

Abstract
Viral infections result in a tremendous burden of neurologic illness worldwide. Although many viral etiologies of neurologic infectious disease have been recently controlled through vaccination and other public health measures, the emergence of new pathogens and the re-emergence of previously controlled pathogens remain significant global public health challenges. One of the fundamental bases for understanding the dynamics of viral nervous system infections is through epidemiology – the science of assessing patterns of disease in populations and the factors that influence these patterns. Neuroepidemiology is a discipline that applies epidemiologic principles and practices to neurologic disease. There are several characteristics of neurologic illness that necessitate the subdiscipline of neuroepidemiology. This chapter reviews some of the basic terms and concepts of neuroepidemiology, including epidemiologic analysis and studies. It then applies these concepts to infections of the nervous system, including encephalitis, aseptic meningitis, acute myelitis, prion diseases, and slow virus infections. The chapter then addresses the epidemiology of emerging and re-emerging viral neurologic diseases as well as the factors driving these emergences. The epidemiology of viral nervous system diseases will undoubtedly continue to change and expand, and as we gain a better understanding of this epidemiology and the pathophysiology of viral infections, this will hopefully lead to improved surveillance, diagnostic, treatment, and prevention strategies for these illnesses.
Keywords: Epidemiology, neuroepidemiology, viral infections, encephalitis, meningitis, myelitis, prion disease
Introduction
Viruses are among the most important causes of infectious neurologic illness worldwide. Recent years have seen the control and near-elimination of neurologic illness due to some viruses, through advancements in community hygiene and public health, societal awareness of preventive measures, and, importantly, vaccines against many viral pathogens. However, these advances have been countered by the emergence and re-emergence of new viral infectious agents; spread of these emerging infectious diseases has been facilitated by increasing global movement and transport of people, animals, and materials, changes in agricultural practices, war and natural disasters, and closer contact between humans and animals, among other things. Thus, viral neurologic infections will continue to be a significant source of ongoing morbidity and mortality worldwide for the foreseeable future.
A fundamental component of understanding viral infections and infectious diseases in general is to define the epidemiology of the pathogen, including how it is transmitted and spread, and how it behaves within a population. An understanding of infectious disease epidemiology places illness in perspective among the population, and aids in assessing the impact of the disease. This chapter will first provide an overview of important concepts of infectious disease epidemiology, particularly as it pertains to neurologic illness, followed by an overview of important epidemiologic principles of specific neurologic syndromes associated with viral infections.
A brief overview of neuroepidemiology
Epidemiology is the study of health and illness and associated factors in populations. Epidemiology is essentially a science that assesses patterns of disease occurrence in human populations and the factors that influence these patterns (Lilienfeld and Stolley, 1994); the field of neuroepidemiology is simply the application of these epidemiologic principles to neurologic disorders. Epidemiologists are primarily interested in the occurrence of diseases as categorized by time, place, and person. Epidemiology attempts to determine whether there has been an increase or a decrease of disease over time, whether one geographic area has a higher frequency of disease than another, and whether the characteristics of persons with a particular disease or condition distinguish them from those without it. The epidemiology of a disease is determined by the fundamental relationships among components of the epidemiologic triad – host, agent, and environment. Attributes of the host, such as age, gender, race, and occupation, often provide insight as to who is at risk for a disease and what exposures may contribute to that risk. Similarly, an understanding of an agent’s biologic, physical, or chemical properties may help focus attention on some of the most likely explanations for disease. Finally, an understanding of where things happened can help identify environmental determinants such as the physical, biologic, social, and economic factors that may have allowed or promoted interaction between host and agent.
There are several features of neurologic disorders that present particular challenges to the traditional epidemiologic methods necessitating the specialized discipline of neuroepidemiology. Neurologic illnesses often progress and change clinical characteristics over time; thus, the likelihood of an accurate diagnosis may be greater in persons with advanced illness, potentially leading to over- or underestimation of cases if evaluated early. Often, the actual timing of onset of a neurologic disease is not known, which may make determination of risk factors difficult. Persons with neurologic disorders may have altered mental status or memory deficits, making recall of past events and potential risk factors problematic. Finally, many neurologic illnesses are syndromes in which diagnosis rests upon an accurate interpretation of a constellation of physical and neurologic signs and symptoms, rather than diagnostic tests. This increases the potential risk of misclassification of cases.
Basic epidemiologic terms and concepts (Lilienfeld and Stolley, 1994)
The fundamental activity of the epidemiologist is counting – the fundamental method of determining the extent of a health event affecting a population is to count cases. It is very important to define what a “case” is, and a case definition must be created. As simple as it may appear, that is not always easy, particularly in the setting of an epidemiologic investigation of an unknown illness. Once the definition of a case is determined, the number of such cases must be assessed in light of the population from which they are obtained. Rates are measures for relating cases to the population; with rate information, it is possible to determine whether one group or another is at increased risk of disease, and by how much. The most commonly used measures of rates are incidence, attack rate, and prevalence.
Incidence or incidence rate measures the risk of new cases occurring in a population over a defined period of time. The numerator is the number of new cases, and the denominator is the population at risk during the specified time period:
An attack rate is the proportion of a defined population that develops a disease usually over the course of an outbreak. Attack rate is often expressed as a percentage.
Prevalence is the proportion of the population that has a disease at a given time. These measures are functions of both the disease incidence and disease duration:
Risk factors are attributes of the agent, host, and environment that increase the risk of a disease. Protective factors are attributes of the agent, host, and environment that decrease risk of the disease. Assessment of risk and protective factors involves the comparison of frequency of disease in a group exposed to those factors with disease frequency in the unexposed group. The relative risk (RR) is the ratio of risk of disease among persons in an exposed population to that in an unexposed population. RRs best reflect the effect of an exposure of interest when the groups being compared are similar in all variables being evaluated that might alter the RR except for the exposure of interest or when the RRs are adjusted to account for the known differences in these variables. An RR of 1.0 indicates identical risk in the compared groups; an RR greater than 1.0 indicates that those with the exposure have a higher risk than those without, and an RR less than 1.0 indicates the opposite – that those with the exposure have a lower risk than those without. It is important to differentiate the RR from the attributable risk (AR) – the absolute amount of risk directly due to exposure. AR is estimated from the difference between the risk of developing a disease in the exposed and unexposed groups. An AR of zero means that there is no difference in risk of disease between the exposed and unexposed groups.
Pragmatically, in epidemiologic studies it is often useful to compare risk factors by looking at odds. The concept of odds is slightly different from that of risk, and is used when the total number of persons who may have been exposed to a situation or agent is not actually known, as is frequently the case in epidemiologic investigations. The odds of an event occurring is the probability that the event will occur divided by the probability that the event will not occur. Odds compare the number of ill persons known to be exposed to a particular factor to the number of non-ill persons known to be exposed to that factor:
By knowing the odds, one can then calculate an odds ratio, which compares the odds in those exposed to the factor to the odds in those not exposed; the odds ratio is essentially an equivalent of an RR when the illness is rare in the population under study.
In the setting of an outbreak, it is useful to assess the number of cases occurring over time, which is generally graphed with a histogram (Gregg, 1996). If the time period of interest represents the duration of the outbreak, the histogram is referred to as an epidemiologic curve (Fig. 3.1 ). The number of cases is specified on the y axis, while the time intervals are represented on the x axis. Such epidemiologic curves may be constructed in ways that stratify case groups by certain variables, such as place (residence, employment) or by personal characteristics (age, gender, race). Incidence rates of disease over time, on the other hand, are generally plotted using a line graph, with the x axis representing the period of time of interest, and the y axis representing the incidence rate of the health event per unit population (usually per 1000 or per 100 000).
Fig. 3.1.
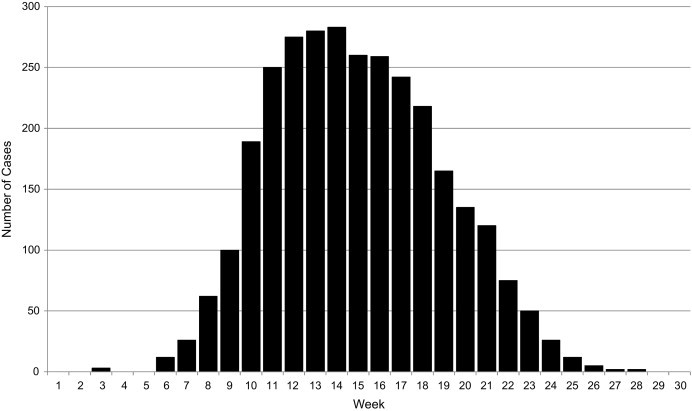
Example of an epidemiologic curve, demonstrating the pattern of a point-source outbreak with a single incubation period.
Epidemiologic studies (Lilienfeld and Stolley, 1994)
There are a number of different epidemiologic methods that are used to assess and characterize diseases. Disease surveillance is an ongoing process which involves the systematic collection, analysis, interpretation, and dissemination of reports on the occurrence of disease within a defined population; surveillance is a key component of public health activities (Teutsch and Churchill, 1994). Surveillance for disease can be conducted on a number of different levels, such as community-based surveillance, hospital-based surveillance, or national surveillance. Syndromic surveillance, which assesses the occurrence of constellations of clinical features, is often used. Surveillance for disease may be either active or passive. Active surveillance utilizes a regular, systematic method to proactively contact reporting sources, review medical records, or otherwise identify cases to assess the occurrence of disease. Passive surveillance relies on the individual clinician, laboratory, or hospital initiating the report of the illness. Unlike with active surveillance, the absence of reported cases in passive surveillance may reflect failures to report rather than absence of identified cases. Active surveillance tends to provide more complete, timely, and accurate data, but is often more resource-intensive. Passive surveillance is often less resource-dependent and easy to implement, but may be less sensitive or complete in case ascertainment.
The two most frequently used study approaches in epidemiologic analysis include cohort and case-control studies. Choice of these study designs is generally dictated by several factors, including the particular study question; the exposure frequency; the prevalence or incidence of the illness; and logistics. In a cohort study, subjects are classified on the basis of presence or absence of exposure to a particular factor, and then followed in time to assess the development of disease. Cohort studies may be either prospective (following subjects actively forward in time) or retrospective (where the outcome of interest has already occurred, and all of the follow-up has occurred in the past and all information is historical). Cohort studies are most useful to study common outcomes that may occur within short periods of time, or to study outcomes following a rare exposure. Cohort studies are useful in documenting the natural history of disease, and identifying particular risk factors for illnesses; population-based cohort studies may allow for determining incidence and prevalence of a disease.
The second main epidemiologic study approach is the case-control study. In case-control studies, subjects with the outcome of interest are identified, and the exposure history is compared with a group (controls) who do not have the outcome of interest. Case-control studies are most useful in studying diseases that are uncommon or have a long period between exposure and development of disease. Case-control studies may be subject to selection bias, since the association is being studied after exposure and disease have already occurred, and subjects are selected on the basis of having the outcome of interest. Case-control studies often involve matching, in which the cases and controls are made as similar as possible based upon various variables (age, sex, occupation) so as to highlight the particular risk factors that might result in disease in some and not in others. Matching of one case with more than one control (e.g., 1:2 matching, 1:4 matching) is often done to increase the power or ability to detect a statistically significant difference between cases and controls.
Another category of epidemiologic assessment is the outbreak investigation. This type of assessment is often employed by public health personnel and combines several different epidemiologic methods. This method is used to investigate apparent sudden increases in numbers of cases of illness over particular periods of time, and is often initiated rapidly. Outbreak investigations may involve several concurrent epidemiologic methods, including surveillance, case series, case-control, and cohort studies. Each of these may be conducted in parallel or sequentially, depending upon the situation. Laboratory testing of biologic or environmental specimens, for etiologic confirmation, is often a critical component of outbreak investigations. The ultimate goals of an outbreak investigation are to determine the cause / etiology, and implement control measures in order to control and prevent further disease (Reingold, 1998).
