

Abstract
Solitary fibrous tumor (SFT) is a rare spindle-cell neoplasm originating from the mesenchyme. It was originally thought to occur exclusively in the intrathoracic region but has been recently described in extrapleural sites including the orbit. SFT of the orbit is a rare lesion, which can be misdiagnosed as hemangiopericytoma, fibrous histiocytoma, meningioma, or neurofibroma. Immunohistochemistry plays an important role. We report an orbital SFT in a 39-year-old female presented with painless, progressive proptosis, and diminished vision in the right eye for the duration of 6 months. Magnetic resonance imaging demonstrated well-defined enhancing mass lesion. The patient underwent complete tumor removal through a right fronto-orbital approach, and a pathological diagnosis of the solitary fibrous tumor was made. Postoperatively, the patient was symptom-free. Clinical and pathological findings including immunohistochemistry are presented along with a brief discussion of literature.
Keywords: Mesenchymal tumor, orbital tumor, solitary fibrous tumor
Introduction
Solitary fibrous tumor (SFT) is an uncommon spindle-cell tumor of mesenchymal origin, which was initially described in the pleura by Klemperer and Rabin in 1931 and thought to be of mesothelial origin.[1] However, the histogenesis of SFT is still controversial. Recently, extrapleural SFTs have been identified including the pericardium, peritoneum, lung, liver, mediastinum, nasal cavities, thyroid, parotid gland, and orbit. The orbital involvement was first described by Dorfman et al. and Westra et al., in 1994.[2,3] SFT of the orbit is an extremely rare lesion. About 60 cases of the orbital SFT have been reported in the literature till date. We report an orbital SFT in a 39-year-old lady with painless, progressive proptosis, and diminution of vision, along with immunohistochemical confirmation.
Case Report
A 39-year-old woman developed slowly progressive, painless proptosis, and diminished vision in the right eye over the duration of 6 months. An ophthalmologic assessment revealed that the patient had right sided eyelid swelling, with restricted extraocular movements. Visual acuity in the right eye was 6/12 and 6/6 in the left eye, pupillary reflexes sluggish in right eye and were well preserved in the left eye. Anterior segment examination of both eyes was normal. Fundus examination revealed disc edema and optic nerve atrophy in the right eye. The intraocular pressure with applanation tonometer was 28 mmHg in the right eye and 15 mmHg in the left eye.
Ultrasonography revealed a large mass superoposteriorly in the right eye with high surface reflectivity. Computed tomography (CT scan) revealed a well-defined intraconal mass in the posterior right orbit with postcontrast heterogenous enhancement causing proptosis and medial displacement of optic nerve [Figure 1]. Magnetic resonance imaging (MRI) demonstrated a retro-orbital lesion of the hypointense signal on T1-weighted and T2-weighted images, with multiple foci of hyperintensity in T2-weighted images. Postcontrast T1-weighted image revealed a heterogenous enhancement of the lesion [Figure 2].
Figure 1.
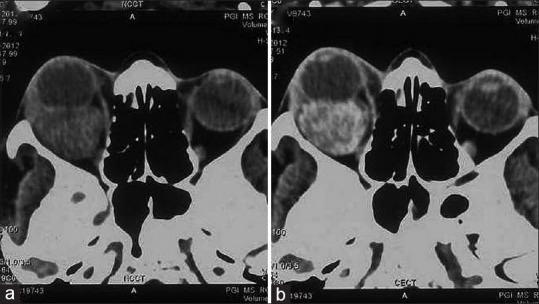
Axial noncontrast (a) computed tomography images shows a well-defined intraconal mass in the posterior right orbit. Postcontrast image (b) heterogenous enhancement of the lesion causing proptosis and medial displacement of optic nerve
Figure 2.
