

Abstract
The authors have carried out an investigation of the two “draft maps of the human proteome” published in 2014 in Nature. The findings include an abundance of poor spectra, low-scoring peptide-spectrum matches and incorrectly identified proteins in both these studies, highlighting clear issues with the application of false discovery rates. This noise means that the claims made by the two papers – the identification of high numbers of protein coding genes, the detection of novel coding regions and the draft tissue maps themselves – should be treated with considerable caution. The authors recommend that clinicians and researchers do not use the unfiltered data from these studies. Despite this these studies will inspire further investigation into tissue-based proteomics. As long as this future work has proper quality controls, it could help produce a consensus map of the human proteome and improve our understanding of the processes that underlie health and disease.
Keywords: Clinical applications, false discovery rates, human proteome, protein coding genes, proteomics
Nature issue number 7502 introduced two large-scale proteomics studies of the human proteome.[,2] They were both advertised as initial drafts of the human proteome and the journal compared the impact of these two mass spectrometry-based analyses to the publication in 2001 of the draft human genome sequence.[,4] The justification for this singular comparison was that they claimed to identify many more proteins than previous experiments while analyzing tissues and body fluids rather than cell lines.
The Wilhelm et al. study [2] introduced a novel large-scale proteomics database, ProteomicsDB, which houses protein, peptide and tissue expression data from 16,857 tandem MS proteomics experiments carried out on human cell lines and tissues. At the time of publication, ProteomicsDB contained 1.1 billion peptide spectrum matches (PSMs) from 49 previously published large-scale MS-based analyses, as well as 24 data sets from the authors’ group (both published and not published). The database uses two tools, Mascot [5] and Andromeda [6], to map the spectra to peptides from UniProt annotation of the human proteome.[7] Most of the spectra in the database were previously published and were reanalyzed for the database, but the authors did carry out experiments on >30 different fluids and tissues especially for the paper. This last set of peptides is a small subset of the data from the Wilhelm analysis and is referred to throughout this work as the “Human_body_map” set. These peptides also form the basis of the tissue-based analysis in the Wilhelm analysis, but again they are only a subset of those peptides used to describe their proteome map.
For the paper, the peptides from ProteomicsDB were refined by filtering out peptides shorter than seven residues and by applying two false discovery rate (FDR) filters. The filtered peptides from ProteomicsDB mapped to 18,097 genes in the UniProt annotation of the human genome. The authors also detected peptide evidence for 430 peptides that mapped to 404 lncRNAs and other likely noncoding genes.
The Kim et al. study [1] analyzed spectra from >80 experiments covering 24 distinct tissues, 6 of which were fetal and 6 hematopoietic. The Kim paper also identified peptides using two different search engines, this time Mascot and Sequest.[8] The authors identified peptides for 17,294 genes from the RefSeq annotation of the human genome,[9] including peptides for >2500 genes that were not previously identified in the main proteomics databases.[,11] There was peptide evidence for 2350 genes in all 30 tissues (“housekeeping genes”), while 1537 genes were identified in just a single tissue. The authors mapped unmatched spectra against noncoding RNA from the translated human genome and detected peptides for 808 “novel” annotations for a variety of noncoding regions. The peptides were deposited in the authors’ own Human Proteome Map database.
Clear contrasts with published studies
Before the two Nature papers were published, the two previous largest studies of the human proteome had reported 11,200 and 11,700 genes,[,13] while the PeptideAtlas database, a reanalysis of spectra from many publically available experiments, had identified peptides for 12,644 genes.[10] The Wilhelm and Kim analyses claimed to identify substantially more genes than all these studies.
The authors published an analysis of large-scale proteomics resources at approximately the same time as the two Nature papers.[14] Several of the data sets the authors reanalyzed overlapped with those in the Wilhelm paper, though analysis was more conservative (e.g., the authors required at least two unique fully tryptic peptides to identify a gene). The authors found that these resources largely identified the same genes and that these genes were the most conserved and those that had the longest evolutionary history. Adding more large-scale analyses did not substantially increase the number of genes the authors identified. In contrast to the Nature papers, which concentrated efforts on finding new genes to annotate as coding, the authors proposed that many automatically annotated protein-coding genes did not code for proteins. More than 1000 coding genes were reclassified as a result.
There was such a clear difference between what the authors found in this paper and what was in the Kim and Wilhelm analysis that they decided to reanalyze the data. As part of the experiment, the authors had looked at the correlation of gene detection rates with UniProt evidence code. The Wilhelm paper carried out the equivalent analysis, and the comparison of the two results is instructive (Figure 1). For those genes with the strongest evidence (“protein evidence”), both analyses identify high numbers of genes (97% in the Wilhelm analysis, 82% in this analysis), but the Wilhelm analysis detects much higher proportions of the four weakest UniProt evidence codes; the paper identifies three-times as many genes with transcript-level evidence and >40-times as many genes that are “predicted” and “uncertain”. Proteins labeled as predicted and uncertain in UniProt are those that are not supported by any experimental protein or transcript evidence.
Figure 1.
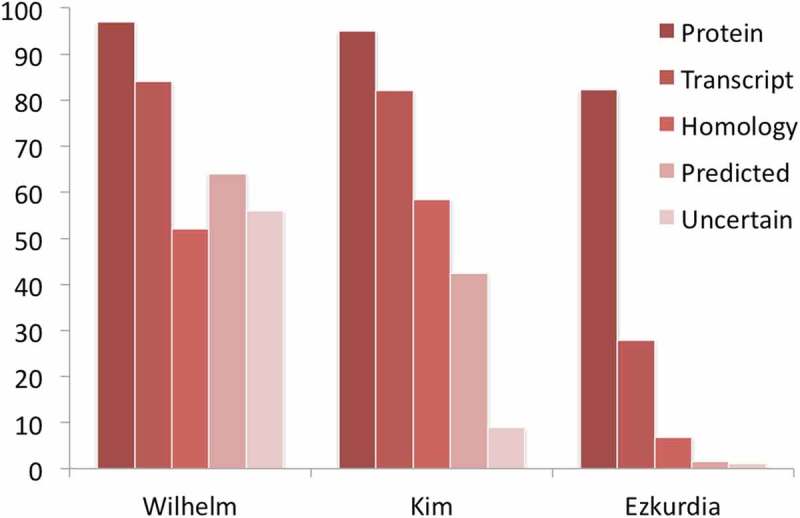
Proportion of human proteins detected by UniProt evidence category. The percentage of proteins identified within each of the five UniProt evidence codes by the Wilhelm analysis,[ 2 ] the Kim analysis [ 1 ] and by the Ezkurdia et al. analysis.[ 14 ] We calculated the evidence codes from the Kim analysis by mapping all 292,000 peptides detected by Kim et al. to the GENCODE annotation [ 15 ] in the same manner as the Kim analysis. The Kim analysis would have identified 18,230 genes if they had searched against the GENCODE annotation in the same way as they searched against the RefSeq database.[ 9 ]
There are two possible explanations for the difference between the two analyses, either the Wilhelm study detects peptides for UniProt predicted and uncertain genes where previous protein and transcript experiments do not or many of the gene identifications in the Wilhelm analyses are simply the result of false positive peptide identifications.
The olfactory receptor test
The authors carried out a simple experiment to test whether the two studies were generating false positive identifications. Olfactory receptors are proteins that should not be detected in standard proteomics experiments since they are integral transmembrane proteins, they have detectable transcript expression levels in very few tissues [16] and the vast majority of them should be limited to a single tissue, the nasal epithelium.[17] The authors did not identify a single olfactory receptor gene in their conservative analysis.[14] Earlier versions of PeptideAtlas [10] identified two olfactory receptors that have been subsequently eliminated in the new human build.[18]
The nasal epithelium was not investigated in either of the two Nature studies. Despite this, there was peptide evidence for 108 olfactory receptors in the Kim data and for 200 olfactory receptors in the Wilhelm analysis.[19] The authors found that some olfactory receptors had been identified from ambiguous peptides (peptides that mapped to more than one gene), but that most were identified from poor quality spectra, spectra with few identifiable fragments. For example of olfactory receptor,[20] see Figure 2.
Figure 2.