When interpreting results of epidemiologic studies, it is important to question whether an observed association between an exposure and outcome reflects a true cause-and-effect relationship, or if it is occurring by chance alone; this is generally determined by various statistical tests. The association can also be the result of bias or by confounding. Bias represents a systematic error in an epidemiologic study that results in an incorrect estimate of the association between exposure and risk of disease. There are several types of bias, including selection bias, in which subjects chosen for a study differ from those who would otherwise be eligible but were not included; and information bias, in which there is a systematic error in the measurement of data on exposure or outcome. For example, if we were to compare the incidence of transverse myelitis (TM) in the United States with that in certain African countries, we might find that TM is far more common in the United States. This may be reflective of a true higher incidence of TM in this setting; however, it may also reflect the fact that advanced diagnostic testing such as magnetic resonance imaging is more widely available in the United States, whereas in Africa the diagnosis is made by neurologic exam alone, which may be less sensitive.
Misclassification, a type of information bias, results when there is an inaccurate assessment of exposure and/or disease status. For example, if disease status is inaccurately determined, this can result in surreptitious findings. This is particularly true in various neurologic disorders, such as Alzheimer’s disease, Parkinson’s disease, or amyotrophic lateral sclerosis, in which illness can be present long before it is clinically apparent, diagnosis is based on a constellation of signs and symptoms, and the validity of a diagnosis may depend upon the clinical skill of the examiner.
Recall bias occurs when subjects with and without outcome of interest report exposures differently. Oftentimes, cases may recall particular events with more clarity than controls, if they feel that particular exposures may have been associated with their illness. In neurologic diseases, cognition is often impaired, there may be difficulty in assessing the validity of exposure history, or history may rely upon information provided by a proxy, who may not have all possible exposure information available.
Confounding occurs when an observed association is due, partially or totally, to the effects of differences between the cases and controls other than the exposure under assessment, which could affect their risk of developing the outcome of interest. It occurs when another factor is associated with both the exposure of interest and the risk of developing the disease and may account for some or all of the association between exposure and disease. Confounding may be reduced, again, by matching for variables that may be associated with exposure and outcome.
The cornerstone of all epidemiologic analyses is the 2 × 2 table (“two-by-two table”), which is often the first analysis performed when looking at epidemiologic data. The basic 2 × 2 table (for a case-control study) appears as such:
| Exposed | Not exposed | ||
|---|---|---|---|
| Cases | a | b | a + b |
| Controls | c | d | c + d |
| a + c | b + d | Total |
where:
a = number of ill people exposed to a factor
c = number of well people exposed to a factor
b = number of ill people not exposed to a factor
d = number of well people who were not exposed
a + b = total number of cases
c + d = total number of controls
a + c = total number who were exposed
b + d = total number who were not exposed
For instance, presume that a group of people at a party consume a particular appetizer; the following day, a number of them develop severe headaches. You would like to know if consuming the appetizer was associated with the headaches. We can try to calculate the odds of the appetizer being associated with the headaches in this group. Let’s say a total of 94 people attended the party; some ate the appetizer, and some did not:
| Ate appetizer | Did not eat appetizer | Total | |
|---|---|---|---|
| Headaches | 6 | 31 | 37 |
| No headaches | 9 | 48 | 57 |
| Total | 15 | 79 | 94 |
The odds of headaches in those eating the appetizer is 6/9 = 0.67, while the odds of headaches in those not eating is 31/48 = 0.65. Thus, the odds ratio for headaches following the appetizer is 0.67 / 0.65 = 1.03. Thus there would appear to be a nearly equal chance of developing a headache, whether the appetizer was consumed or not.
Additional statistical methods are needed to further assess significance, and to analyze more complex data sets; however, the 2 × 2 table is an effective and simple method of assessing basic epidemiologic data.
Epidemiology of viral infections of the nervous system
Mechanisms of viral-mediated neurologic disease
Viruses may lead to nervous system disorders in multiple ways (Johnson, 1998). They may directly infect and replicate within nerve cells, leading to neural inflammation, necrosis, and damage (“neuroinvasive disease”), as is the case with viral encephalitis and myelitis. They may cause infection and inflammation limited to the meninges, leading to aseptic meningitis. In some cases, viruses may serve as an antigenic stimulus to the immune system and produce an indirect, immune-mediated attack on the central or peripheral nervous system, as is the case in acute inflammatory demyelinating polyradiculoneuropathy or acute disseminated encephalomyelitis (Hughes et al., 1999, Young et al., 2008). In rare cases, viruses may produce latent or persistent infections within nerve cells, resulting in episodic recurrences of clinical illness (e.g., herpes zoster with varicella-zoster virus (VZV)) (Guess et al., 1986), or progressive neurodegenerative illness (subacute sclerosing panencephalitis (SSPE) following measles infection) (Gutierrez et al., 2010). Some viruses may be acquired congenitally and result in developmental neurologic illness (e.g., congenital cytomegalovirus and rubella neurodevelopmental disorders) (Bale and Murph, 1992, Griffith and Booss, 1994). Some viral infections may initiate a vasculitis, producing central nervous system (CNS) effects through brain infarcts or focal neurologic deficits (Nagel et al., 2008). In some cases, a combination of specific host factors and an infecting virus may produce indirect neurologic disease: this is the case for Reye syndrome that occurs shortly after salicylate treatment of children with various viral illnesses, particularly influenza and varicella-zoster infection (Glasgow and Middleton, 2001). Although not the case for viruses, some infectious agents elaborate neurotoxins that can result in neurologic disease, as is the case for tetanus and dipththeria toxins produced by the bacteria Clostridium tetani and Corynebacterium diphtheriae, respectively. Finally, other end-organ damage from a viral infection may produce a concomitant neurologic illness indirectly (e.g., encephalopathy in the setting of viral hepatitis-associated liver failure). Neurologic illness with any particular virus may be a result of one or several of these mechanisms. The epidemiology of viral-associated neurologic illness is in part influenced by which of the mechanisms above is associated with a particular virus.
Factors influencing the epidemiology of viral CNS disease
There are several key factors that influence the epidemiology of viral infections of the nervous system. Of critical importance is the host–agent relationship, which is dependent upon various features and characteristics of both the infecting agent, as well as the particular properties or characteristics of the person who is infected (host) (Mandell et al., 2005). Both of these factors influence the occurrence and pattern of human infections, and need to be considered in tandem. Various features of the infecting agent may influence the occurrence of disease (Mandell et al., 2005). Infectiousness describes the relative ease or difficulty by which an agent may be transmitted to other hosts; respiratory or droplet-spread agents tend to be more infectious than those spread by close person-to-person contact, for instance. Infectivity relates to the ability of an infectious agent to enter, survive, and multiply in a host, and is generally estimated by assessing the number of hosts infected in the context of the number of susceptible and exposed individuals. Pathogenicity is the ability of an agent to cause clinical disease in a host once infection occurs; although this is in large part related to specific characteristics of the infectious agent, host susceptibility may also play a large role in pathogenicity. Similarly, virulence refers to the relative severity of infection caused by a pathogenic agent, and refers to the numbers of serious or disability-producing infections compared to the total number of infected persons. Immunogenicity is the ability of an infecting agent to stimulate an immunologic response in the host; this in part determines the pathogenicity and virulence of a particular agent, and is an important determinant of whether infection confers long-lasting immunity to the agent. Nearly all of these agent factors are substantially influenced by concomitant host factors. For instance, the pathogenicity of a particular agent may be greatly increased in persons with particular risk factors, such as weakened immunity or extremes of age. The virulence of a particular agent may similarly be influenced by particular host risk factors.
Another important influential determinant of virus epidemiology is the route of transmission; this is the pathway by which an infectious agent is spread through the environment or to another person (Mandell et al., 2005). Direct-contact spread involves passing an infectious agent directly from one person to another by way of respiratory droplets, touching, kissing, or sexual intercourse, or by exposure of susceptible tissue to the agent, for instance through a rabid animal bite. Indirect transmission includes vehicle, vector-borne, and air-borne transmission. Vehicle transmission involves a material that serves as an intermediate by which an infectious agent can gain access to the host, for instance by food, water, biological tissues (blood, urine), or objects (fomites). Vector-borne infections are spread when an insect vector transmits an infectious agent mechanically, as in a bite, directly to the host. Air-borne transmission occurs when aerosols containing infectious agents disseminate to a host and gain entry to the host through some portal, generally the respiratory tract. Air-borne transmission requires infectious particles to be 1–5 μm in diameter, smaller than respiratory droplets (Polymenakou et al., 2008).
There are various epidemiologic patterns associated with viral neurologic infections. Some viral infections result in sporadic cases of neurologic disease, with no specific temporal or geographic pattern. Infections may also have an endemic pattern, in which cases of illness predictably occur among members of the population at a certain frequency. In some cases, infections may result in regular, predictable seasonal increases in numbers of cases, due to various geoclimactic or population factors. Others may result in epidemics, in which large numbers of cases of illness occur in a particular geographic area and over a specific period of time; these epidemics may occur on a predictable basis, but more commonly occur in an unpredictable fashion. An epidemic occurring within a large geographic area, typically affecting many countries in multiple continents, is referred to as a pandemic. Following a certain period of time, an infectious agent that initially resulted in epidemic disease may become endemic in a population (e.g., occurring regularly within the native population); this shift from epidemic to endemic disease has most recently been demonstrated by West Nile virus (WNV) in the United States.
Epidemiology of viral neurologic infections (Table 3.1)
Table 3.1.
Common causes, distribution, and epidemiology of viral neurologic infections
| Virus | Distribution, epidemiology | Clinical neurologic manifestations |
|---|---|---|
| Adenoviruses | Worldwide, sporadic | Uncommon cause of meningitis, encephalitis, anterior myelitis; sometimes associated with immunosuppression; has been associated with Reye syndrome |
| Arenaviruses | ||
| Lymphocytic choriomeningitis virus | Worldwide, sporadic | Uncommon cause of meningitis / encephalitis, mainly in immunosuppressed individuals |
| “New world”* | South America, sporadic with epidemics | Primarily results in hemorrhagic fever; may occasionally result in encephalitis |
| Cytomegalovirus | Worldwide, sporadic | Associated with several neurologic manifestations, including congenital neurodevelopmental disorders, meningitis, encephalitis, polyradiculitis; association with Guillain–Barré syndrome; illness associated with immunosuppression |
| Enteroviruses | Worldwide, endemic with occasional large epidemics | Multiple different serotypes; common causes of aseptic meningitis worldwide. Occasional cause of encephalitis, anterior myelitis, particularly in the setting of large epidemics |
| Poliovirus† | Africa, Asia, endemic with epidemics | Most common cause of anterior myelitis; eradicated in the western hemisphere, but continued epidemic disease in several African and Asian countries; may result in meningitis, encephalitis |
| Epstein–Barr virus | Worldwide, sporadic | Occasional cause of meningitis, encephalitis; association with Guillain–Barré syndrome, brachial plexopathy; associated with primary central nervous system lymphoma |
| Hendra virus | Australia, sporadic | Recently recognized cause of severe encephalitis in northern Australia |
| Herpes simplex virus (1 and 2) | Worldwide, sporadic | Most common cause of sporadic encephalitis with identified etiology worldwide; can cause aseptic meningitis, anterior myelitis, radiculomyelitis |
| Human herpesvirus-6 | Worldwide, sporadic | Occasional cause of encephalitis, particularly in immunosuppressed individuals |
| Human herpesvirus-7 | Worldwide, sporadic | Occasional cause of encephalitis, particularly in immunosuppressed individuals |
| Human immunodeficiency virus | Worldwide, currently largest epidemics in Africa, Asia | Wide range of neurologic manifestations; may include aseptic meningitis, subacute encephalitis, dementia, peripheral neuropathy, myelopathy |
| Human T-cell lymphotropic virus (HTLV) | Worldwide (higher incidence in Japan, warmer equatorial regions), sporadic | Neurological manifestations include tropical spastic paraparesis (TSP), chronic HTLV-1 myelitis, and HTLV-1-associated myelopathy (HAM) |
| Influenza (A, B) | Worldwide, epidemic | Primary illness is respiratory; may result in influenza-associated encephalopathy (uncommon); association with Reye syndrome |
| Japanese encephalitis virus | Asia, Pacific; endemic with large epidemics | Most common cause of encephalitis in Asia; may result in meningitis, encephalitis, anterior myelitis |
| JC virus | Worldwide, sporadic | Causative agent of progressive multifocal leukoencephalopathy (PML), resulting in progressive demyelinating syndrome; primarily seen in the setting of immunosuppression |
| Measles virus | Worldwide, endemic with epidemics | May uncommonly cause meningitis, encephalitis; persistent infection results in subacute sclerosing panencephalitis (SSPE) |
| Mumps virus | Worldwide, endemic with epidemics | May occasionally cause meningitis, less commonly, encephalitis |
| Nipah virus | South Asia, Pacific, epidemic | Epidemics of encephalitis; incidence of meningitis unknown; may result in relapsing neurologic disease |
| Rabies virus | Worldwide, endemic | Cause of severe, fatal encephalitis; rare in developed world, still common in developing world; less commonly, may produce a paralytic illness without encephalitis |
| Rotavirus | Worldwide, endemic | Uncommon cause of meningitis, encephalitis in children |
| Rubella virus | Worldwide, endemic with epidemics | May cause congenital neurologic disease; uncommon cause of meningitis, encephalitis; rare cases of progressive rubella panencephalitis |
| Tick-borne encephalitis virus | Europe, Asia, North America; endemic | Important cause of meningitis, encephalitis in central Europe; uncommon cause of illness in other endemic areas |
| Varicella-zoster virus | Worldwide, endemic | May cause meningitis, encephalitis, cerebellitis, anterior myelitis. Associated with granulomatous arteritis. Reactivation may produce ganglionitis, radiculitis (shingles), postherpetic neuralgia |
| West Nile virus | The Americas, Europe, Middle East, Africa, endemic with epidemics | Most common cause of epidemic encephalitis in North America; may result in meningitis, encephalitis, anterior myelitis |
| Other epidemiologically important arboviruses | ||
| Togaviruses | ||
| Eastern equine encephalitis virus | Americas, epidemics | Cause of sporadic cases of encephalitis with occasional geographically limited epidemics |
| Western equine encephalitis virus | Americas, epidemics | Currently rarely reported cause of sporadic and epidemic encephalitis in North America |
| Venezuelan equine encephalitis virus | Americas, epidemics | Cause of epidemic encephalitis in Central and South America |
| Chikungunya virus | Africa, Asia, Pacific, epidemics | Recent emergence and geographic spread; associated with febrile illness/arthritic features, but occasional association with encephalitis |
| Flaviviruses | ||
| St. Louis encephalitis virus | Americas, epidemics | Cause of seasonal sporadic cases of encephalitis with occasional large epidemics |
| Murray Valley encephalitis virus | Australia | Important cause of epidemic encephalitis in Australia |
| Bunyaviridae | ||
| La Crosse encephalitis virus | North America | Cause of seasonal sporadic cases of encephalitis in North America |
| Other California encephalitis serogroup viruses (excluding La Crosse virus) | North America, Europe, Asia | Cause of seasonal sporadic cases of encephalitis; occasional geographically limited epidemics |
Terminology:
Sporadic: Resulting in occasional cases of illness on an irregular and unpredictable pattern.