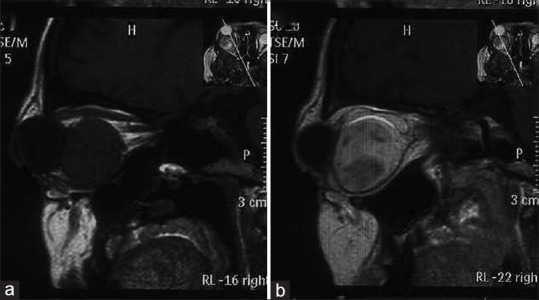
Magnetic resonance sagittal images shows a retro-orbital lesion of hypointense signal on T1-weighted (a) with postcontrast T1-weighted image (b) revealing heterogenous enhancement of the lesion
Clinically, a probable diagnosis of hemangioma was made.
The patient underwent right fronto-orbital craniotomy with unroofing of the orbit. Peroperatively a well-encapsulated, firm tumor mass was identified superoposteriorly near the orbital apex and was adherent with nerve root at one place. The tumor was removed completely under a microscope. Possibilities of neurofibroma and meningioma were kept.
Gross examination revealed a tan-colored tumor in multiple pieces measuring together 4 cm × 3 cm × 2 cm. Microscopically tumor revealed patternless fibroblastic proliferation with hypo- and hyper-cellular areas alternating with each other and bands of collagen fibers. Vascular channels with pericytomatous arrangement were also seen. There was no significant cellular atypia and mitosis was 1–2/10 high-power fields [Figure 3]. On immunohistochemistry, the tumor cells had strong and diffuse positivity to CD34, vimentin and bcl-2 but negativity for S-100, progesterone receptor, smooth-muscle actin (SMA), and cytokeratin [Figure 4]. With these findings, a final diagnosis of SFT was made. Postoperatively, proptosis was completely resolved with improved ocular movement.
Figure 3.
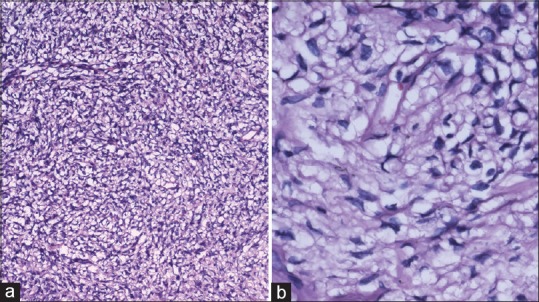
(a) Tumor revealing patternless fibroblastic proliferation and vascular channels with pericytomatous arrangement (H and E, ×100) (b) spindle cells growing in a haphazard manner in a variable cellular stroma described as “patternless pattern” (H and E, ×400)
Figure 4.
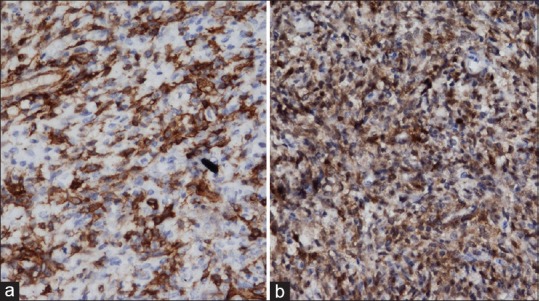
Tumor cells with strong and diffuse positivity to CD34 (a) but negativity for S-100 (b)
Discussion
SFT is an uncommon, benign, slowly progressive spindle cell tumor. Histogenesis of SFT is controversial. It was initially described on the mesothelial surface of pleura by Klemperer and Rabin in 1931.[1] Because of hypothesized origin of the tumor from mesenchymal cells with the fibroblastic origin, term “localized mesothelioma” was given.[4] Recently, this tumor has been described in extrapleural sites such as the upper airway tract, nasal and paranasal sinuses, parotid and salivary glands, thyroid, lung, mediastinum, pericardium, peritoneum, liver, spine, soft-tissue, and orbit.[4]
It was described by Stout and Murray in 1942 as a soft-tissue tumor characterized by cells surrounding thick-walled, branching staghorn blood vessels, hence, thought to be derived from pericytes, and the term “hemangiopericytoma” was introduced.[5] Regardless, it is accepted that the neoplastic cells are not derived from pericytes, and the term hemangiopericytoma is best thought of as a descriptive term rather than a diagnostic entity. The orbital involvement was first described independently by Dorfman et al. and Westra et al. in 1994.[2,3] About 60 cases of the orbital SFT have been reported in the literature till date.
