz A., Schwer B., Smith P., and Shuman S. (2014) Crystal structure, mutational analysis and RNA-dependent ATPase activity of the yeast DEAD-box pre-mRNA splicing factor Prp28. Nucleic Acids Res. 42, 12885–12898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang Z. M., Yang F., Zhang J., Tang Q., Li J., Gu J., Zhou J., and Xu Y. Z. (2013) Crystal structure of Prp5p reveals interdomain interactions that impact spliceosome assembly. Cell Rep 5, 1269–1278 [DOI] [PubMed] [Google Scholar]
- 55. Buchwald G., Schüssler S., Basquin C., Le Hir H., and Conti E. (2013) Crystal structure of the human eIF4AIII-CWC22 complex shows how a DEAD-box protein is inhibited by a MIF4G domain. Proc. Natl. Acad. Sci. U.S.A. 110, E4611–E4618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kim K. M., Cho H., Choi K., Kim J., Kim B. W., Ko Y. G., Jang S. K., and Kim Y. K. (2009) A new MIF4G domain-containing protein, CTIF, directs nuclear cap-binding protein CBP80/20-dependent translation. Genes Dev. 23, 2033–2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Montpetit B., Thomsen N. D., Helmke K. J., Seeliger M. A., Berger J. M., and Weis K. (2011) A conserved mechanism of DEAD-box ATPase activation by nucleoporins and InsP6 in mRNA export. Nature 472, 238–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Craig A. W., Haghighat A., Yu A. T., and Sonenberg N. (1998) Interaction of polyadenylate-binding protein with the eIF4G homologue PAIP enhances translation. Nature 392, 520–523 [DOI] [PubMed] [Google Scholar]
- 59. Marcotrigiano J., Lomakin I. B., Sonenberg N., Pestova T. V., Hellen C. U., and Burley S. K. (2001) A conserved HEAT domain within eIF4G directs assembly of the translation initiation machinery. Mol. Cell 7, 193–203 [DOI] [PubMed] [Google Scholar]
- 60. Collins R., Karlberg T., Lehtiö L., Schütz P., van den Berg S., Dahlgren L. G., Hammarström M., Weigelt J., and Schüler H. (2009) The DEXD/H-box RNA helicase DDX19 is regulated by an α-helical switch. J. Biol. Chem. 284, 10296–10300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mallam A. L., Del Campo M., Gilman B., Sidote D. J., and Lambowitz A. M. (2012) Structural basis for RNA-duplex recognition and unwinding by the DEAD-box helicase Mss116p. Nature 490, 121–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Del Campo M., and Lambowitz A. M. (2009) Structure of the Yeast DEAD box protein Mss116p reveals two wedges that crimp RNA. Mol. Cell 35, 598–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schmitz R., Young R. M., Ceribelli M., Jhavar S., Xiao W., Zhang M., Wright G., Shaffer A. L., Hodson D. J., Buras E., Liu X., Powell J., Yang Y., Xu W., Zhao H., Kohlhammer H., Rosenwald A., Kluin P., Müller-Hermelink H. K., Ott G., Gascoyne R. D., Connors J. M., Rimsza L. M., Campo E., Jaffe E. S., Delabie J., Smeland E. B., Ogwang M. D., Reynolds S. J., Fisher R. I., Braziel R. M., Tubbs R. R., Cook J. R., Weisenburger D. D., Chan W. C., Pittaluga S., Wilson W., Waldmann T. A., Rowe M., Mbulaiteye S. M., Rickinson A. B., and Staudt L. M. (2012) Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 490, 116–120 [DOI] [PMC free article] [PubMed] [Google Scholar]