Endemic: Prevalent or persistently present in population, regularly causing illness.
Epidemic: Affecting large numbers of persons in a geographic area over a particular time period, above expected rates.
Includes Junin, Machupo, Guanarito, Sabia viruses.
Poliovirus included as a separate subset of enterovirus.
Encephalitis
Encephalitis is defined as an acute or subacute cerebral inflammatory condition (Sejvar et al., 2011). Clinically, it is characterized by the acute onset of headache, encephalopathy or alteration of mental status, focal neurologic signs, and seizures. Cerebrospinal fluid (CSF) is characterized by moderate pleocytosis and protein elevation. There are many different etiologies that may lead to encephalitis, including toxic agents, neoplastic causes, and immune-mediated disorders; however, the most commonly identified causes of encephalitis are infectious agents, particularly viruses. In many cases, however, a definitive underlying etiology for encephalitis can be elusive; even with extensive testing for infectious or chemical etiologies, up to 60% of cases of encephalitis remain without a definitive underlying etiology (Glaser et al., 2003, Granerod et al., 2010). This likely attests to both the wide range of potential infectious pathogens, toxic substances, and environmental factors that may result in encephalitis, as well as limitations on the sensitivity and repertoire of laboratory testing currently available to identify potential etiologies.
The epidemiology of encephalitis is complex, and estimates of incidence rates vary widely. This is due in part to differences in case definitions between studies, differences in diagnostic capabilities, and geographic location in which studies have been conducted. Most studies of encephalitis have been hospital-based, and assess endemic disease. Estimated overall rates among all ages have been between 1 and 6 cases / 100 000 persons per year in most hospital-based studies (Klemola et al., 1965, Ponka and Pettersson, 1982, Radhakrishnan et al., 1987); one population-based study estimated an incidence of 7.4 cases / 100 000 persons per year (Nicolosi et al., 1986). Most studies suggest that encephalitis is more frequently observed in younger persons; in children and adolescents, incidence estimates range from 1 to 16 cases per 100 000 per year in children (Rantakallio et al., 1986, Koskiniemi et al., 1991). It is important to keep in mind that most studies of the incidence and epidemiology of encephalitis have been conducted in industrialized countries, and that the epidemiology of encephalitis in the developing world is less well described. Case fatality rates and estimates of neurologic sequelae also vary, depending upon study methods and underlying etiologic agent. Population- and hospital-based estimates of neurologic sequelae following encephalitis have ranged from 0.35 to 2.7 per 100 000 population (Koskiniemi et al., 1991, Rantala et al., 1991, Sejvar et al., 2008a), with case fatality rates ranging from 1% to 10% (Rautonen et al., 1991). However, certain agents are associated with higher case fatality rates; mortality of up to 33% has been reported with herpes simplex encephalitis (HSE) (Skoldenberg and Forsgren, 1985).
The etiology and epidemiology of viral encephalitis will vary based upon a number of variables, including age group affected, geographic location, and underlying immune status of affected individuals. Some viruses lead to sporadic cases of encephalitis. This is particularly true in the case of alpha-herpesviruses, which include herpes simplex virus-1 (HSV-1), HSV-2, and VZV. Herpesviruses are double-stranded DNA viruses and are ubiquitous throughout the world (Skoldenberg, 1996, Hjalmarsson et al., 2007, Steiner et al., 2007). HSV-1 is the most common cause of identified sporadic viral encephalitis worldwide, and in the United States accounts for 20% of all cases with a defined etiology, with an incidence of approximately 2 cases per million population (Steiner et al., 2007). HSV-2 more typically produces genital cutaneous lesions, but may also cause encephalitis, accounting for approximately 5% of HSE overall (Johnson, 1998). Less commonly, it may result in aseptic meningitis. HSE occurs worldwide and throughout the year, with no seasonal variation. Spread of the virus is through direct close contact, and HSV-1 infection is influenced by socioeconomic factors, with lower socioeconomic status associated with earlier and more frequent infection (Skoldenberg, 1996). The age distribution of HSE appears to be bimodal, with peaks at ages 5–30 years, and then over 50 years. Currently, specific risk factors for the development of HSE are not known.
VZV is the third member of the alpha-herpesvirus group. Primary infection with VZV results in a febrile rash illness with a vesicular exanthema (“chickenpox” or varicella). Susceptible individuals become infected through respiratory exposure, and VZV is highly infectious, with attack rates among susceptible household contacts of over 80%. Although predominantly a febrile rash illness, neurologic disease with varicella is not uncommon, with the overall incidence of CNS disease estimated to be 1–3/10 000 clinical cases (Guess et al., 1986). The most commonly reported CNS manifestations are acute encephalitis and cerebellar ataxia. Encephalitis may occur in approximately 1–2 per 10 000 cases of clinical varicella (Choo et al., 1995); although most total varicella cases occur in children, incidence of neurologic disease associated with varicella illness is highest in adults over age 20, and infants < 1 year (Guess et al., 1986). Cerebellar ataxia is somewhat more common, affecting approximately 1 in 4000 cases (Guess et al., 1986). Case fatality from varicella encephalitis varies considerably, ranging from 5% to 35%, and long-term sequelae may be seen in 10–20% of survivors (Guess et al., 1986).
More commonly, viral encephalitis occurs in epidemics, in which a large number of cases occur in a geographically and temporally clustered fashion. Enteroviral infections, while more commonly associated with aseptic meningitis (see below), may result in seasonal epidemics of viral encephalitis in temperate areas. Enteroviruses have a worldwide distribution, and may produce endemic viral encephalitis, or large outbreaks of encephalitis affecting large populations. Recently, large outbreaks of enterovirus-71-associated illness have occurred and resulted in large numbers of cases of severe neurologic illness, primarily in children (Solomon et al., 2010). Arthropod-borne viruses (arboviruses) are among the most common causes of epidemic encephalitis worldwide (Gubler, 2001). Arboviruses refer to a class of viruses that are transmitted by arthropod vectors, generally mosquitoes or ticks (Calisher, 1994, Gubler, 2001). They all have complex life cycles involving enzootic transmission between vertebrate and invertebrate hosts, which allows for the virus to become increasingly prevalent in the environment (amplification). Humans become infected following the bite of an infected arthropod vector. While most human infections with arboviruses result in clinically silent illness or mild febrile illness, more severe manifestations, including hemorrhagic fever, aseptic meningitis, and encephalitis, may result.
There are over 20 arboviruses recognized to cause encephalitis, constituting several different viral families (Gubler, 2002). Most arboviruses produce endemic illness with periodic large outbreaks; these outbreaks are dependent upon a complex interaction of geoclimactic factors that favor increased populations of arthropod vectors, sufficient populations of amplifying hosts, and susceptible human populations. For instance, St. Louis encephalitis virus, a mosquito-borne flavivirus found in North America and, to a limited extent, South America, results in sporadic cases of encephalitis yearly, predominantly in late summer or fall. However, periodic focal outbreaks involving hundreds of cases have occurred at intervals in various regions of the United States (Luby et al., 1969, Zweighaft et al., 1979). Japanese encephalitis virus (JEV), another flavivirus transmitted by mosquito vectors, is the most important cause of encephalitis throughout Asia (Misra and Kalita, 2010). In areas with endemic transmission, incidence of Japanese encephalitis in children aged < 15 years is approximately 2.5 / 100 000 population, with case fatality estimated at 25% (Solomon et al., 2000). However, in temperate areas, Japanese encephalitis may result in frequent and large epidemics, with attack rates of up to 20 cases per 100 000.
The emergence of a pathogen into a new setting, by importation or geographic spread, may initially result in large epidemics; over time, the pathogen may then become endemic in this new region. An example of this shift from epidemic to endemic disease has been the epidemiologic pattern of WNV in North America. WNV, an arbovirus of the flavivirus family, was first identified in North America in 1999, and by 2002, resulted in an explosive epidemic of encephalitis covering much of continental United States and parts of Canada (Granwehr et al., 2004, Kramer et al., 2007). By 2006, however, case counts declined, and in subsequent years, WNV appears to have become endemic in various parts of the United States (Centers for Disease Control and Prevention). The possibility of subsequent large outbreaks, however, cannot be excluded.
Aseptic meningitis
Aseptic meningitis is commonly defined as a syndrome consisting of acute onset of meningeal signs and symptoms, CSF pleocytosis, and absence of microorganisms on Gram stain or culture (Tapiainen et al., 2007). Clinically, aseptic meningitis presents with the abrupt onset of fever, headache, and meningeal signs, including nuchal rigidity, photo- or phonophobia, and nausea/vomiting. CSF is characterized by a moderate pleocytosis and elevation in protein, but negative bacterial studies. It is characterized by a relatively benign clinical course, and the absence of features of encephalitis or myelitis. Like encephalitis, there are numerous etiologies of aseptic meningitis, including toxic, immune-mediated, neoplastic, and chemical causes, as well as a host of infectious pathogens. However, the most common causes of aseptic meningitis are viral etiologies (Tapiainen et al., 2007).
The various epidemiologic features described for encephalitis are largely true for aseptic meningitis as well. Despite advanced testing, a large proportion of cases remain without a definitive identified etiology. Most cases are sporadic or endemic, which may or may not have a regular expected seasonal fluctuation, with some pathogens associated with epidemic disease.