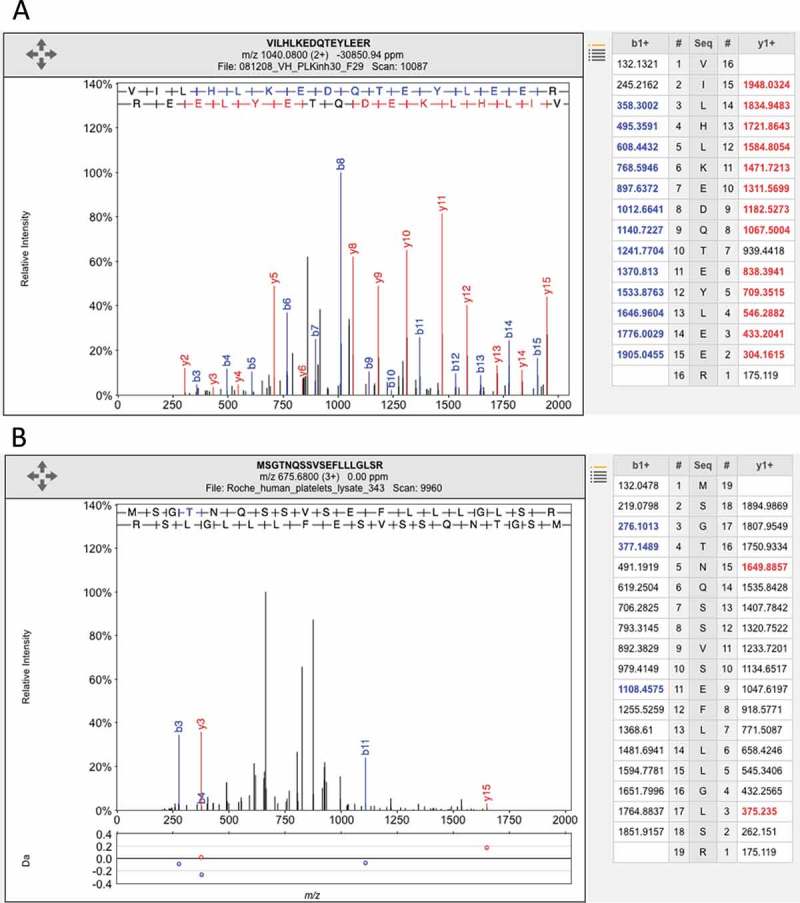
Illustrating the difference between a good and a poor peptide-spectrum match. (A) A good peptide-spectrum match for the peptide VILHLKEDQTEYLEER, a peptide shared by HSP90AB1 and by several other genes. Note that almost all the b-series ions and the y-series ions in the image and in the legend on the right have been correctly identified (correct identification is indicated by the colo r and by the label in the image). (B) A poor peptide-spectrum match for the peptide MSGTNQAAVSEFLLLGLSR, a peptide that maps to the olfactory receptor OR1F1. Just three of the b-series ions and two of the y-series ions have been correctly mapped (again, shown by the label in the image and the colo r in the image and the legend on the right) and none of the correct mappings were consecutive. Both spectra came from ProteomicsDB.
Nondiscriminating peptides
The Kim study was considerably smaller than the Wilhelm study (292,500 peptides vs. 598,845 peptides) yet it identified only 800 fewer genes. The reason for this is that the Kim paper identifies genes using nondiscriminating peptides. Nondiscriminating peptides map promiscuously to two or more genes either because the two genes products are highly similar or because the peptide is very short.
In the Kim study, if a nondiscriminating peptide mapped to two genes, the peptide was used to identify both genes. As an example, the short peptide “DISPVLK” is used in the Kim study to identify both olfactory receptor OR10K1 and gene APRT (adenine phosphoribosyl transferase). In the case of OR10K1, the peptide would be semi-tryptic, it is the only peptide that maps to this gene and it is used to demonstrate that this olfactory receptor is most highly expressed in platelets and fetal liver. “DISPVLK” in APRT is fully tryptic and is one of many peptides that identifies APRT – in fact, APRT has practically 100% peptide coverage. It is clearly much more likely that this peptide is from the gene APRT. From a methodological point of view, identifying multiple genes from a single peptide is highly irregular; instead, nondiscriminating peptides should be discarded because they cannot properly distinguish one gene from another.
The Kim study used trypsin as the cleavage enzyme, but they identified semi-tryptic peptides too (peptides that are not fully cleaved by trypsin). This made it even easier for nondiscriminating peptides to map to multiple genes. One nondiscriminating peptide (“EEELRK”) maps to 21 genes in the GENCODE 20 annotation of the human genome, [15] and the number of RefSeq genes identified by this peptide would have been similar. Approximately 3000 genes in the Kim study were identified solely by nondiscriminating promiscuous peptides. Without these promiscuous peptides, the Kim study identifies only 14,286 genes from the GENCODE 20 human gene set.
In addition, the two studies have problems with the way they treat leucine and isoleucine. Search engines cannot distinguish leucine from isoleucine because they have almost the same mass, so they should be treated as if they were the same amino acid. For example, the peptides “ILVAIMK” and “LIVALMK” belong to different proteins, but they should be treated as if they were the same peptide. The Wilhelm and Kim studies both chose to identify the peptide “ILVAIMK” and map it to the olfactory receptor OR1M1. According to the Kim study, the peptide places this olfactory receptor in placenta, while in the Wilhelm study OR1M1 is found in lung and ovary. However, the spectrum can also be explained by the peptide, “LIVALMK,” which maps to the highly expressed gene ANXA5 (placental anticoagulant protein 4). The authors do not know which one of the two peptides the spectrum belongs to, although given the evidence it seems much more likely that the two studies identified the highly expressed ANXA5. In total, 40 of the 108 olfactory receptors identified in the Kim analysis were identified solely by nondiscriminatory or isobaric peptides.
Calculating the FDR
Perhaps the biggest single problem in these two studies is the way they tackle the difficult issue of FDRs .[– 23] FDRs allow researchers to determine where to draw a line that limits the number of incorrect peptide matches in a study. If the study is small-scale and can be followed up by experimental verifications, researchers might use a high FDR to improve coverage. Generally in larger-scale studies, a stricter FDR cutoff is used, often 1%, because researchers cannot follow up their predictions with experimental confirmation.
FDRs can be calculated at the PSM, peptide and protein levels. For the first two levels, the calculation of FDRs is well described. The standard target-decoy strategy involves generating decoy peptides with the same composition as known peptides and searching against a joint database of known and decoy peptides. The decoy peptides simulate the false positive mappings and the FDR rate is then calculated based on the proportion of random peptides that are detected in the search.[21] Protein FDRs are harder to estimate and there are many ways to do this.[23] What is known is decoy hits (and, therefore, false positive matches) are distributed randomly,[24] while correctly mapped PSM tend to map to the most highly expressed proteins. This means that a 1% FDR at the PSM level translates into a much higher PSM at the peptide level, and a 1% FDR at the peptide level means that the protein-level FDR is substantially higher. For example, the PeptideAtlas database [18] has a 0.00009% PSM-level FDR, a 0.0003% peptide-level FDR and a 1% protein-level FDR. The current version of PeptideAtlas identifies 14,629 proteins from the NextProt annotation of the human genome [25] and includes the spectra from the Kim analysis and many (but not all) of the experiments included in the Wilhelm analysis.[18]
The calculation of FDRs in large-scale experiments is particularly complicated because large-scale proteomics experiments are generally made up of many smaller experiments. The peptide FDR for an individual experiment may be correctly estimated, but this FDR will increase when the experiments are concatenated because correctly identified peptides will appear in multiple experiments, while false positive matches tend to be random in nature and different in each experiment (Figure 3).[24]
Figure 3.
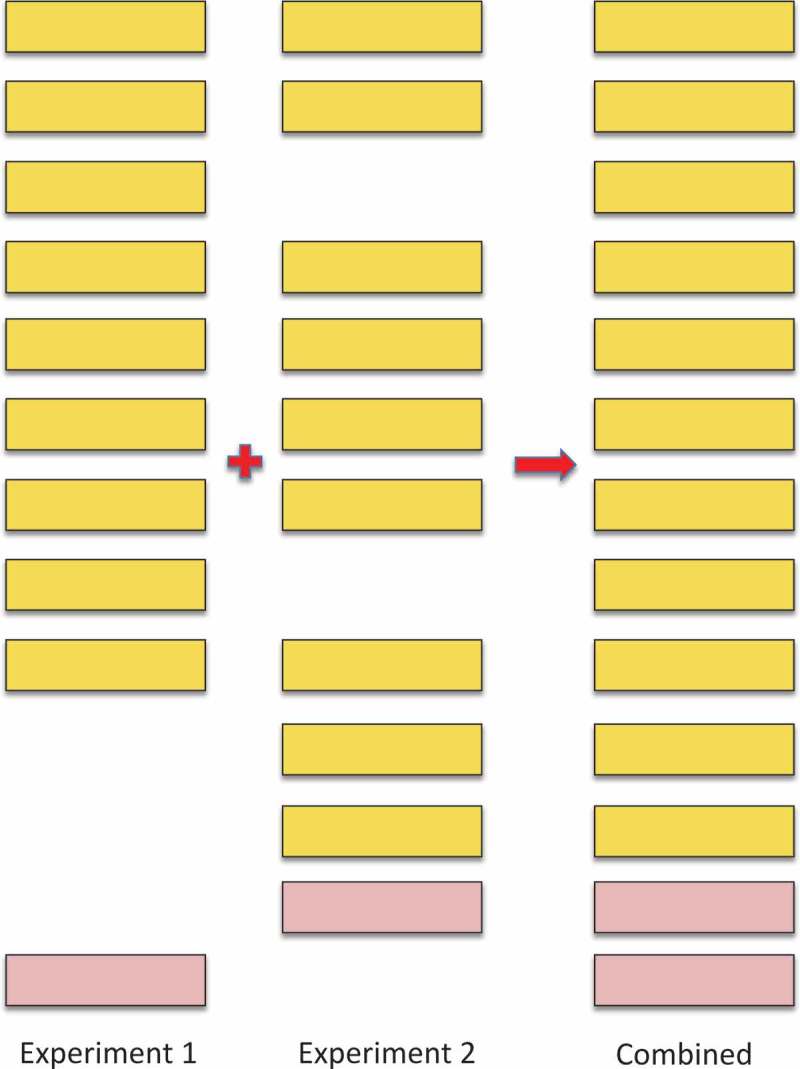
Illustrating how combining experiments increments the false discovery rate. The illustration shows the effect of combining two imaginary experiments, experiment s 1 and 2. In the figure , the yellow boxes represent true positive peptide hits, the pink boxes represent false positive peptide identifications. The real peptide false positive rate for both experiment s 1 and 2 is 10% (one false positive event in 10). However, when the two imaginary experiments are combined , the number of true positive hits only rises to 11 because 7 of the peptides were identified in both experiments. The false positive identifications were not the same in both experiments, so the real peptide false positive rate rises to 15.39% ( 2 in 13). In general , many of the true positive peptide hits are repeated across experiments and few of the false positive identifications are repeated, so the false discovery rate will always go up when experiments are combined – and the more experiments that are combined, the greater the effect as it gets harder and harder to identify peptides that have not previously been identified in another experiment.