Orbital SFTs can affect any orbital space including lacrimal gland fossa, lacrimal sacs, conjunctivae, and sclera. Age of presentation varies from 20 to 76 years, predominating in middle-aged adults, with no sex predilection. The most common clinical presentation is unilateral, slowly progressive, painless proptosis with well-defined, strongly enhancing mass lesion on CT and MRI. Occasionally visual disturbances, ocular motility restriction, palpable mass, or blepharoptosis can be found. Tumor invasion into adjacent bone or soft tissue is also uncharacteristic. Rarely, intracranial extension and invasion into sinus cavities can produce associated symptoms.
Most of SFTs behaves in a benign fashion, but a local invasion, recurrence, and distant metastases have been reported in pleural cases.[6] Only one case of malignant SFT of orbit has been reported so far.[7] Local recurrences usually follows an incomplete initial excision.
The classic histopathological feature of SFT is the presence of spindle cells growing in a haphazard manner in a variable cellular stroma described as “patternless pattern” or keloid-like hyalinization. Other features are thick bands of collagen interspersed between the tumor cells, alternating hypo- and hyper-cellular areas, branching thin walled vessels of varying caliber and “staghorn” vascular pattern, similar to hemangiomapericytoma.[2,3,8] These features are consistent with our findings.
A wide range of mesenchymal tumors occurs in the orbit. The differential diagnosis of orbital SFT includes fibrous histiocytoma (the most common mesenchymal orbital tumor in adults), rare fibroblastic tumors such as the juvenile fi juvenile tu, hemangiopericytoma, angiofibroma, nerve sheath tumors, meningioma, and synovial sarcoma. Previously, orbital SFTs have been misdiagnosed because of similar clinical presentation and microscopic picture. Recently advanced immunohistochemical techniques have facilitated a clear distinction.
Immunohistochemical studies show SFTs have strong and diffuse positivity to CD34, vimentin, bcl-2, and nonspecific reactivity to CD99. SFTs are negative to desmin, cytokeratin, factor VIII-related antigen, S-100, SMA, and muscle-specific actin.[3,9] Differentiation of SFT from hemangioperictyoma is mandatory because hemangiopericytoma has aggressive behavior with 83% recurrence rate, 27% distant metastasis, and 22% mortality rate, as against the usually benign nature of SFT.[10] Strong and consistent positivity to CD34 is an important diagnostic clue favoring SFT. Hemangiopericytoma shows inconsistent and weak positivity to CD34. In one of the largest studies by Furusato et al. (n = 41), CD34 positivity was seen in all cases, CD99 in 67.5%, and bcl-2 in 47.5% of the cases.[11]
Smooth muscle tumors, on the other hand, show positivity to desmin and actin, and negativity to bcl-2 and CD34. Neural tumors show focal positivity to bcl-2 and CD34, and strong positivity to S-100 protein.[9]
The mainstay of treatment of orbital SFTs is surgical resection with long-term follow-up. In general, orbital SFTs represent a benign disease, and most cases have been treated by local excision. The possibility for malignant transformation is closely associated with histologic features.[4] The findings, such as nuclear atypia, increased cellularity, necrosis, and greater than 4 mitoses/10 high-power fields, are suggestions of the malignant potential of SFT.[6] Various surgical approaches are available for resecting orbital tumors such as fronto-orbital and pterional approach; lateral orbitotomy; and medial orbitotomy. Lesions with intracranial extension, involving the optic canal, or medial to the optic nerve in the apex, are addressed through the transcranial fronto-orbital approach.[12]