Although there are a number of different viral etiologies of aseptic meningitis, the most important cause worldwide of aseptic meningitis are the enteroviruses, with non-poliovirus enteroviruses accounting for up to 90% of all cases of aseptic meningitis in which an etiologic agent is detected (Tapiainen et al., 2007). Enteroviruses are made up of over 70 different serotypes within the family Picornaviridae, and many of these different serotypes may result in aseptic meningitis. Enteroviruses are distributed worldwide, and the epidemiologic pattern varies with geographic location. In temperate areas, enteroviral infections occur with a distinct summer / fall seasonal distribution; in tropical and subtropical areas, there is a higher year-round incidence of infection. Enteroviruses are spread from person to person by a fecal–oral route; transmission may also occur through the respiratory route, usually through direct contact with nose and throat discharges or through aerosol droplets or through contaminated food or water (Lee and Davies, 2007). Humans are thought to be the only natural reservoir of enteroviruses (Mandell et al., 2005). While there are many different serotypes of enteroviruses that may result in aseptic meningitis, in any particular geographic location infection is generally dominated by only a few of these serotypes (Glass et al., 2001). It appears that certain enterovirus serotypes are more commonly associated with aseptic meningitis.
Population-based estimates in the United States have suggested an overall incidence of aseptic meningitis of 10 / 100 000 persons among all ages (Beghi et al., 1984). Children are most likely to develop aseptic meningitis; estimates in Finland suggest an annual incidence of viral meningitis of 219 / 100 000 population in children less than 1 year of age (Rantakallio et al., 1986); this incidence dropped to 19 / 100 000 population in children between 1 and 4 years, and continued to decline with increasing age. Similar findings suggesting higher childhood incidence have been obtained by other studies (Khetsuriani et al., 2003, Khetsuriani et al., 2006, Lee et al., 2005, Lee and Davies, 2007).
Some viruses more commonly associated with more severe CNS infection may also result in endemic or epidemic aseptic meningitis. Arboviruses, in addition to causing endemic and epidemic encephalitis, may cause aseptic meningitis, particularly in older persons (Calisher, 1994). Herpesviruses, while more commonly associated with encephalitis, may result in aseptic meningitis as well (Skoldenberg, 1996).
Anterior myelitis
Anterior myelitis is the most common cause of acute flaccid paralysis worldwide. It is a syndrome characterized by acute, areflexic limb weakness or paralysis due to viral damage to the anterior horn cells or lower motor neurons of the spinal cord. Prior to the advent of vaccination, far and away the most common cause of anterior myelitis was infection with poliovirus, a small enterovirus of the family Picornaviridae (Mandell et al., 2005). Poliovirus has historically been so strongly associated with the syndrome of anterior myelitis that the term “poliomyelitis” has become synonymous with the syndrome. There are, however, a number of other viruses that are important causes of anterior myelitis, including some other enteroviruses, arboviruses, and herpesviruses (Solomon and Willison, 2003).
The epidemiology of poliovirus anterior myelitis has undergone significant changes in the past decades. The syndrome has been recognized for centuries; however, detailed descriptions of the clinical illness did not begin to appear until the 1700 s. Until the late 19th century, poliomyelitis was only associated with sporadic cases of acute limb weakness (Nathanson and Kew, 2010). However, beginning in the late 1800 s, large summertime outbreaks of poliomyelitis began occurring in northern Europe and the United States (Mandell et al., 2005). This shift from sporadic disease to large epidemics has largely been attributed to improved hygiene in these areas, which resulted in lack of exposure to the virus in infancy. Exposure in early childhood generally produced mild infections due to the presence of passively acquired maternal antibody; these mild infections, however, resulted in the development of inherent immunity, affording ongoing protection. Infections later in life result in more severe neurotropic disease, thus leading to increasing burden from poliovirus infections (Nathanson and Kew, 2010).
Following the recognition in the early 1900s that the cause of poliomyelitis was a small enterovirus, and that there were three different serotypes of poliovirus, successful production of both an inactivated (killed) polio vaccine and a live attenuated oral poliovirus vaccine was possible. Development and implementation of these effective poliovirus vaccines had a dramatic effect on epidemic poliomyelitis. In the United States, attack rates of poliomyelitis decreased from over 17 cases per 100 000 population in 1955 to 0.4 cases per 100 000 population in 1962 (Nathanson and Kew, 2010). By 1994, the World Health Organization declared the Americas as “polio free” (Fig. 3.2 ) (Alexander et al., 2004).
Fig. 3.2.
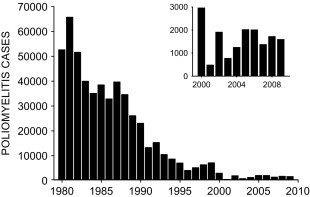
Global incidence of poliomyelitis, reported as virologically confirmed cases of paralytic poliomyelitis, from 1980 to 2009. Cases for 2000–2009 have been replotted in inset to demonstrate recent increase.
(Reproduced from Nathanson and Kew (2010), with permission.)
Even after the introduction of polio vaccine, poliomyelitis was widely regarded as an epidemic disease of the developed world, and largely ignored in developing countries. However, acute flaccid paralysis surveillance during the 1960s and 1970s suggested that the prevalence of limb paralysis, mostly attributable to poliovirus infection, in populations of the developing world was between 2 and 11/1000 population (Henderson, 1984), a rate exceeding that of poliomyelitis in peak epidemic years in the United States. Most cases of poliomyelitis in developing countries occur in young children between 6 months and 2 years. Because poliomyelitis in developing countries is largely endemic rather than epidemic, and there is a high incidence of other causes of severe physical disabilities in these areas, poliomyelitis had received less public concern. However, beginning in 1974, the World Health Organization began making concerted efforts toward the worldwide control of vaccine-preventable infectious diseases, including poliomyelitis. By 2002, fewer than 1900 cases of poliomyelitis were reported worldwide, strictly among a handful of countries in sub-Saharan Africa, the Middle East, and south Asia (Centers for Disease Control and Prevention, 2009a). However, in recent years, there have been several setbacks in this goal, with several large outbreaks of poliomyelitis occurring in Tajikistan, the Republic of Congo, and elsewhere. Such setbacks have been due to a variety of problems, including a less developed infrastructure in developing countries contributing to limitations in both the effectiveness of the oral poliovirus vaccines themselves (e.g., more potentially interfering enteric viruses in recipients) and to decreased capacity to implement the multiple vaccination campaigns required to maintain high population levels of immunity (Centers for Disease Control and Prevention, 2009b). New strategies are not being introduced to reach the elusive goal of poliomyelitis eradication.
As poliovirus poliomyelitis has been controlled in much of the developed and developing world, however, other causes of infectious myelitis have become increasingly recognized. Other non-poliovirus enteroviruses, in particular coxsackie A and B viruses (Santhanam and Choudhury, 1985, Jiang et al., 2007, Dhole et al., 2009), have been associated with sporadic cases of pediatric and adult anterior myelitis. Cases of anterior myelitis have occurred in several notable large outbreaks of enterovirus-71 in Malaysia, Taiwan, and other Asian countries (Solomon et al., 2010), although the usual neurologic complication of this virus is acute encephalitis. Arthropod-borne flaviviruses have recently emerged as an important cause of infectious myelitis, with large outbreaks of Japanese encephalitis associated with cases of myelitis in endemic and epidemic regions (Solomon and Vaughn, 2002, Solomon and Willison, 2003); during the North American WNV epidemic, incidence of WNV myelitis was estimated to be up to 3.7 / 100 000 population (Sejvar et al., 2005). Herpesviruses, particularly VZV and HSV-1, have been associated with sporadic cases of myelitis worldwide (Guess et al., 1986). The incidence of these other causes of infectious myelitis does not approach those historically seen for poliovirus infection, however, and fortunately the ongoing control of poliovirus infection through vaccination has resulted in a continued dramatic decline in infectious myelitis worldwide.
Myelopathy
In addition to anterior myelitis, several viruses may be associated with a more diffuse myelopathy or myeloneuropathy. This is particularly true for several retroviruses, including human T-cell lymphotrophic virus (HTLV), types I and II, and human immunodeficiency virus (HIV). HTLV-I infection is associated with adult T-cell leukemia; however, it also may cause tropical spastic paraparesis (TSP) and HTLV-I-associated myelopathy (HAM) (Araujo and Silva, 2006, Cooper et al., 2009). HTLV-I has a worldwide distribution, but has focal concentrations in certain populations; seroprevalence for the virus is particularly high in certain districts in Japan and in the Caribbean (Araujo and Silva, 2006). The virus is transmitted primarily through sexual contact, contaminated blood, or perinatally through breast milk. Onset of TSP-HAM is most frequent in the fourth and fifth decades, although younger ages of onset may occur, and it is more common in females. The onset of illness is slow and progressive, with the development of backache, leg stiffness, and dysesthesias, as well as weakness, spasticity, hyperreflexia, and extensor plantar responses. Upper extremities are generally spared. Sensory loss, particularly to vibration and position sense, may occur, and in some cases a concomitant peripheral neuropathy may develop (Grindstaff and Gruener, 2005). The illness is generally progressive, leading to gait difficulties. An illness resembling TSP-HAM has been observed with infection with HTLV-II, which appears to be particularly prevalent among Native Americans (Roucoux and Murphy, 2004). Among the various neurologic manifestations that it may cause, HIV can be associated with a progressive vacuolar myelopathy (Berger and Sabet, 2002). Herpesviruses and enteroviruses can also uncommonly be associated with myelopathy (Berger and Sabet, 2002).
Chronic viral CNS infections/prion disease
A group of CNS viral infections have been recognized as having unusual characteristics for infectious diseases (Asher, 1997). These include long, asymptomatic incubation periods in the range of years or decades in humans, long durations of overt clinical illness, and clinical manifestation as neurodegenerative conditions. Sometimes referred to as “slow viruses,” these disorders may be caused by conventional viruses – those more commonly associated with acute neurologic infectious illness. Others are caused by unconventional transmissible agents, in which the mechanism of pathology is less clear.
Subacute sclerosing panencephalitis
SSPE is a syndrome caused by persistent CNS infection with measles virus (Johnson, 1998). It is thought that various mutations in the infecting measles virus allow for establishment of a persistent infective state within the nervous system; various pathophysiologic mechanisms for this persistent infection have been hypothesized, but none has been proven. SSPE primarily affects children and adolescents, with more than 85% of cases being between 5 and 15 years of age (Gutierrez et al., 2010). Typically there is a history of measles rash illness with uneventful recovery; several years later, affected persons develop insidious onset of behavioral changes, neurocognitive impairment, and movement disorders followed by frank dementia; signs of acute encephalitis are not present. The illness is generally fairly rapidly progressive, with death occurring between several months and 3 years after onset, generally due to secondary complications.
Epidemiologic studies of SSPE suggest that males are twice as likely to develop the syndrome as females; cases are more common in children from rural areas than from urban settings (Modlin et al., 1979). There has been some evidence for geographic clustering of cases of SSPE, with incidence in the United States being highest in southeastern and midwestern states (Modlin et al., 1979). In the United States, the incidence of SSPE has decreased dramatically since the 1960s: the mean annual incidence of SSPE in 1960 was 0.61 per million persons under age 20, compared with 0.06 cases per million in 1980 (Modlin et al., 1977, Modlin et al., 1979). This decrease in SSPE correlates with the use of live attenuated measles vaccine starting in 1963. Despite concerns that the attenuated measles vaccine may itself represent a risk for development of SSPE, the overall risk for SSPE following measles infection has been estimated to be 29 times greater than that following measles vaccination (Halsey et al., 1980). Earlier measles infection appears to carry a greater risk, with the risk of SSPE following measles infection before age 18 months greater than infection at older ages. Despite a dramatic decline in cases of SSPE in the developed world, SSPE continues to occur in the developing world and areas where measles vaccine is not widely used (Bellini et al., 2005, Campbell et al., 2007).