The authors of the Wilhelm paper, which brought together a huge number of published and unpublished experiments, recognized this problem and carried out their own detailed investigations into the effects of combining many experiments in their “supplementary material”. They suggested that the result of combining peptides from many experiments under the standard FDR calculations would be a huge increase of the numbers of false positive mappings from the decoy database. They made two further pertinent points, namely that the classical protein FDR calculations are only “appropriate for relatively small studies” and that the current protein-level FDR model is not valid. The authors even suggested a number of possible strategies to get around this problem,[24] including one of their own,[26] but finally did not apply any protein-level FDR at all to their data.[23] Instead, the Wilhelm paper calculated a 1% FDR for the peptides in each individual experiment and a length-dependent 5% FDR for all the peptides once the experiments have all been combined.
The justification for the FDR limits in the Wilhelm paper is that they are in line with results from the individual large-scale experiments that make up the analysis. However, the supplementary material from the paper shows that this is not so. Even though the number of genes identified by the Wilhelm analysis is similar to those identified in each individual experiment, the Wilhelm paper uses two search engines, not one. For example, the Shiromizu et al. experiment [12] reported 11,278 proteins. The Wilhelm analysis of the Shiromizu spectra identified 11,204 genes using the Mascot search engine, and also 11,703 genes using Andromeda. However, the total number of genes identified from the Shiromizu spectra will be substantially higher because the Wilhelm paper adds the peptides identified by Mascot to those found by Andromeda. The overlap between the peptides identified by the two search engines is likely to be between 50 [27] and 75%,[28] which means that a substantial number of peptides are gained by using two search engines. Using both Mascot and Andromeda to reanalyze the large-scale experiments means that the paper is effectively doubling the number of large-scale experiments that are being analyzed. Each of these >150 separate large-scale analyses will have their own set of unique false positive matches.
The Wilhelm analysis addresses the issue of false positive matches from multiple large-scale analyses by using a pre-calculated length-dependent 5% FDR filter at the peptide level. As argued above, the real peptide FDR is likely to be much higher than 5% because of the effect of combining multiple large-scale experiments. But even if the pre-calculated global 5% length-dependent FDR rate for the combined peptides is accurate, this suggests that approximately 30,000 (5%) of the 598,845 peptides identified in the analysis are false positive identifications. That is, 1.66 falsely identified peptides for every one of the 18,097 proteins detected in the analysis, a staggeringly high number. In other words, the 5% FDR used in the Wilhelm analysis implies that any gene identified by just one or two peptides has a very high probability of being a false positive identification.
This is almost certainly part of the reason why the Wilhelm paper identifies peptides for 200 olfactory receptors.[19] Most of these olfactory receptors (127) were identified with a single peptide, 62 were identified with two peptides and none by more than four peptides.
In fact, the Wilhelm and Kim studies include a large number of very poor spectra and consequent false positive mappings. For example, the Kim study reports two peptide-spectrum matches in adult liver that supposedly identify the olfactory receptor OR4F6. The peptide “TISFGGCVVQIFFIHAVGGTEMVLLIAMAFDRYVAICKPLHYLTIMNPQR” covers 16% of the protein sequence, but the corresponding fragmentation spectrum is of very poor quality (Figure 4). The Mascot scores of the two matches are very low, 3.22 and 2.57. Another example is the peptide “DVAVVFTEEELELLDSTQRQLYQDVMQENFR.” This peptide is the only discriminating peptide that identifies the gene ZNF229, but the spectrum is poor and the Mascot score is 1.22, very close to zero (Figure 4).
Figure 4.
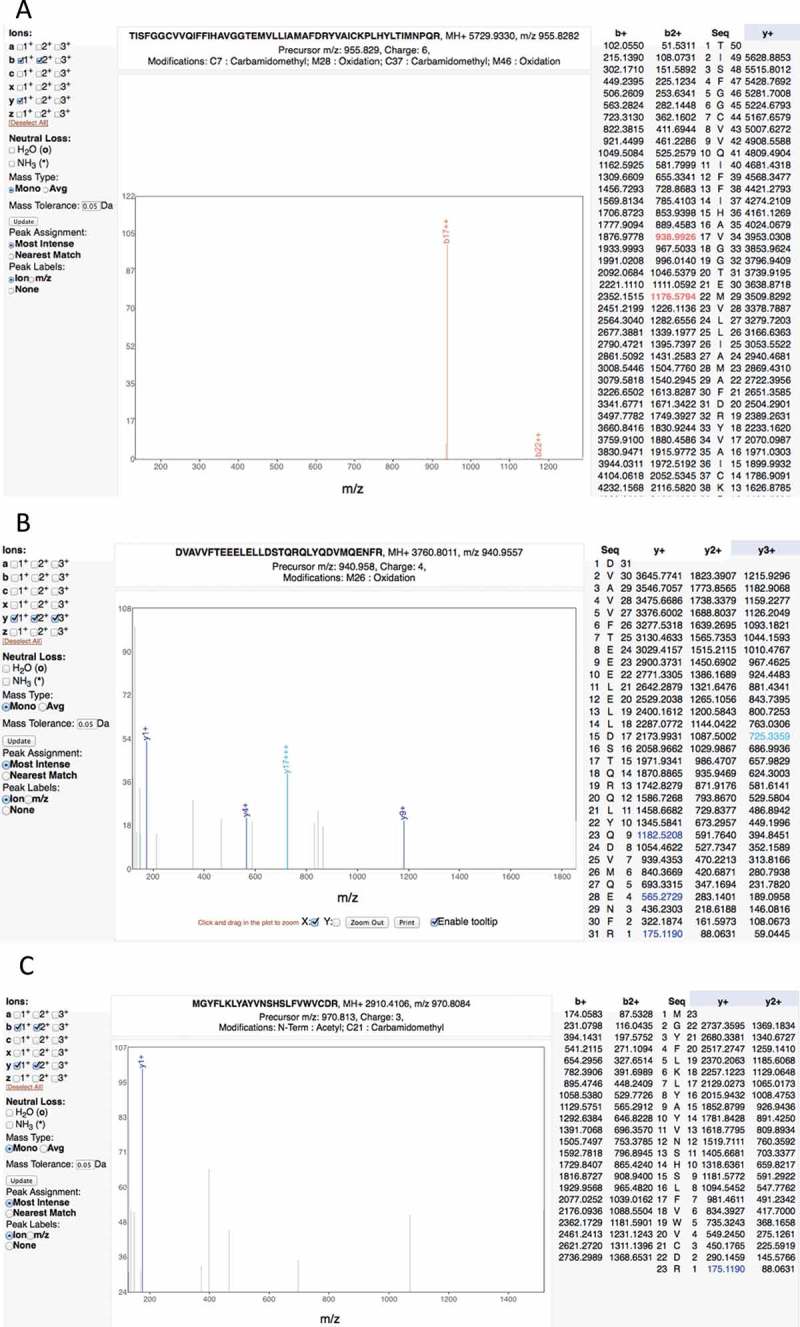
Examples of the many poor spectra from the Kim analysis. (A) One of the two very poor spectra used to identify peptide TISFGGCVVQIFFIHAVGGTEMVLLIAMAFDRYVAICKPLHYLTIMNPQR for gene OR4F6. The Mascot scores of the two matches are very low, 3.22 and 2.57, only a handful of ions are properly identified. (B) A very poor spectrum for the peptide DVAVVFTEEELELLDSTQRQLYQDVMQENFR, which is the only peptide that identifies gene ZNF229. Only the y-series is shown for this +4 charge spectrum, very few y-series ions are identified. (C) A very poor spectrum for peptide MGYFLKLYAYVNSHSLFVWVCDR, which is used to identify EBLN2. Here just a single ion is identified. It is worth noting that this peptide is supposed to have both an N -terminal acetylation. All these spectra are from the Human Proteome Map from the Kim analysis.
The predicted Ensembl gene LINC00346 is a good example of how the analysis and data quality problems can accumulate. LINC00346 is coded from a single poorly conserved exon, had no protein features, and UniProt annotates it as “Uncertain,” “product of a dubious CDS prediction” and “may be a noncoding RNA”.[7] Indeed, in recent versions of the database it is no longer annotated as protein coding. There are eight peptides in ProteomicsDB that map uniquely to gene LINC00346 and six of these peptides pass the local 5% FDR filter for the paper. If the statistical calculations for peptide identity and filtering were correctly carried out, the eight peptides detected for this gene would mean that the identification of this gene would never be in doubt; for most analyses, just two peptides are usually sufficient to be sure of a positive identification. However, the spectra for all eight peptides are poor, only a couple of fragments are identified for each spectrum. Even though the analysis identifies eight separate peptides for this gene, the identification is absolutely untrustworthy (Figure 5). Another example is the gene EBLN2, which is identified by 4 discriminating peptides in the Kim analysis, and another 9 discriminating peptides in the ProteomicsDB, and the PSMs that identify all 13 peptides are very poor (Figure 5).