Conclusion
The SFT of the orbit is very rare. It must be considered in the differential diagnosis of other spindle cell tumor of orbit. A clinical presentation such as painless proptosis and CD34 immunoreactivity plays an important role. The treatment is complete surgical excision with continuous postoperative follow-up as recurrence may appear long after excision.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Klemperer P, Rabin CB. Primary neoplasma of the pleura: A case report of 5 cases. Arch Pathol. 1931;11:385–412. [Google Scholar]
- 2.Dorfman DM, To K, Dickersin GR, Rosenberg AE, Pilch BZ. Solitary fibrous tumor of the orbit. Am J Surg Pathol. 1994;18:281–7. doi: 10.1097/00000478-199403000-00008. [DOI] [PubMed] [Google Scholar]
- 3.Westra WH, Gerald WL, Rosai J. Solitary fibrous tumor. Consistent CD34 immunoreactivity and occurrence in the orbit. Am J Surg Pathol. 1994;18:992–8. doi: 10.1097/00000478-199410000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Bernardini FP, de Conciliis C, Schneider S, Kersten RC, Kulwin DR. Solitary fibrous tumor of the orbit: Is it rare?. Report of a case series and review of the literature. Ophthalmology. 2003;110:1442–8. doi: 10.1016/S0161-6420(03)00459-7. [DOI] [PubMed] [Google Scholar]
- 5.Stout AP, Murray MR. Hemangiopericytoma: A vascular tumor featuring Zimmermann's pericytes. Ann Surg. 1942;116:26–33. doi: 10.1097/00000658-194207000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vallat-Decouvelaere AV, Dry SM, Fletcher CD. Atypical and malignant solitary fibrous tumors in extrathoracic locations: Evidence of their comparability to intra-thoracic tumors. Am J Surg Pathol. 1998;22:1501–11. doi: 10.1097/00000478-199812000-00007. [DOI] [PubMed] [Google Scholar]
- 7.Carrera M, Prat J, Quintana M. Malignant solitary fibrous tumour of the orbit: Report of a case with 8 years follow-up. Eye (Lond) 2001;15(Pt 1):102–4. doi: 10.1038/eye.2001.24. [DOI] [PubMed] [Google Scholar]
- 8.Mentzel T, Bainbridge TC, Katenkamp D. Solitary fibrous tumor: Clinicopathologic, immunohistochemical, and ultrastructural analysis of 12 cases arising in soft tissues, nasal cavity and naso-pharynx, urinary bladder and prostate. Virchows Arch. 1997;430:445–53. doi: 10.1007/s004280050054. [DOI] [PubMed] [Google Scholar]
- 9.Suster S, Fisher C, Moran CA. Expression of bcl-2 oncoprotein in benign and malignant spindle cell tumors of soft tissue, skin, serosal surfaces, and gastrointestinal tract. Am J Surg Pathol. 1998;22:863–72. doi: 10.1097/00000478-199807000-00008. [DOI] [PubMed] [Google Scholar]
- 10.Tihan T, Viglione M, Rosenblum MK, Olivi A, Burger PC. Solitary fibrous tumors in the central nervous system. A clinicopathologic review of 18 cases and comparison to meningeal hemangiopericytomas. Arch Pathol Lab Med. 2003;127:432–9. doi: 10.5858/2003-127-0432-SFTITC. [DOI] [PubMed] [Google Scholar]
- 11.Furusato E, Valenzuela IA, Fanburg-Smith JC, Auerbach A, Furusato B, Cameron JD, et al. Orbital solitary fibrous tumor: Encompassing terminology for hemangiopericytoma, giant cell angiofibroma, and fibrous histiocytoma of the orbit: Reappraisal of 41 cases. Hum Pathol. 2011;42:120–8. doi: 10.1016/j.humpath.2010.05.021. [DOI] [PubMed] [Google Scholar]
- 12.Bejjani GK, Cockerhan K. Diagnosis and management of orbital tumors. Contemp Neurosurg. 2003;25:1–8. [Google Scholar]