Progressive multifocal leukoencephalopathy (PML)
PML is a rare, progressive demyelinating neurologic disorder due to persistent infection with JC virus (JCV), a human polyomavirus (Scheld et al., 1997). Infection with JCV appears to be extremely common, with infection occurring in childhood, but in most individuals is asymptomatic. Estimates have suggested that nearly 80% of adults in the United States and Europe have antibodies to JCV, with seropositivity increasing during childhood. The overall pathogenesis of JCV and how it leads to PML is not understood, but it is hypothesized that viremia during primary infection results in persistent latent infection of the kidneys and, possibly, mononuclear cells (Mandell et al., 2005). During periods of immunosuppression, it is thought that latent virus within kidney cells reactivates. Clinically, PML most often presents with progressive focal neurologic deficits, including hemiparesis, visual field deficits, and ataxia, as well as neurocognitive impairment. Most involvement is within the cerebral white matter, with progressive patchy or confluent demyelination. Patients generally undergo rapid deterioration, with death occurring within 6 months of onset. Definitive diagnosis of PML is based upon detection of JCV in brain tissue or CSF by polymerase chain reaction or virus isolation.
Prior to the emergence of HIV, PML was uncommon, occurring mostly in older individuals with underlying hematologic malignancies or other severe immunosuppression. In the HIV era, the epidemiology of PML has undergone substantial and interesting changes. Subsequent to the emergence of HIV, reported PML deaths rose considerably; the age-adjusted death rate from PML in the United States increased 20-fold, from 0.2 per million persons in 1984 to 3.3 per million persons in 1994 (Holman et al., 1998). PML-associated death rates peaked in the mid-1990s, however, and subsequently there has been a decrease in PML-associated deaths, from 2.76 deaths per million persons in 1992–1995 to 0.66 in 2002–2005 (Christensen et al., 2010). This decrease has corresponded to the widespread use of highly active antiretroviral treatment (HAART) in the treatment of HIV/acquired immunodeficiency syndrome (AIDS) in the United States (Clifford et al., 1999, Christensen et al., 2010).
Similar findings were obtained from a Swiss cohort study, which found a decrease in incidence rate of PML from 0.24 per 100 person-years from 1993 to 1995 to 0.06 per 100 person-years from 1996 onward (Khanna et al., 2009). HAART also appears to have improved survival among persons with HIV-associated PML, with mortality decreasing to approximately 50% (Clifford et al., 1999). More recently, reports of PML associated with the use of immune-modulating monoclonal antibodies such as natalizumab to treat multiple sclerosis and Chron’s disease have suggested that the use of these medications may be a contributing risk factor (Clifford et al., 2010). Currently, it is estimated that more than half of deaths attributable to PML are still associated with HIV infection, however.
Creutzfeld–Jakob disease (CJD) and other prionopathies
The prionopathies constitute an unusual group of syndromes caused by novel infectious agents that result in rapidly progressive neurologic deterioration and ultimately death. The causative agent of these illnesses is thought to be a transmissible proteinaceous particle devoid of nucleic acid (a misfolded prion protein, PrPsc) (Belay, 1999, Johnson, 2005). The normal form of the prion protein is a normal cellular protein with an unclear function (cellular PrP, PrPc). A transformation of the normal prion protein into an abnormal configuration (PrPsc, see below) changes the physicochemical properties of the protein; the resultant aberrant protein is extremely resistant to typical methods of denaturation such as heat, chemical, or radiation exposure. This form of the protein has the capacity for self-replication; transformation of PrPc to PrPsc may occur sporadically but rarely, in genetically normal individuals, or the transformation may be facilitated and occur commonly in persons with certain genetic mutations that alter the amino acid sequence of the prion protein. It is unclear how PrP replicates, but it has been proposed that the combination of PrPsc with a PrPc molecule results in the formation of a heterodimer intermediate that subsequently forms two PrPsc molecules; this becomes a self-perpetuating process in which exponentially increasing numbers of PrPc are converted to abnormal PrPsc (Hu et al., 2008). The accumulation of PrPsc in nervous system tissue results in neuronal damage characterized by vacuolar changes, resulting in a spongiform appearance of neural parenchyma at histopathology, and giving the prionopathies the nomenclature of “transmissible spongiform encephalopathies” (TSEs) (Belay, 1999).
Prionopathies may affect both animals and humans. The prototypical animal prionopathy is scrapie, a TSE affecting sheep and goats, from which the term PrPscrapie (PrPsc) is derived. Other animal TSEs include transmissible mink encephalopathy, chronic wasting disease in North American elk and deer, and bovine spongiform encephalopathy (BSE) in cattle (Sejvar et al., 2008b). Human prionopathies include kuru, fatal familial insomnia, Gerstman–Straussler–Scheinker syndrome, and CJD (Brown and Mastrianni, 2010).
The epidemiology of CJD is the best understood of the human prionopathies. First described in the early 1920s, there are several subtypes of CJD. In all subtypes, CJD generally begins clinically with cognitive deterioration, including dementia, behavioral problems, and other cortical signs (Johnson, 2005). During the course of disease, additional neurologic signs will develop, including myoclonus, ataxia, and extrapyramidal features. Cognitive decline is progressive, and although longer survival times have been reported, most patients die within 6–9 months of diagnosis. The most common form of CJD is sporadic CJD (sCJD), in which by definition there is neither a mutation in the prion protein nor a clear environmental source of infection. sCJD is uncommon, and occurs worldwide, with an incidence of about 1 per million population per year in North America and Europe (Brown and Mastrianni, 2010). The incidence may be slightly less in Asia (Shi et al., 2008, Lu et al., 2010, Nozaki et al., 2010), and little is known about the epidemiology of sCJD in sub-Saharan Africa. sCJD is an illness of advanced age, with a mean age at onset of approximately 62 years; the age-adjusted incidence of sCJD in persons 65 years and older is approximately 5 per million per year. There is no gender difference; in the United States, the age-adjusted incidence of sCJD is 2.7 times higher in whites than blacks (Holman et al., 2010). A number of epidemiologic studies have been conducted to assess for possible risk factors for sCJD; however, aside from age and a family history of CJD, no consistent risk factors have been identified (Belay and Schonberger, 2005, Ward et al., 2008, Ruegger et al., 2009).
Familial CJD (fCJD) results from genetic mutations in the gene encoding the PrP (PRNP gene), and accounts for approximately 10% of CJD cases overall. At least 30 different mutations in the PRNP gene are associated with inherited prion diseases, including fCJD; the most common of these mutations (E200K) has been associated with geographic clusters of fCJD in various regions, including Slovakia, Chile, and among Jews in Greece and Israel (Johnson, 2005). fCJD is frequently associated with a younger age at onset. Due to its transmissible nature, various cases of iatrogenic CJD have occurred, through various mechanisms, including contaminated neurosurgical instruments, dura mater grafts, corneal transplant, and human pituitary hormone administration (Hamaguchi et al., 2009a). Transmission through neurosurgical instruments has been based upon the high infectivity of neural tissue with PrPsc, and the resistance of PrPsc to typical decontamination and sterilization methods that would typically be used on such instruments (Hamaguchi et al., 2009b). However, despite the millions of neurosurgeries performed each year, there have been only a handful of documented cases of CJD being associated with contaminated neurosurgical instruments or implanted electroencephalogram electrodes.
Beginning in 1995, several cases of CJD with atypical features were noted in the United Kingdom; these cases had illness onset at a much younger age (mean age at onset of 26 years), a longer duration of illness before death, and atypical initial symptoms, including psychiatric manifestations and prominent sensory disturbances (Collinge and Rossor, 1996, Will et al., 1996). Early on, a link between this “new variant” CJD (now variant CJD, vCJD) and BSE in cattle was suspected, based upon several lines of epidemiologic and pathologic data, and suggested transmission of the BSE prion agent from cattle to humans (Collinge, 1997). The BSE epidemic began in 1985, and peaked during 1992–1993, with a decline following the prohibition of using ruminant protein, which presumably contained BSE-infected brain and spinal cord, as feed for other ruminants.
The identification of the first cases of vCJD in 1995 would roughly correspond to what is understood about the incubation period for prions. The neuropathologic features of vCJD are quite different from those of sCJD, and the PrPsc of vCJD has a similar mobility pattern on electrophoresis to that of BSE PrPsc (Bradley, 2002). These data, along with experimental animal inoculation studies, and the epidemiology of vCJD in relation to BSE, suggest that vCJD was the result of human infection with the BSE agent, with the suspected route of transmission being the human consumption of meat or meat products contaminated with neural tissue harboring the BSE agent. Presumably, exportation of either contaminated feed or contaminated beef products prior to implementation of the feed ban allowed for spread of the BSE agent outside the United Kingdom.
As of January 2011, there had been 219 cases of definite or probable vCJD reported worldwide from 13 countries, with the vast majority occurring within the United Kingdom (University of Edinburgh, 2011). As opposed to sCJD, there is evidence that the vCJD agent may be transmitted from person to person through blood or blood products (MacGregor and Prowse, 2004); this fact has led to restrictions on blood donation from persons spending specific periods of time in the United Kingdom. To date, the vCJD epidemic appears to have peaked in 1999, with a steady decline in cases since then; the future epidemiology of vCJD remains unknown at present, however.
Epidemiology of emerging viral neurologic diseases
By the middle of the 20th century, the impact of infectious diseases on human health was widely viewed to be in its final phases. The infectious disease pioneer Sir McFarland Burnett wrote, in 1962, “One can think of the middle of the 20th century as the end of one of the most important social revolutions in history – the virtual elimination of the infectious disease as a significant factor in social life” (Davis and Lederberg, 2001). This view had been bolstered by recent successes, including the worldwide eradication of smallpox, the development of effective antimicrobial and antiviral drugs, and dramatic improvements in public health and sanitation; the possibility that infectious diseases would no longer pose a continued threat to humans seemed quite promising. Unfortunately, recent decades have seen the emergence and re-emergence of infectious diseases on a wide scale (Table 3.2 ). The emergence of HIV and AIDS in the early 1980s, the emergence of BSE and the subsequent breach of the species barrier to cause vCJD, and, more recently, the emergence of WNV in North America all serve as recent examples.
Table 3.2.
Emerging and re-emerging infectious diseases worldwide since 1980. Viral infectious diseases and prionopathies are given in bold text
| Year | Natural epidemics | Intentional release |
|---|---|---|
| 1981 | Human immunodeficiency virus (worldwide) | |
| 1982 | Lyme disease (neuroborrreliosis) (northeastern United States) | |
| 1984 | Cryptosporidiosis (Texas, NM) | Salmonella (salad bars, United States) |
| 1985 -86 | Bovine spongiform encephalopathy emerges (United Kingdom) | |
| 1987 | Multidrug-resistant tuberculosis (US prisons) | |
| 1991 | Guanarito virus (Venezuela) | |
| 1992 | Vibrio cholerae 0:139 (India) | |
| 1993 | Escherichia coli O:157:H7 (United States) | |
| Hantavirus pulmonary syndrome (southwestern United States) | ||
| 1994 | Variant Creutzfeldt–Jakob disease (United Kingdom) | |
| Hendra virus (Australia) | ||
| 1995 | Recurrence of Ebola hemorrhagic fever (Democratic Republic of the Congo) | |
| 1996 | Poliovirus vaccine virus reversion to neurovirulent strain (Dominican Republic) | Shigella (baked goods, United States) |
| 1997 | Avian influenza (H5N1) (Hong Kong) | |
| Vancomycin-resistant enterococcus (United States) | ||
| Enterovirus-71 encephalitis (Asia) | ||
| 1998 | Nipah virus encephalitis (Malaysia) | |
| 1999 | West Nile virus (New York City) | |
| 2000 | Rift Valley fever (Yemen, Saudi Arabia) | |
| 2001 | Anthrax (US postal facilities, Eastern United States) | |
| 2002 | Severe acute respiratory syndrome (SARS) (Asia, Canada) | |
| 2003 | Monkeypox (midwestern United States) | |
| 2004 | Marburg hemorrhagic fever (Angola) | |
| Nipah virus (Bangladesh) | ||
| 2005 | Multidrug-resistant Salmonella (United States, pet rodents) | |
| 2006 | E. coli (salad greens, multiple US states) | |
| 2007 | Rift Valley fever (Kenya) | |
| 2009 | Salmonella typhi with neurologic illness (Malawi/Mozambique) | |
| Influenza A H1N1 (worldwide) | ||
| 2010 | Vibrio cholerae (Haiti) |
There are a number of social, cultural, and epidemiologic factors that are thought to play an ever-increasing role in the emergence and re-emergence of various infectious diseases (Davis and Lederberg, 2001):
-
1.