Figure 5.
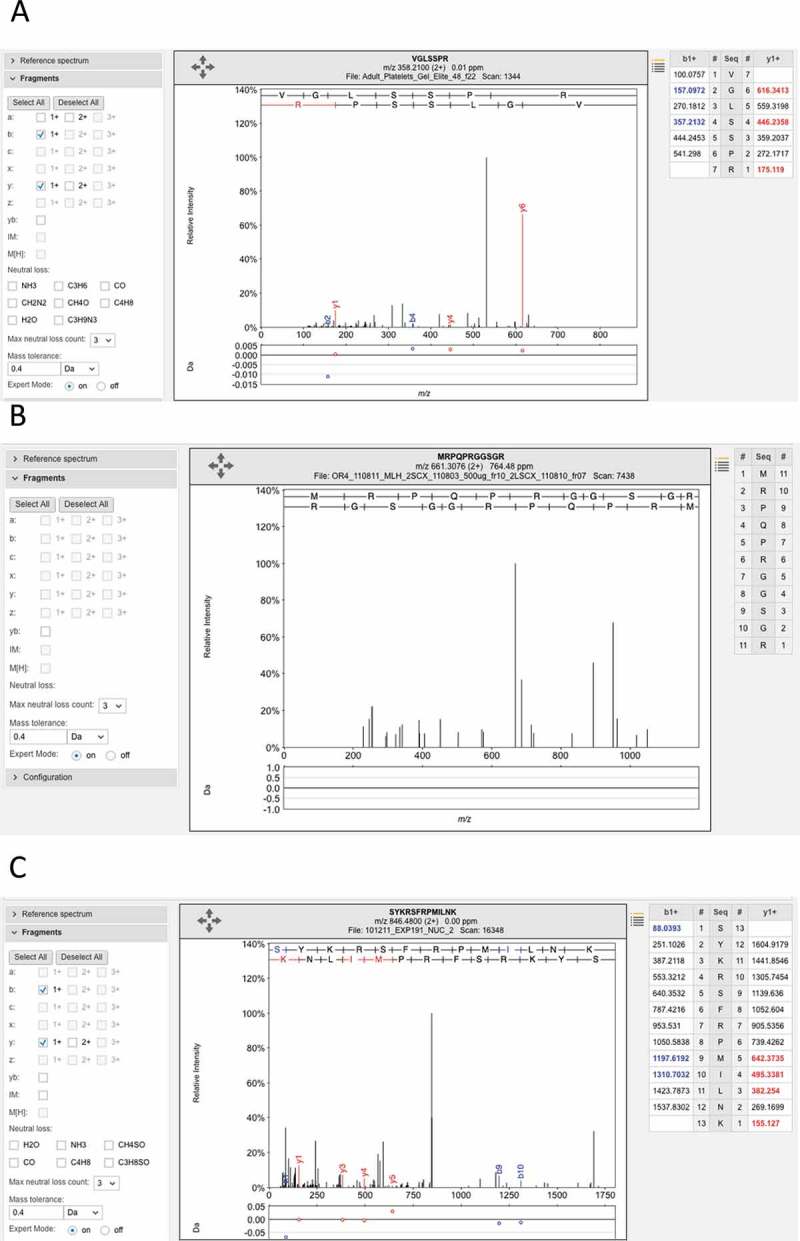
Examples of the many poor spectra from the Wilhelm analysis. (A) One of the three poor spectra used to identify peptide VGLSSPR for gene LINC00346. This peptide was identified with an Andromeda score of 71.95. No consecutive ions in the series were identified. (B) The very poor spectrum for peptide MRPQPRGGSGR, which maps to gene LINC00346. The peptide is supposed to be N -terminal acetylated. None of the fragments are identified. C. One of three poor spectra for peptide SYKRSFRMILNK, which is used to identify EBLN2. Again very few fragments are identified. All these spectra are from the ProteomicsDB and from the Wilhelm analysis.
It has long been standard practice to require that proteins in large-scale proteomics experiments be identified by at least two distinct peptides.[,30] This requirement is essential for large-scale experiments because no matter how well filtered the peptides are, a protein with a single peptide hit is more likely to be a false positive identification. False positive hits will be randomly distributed among the genes in the genome, so proteins identified with just a single peptide will be rich in false positive matches (see Figure 3).
Despite this, both the Kim and Wilhelm analyses identified many proteins with a single peptide. The authors calculated that if the Kim study had required two nondiscriminating peptides to identify proteins they would have identified just 12,006 genes from the GENCODE 20 gene set. The supplementary material from the Wilhelm paper suggests that the number of proteins identified by just one peptide is 1259, which means that the authors identified <17,000 genes with two or more peptides.
As described above, both the Kim and Wilhelm analyses used two search engines to map the peptides to their search databases. It has been shown that where two or more search engines agree on a PSM the identification is more likely to be reliable than when the identification comes from a single search engine.[31] This suggests that when search engines are used in tandem as in the Kim and Wilhelm analyses it is better to take the intersection of their PSMs, those PSM where both engines agree on the identified peptide, rather than the union of the PSMs, where peptides need only to be identified by a single search engine. Unfortunately, both the Kim and Wilhelm papers take the union of the PSMs. This decision clearly improves coverage, the numbers of genes identified, but will come at the expense of a higher false positive rate. The studies give no figures for the numbers of peptides identified by just a single search engine, nor do they explain what happens when they map two different peptides to the same spectrum, but we can estimate how many genes were identified by a single peptide. The supplementary material of the Wilhelm analysis suggests that at least 1632 genes were supported by peptides from a single search engine, while the Kim study identified 1248 GENCODE 20 genes from peptides detected by just one of the two single search engines.
Identifying genes with more than just a single peptide and using the intersection rather than the union of the search engines are two very simple strategies that could have been used to limit the worst of the effects of combining multiple large-scale experiments. If the Kim analysis had required genes to be identified by two discriminating peptides that in turn were identified in the same spectrum by both the Mascot and Sequest search engines, the Kim study would have identified just 11,229 genes from the GENCODE 20 human gene set, in line with previous studies.[– 14] This is still a substantial number, but it is a full 6000 fewer genes than the authors claimed to have identified in their paper.
The supplementary material from the Wilhelm analysis suggests that combining these two strategies would have decreased the number of identified proteins by 1854. However, the supplementary material underestimates the effect of the combination of the two strategies. For example, according to the supplementary material the gene LINC00472 was identified with six peptides found by the Andromeda and Mascot search engines. However, close inspection shows that Mascot and Andromeda do not identify the same PSM for any peptide; so even LINC00472 would not have been identified with these filters. This case demonstrates that these two simple filters would have removed substantially >1854 proteins from the list of identifications.
For researchers, the real problem is that FDR calculations have been complicated by improvements in proteomics technology.[– 34] The result is that false positive rates cannot be calculated correctly. Something is clearly wrong with the FDR calculations in these papers if the spectra in Figures 4 and 5 are able to pass a standard 1% FDR. There is still no agreed solution to this problem,[,25] but in the interim there are simple quality filters that can be put in place to reduce the false positives.
Applying the intersection of the search engines and a minimum of two discriminating peptides per gene is not a panacea, but the Wilhelm data show the importance of even these two minimal rules. With both filters in place, ProteomicsDB would only have identified two olfactory receptors. And both of these would have fallen foul of the 5% length-dependent FDR used in the Wilhelm analysis.
The Human Proteome Project
The Human Proteome Project (HPP) was instigated with the goal of experimentally observing all human proteins.[35] They have recently produced three papers that deal with the Kim and Wilhelm analyses in some detail.[,35,36] Horvatovich et al. [36] discussed the importance of error analysis in large-scale data projects in the light of the Kim and Wilhelm analyses. They advocate the use of protein-level FDR when proteins are being identified; a 1% peptide FDR will dramatically underestimate the number of false positive proteins identified since false positive peptides will be randomly distributed among the proteins in the search database – and the larger the search database, the more likely these false positive peptides will identify sequences that are not present in the experimental sample. The update of the PeptideAtlas database [18] did use a 1% protein FDR and found that the spectra from the Kim and Wilhelm studies added <500 genes to those that were already identified by proteomics. The main HPP paper [35] also looked into the studies, finding that an independent analysis of the Kim spectra suggested that the authors had identified 11,000, not 17,000, genes. They reported that the GPM proteomics database [11] used the same lax filters as the Wilhelm analysis to analyze their own spectra, and they were able to map the peptides they found to 97% of the human genome. The HPP paper concluded by calling for a 1% protein FDR and a minimum of two uniquely mapping peptides to be used in large-scale analyses.
Identification of noncoding genes
Proteogenomics [37] is the use of proteomics to detect novel coding regions. In essence, proteogenomics is a standard proteomics search except that the search is carried out against a combined database of known genes and known noncoding regions. It has most value when genomes are poorly annotated: for well-annotated genomes, the distributions of the novel and decoy hits are almost identical,[38] and the human genome is the best-annotated higher eukaryote genome.
Between them the two studies identified more than a thousand novel protein-coding regions from proteogenomics studies. The Wilhelm study identified 430 novel peptides using just 1% of the spectra from ProteomicsDB and mapped them to 404 noncoding genes. The Kim paper identified 808 novel coding regions. They found 44 new coding genes and 216 events that added exons to known gene models.
Nesvizhkii recently wrote that the identification of peptides that map to novel coding sequences requires a higher burden of proof.[37] The only filter used in the Wilhelm study was that each PSM had to have a minimum delta score of 10. Delta scores measure the difference between the best scoring peptide and the next best scoring peptide in a spectrum. This unfortunately falls short of filtering all poor quality spectra as Figure 6 shows because delta scores do not have the same discriminating power when the number of candidate peptides is small, as will be the case with many poor spectra.[34] Almost all the novel coding genes were identified by a single peptide and most were pseudogenes. Practically all of the novel peptides identifications in this study were identified from poor spectra or were probable single amino acid variants or were misidentified post-translational modifications. Indeed, the authors have observed that there is an over-representation of modified peptides among those that identify noncoding regions in both studies (see supplementary material). With the exception of ORF1, a retroviral open reading frame with many copies in the human genome, there is not enough evidence to support the identification of any of the 404 noncoding genes in the Wilhelm paper.
Figure 6.
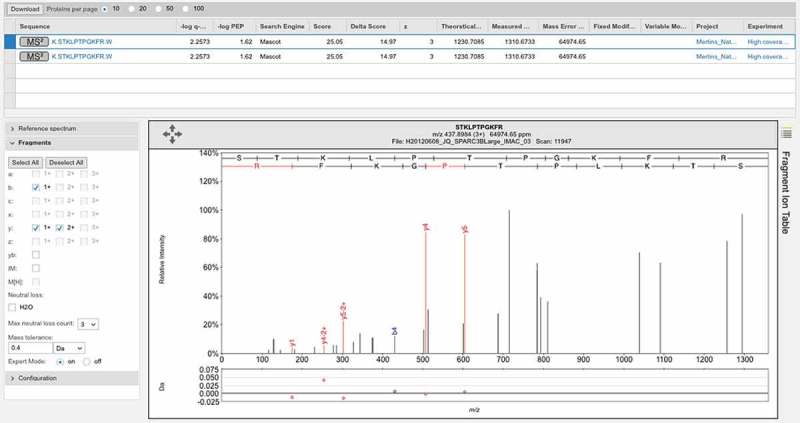
Poor spectrum with a delta score of>10. One of two poor spectra that identify peptide GQGVPISCK for gene LINC00346. This peptide was identified with Mascot delta score of 17.95, but the Mascot score was just 24.02, a score that is worse than the 5% local peptide cut off used in the main study. Again few ions in the two series are identified.
While the Kim study did not require a higher burden of proof to identify novel coding peptides, they did compare the spectra for the best scoring peptides with spectra from synthetic peptides. The Kim study interrogated almost three-times as many spectra as the Wilhelm study, and some of the candidate novel coding regions are plausible. However, the authors chose to search against the RefSeq annotation of the human genome, and many of the “new” coding regions they found have been annotated as coding in other databases for a number of years. For example, the gene MYO1B, a pseudogene in RefSeq, has been annotated as coding by UniProt since 2007. The “new” uORF in CHTF8 highlighted in the paper has been annotated as a splice variant in both the UniProt and Ensembl/GENCODE databases since 2010. These are interesting for RefSeq because they are missing in their annotation, but they are not new coding regions. Despite this, the Kim study did map two or more peptides to 27 coding regions that were novel to all annotations. It is notable that, 25 of these 27 potential novel coding regions are extensions to existing gene models rather than new coding genes (see Table 1).
Table 1.
A list of the novel coding regions identified from the Pandey analysis by more than one peptide that is not already annotated in one of the main genome databases.
| Gene | Type | Peptides | Notes | |||
|---|---|---|---|---|---|---|
| AAK1 | Gene extension | 10 | Extended version now in Ensembl | |||
| ABR | Gene extension | 2 | ||||
| AIM1 | Gene extension | 2 | ||||
| BEGAIN | Gene extension | 2 | ||||
| EIF4G3 | Gene extension | 2 | ||||
| GLUL | Gene extension | 2 | ||||
| HECTD4 | Gene extension | 2 | ||||
| HNRNPA2B1 | Gene extension | 3 | ||||
| KHDRBS3 | Gene extension | 4 | ||||
| KSR1 | Gene extension | 2 | ||||
| NPLOC4 | Gene extension | 2 | ||||
| TPST2 | Gene extension | 2 | ||||
| VKORC1L1 | Gene extension | 2 | ||||
| ADPRHL1 | Novel coding exons | 3 | ||||
| EPB41 | Novel coding exons | 4 | ||||
| EPB41L1 | Novel coding exons | 2 | ||||
| EPB41L3 | Novel coding exons | 7 | ||||
| MYO18A | Novel coding exons | 6 | ||||
| NCAM1 | Novel coding exons | 5 | ||||
| SORBS1 | Novel coding exons | 11 | ||||
| SORBS2 | Novel coding exons | 2 | ||||
| GSG1 | Other ORF | 2 | ||||
| HNRNPUL1 | Other ORF | 3 | ||||
| HNRNPUL2 | Other ORF | 2 | ||||
| IMPDH1 | Other ORF | 2 | ||||
| LOC100421372 | Pseudogene | 2 | ||||
We were not able to check the spectra for the peptides that identified these regions or whether these new regions have other evidence to support their coding potential.
ORF: Open reading frame.
The use of synthetic peptides
Synthetic peptides have been used in both these studies to validate peptide identifications for subsets of potential coding genes. Although the use of synthetic peptides for validation is to be applauded, there are pitfalls with their use in large-scale studies. First of all if the synthetic peptides are just a sample of the subset to be validated, it should be made clear whether the sample is representative or is made up of the peptide most likely to be positively validated from the subset. The samples used in both these studies are of the second type – those most likely to be validated (because they either have higher scores or more PSM), but this is not clear from their context in the two papers. Second, the spectra from the synthetic peptide are generally compared to the experimental spectra by the human eye and this is open to interpretation. The authors’ interpretation of the spectra from the two analyses differs from those of the other authors (see supplementary material).
After manual comparison of the spectra from these synthetic peptides against the experimental data, the authors found several problems. In many cases, the information obtained from the spectra generated from the synthetic peptides could not be used for experimental validation due to a very poor correlation and fragmentation quality.
In other cases, the synthetic peptides were acquired without taking into account all post-translational modifications or ITRAQ labeling [39] was found in the experimental identification, which thus impaired the ability to make a correct assessment.
The authors validated 89 of the 98 comparisons between experimental and synthetic spectra from the Kim analysis. Just 30 of these peptides were not previously identified by UniProt or GENCODE and were reported in the supplementary material. In the Wilhelm analysis, the authors validated just 18 of 53 synthetic-experimental spectra comparisons that were supposed to confirm the identification of “uncertain” annotated UniProt coding genes. If isobaric peptides were taken into account, 8 of these 18 peptides also mapped to known coding genes and 4 of the remaining 10 peptides were just 7 residues in length and may be explained by SNPs.
The draft maps of the human proteome
Both the Wilhelm and Kim papers claimed to have a draft map of the human proteome. The Wilhelm analysis combined results from their Human_body_map tissue and fluid experiments with a number of experiments on cell lines to determine that each “organ” had its own specific pattern of gene expression. This pattern matched the specific biology of the organ according to the gene ontology (GO) analyses they carried out (e.g., platelets were enriched in platelet growth factors).
One problem with the Wilhelm data is that the numbers of tissues and cell lines that were used to build the map and how they were combined is not fully clear. Within the same paragraph, the authors state that they used 42 tissues and cell lines for the principal component analysis and investigated 47 “organs and fluids” to analyze the 100 most expressed proteins. This in turn contrasts with the 45 “tissues” listed in the supplementary material and the 43 organs/fluids illustrated in extended Figure 6. The authors then describe how they complemented the 27 tissues and fluids from their Human_body_map set with data from other experiments, but in the supplementary material the Human_body_map set is made up of 30 tissues and fluids, while the peptides deposited from the Human_body_map experiments come from 34 different tissues and fluids.
As well as the confusion with the numbers, there are two problems with the use of the Wilhelm data as a map of the human proteome. The first problem is that none of the experiments from the Human_body_map tissue data had any replicates. Individual proteomics experiments rarely have high peptide coverage, and this sparseness of peptide coverage in individual experiments means that it is difficult to draw many conclusions from single tissue-specific experiments. Peptides may be detected for a gene in one replicate, but not in another. For example, the authors compared two replicates from the Kim study that had undergone the same experimental procedure (both from adult heart); just 29% of the peptides identified were found in both studies. Without multiple replicates, the authors cannot be sure whether a protein is expressed; the absence of peptides could be because the protein is expressed in limited quantities or not at all, or because the peptides were present in the mixture, but not detected.
Secondly, the experiments that supplemented the Human_body_map tissue data came from experiments on cell lines rather than tissues. Although the Wilhelm paper carried out comparisons between tissues and cell lines to show that they are similar, these extra experiments were not only carried out on cell lines but also by a number of different groups and using a range of different instruments.