Demographics—the world’s population is now estimated to increase by over 350 000 persons per day (http://www.census.gov/cgi-bin/ipc/pcwe). In certain regions of the world, this population growth rate is even higher. Such population growth has numerous effects, including an increase in population and housing density with the associated breakdown in hygienic infrastructure; increased migration within and between populations; and encroachment of human populations upon wildlife habitats, increasing the chances for zoonotic transmission of infectious agents.
-
2.
Social/behavioral changes – several aspects of human behavior directly or indirectly foster the emergence of infectious diseases. Sexual behavioral practices can lead to the spread of sexually transmitted disease (STDs), and foster the spread of emerging and re-emerging STDs, with HIV and the re-emergence of syphilis serving as prime examples. The increase in world travel, and the speed in which that travel may be accomplished, facilitates the spread of agents around the globe, as has been seen with the rapid spread of severe acute respiratory syndrome (SARS). Even activities such as keeping exotic pets have been linked to emerging zoonoses, as was seen in an outbreak of monkeypox in the midwestern United States.
-
3.
Advances in healthcare – recent beneficial accomplishments in human healthcare have had implications on infectious disease emergence. Advances in chemotherapy and immunomodulation, while of tremendous benefit, have set the stage for a surge in opportunistic infections. Transplantations themselves have been associated with several cases of transmission of encephalitic agents, including recent cases of transplant-associated rabies, WNV, and lymphocytic choriomeningitis virus. Invasive procedures have similarly carried with them the risk of introduction of infectious pathogens in unusual settings.
-
4.
Changes in treatment of water/food – as the food and agricultural industries shift more and more to mass-production paradigms, the potential for food-borne infectious disease outbreaks to occur on a large scale has increased. What in the past would have been limited to local outbreaks of food-borne illness have now become multistate or multinational epidemics, leading to hundreds or thousands of cases of illness spread over vast geographic areas. This has been seen recently in outbreaks of Escherichia coli associated with salad greens, and with cases of bacterial infections from tainted meat products. Sometimes animal feed practices have been associated with continued spread of an epidemic; the practice of feeding meat-and-bone meal from rendered cattle presumably infected with the BSE agent is thought to be the mechanism of amplification of the outbreak of mad cow disease in the United Kingdom.
-
5.
Microbial evolution – the emergence of a new disease often represents natural evolution and change of the agent in question, leading to increased virulence in humans. In some cases, the use of antimicrobials leads to selection for microbes that are resistant to the agents, and may select for strains that have increased virulence. Increased antibiotic resistance among some of the more common pathogenic agents is likely to continue to be a problem.
-
6.
War / natural disasters – wars and conflicts lead to a breakdown of public health infrastructure, increasing the likelihood of outbreaks of various diseases. Similarly, natural disasters may lead to similar public health breakdowns, increasing the likelihood of infectious disease outbreaks.
-
7.
Deliberate release of pathogens – although biowarfare and bioterrorism have existed since antiquity, the potential for mass casualties due to the deliberate use of biologic agents in war or terrorism has grown concomitant with the dynamics and changing demographics of modern urban society.
Thus, there are multiple factors of modern society that, perhaps more than at any time in history, appear to be able to facilitate the emergence, re-emergence, and spread of infectious agents. In considering the neurologic manifestations of the emergence of an infectious agent, there are several notable examples that serve as templates for possible future occurrences.
The introduction or emergence of an infectious agent outside its endemic area
West Nile virus
The emergence of WNV in North America and its subsequent spread throughout the western hemisphere serve as good examples of a neurotropic human virus emerging in an unsuspected setting. WNV historically had been associated with infrequent outbreaks of mild, non-specific febrile illness, mainly among children and young adults, in areas throughout Africa and the Middle East (Hayes, 1989, Solomon et al., 2000, Thakare et al., 2002), with several larger outbreaks occurring in Romania, Russia, and Israel in the mid- and late 1990s. The first relatively large outbreak of WNV with significant numbers of neurologic illness occurred in Romania in the summer of 1996 (Ceausu et al., 1997, Campbell et al., 2001); until that time, neurologic illness with WNV was infrequently reported. In 1998, however, a large outbreak of WNV with significant numbers of neurologic cases occurred in Israel (Chowers et al., 2001). In the summer of 1999, however, an unusual clustering of cases of encephalitis was identified in New York City; due to serologic testing difficulties, the outbreak was initially attributed to St. Louis encephalitis virus, an arbovirus closely related to WNV. However, subsequent testing identified the agent as WNV, the first time it had appeared in North America. Since that time, WNV has spread dramatically throughout North America, producing the largest consecutive outbreaks of viral encephalitis in the western hemisphere, with activity now reported in all 48 contiguous US states, and in parts of southern Canada. The epidemic peaked in 2003, and WNV activity has subsequently declined; however, the future epidemiologic pattern of the virus remains unclear. Mosquito, avian, and equine circulation has also been seen in the Caribbean and in Central and South America. As of January 2011, there have been over 30 000 human cases in the United States, including > 12 000 neuroinvasive disease cases and > 1200 deaths.
The emergence of WNV in North America also witnessed the recognition of previously unrecognized modes of transmission of the virus. In 2003, cases of WNV acquired through blood transfusion were identified; recognition of this mode of transmission led to the rapid development of a screening nucleic acid amplification test allowing for the screening of all blood donors for WNV to minimize this risk. Also during that year, a cluster of cases of WNV acquired through solid-organ transplantation was identified, and since that time several other episodes of this mode of transmission have occurred.
Monkeypox
In 2003, an outbreak of rash illness was detected in the midwestern United States. The etiology of the illness was initially uncertain, but eventually was determined to be monkeypox, a rare viral disease occurring in central and West Africa. Monkeypox is an orthopoxvirus with clinical features similar to smallpox, though human disease from monkeypox is generally less severe and associated with lower mortality than that of smallpox. It is endemic in several central and West African countries, and causes human illness through contact with infected primates or small mammals (Parker et al., 2007). Traceback investigation of the outbreak in the United States determined that the virus had been introduced through a shipment of animals from Ghana which included monkeypox-infected rodents (Kile et al., 2005). These African rodents, imported as exotic pets, then infected prairie dogs in the United States during transport and holding; humans became infected through contact with the infected prairie dogs. Overall, 72 cases of illness were reported from six midwestern states, including a young child who developed severe monkeypox encephalitis (Sejvar et al., 2004). This outbreak highlighted the potential threat of introduction of non-indigenous pathogens from exotic animals, and illustrated the possible emergence of new zoonotic diseases through animal transport. As a result of this outbreak, new rules and guidelines regarding the shipment and sale of exotic animals have been drafted in the United States and Europe.
The emergence of a novel infectious agent
The description of the emergence of BSE in the United Kingdom serves as a good example of this type of emergence (see above). Another recent example is the emergence of Nipah virus in South Asia. In 1998 a large outbreak of encephalitis occurred in Malaysia, mainly among abattoir workers. The outbreak was quickly recognized as being associated with exposure to swine, and as a result the outbreak was initially attributed to JEV, an arbovirus in which pigs serve as intermediate hosts, and which was known to cause illness in the area. Subsequent laboratory testing, however, excluded JEV, and the infecting agent eventually was discovered to be a new paramyxovirus; this virus was closely related to the recently described hendraviruses, and to measles virus (Dixon et al., 1999, Bhangoo et al., 2005). The virus was named Nipah virus (NiV), based upon the location of its first isolation. Since its recognition, Nipah virus has caused outbreaks of severe encephalitis in a number of countries in South Asia, most notably Singapore, Indonesia, and Bangladesh (Anonymous, 2004, Hsu et al., 2004, Bhangoo et al., 2005, Reynes et al., 2005). Nipah virus is a zoonotic agent, and is transmitted to humans through exposure to secretions from animals, primarily pigs and bats of the Pteropus genus (Kang et al., 2003, Wacharapluesadee et al., 2005). Human-to-human transmission has been thought to be uncommon, but evidence from recent outbreaks suggests that it may occur. To date, outbreaks of Nipah virus have been infrequent and have remained geographically self-limited. The possible widespread transmission of this emerging zoonotic disease is at present unclear.
Deliberate release of an otherwise uncommon but virulent agent in an act of bioterrorism or biowarfare
There are multiple infectious agents, including several viruses, which have the potential to be used in a deliberate attack. Fortunately, in the recent past such intentional attacks have been infrequent. Perhaps the most notable recent event has been the anthrax attacks in the northeastern United States of 2001 (Jernigan et al., 2001). The greatest concern for weaponization of viral agents centers on variola virus, the causative agent of smallpox, Venezuelan equine encephalitis virus, an arbovirus of the Togavirus family, and the viral hemorrhagic fevers, including Ebola and Marburg viruses, filoviruses that have been associated with periodic outbreaks of severe hemorrhagic fever. Although the weaponization of these viruses remains difficult, advances in technology and human creativity unfortunately render this plausible.
Thus, in addition to the more typical factors influencing the epidemiology of infectious diseases, these various other ancillary factors influencing the emergence and re-emergence of novel pathogens and means of transmission are likely to continue to play an important and increasing role in the epidemiology of nervous system infections.
Summary
The field of neurovirology will undoubtedly experience evolution and change in the years to come. The epidemiology of viral CNS diseases continues to change, and as our understanding of the pathogenesis and pathophysiology associated with viral agents grows, so does our understanding of the behavior of these pathogens among populations. The appearance of viral pathogens in new settings, new or unrecognized modes of transmission, and the emergence of previously unrecognized pathogens will continue to challenge our laboratory diagnostic and epidemiologic capabilities. However, each lesson that is learned from this evolving epidemiology will hopefully result in improved surveillance, diagnostic, and treatment and prevention capabilities.