By way of contrast, the Kim paper does provide replicates for the tissues, and only interrogates tissues and hemopoietic cells. There is a serious issue with the validity of many of the peptide identifications, four of the experiments they deposited do not identify any peptides, one of the “monocyte” experiments is an identical copy of another and the third “monocyte” experiment seems to not have been properly purified, but the Kim study could be described as a draft of the human proteome. The authors have generated a filtered set of peptides based on the advice in the paper; they are available as supplementary material (Table S1).
What we have learned from the publication of the two drafts
Both these collaborations are the fruits of a huge amount of time and effort, and the fact that the data they have generated is available to the wider community is to be applauded. Despite the noise in the data, the two studies did identify peptides for known coding genes that have not previously been identified in proteomics experiments and that are likely to be tissue-specific or at least not usually identifiable in cell lines. The authors recently carried out an analysis of the data from Kim experiment and the Human_body_map set from the Wilhelm experiment and compared that with the peptide data from six large-scale analyses that largely identified peptides in cell lines rather than tissues.[,41] The authors generated conservative subsets of peptides from all eight studies in order to remove as much noise as possible and compared genes identified by the six large-scale cell line studies with genes detected in the Human_body_map set and the Kim study. They found genes that were almost exclusively found in the Kim study and the Human_body_map set (Table S2) and 237 genes that were almost exclusively found in the six large-scale cell-line-based proteomics studies (Table S3). These two sets of genes have been made available in the supplementary material.
For the most part, the genes found in the Kim and Wilhelm data sets but not in any of the cell line experiments tended to be tissue specific. For example, CYP17A1 (steroid 17-alpha-hydroxylase/17,20 lyase) is known to be adrenal and testis specific and there was reliable peptide evidence for CYP17A1 in adult and fetal ovary and testis, along with adult adrenal glands, strongly suggesting that CYP17A1 is ovary-specific too. The gene TG (thyroglobulin) is annotated as thyroid specific, but there was peptide evidence in many tissues in the Kim analysis, especially in kidney, esophagus, heart and ovary. The Kim analysis did not include thyroid tissue, but the Human_body_map set from the Wilhelm paper did and found 61 reliably identified peptides for TG found in thyroid. CADPS2 (calcium-dependent secretion activator 2) is supposed to be widely expressed, but most reliable peptide evidence was in frontal cortex. There were also genes identified with many peptides solely in the Kim experiment such as ABCA4 (retinal-specific ATP-binding cassette transporter) for which the authors identified 48 reliable peptides, all in retina samples, and SMC1B (structural maintenance of chromosomes protein 1B), a meiosis-specific component of the cohesin complex for which there were 38 peptides in ovary and testis.
The authors carried out a GO term analysis using the DAVID functional analysis tool [42] (Table 2) for the enriched peptides. The most significantly enriched terms for the 322 tissue-expressed genes (one term from each of the most enriched clusters) were “sensory perception of light stimulus” (27 genes) and “neurological system process” (48 genes). Among the 237 genes identified in cell line experiments, but not in tissues were cell cycle-specific proteins such as BRCA2 (breast cancer type 2 susceptibility protein), BRCA1 (breast cancer type 1 susceptibility protein), AURKA (aurora kinase A) and ERCC6L (DNA excision repair protein ERCC-6-like), and DNA repair proteins such as KDM4A (lysine-specific demethylase 4A). The significantly enriched terms for the cell line-expressed genes included “cell cycle” (58 genes), “DNA metabolic process” (45 genes) and “chromosome organization” (36 genes).
Table 2.
A list of the most significant GO terms from those genes that appeared only in cell line experiments and those that appeared in the Kim and Wilhelm tissue-based analyses.
| Tissue-detected genes | Numbers | Benjamini |
|---|---|---|
| Sensory perception of light stimulus | 27 | 1.30E-13 |
| Neurological system process | 48 | 6.40E-08 |
| Sexual reproduction | 22 | 4.10E-04 |
| Plasma membrane part | 60 | 7.90E-04 |
| Cell–cell signaling | 24 | 1.70E-03 |
| Gated channel activity | 18 | 1.70E-03 |
| Cell line detected genes | ||
| Cell cycle | 58 | 5.30E-18 |
| DNA metabolic process | 45 | 1.60E-16 |
| Chromosome organization | 36 | 1.70E-10 |
| Spindle | 19 | 8.40E-10 |
| Nuclear lumen | 56 | 8.40E-09 |
| Helicase activity | 16 | 7.90E-07 |
| Nucleotide binding | 73 | 8.70E-07 |
| Regulation of cell cycle | 22 | 4.40E-05 |
| Chromatin organization | 23 | 9.00E-05 |
A complete list of the experiments is in references [] and [39]. Only one GO term is taken from each of the clusters generated by DAVID. Numbers shows the numbers of genes that were annotated with the GO term and Benjamini shows the Benjamini value associated to that GO term.
GO: Gene ontology.
The potential impact of the Kim and Wilhelm papers on medical and clinical proteomics
With the improvement in proteomics technologies, MS has become an increasingly useful tool for biomedical research, although there is still some way to go before proteomics will be as widely used as gene expression studies. MS-based proteomic studies can help in diagnosis by finding disease-related mutations and biomarkers, such as alternative splice variants. How genomic alterations in tumors and other diseases correlate with proteomics data is still a major unexplored question.
The two Nature studies should have been an important advance because they generated freely available tissue-based maps of the human proteome. If these had been of higher quality, they might have provided a baseline of healthy protein expression, which might then have been used by researchers in conjunction with other large-scale studies such as the Human Proteome Atlas [43] and the Clinical Proteomics Tumor Analysis Consortia ([44]) to find patterns typical of cancers and other diseases and to help make sense of predictions from genome-wide association studies.
Currently, just one group has used the data to help make clinical predictions.[,46] The first paper used the Human Proteome Map from the Kim analysis to predict tissue specificity for 10 possible previously uncharacterized pancreatic cancer targets. The tissue specificity of four of these was supported only by peptides erroneously identified from dubious PSM. The second paper found proteomics evidence for 20 novel Ebola virus-associated proteins from the Kim and Wilhelm studies. At least eight proteins that were supposed to be present in fluids or in tissues related to Ebola (skin, kidney, liver and retina) were identified via poor spectra. Two examples stand out. The expression of a killer cell immunoglobulin-like receptor (KIR2DL4) was highest in urine in ProteomicsDB (from the Wilhelm analysis). But this result is supported by just a single peptide from a dubious PSM, while KIR2DL4 is not detected anywhere else, not even in NK cells. BORA, protein aurora borealis, was found uniquely in platelets. BORA is required for the activation of aurora kinase at the onset of mitosis, yet platelets have no cell nucleus. It should be noted that the authors in these two papers used ProteomicsDB without even the extra filtering step that was introduced in the Wilhelm analysis.
One of the main objectives of medical proteomics is the identification of disease biomarkers, peptides that are specific to a disorder or a number of disorders.[,48] Here proteomics searches of healthy and diseased tissues can help identify peptides unique to the condition being interrogated. Clearly a reliable tissue-based map of the human proteome would be a first step to understand functional differences between diseased and healthy tissues. An understanding of tissue specificity from such a catalogue of the human proteome would be particularly valuable when narrowing down candidate disease biomarkers from large-scale proteomics studies. This catalogue is clearly not yet in place, but these two papers are likely to inspire further research in this direction and the Biology/Disease-driven HPP [49] may provide the mechanisms to support this research. A catalogue of genes and peptides for known healthy tissues would open the door to proteomics searches for biomarkers for a multitude of diseases and cancers.
Differences in gene expression at the protein level, such as changes in signaling pathways that are only found in disease tissues, might also be exploited in the search for biomarkers. In this analysis, the authors found 237 genes that standard proteomics experiments detected in immortal cells, but not in healthy tissue. One example is ERCC6L, a known cell cycle protein. RNAseq data from the Human Protein Atlas [43] backs this up – there is ample RNAseq evidence for ERCC6L in cell lines, but very little evidence in healthy tissues. It may be that proteomics experiments can be used not only to detect biomarker peptides for disease but also to detect the overexpression of a panel of genes specific for a certain condition.
Proteogenomics studies could also identify novel peptide biomarkers such as single-nucleotide variants, aberrant gene fusions, alternative splice variants, post-translational modifications and even noncoding genes. Unless they are designed with care,[,38,50] proteogenomics studies can generate large numbers of false positive identifications,[,52] but they can still have value for identifying biomarkers especially when combined with targeted proteomics,[53] and large-scale proteogenomics studies claim to have identified new cancer-specific peptides.[44]
Expert commentary
The high-profile nature of the publications means that they have generated a lot of interest (between them they have >450 references in little more than a year). The two associated databases provide almost unrestricted access to peptide, protein and tissue data, and ProteomicsDB also integrates RNAseq data and useful analysis tools.
However, the lax use of FDR filters in the two Nature studies leads both papers to vastly overestimate the numbers of coding and noncoding genes that were identified from the experiments. This will have far-reaching consequences including the wasting of annotators and researchers time and resources, and the propagation of false positive identifications in databases. The data will obscure real biological insights, such as the >300 proteins identified solely in tissue-based proteomics studies and has already been used to justify unjustifiable scientific hypotheses.[,54] In the long term, this will undermine confidence in large-scale proteomics data.[32]
Much of the data generated by these two experiments is of poor quality and the danger is that users of the ProteomicsDB and Human Proteome Map databases will be unaware that many thousands of gene identifications and as many as 50% of the peptide identifications are dubious and will use the data without any knowledge of its origin or quality.
Beyond the two papers, the most important issue in large-scale proteomics experiments is the inappropriate use of the standard target-decoy strategy to effectively estimate false positive rates. There are substantial issues with the narrowness of the mass precursor windows used by modern high-resolution mass spectrometers [33,34] and with the identification of post-translational modifications.[34] In large-scale experiments, this is exaggerated by the way in which multiple smaller experiments are combined to make one large-scale experiment [24,25] and in proteogenomics analyses by the size of the search databases.[37] These problems are not limited to the two Nature papers discussed in this review and ideally the community should come up with standards to deal with the growing level of false positive identifications in large-scale experiments. Some work has been carried out in this area,[,25] but there is still no agreed simple way to do this.
Until the proteomics community agrees on a solution to the problem, we would suggest that large-scale proteomics studies (and the researchers that review the papers) follow these simple recommendations. The study should use at least two search engines and include the intersection of the PSMs identified, not the union.[31] If trypsin is used as the cleavage enzyme, the authors should not allow non-tryptic or semi-tryptic peptides (except to identify signal peptide cleavage sites). Nondiscriminating peptides should be discarded, bearing in mind that leucine and isoleucine cannot be discriminated by weight. Genes should be identified by at least two peptides unless it can be shown that detecting more than a single tryptic peptide is unlikely for that gene. Researchers should take care with post-translational modifications [55]; we have found that acetyl and deamidyl modifications in particular cannot be reliably identified. Search databases for noncoding proteins should be of limited size – the greater the size of the noncoding database the more likely it is to find random (incorrect) matches.[,50] In addition, peptides that identify new coding genes should be subject to more robust measures,[37] including the manual checking of all individual spectra.
Five-year view
The publication of these two papers has galvanized the proteomics and annotation communities alike into a series of meetings to discuss the potential implications. There are a series of initiatives underway in the proteomics community to discuss how best to deal with the clear problems in FDR calculations for large-scale experiments, while the claims made for the novel coding regions provided the impetus for bringing together representatives of proteomics and gene annotation communities in a conference in Cambridge with a view to drawing up guidelines for future large-scale proteomics experiments. The conference was a first step in providing a guide as to how proteomics experiments could be used to support the annotation of coding genes and transcript across for the whole annotation community.
Which genes and transcripts code for proteins and which do not is a fundamental scientific question that is still not fully answered. Although the catalogue of standard protein coding genes is likely to be close to completion, there is some debate about how many short open reading frames are protein coding [56] and whether or not there are protein coding regions in long intergenic RNA.[,58] While these are interesting questions, it seems unlikely that current proteomics techniques can provide much help since few of these potential new coding regions are conserved. Standard proteomics experiments detect few peptides for proteins that have evolved recently.[14]
While proteomics techniques will identify few new human protein coding genes, they still have an important role to play in improving gene models. It is interesting to note that all but three of the possible new coding regions we identified from the Kim analysis would extend the gene model by adding coding exons, rather than adding a whole new gene with coding potential. Large-scale proteomics studies also have a role to play in identifying common single amino acid variants and minor annotation errors,[18] while we have shown that proteomics can help determine the dominant cellular isoform for many genes.[40]
The two papers, particularly the Kim analysis, are a first attempt to determine protein tissue specificity via proteomics technology. A proper draft map of the human proteome would clearly require a more rigorous study, but the high-profile nature of these two studies will almost certainly inspire further large-scale tissue-based experiments. It is to be hoped that these studies are more rigorous than the Nature studies, include replicates to avoid the problem of sparse peptide coverage and come with sufficient metadata to allow easy data mining across the different experiments.[59]
Key Issues
The two “drafts of the human proteome” published by Nature substantially overestimate the numbers of protein coding genes they identify.
We believe that the Kim et al. study may have reliably identified <12,000 genes, it is not clear how many the (larger) Wilhelm et al. analysis might have identified.
The reasons for the inflated gene numbers are the incorrect use of nondiscriminating peptides and multiple problems with the calculation of false discovery rates.
A surprisingly large number of peptides in both studies are identified from poor spectra, and these identifications are likely to be incorrect.
This noise from the individual experiments that make up the two studies is amplified when the individual experiments that make up each study are combined.
Few of the novel coding regions identified by the Kim et al. study and almost none of the noncoding genes identified by the Wilhelm et al. study are supported by sufficient peptide evidence to warrant further investigation by manual annotators.
The use of synthetic peptides to confirm peptide identifications requires very careful manual assessment.
Despite the abundant noise in these two analyses, we find that they do identify potentially interesting signals from the tissues they interrogate.
The two papers make their data freely available and the tissue-based nature of the experiments is likely to inspire further research.
The delineation of a baseline of healthy tissue-based expression for the human proteome could have profound implications for biomedical researchers.
Supplementary Material
Financial & competing interests disclosure
This paper is supported by a National Institutes of Health grant [U41 HG007234] and by the Spanish Ministry of Economics and Competitiveness [BIO2012-40205, BIO2012-37926 PRB2-ProteoRed-PT13/0001/0017 RD07-0067-0014-COMBIOMED, RETICS-RD12-0042-0056]. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Supplementary material available online
References
- Papers of special note have been highlighted as:
- • of interest
- •• of considerable interest
- Kim MS, Pinto SM, Getnet D. A draft map of the human proteome. Nature. 2014;509:575–581. doi: 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
•• One of the two papers studied in depth for this article. A proteomics analysis carried out wholly on tissues and hematopoietic cells.
- Wilhelm M, Schlegl J, Hahne H. Mass-spectrometry-based draft of the human proteome. Nature. 2014;509:582–587. doi: 10.1038/nature13319. [DOI] [PubMed] [Google Scholar]
•• The other paper that is the subject of this article. The tissue and fluid proteomics experiments were only a small part of this study.
- Venter JC, Adams MD, Myers EW. The sequence of the human genome. Science. 2001;291 [Google Scholar]
- International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409 [Google Scholar]
- Koenig T, Menze BH, Kirchner M. Robust prediction of the MASCOT score for an improved quality assessment in mass spectrometric proteomics. J Proteome Res. 2008;7:3708–3717. doi: 10.1021/pr700859x. [DOI] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res. 2011;10:1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- UniProt Consortium UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Pruitt KD, Brown GR, Hiatt SM. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 2014;42:D756–763. doi: 10.1093/nar/gkt1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrah T, Deutsch EW, Hoopmann MR. The state of the human proteome in 2012 as viewed through PeptideAtlas. J Proteome Res. 2013;12:162–171. doi: 10.1021/pr301012j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenyö D, Eriksson J, Beavis R. Mass spectrometric protein identification using the global proteome machine. Methods Mol Biol. 2010;673:189–202. doi: 10.1007/978-1-60761-842-3_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiromizu T, Adachi J, Watanabe S. Identification of missing proteins in the neXtProt database and unregistered phosphopeptides in the PhosphoSitePlus database as part of the chromosome-centric human proteome project. J Proteome Res. 2013;12:2414–2421. doi: 10.1021/pr300825v. [DOI] [PubMed] [Google Scholar]
- Geiger T, Wehner A, Schaab C. Comparative proteomic analysis of eleven common cell lines reveals ubiquitous but varying expression of most proteins. Mol Cell Proteomics. 2012;11 doi: 10.1074/mcp.M111.014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezkurdia I, Juan D, Rodriguez JM. Multiple evidence strands suggest that there may be as few as 19,000 human protein-coding genes. Hum Mol Genet. 2014;23:5866–5878. doi: 10.1093/hmg/ddu309. [DOI] [PMC free article] [PubMed] [Google Scholar]
•• A paper that is a counterpoint to the twoNature articles. The authors found that proteomics analyses detect peptides from the most ancient genes and very few from recently evolved genes. Proteins the two Nature studies claimed to have detected will have been removed from the reference genome as a result of this article
- Harrow J, Frankish A, Gonzalez JM. GENCODE: the reference human genome annotation for the ENCODE project. Genome Res. 2012;22:760–774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCBI Resource Coordinators Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2015;43:D16–17. doi: 10.1093/nar/gkz899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeurgt C, Wilkin F, Tarabichi M. Profiling of olfactory receptor gene expression in whole human olfactory mucosa. PLoS One. 2014;9:e96333. doi: 10.1371/journal.pone.0096333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch EW, Sun Z, Campbell D. The state of the human proteome in 2014/2015 as viewed through PeptideAtlas: enhancing accuracy and coverage through the AtlasProphet. J Proteome Res. 2015 doi: 10.1021/acs.jproteome.5b00500. [DOI] [PMC free article] [PubMed] [Google Scholar]
•• Another contrast to the two Nature papers. The PeptideAtlas update very elegantly finds that the two studies add no more than 500 proteins to those already identified in experiments on cell lines.