References
- Alexander L.N., Seward J.F., Santibanez T.A. Vaccine policy changes and epidemiology of poliomyelitis in the United States. JAMA. 2004;292:1696–1701. doi: 10.1001/jama.292.14.1696. [DOI] [PubMed] [Google Scholar]
- Anonymous Nipah virus outbreak(s) in Bangladesh, January–April 2004. Wkly Epidemiol Rec. 2004;79:168–171. [PubMed] [Google Scholar]
- Araujo A.Q., Silva M.T. The HTLV-1 neurological complex. Lancet Neurol. 2006;5:1068–1076. doi: 10.1016/S1474-4422(06)70628-7. [DOI] [PubMed] [Google Scholar]
- Asher D.M. Slow viral infections. In: Scheld W.M., Whitley R.J., Durack D.T., editors. Infections of the central nervous system. Lippincott-Raven; Philadelphia: 1997. [Google Scholar]
- Bale J.F., Jr., Murph J.R. Congenital infections and the nervous system. Pediatr Clin North Am. 1992;39:669–690. doi: 10.1016/s0031-3955(16)38370-5. [DOI] [PubMed] [Google Scholar]
- Beghi E., Nicolosi A., Kurland L.T. Encephalitis and aseptic meningitis, Olmsted County, Minnesota, 1950–1981: I Epidemiology. Ann Neurol. 1984;16:283–294. doi: 10.1002/ana.410160304. [DOI] [PubMed] [Google Scholar]
- Belay E.D. Transmissible spongiform encephalopathies in humans. Annu Rev Microbiol. 1999;53:283–314. doi: 10.1146/annurev.micro.53.1.283. [DOI] [PubMed] [Google Scholar]
- Belay E.D., Schonberger L.B. The public health impact of prion diseases. Annu Rev Public Health. 2005;26:191–212. doi: 10.1146/annurev.publhealth.26.021304.144536. [DOI] [PubMed] [Google Scholar]
- Bellini W.J., Rota J.S., Lowe L.E. Subacute sclerosing panencephalitis: more cases of this fatal disease are prevented by measles immunization than was previously recognized. J Infect Dis. 2005;192:1686–1693. doi: 10.1086/497169. [DOI] [PubMed] [Google Scholar]
- Berger J.R., Sabet A. Infectious myelopathies. Semin Neurol. 2002;22:133–142. doi: 10.1055/s-2002-36536. [DOI] [PubMed] [Google Scholar]
- Bhangoo S., Chua R., Hammond C. Focal neurological injury caused by West Nile virus infection may occur independent of patient age and premorbid health. J Neurol Sci. 2005;234:93–98. doi: 10.1016/j.jns.2005.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley R. Bovine spongiform encephalopathy. Update. Acta Neurobiol Exp (Wars) 2002;62:183–195. doi: 10.55782/ane-2002-1437. [DOI] [PubMed] [Google Scholar]
- Brown K., Mastrianni J.A. The prion diseases. J Geriatr Psychiatry Neurol. 2010;23:277–298. doi: 10.1177/0891988710383576. [DOI] [PubMed] [Google Scholar]
- Calisher C.H. Medically important arboviruses of the United States and Canada. Clin Microbiol Rev. 1994;7:89–116. doi: 10.1128/cmr.7.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell G.L., Ceianu C.S., Savage H.M. Epidemic West Nile encephalitis in Romania: waiting for history to repeat itself. Ann N Y Acad Sci. 2001;951:94–101. [PubMed] [Google Scholar]
- Campbell H., Andrews N., Brown K.E. Review of the effect of measles vaccination on the epidemiology of SSPE. Int J Epidemiol. 2007;36:1334–1348. doi: 10.1093/ije/dym207. [DOI] [PubMed] [Google Scholar]
- Ceausu E., Erscoiu S., Calistru P. Clinical manifestations in the West Nile virus outbreak. Rom J Virol. 1997;48:3–11. [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention Progress toward interruption of wild poliovirus transmission – worldwide, 2008. MMWR Morb Mortal Wkly Rep. 2009;58:308–312. [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention Wild poliovirus type 1 and type 3 importations – 15 countries, Africa, 2008–2009. MMWR Morb Mortal Wkly Rep. 2009;58:357–362. [PubMed] [Google Scholar]
- Choo P.W., Donahue J.G., Manson J.E. The epidemiology of varicella and its complications. J Infect Dis. 1995;172:706–712. doi: 10.1093/infdis/172.3.706. [DOI] [PubMed] [Google Scholar]
- Chowers M.Y., Lang R., Nassar F. Clinical characteristics of the West Nile fever outbreak, Israel, 2000. Emerg Infect Dis. 2001;7:675–678. doi: 10.3201/eid0704.010414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen K.L., Holman R.C., Hammett T.A. Progressive multifocal leukoencephalopathy deaths in the USA, 1979–2005. Neuroepidemiology. 2010;35:178–184. doi: 10.1159/000311014. [DOI] [PubMed] [Google Scholar]
- Clifford D.B., Yiannoutsos C., Glicksman M. HAART improves prognosis in HIV-associated progressive multifocal leukoencephalopathy. Neurology. 1999;52:623–625. doi: 10.1212/wnl.52.3.623. [DOI] [PubMed] [Google Scholar]
- Clifford D.B., De Luca A., Simpson D.M. Natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: lessons from 28 cases. Lancet Neurol. 2010;9:438–446. doi: 10.1016/S1474-4422(10)70028-4. [DOI] [PubMed] [Google Scholar]
- Collinge J. Human prion diseases and bovine spongiform encephalopathy (BSE) Hum Mol Genet. 1997;6:1699–1705. doi: 10.1093/hmg/6.10.1699. [DOI] [PubMed] [Google Scholar]
- Collinge J., Rossor M. A new variant of prion disease. Lancet. 1996;347:916–917. doi: 10.1016/s0140-6736(96)91407-5. [DOI] [PubMed] [Google Scholar]
- Cooper S.A., van der Loeff M.S., Taylor G.P. The neurology of HTLV-1 infection. Pract Neurol. 2009;9:16–26. doi: 10.1136/jnnp.2008.167155. [DOI] [PubMed] [Google Scholar]
- Davis J.R., Lederberg J. National Academy Press; Washington, DC: 2001. Emerging infectious diseases from the global to the local perspective: a summary of a workshop of the Forum on Emerging Infections. [PubMed] [Google Scholar]
- Dhole T.N., Ayyagari A., Chowdhary R. Non-polio enteroviruses in acute flaccid paralysis children of India: vital assessment before polio eradication. J Paediatr Child Health. 2009;45:409–413. doi: 10.1111/j.1440-1754.2009.01529.x. [DOI] [PubMed] [Google Scholar]
- Dixon T.C., Meselson M., Guillemin J. Anthrax. N Engl J Med. 1999;341:815–826. doi: 10.1056/NEJM199909093411107. [DOI] [PubMed] [Google Scholar]
- Glaser C.A., Gilliam S., Schnurr D. In search of encephalitis etiologies: diagnostic challenges in the California Encephalitis Project, 1998–2000. Clin Infect Dis. 2003;36:731–742. doi: 10.1086/367841. [DOI] [PubMed] [Google Scholar]
- Glasgow J.F., Middleton B. Reye syndrome – insights on causation and prognosis. Arch Dis Child. 2001;85:351–353. doi: 10.1136/adc.85.5.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass R.I., Bresee J., Jiang B. Gastroenteritis viruses: an overview. Novartis Found Symp. 2001;238:5–19. doi: 10.1002/0470846534.ch2. discussion 19–25. [DOI] [PubMed] [Google Scholar]
- Granerod J., Cunningham R., Zuckerman M. Causality in acute encephalitis: defining aetiologies. Epidemiol Infect. 2010;138:783–800. doi: 10.1017/S0950268810000725. [DOI] [PubMed] [Google Scholar]
- Granwehr B.P., Lillibridge K.M., Higgs S. West Nile virus: where are we now? Lancet Infect Dis. 2004;4:547–556. doi: 10.1016/S1473-3099(04)01128-4. [DOI] [PubMed] [Google Scholar]
- Gregg M.B. Oxford University Press; New York: 1996. Field Epidemiology. [Google Scholar]
- Griffith B.P., Booss J. Neurologic infections of the fetus and newborn. Neurol Clin. 1994;12:541–564. [PubMed] [Google Scholar]
- Grindstaff P., Gruener G. The peripheral nervous system complications of HTLV-1 myelopathy (HAM/TSP) syndromes. Semin Neurol. 2005;25:315–327. doi: 10.1055/s-2005-917668. [DOI] [PubMed] [Google Scholar]
- Gubler D.J. Human arbovirus infections worldwide. Ann N Y Acad Sci. 2001;951:13–24. doi: 10.1111/j.1749-6632.2001.tb02681.x. [DOI] [PubMed] [Google Scholar]
- Gubler D.J. The global emergence/resurgence of arboviral diseases as public health problems. Arch Med Res. 2002;33:330–342. doi: 10.1016/s0188-4409(02)00378-8. [DOI] [PubMed] [Google Scholar]
- Guess H.A., Broughton D.D., Melton L.J. Population-based studies of varicella complications. Pediatrics. 1986;78:723–727. [PubMed] [Google Scholar]
- Gutierrez J., Issacson R.S., Koppel B.S. Subacute sclerosing panencephalitis: an update. Dev Med Child Neurol. 2010;52:901–907. doi: 10.1111/j.1469-8749.2010.03717.x. [DOI] [PubMed] [Google Scholar]
- Halsey N.A., Modlin J.F., Jabbour J.T. Risk factors in subacute sclerosing panencephalitis: a case-control study. Am J Epidemiol. 1980;111:415–424. doi: 10.1093/oxfordjournals.aje.a112916. [DOI] [PubMed] [Google Scholar]
- Hamaguchi T., Noguchi-Shinohara M., Nozaki I. Medical procedures and risk for sporadic Creutzfeldt–Jakob disease, Japan, 1999–2008. Emerg Infect Dis. 2009;15:265–271. doi: 10.3201/eid1502.080749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaguchi T., Noguchi-Shinohara M., Nozaki I. The risk of iatrogenic Creutzfeldt–Jakob disease through medical and surgical procedures. Neuropathology. 2009;29:625–631. doi: 10.1111/j.1440-1789.2009.01023.x. [DOI] [PubMed] [Google Scholar]
- Hayes C. West Nile fever. In: Monath T., editor. The arboviruses: epidemiology and ecology. CRC Press; Boca Raton: 1989. pp. 59–88. [Google Scholar]
- Henderson R.H. The expanded programme on immunization of the World Health Organization. Rev Infect Dis. 1984;6(Suppl 2):S475–S479. doi: 10.1093/clinids/6.supplement_2.s475. [DOI] [PubMed] [Google Scholar]
- Hjalmarsson A., Blomqvist P., Skoldenberg B. Herpes simplex encephalitis in Sweden, 1990–2001: incidence, morbidity, and mortality. Clin Infect Dis. 2007;45:875–880. doi: 10.1086/521262. [DOI] [PubMed] [Google Scholar]
- Holman R.C., Torok T.J., Belay E.D. Progressive multifocal leukoencephalopathy in the United States, 1979–1994: increased mortality associated with HIV infection. Neuroepidemiology. 1998;17:303–309. doi: 10.1159/000026184. [DOI] [PubMed] [Google Scholar]
- Holman R.C., Belay E.D., Christensen K.Y. Human prion diseases in the United States. PLoS One. 2010;5:e8521. doi: 10.1371/journal.pone.0008521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu V.P., Hossain M.J., Parashar U.D. Nipah virus encephalitis reemergence, Bangladesh. Emerg Infect Dis. 2004;10:2082–2087. doi: 10.3201/eid1012.040701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W., Kieseier B., Frohman E. Prion proteins: physiological functions and role in neurological disorders. J Neurol Sci. 2008;264:1–8. doi: 10.1016/j.jns.2007.06.019. [DOI] [PubMed] [Google Scholar]
- Hughes R.A., Hadden R.D., Gregson N.A. Pathogenesis of Guillain–Barré syndrome. J Neuroimmunol. 1999;100:74–97. doi: 10.1016/s0165-5728(99)00195-2. [DOI] [PubMed] [Google Scholar]
- Jernigan J.A., Stephens D.S., Ashford D.A. Bioterrorism-related inhalational anthrax: the first 10 cases reported in the United States. Emerg Infect Dis. 2001;7:933–944. doi: 10.3201/eid0706.010604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P., Faase J.A., Toyoda H. Evidence for emergence of diverse polioviruses from C-cluster coxsackie A viruses and implications for global poliovirus eradication. Proc Natl Acad Sci U S A. 2007;104:9457–9462. doi: 10.1073/pnas.0700451104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R.T. 2nd edn. Lippincott-Raven; Philadelphia, PA: 1998. Viral Infections of the Nervous System. [Google Scholar]
- Johnson R.T. Prion diseases. Lancet Neurol. 2005;4:635–642. doi: 10.1016/S1474-4422(05)70192-7. [DOI] [PubMed] [Google Scholar]
- Kang S.C., Li R., Wang C. Guanidine hydrochloride extraction and detection of prion proteins in mouse and hamster prion diseases by ELISA. J Pathol. 2003;199:534–541. doi: 10.1002/path.1294. [DOI] [PubMed] [Google Scholar]