- Ezkurdia I, Vázquez J, Valencia A. Analyzing the first drafts of the human proteome. J Proteome Res. 2014;13:3854–3855. doi: 10.1021/pr500572z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezkurdia I, Vázquez J, Valencia A. Correction to “Analyzing the first drafts of the human proteome”. J Proteome Res. 2015;14:1991. doi: 10.1021/acs.jproteome.5b00186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
• One of the first papers to propose the calculation of false positive rates using decoy peptides.
- Nesvizhskii AI. A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. J Proteom. 2010;73:2092–1223. doi: 10.1016/j.jprot.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
• A detailed review of the use of false discovery rates in proteomics experiments, showing how errors are amplified when going from peptide to protein level.
- Serang O, Käll L. Solution to statistical challenges in proteomics is more statistics, not less. J Proteome Res. 2015 doi: 10.1021/acs.jproteome.5b00568. [DOI] [PubMed] [Google Scholar]
- Reiter L, Claassen M, Schrimpf SP. Protein identification false discovery rates for very large proteomics data sets generated by tandem mass spectrometry. Mol Cell Proteomics. 2009;8:2405–2417. doi: 10.1074/mcp.M900317-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitski MM, WIlhelm M, Hahne H. A scalable approach for protein false discovery rate estimation in large proteomic data sets. Mol Cell Proteomics. 2015 doi: 10.1074/mcp.M114.046995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudet P, Michel PA, Zahn-Zabal M. The neXtProt knowledgebase on human proteins: current status. Nucleic Acids Res. 2015;43:D764–770. doi: 10.1093/nar/gku1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colaert N, Van Huele C, Degroeve S. Combining quantitative proteomics data processing workflows for greater sensitivity. Nat Methods. 2011;8:481–483. doi: 10.1038/nmeth.1604. [DOI] [PubMed] [Google Scholar]
- Paulo JA. Practical and efficient searching in proteomics: a cross engine comparison. Webmedcentral. 2013;4:WMCPLS0052. doi: 10.9754/journal.wplus.2013.0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr S, Aebersold R, Baldwin M. The need for guidelines in publication of peptide and protein identification data: working group on publication guidelines for peptide and protein identification data. Mol Cell Proteomics. 2004;3:531–533. doi: 10.1074/mcp.T400006-MCP200. [DOI] [PubMed] [Google Scholar]
- Omenn GS, States DJ, Adamski M. Overview of the HUPO plasma proteome project: results from the pilot phase with 35 collaborating laboratories and multiple analytical groups, generating a core dataset of 3020 proteins and a publicly-available database. Proteomics. 2005;5:3226–3245. doi: 10.1002/pmic.200500358. [DOI] [PubMed] [Google Scholar]
- Shteynberg D, Nesvizhskii AI, Moritz RL. Combining results of multiple search engines in proteomics. Mol Cell Proteomics. 2013;12:2383–2393. doi: 10.1074/mcp.R113.027797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White FM. The potential cost of high-throughput proteomics. Sci Signal. 2011;4:pe8. doi: 10.1126/scisignal.2001813. [DOI] [PubMed] [Google Scholar]
• This paper sets out the potential harmful effects of combining large-scale high-throughput proteomics and insufficiently validated data.
- Cooper B. The problem with peptide presumption and the downfall of target-decoy false discovery rates. Anal Chem. 2012;84:9663–9667. doi: 10.1021/ac303051s. [DOI] [PubMed] [Google Scholar]
• Explains how recent advances in high-throughput proteomics can easily lead to identifying peptides that do not exist.
- Bonzon-Kulichenko E, Garcia-Marques F, Trevisan-Herraz M. Revisiting peptide identification by high-accuracy mass spectrometry: problems associated with the use of narrow mass precursor windows. J Proteome Res. 2015;14:700–710. doi: 10.1021/pr5007284. [DOI] [PubMed] [Google Scholar]
• The authors set out solutions for the problems identified in Ref. [33]
- Omenn GS, Lane L, Lundberg EK. Metrics for the human proteome project 2015: progress on the human proteome and guidelines for high-confidence protein identification. J Proteome Res. 2015 doi: 10.1021/acs.jproteome.5b00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
•• Details many of the shortcomings of the two Nature analyses and addresses the state of the art in protein detection.
- Horvatovich P, Lundberg EK, Chen YJ. Quest for missing proteins: update 2015 on chromosome-centric human proteome project. J Proteome Res. 2015 doi: 10.1021/pr5013009. [DOI] [PubMed] [Google Scholar]
- Nesvizhskii AI. Proteogenomics: concepts, applications and computational strategies. Nat Methods. 2014;11:1114–1125. doi: 10.1038/nmeth.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
• The paper discusses the concepts and potential pitfalls of proteogenomics studies in considerable detail.
- Krug K, Carpy A, Behrends G. Deep coverage of the Escherichia coli proteome enables the assessment of false discovery rates in simple proteogenomic experiments. Mol Cell Proteomics. 2013;12:3420–3430. doi: 10.1074/mcp.M113.029165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross PL, Huang YN, Marchese JN. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- Ezkurdia I, Rodriguez JM, Carrillo-de Santa Pau E. Most highly expressed protein-coding genes have a single dominant isoform. J Proteome Res. 2015;14:1880–1887. doi: 10.1021/pr501286b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abascal F, Ezkurdia I, Rodriguez-Rivas J. Alternatively spliced homologous exons have ancient origins and are highly expressed at the protein level. PLoS Comput Biol. 2015;11:e1004325. doi: 10.1371/journal.pcbi.1004325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Zhang B, Wang J, Wang X. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014;513:382–387. doi: 10.1038/nature13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
• This large-scale study concentrates on cancer cells instead of tissues. Combining large-scale proteomics analysis of healthy and diseased cells has promise for the detection of biomarkers.
- Narayanan R. Phenome-genome association studies of pancreatic cancer: new targets for therapy and diagnosis. Cancer Genom Proteom. 2015;12:9–19. [PubMed] [Google Scholar]
- Narayanan R. Ebola-associated genes in the human genome: implications for novel targets. MOJ Proteom Bioinform. 2015;1:00032. [Google Scholar]
- Shao S, Guo T, Aebersold R. Mass spectrometry-based proteomic quest for diabetes biomarkers. Biochim Biophys Acta. 2015;1854:519–527. doi: 10.1016/j.bbapap.2014.12.012. [DOI] [PubMed] [Google Scholar]
• In this work, the authors review the current status of diabetes mellitus biomarker discovery through different mass spectrometry techniques.
- Hathout Y. Proteomic methods for biomarker discovery and validation. Are we there yet? Expert Rev Proteom. 2015;12:329–331. doi: 10.1586/14789450.2015.1064771. [DOI] [PubMed] [Google Scholar]
• A review detailing recent advances in the discovery of protein biomarkers via proteomics and the difficulties of validating these biomarkers.
- Aebersold R, Bader GD, Edwards AM. The biology/disease-driven human proteome project (B/D-HPP): enabling protein research for the life sciences community. J Proteome Res. 2013;12:23–27. doi: 10.1021/pr301151m. [DOI] [PubMed] [Google Scholar]
- Zhang K, Fu Y, Zeng WF. A note on the false discovery rate of novel peptides in proteogenomics. Bioinformatics. 2015;31:3249–3253. doi: 10.1093/bioinformatics/btv340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Ward CC, Jungreis I. Discovery of human sORF-encoded polypeptides (SEPs) in cell lines and tissue. J Proteome Res. 2014;13:1757–1765. doi: 10.1021/pr401280w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderperre B, Lucier JF, Bissonnette C. Direct detection of alternative open reading frames translation products in human significantly expands the proteome. PLoS One. 2013;8:e70698. doi: 10.1371/journal.pone.0070698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brusniak MY, Chu CS, Kusebauch U. An assessment of current bioinformatic solutions for analyzing LC-MS data acquired by selected reaction monitoring technology. Proteomics. 2012;12:1176–1184. doi: 10.1002/pmic.201100571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Colak R, Teyra J. Semi-supervised learning predicts approximately one third of the alternative splicing isoforms as functional proteins. Cell Rep. 2015;12:183–189. doi: 10.1016/j.celrep.2015.06.031. [DOI] [PubMed] [Google Scholar]
- Fu Y, Qian X. Transferred subgroup false discovery rate for rare post-translational modifications detected by mass spectrometry. Mol Cell Proteomics. 2014;13:1359–1368. doi: 10.1074/mcp.O113.030189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Q, Ma J, Saghatelian A. Identification and characterization of sORF-encoded polypeptides. Crit Rev Biochem Mol Biol. 2015;50:134–141. doi: 10.3109/10409238.2015.1016215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Russell P, Ingolia NT. Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins. Cell. 2013;154:240–251. doi: 10.1016/j.cell.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
• The authors find that lincRNA behave differently from protein coding transcripts when passing through the ribosome
- Ruiz-Orera J, Messeguer X, Subirana JA. Long non-coding RNAs as a source of new peptides. Elife. 2014;3:e03523. doi: 10.7554/eLife.03523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griss J, Perez-Riverol Y, Hermjakob H. Identifying novel biomarkers through data mining-a realistic scenario? Proteom Clin Appl. 2015;9:437–443. doi: 10.1002/prca.201400107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatun J, Yu Y, Wrobel JA. Whole human genome proteogenomic mapping for ENCODE cell line data: identifying protein-coding regions. BMC Genom. 2013;14:141. doi: 10.1186/1471-2164-14-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.