- Khanna N., Elzi L., Mueller N.J. Incidence and outcome of progressive multifocal leukoencephalopathy over 20 years of the Swiss HIV cohort study. Clin Infect Dis. 2009;48:1459–1466. doi: 10.1086/598335. [DOI] [PubMed] [Google Scholar]
- Khetsuriani N., Quiroz E.S., Holman R.C. Viral meningitis-associated hospitalizations in the United States, 1988–1999. Neuroepidemiology. 2003;22:345–352. doi: 10.1159/000072924. [DOI] [PubMed] [Google Scholar]
- Khetsuriani N., Lamonte-Fowlkes A., Oberst S. Enterovirus surveillance – United States, 1970–2005. MMWR Surveill Summ. 2006;55:1–20. [PubMed] [Google Scholar]
- Kile J.C., Fleischauer A.T., Beard B. Transmission of monkeypox among persons exposed to infected prairie dogs in Indiana in 2003. Arch Pediatr Adolesc Med. 2005;159:1022–1025. doi: 10.1001/archpedi.159.11.1022. [DOI] [PubMed] [Google Scholar]
- Klemola E., Kaeaeriaeinen L., Ollila O. Studies on viral encephalitis. Acta Med Scand. 1965;177:707–716. doi: 10.1111/j.0954-6820.1965.tb01881.x. [DOI] [PubMed] [Google Scholar]
- Koskiniemi M., Rautonen J., Lehtokoski-Lehtiniemi E. Epidemiology of encephalitis in children: a 20-year survey. Ann Neurol. 1991;29:492–497. doi: 10.1002/ana.410290508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer L.D., Li J., Shi P.Y. West Nile virus. Lancet Neurol. 2007;6:171–181. doi: 10.1016/S1474-4422(07)70030-3. [DOI] [PubMed] [Google Scholar]
- Lee B.E., Davies H.D. Aseptic meningitis. Curr Opin Infect Dis. 2007;20:272–277. doi: 10.1097/QCO.0b013e3280ad4672. [DOI] [PubMed] [Google Scholar]
- Lee K.Y., Burgner D., Lee H.S. The changing epidemiology of pediatric aseptic meningitis in Daejeon, Korea from 1987 to 2003. BMC Infect Dis. 2005;5:97. doi: 10.1186/1471-2334-5-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilienfeld D.E., Stolley P.D. 3rd edn. Oxford University Press; New York: 1994. Foundations of Epidemiology. [Google Scholar]
- Lu C.J., Sun Y., Chen S.S. Incidence of Creutzfeldt–Jakob disease in Taiwan: a prospective 10-year surveillance. Eur J Epidemiol. 2010;25:341–347. doi: 10.1007/s10654-010-9446-4. [DOI] [PubMed] [Google Scholar]
- Luby J.P., Sulkin S.E., Sanford J.P. The epidemiology of St. Louis encephalitis: a review. Annu Rev Med. 1969;20:329–350. doi: 10.1146/annurev.me.20.020169.001553. [DOI] [PubMed] [Google Scholar]
- MacGregor I.R., Prowse C.V. Impacts and concerns for vCJD in blood transfusion: current status. Curr Mol Med. 2004;4:361–473. doi: 10.2174/1566524043360591. [DOI] [PubMed] [Google Scholar]
- Mandell G.L., Bennett J.E., Dolin R. 6th edn. Elsevier Churchill Livingstone; Philadelphia, PA: 2005. Principles and Practice of Infectious Disease. [Google Scholar]
- Misra U.K., Kalita J. Overview: Japanese encephalitis. Prog Neurobiol. 2010;91:108–120. doi: 10.1016/j.pneurobio.2010.01.008. [DOI] [PubMed] [Google Scholar]
- Modlin J.F., Jabbour J.T., Witte J.J. Epidemiologic studies of measles, measles vaccine, and subacute sclerosing panencephalitis. Pediatrics. 1977;59:505–512. [PubMed] [Google Scholar]
- Modlin J.F., Halsey N.A., Eddins D.L. Epidemiology of subacute sclerosing panencephalitis. J Pediatr. 1979;94:231–236. doi: 10.1016/s0022-3476(79)80829-x. [DOI] [PubMed] [Google Scholar]
- Nagel M.A., Cohrs R.J., Mahalingam R. The varicella zoster virus vasculopathies: clinical, CSF, imaging, and virologic features. Neurology. 2008;70:853–860. doi: 10.1212/01.wnl.0000304747.38502.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathanson N., Kew O.M. From emergence to eradication: the epidemiology of poliomyelitis deconstructed. Am J Epidemiol. 2010;172:1213–1229. doi: 10.1093/aje/kwq320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolosi A., Hauser W.A., Beghi E. Epidemiology of central nervous system infections in Olmsted County, Minnesota, 1950–1981. J Infect Dis. 1986;154:399–408. doi: 10.1093/infdis/154.3.399. [DOI] [PubMed] [Google Scholar]
- Nozaki I., Hamaguchi T., Sanjo N. Prospective 10-year surveillance of human prion diseases in Japan. Brain. 2010;133:3043–3057. doi: 10.1093/brain/awq216. [DOI] [PubMed] [Google Scholar]
- Parker S., Nuara A., Buller R.M. Human monkeypox: an emerging zoonotic disease. Future Microbiol. 2007;2:17–34. doi: 10.2217/17460913.2.1.17. [DOI] [PubMed] [Google Scholar]
- Polymenakou P.N., Mandalakis M., Stephanou E.G. Particle size distribution of airborne microorganisms and pathogens during an intense African dust event in the eastern Mediterranean. Environ Health Perspect. 2008;116:292–296. doi: 10.1289/ehp.10684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponka A., Pettersson T. The incidence and aetiology of central nervous system infections in Helsinki in 1980. Acta Neurol Scand. 1982;66:529–535. doi: 10.1111/j.1600-0404.1982.tb03139.x. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan K., Maloo J.C., Poddar S.K. Central nervous system infections in Benghazi, Libya: experience from a community-based adult medical neurology set-up. J Trop Med Hyg. 1987;90:123–126. [PubMed] [Google Scholar]
- Rantakallio P., Leskinen M., von Wendt L. Incidence and prognosis of central nervous system infections in a birth cohort of 12 000 children. Scand J Infect Dis. 1986;18:287–294. doi: 10.3109/00365548609032339. [DOI] [PubMed] [Google Scholar]
- Rantala H., Uhari M., Saukkonen A. Outcome after childhood encephalitis. Dev Med Child Neurol. 1991;33:858–867. doi: 10.1111/j.1469-8749.1991.tb14794.x. [DOI] [PubMed] [Google Scholar]
- Rautonen J., Koskiniemi M., Vaheri A. Prognostic factors in childhood acute encephalitis. Pediatr Infect Dis J. 1991;10:441–446. doi: 10.1097/00006454-199106000-00005. [DOI] [PubMed] [Google Scholar]
- Reingold A.L. Outbreak investigations – a perspective. Emerg Infect Dis. 1998;4:21–27. doi: 10.3201/eid0401.980104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynes J.M., Counor D., Ong S. Nipah virus in Lyle's flying foxes, Cambodia. Emerg Infect Dis. 2005;11:1042–1047. doi: 10.3201/eid1107.041350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roucoux D.F., Murphy E.L. The epidemiology and disease outcomes of human T-lymphotropic virus type II. AIDS Rev. 2004;6:144–154. [PubMed] [Google Scholar]
- Ruegger J., Stoeck K., Amsler L. A case-control study of sporadic Creutzfeldt–Jakob disease in Switzerland: analysis of potential risk factors with regard to an increased CJD incidence in the years 2001–2004. BMC Public Health. 2009;9:18. doi: 10.1186/1471-2458-9-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santhanam S., Choudhury D.S. Coxsackie A-9 in the etiology of poliomyelitis-like diseases. Indian J Pediatr. 1985;52:405–408. doi: 10.1007/BF02806628. [DOI] [PubMed] [Google Scholar]
- Scheld W.M., Whitley R.J., Durack D.T., editors. Infections of the central nervous system. 2nd edn. Lippincott-Raven; Philadelphia, PA: 1997. [Google Scholar]
- Sejvar J.J., Chowdary Y., Schomogyi M. Human monkeypox infection: a family cluster in the midwestern United States. J Infect Dis. 2004;190:1833–1840. doi: 10.1086/425039. [DOI] [PubMed] [Google Scholar]
- Sejvar J.J., Bode A.V., Marfin A.A. West Nile virus-associated flaccid paralysis. Emerg Infect Dis. 2005;11:1021–1027. doi: 10.3201/eid1107.040991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sejvar J.J., Curns A.T., Welburg L. Neurocognitive and functional outcomes in persons recovering from West Nile virus illness. J Neuropsychol. 2008;2:477–499. doi: 10.1348/174866407x218312. [DOI] [PubMed] [Google Scholar]
- Sejvar J.J., Schonberger L.B., Belay E.D. Transmissible spongiform encephalopathies. J Am Vet Med Assoc. 2008;233:1705–1712. doi: 10.2460/javma.233.11.1705. [DOI] [PubMed] [Google Scholar]
- Sejvar J.J., Kohl K.S., Gidudu J. Guillain–Barré syndrome and Fisher syndrome: case definitions and guidelines for collection, analysis, and presentation of immunization safety data. Vaccine. 2011;29:599–612. doi: 10.1016/j.vaccine.2010.06.003. [DOI] [PubMed] [Google Scholar]
- Shi Q., Gao C., Zhou W. Surveillance for Creutzfeldt–Jakob disease in China from 2006 to 2007. BMC Public Health. 2008;8:360. doi: 10.1186/1471-2458-8-360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoldenberg B. Herpes simplex encephalitis. Scand J Infect Dis Suppl. 1996;100:8–13. [PubMed] [Google Scholar]
- Skoldenberg B., Forsgren M. Acyclovir versus vidarabine in herpes simplex encephalitis. Scand J Infect Dis Suppl. 1985;47:89–96. [PubMed] [Google Scholar]
- Solomon T., Vaughn D.W. Pathogenesis and clinical features of Japanese encephalitis and West Nile virus infections. Curr Top Microbiol Immunol. 2002;267:171–194. doi: 10.1007/978-3-642-59403-8_9. [DOI] [PubMed] [Google Scholar]
- Solomon T., Willison H. Infectious causes of acute flaccid paralysis. Curr Opin Infect Dis. 2003;16:375–381. doi: 10.1097/00001432-200310000-00002. [DOI] [PubMed] [Google Scholar]
- Solomon T., Dung N.M., Kneen R. Japanese encephalitis. J Neurol Neurosurg Psychiatry. 2000;68:405–415. doi: 10.1136/jnnp.68.4.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon T., Lewthwaite P., Perera D. Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect Dis. 2010;10:778–790. doi: 10.1016/S1473-3099(10)70194-8. [DOI] [PubMed] [Google Scholar]
- Steiner I., Kennedy P.G., Pachner A.R. The neurotropic herpes viruses: herpes simplex and varicella-zoster. Lancet Neurol. 2007;6:1015–1028. doi: 10.1016/S1474-4422(07)70267-3. [DOI] [PubMed] [Google Scholar]
- Tapiainen T., Prevots R., Izurieta H.S. Aseptic meningitis: case definition and guidelines for collection, analysis and presentation of immunization safety data. Vaccine. 2007;25:5793–5802. doi: 10.1016/j.vaccine.2007.04.058. [DOI] [PubMed] [Google Scholar]
- Teutsch S.M., Churchill R.E. Oxford University Press; New York: 1994. Principles and Practice of Public Health Surveillance. [Google Scholar]
- Thakare J.P., Rao T.L., Padbidri V.S. Prevalence of West Nile virus infection in India. Southeast Asian J Trop Med Public Health. 2002;33:801–805. [PubMed] [Google Scholar]
- University of Edinburgh . 2011. The National Creutzfeldt–Jakob Disease Research & Surveillance Unit (NCJDRSU)http://www.cjd.ed.ac.uk/ Available online at: [cited 2011 May] [Google Scholar]
- Wacharapluesadee S., Lumlertdacha B., Boongird K. Bat Nipah virus, Thailand. Emerg Infect Dis. 2005;11:1949–1951. doi: 10.3201/eid1112.050613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward H.J., Everington D., Cousens S.N. Risk factors for sporadic Creutzfeldt–Jakob disease. Ann Neurol. 2008;63:347–354. doi: 10.1002/ana.21294. [DOI] [PubMed] [Google Scholar]
- Will R.G., Ironside J.W., Zeidler M. A new variant of Creutzfeldt–Jakob disease in the UK. Lancet. 1996;347:921–925. doi: 10.1016/s0140-6736(96)91412-9. [DOI] [PubMed] [Google Scholar]
- Young N.P., Weinshenker B.G., Lucchinetti C.F. Acute disseminated encephalomyelitis: current understanding and controversies. Semin Neurol. 2008;28:84–94. doi: 10.1055/s-2007-1019130. [DOI] [PubMed] [Google Scholar]
- Zweighaft R.M., Rasmussen C., Brolnitsky O. St. Louis encephalitis: the Chicago experience. Am J Trop Med Hyg. 1979;28:114–118. doi: 10.4269/ajtmh.1979.28.114. [DOI] [PubMed] [Google Scholar]