

Abstract
Rationale
The let-7 family of microRNAs (miRs) regulates critical cell functions, including survival signaling, differentiation, metabolic control and glucose utilization. These functions may be important during myocardial ischemia. MiR-let-7 expression is under tight temporal and spatial control through multiple redundant mechanisms that may be stage-, isoform- and tissue-specific.
Objective
To determine the mechanisms and functional consequences of miR-let-7 regulation by hypoxia in the heart.
Methods and Results
MiR-let-7a, -7c and -7g were downregulated in the adult mouse heart early after coronary occlusion, and in neonatal rat ventricular myocytes subjected to hypoxia. Let-7 repression did not require glucose depletion, and occurred at a post-transcriptional level. Hypoxia also induced the RNA binding protein Lin28, a negative regulator of let-7. Hypoxia induced neither Lin28 induction nor miR-let-7 repression in cardiac fibroblasts. Both changes were abrogated by treatment with the histone deacetylase inhibitor trichostatin A. Restoration of let-7g to hypoxic myocytes and to ischemia-reperfused mouse hearts in vivo via lentiviral transduction potentiated the hypoxia-induced phosphorylation and activation of Akt, and prevented hypoxia-dependent caspase activation and death. Mechanistically, phosphotidyl inositol 3’kinase interacting protein 1 (PIK3IP1), a negative regulator of PI3K, was identified as a novel target of miR-let-7 by a crosslinking technique showing that miR-let-7g specifically targets PI3KIP1 to the cardiac myocyte Argonaute complex RISC. Finally, in non-failing and failing human myocardium, we found specific inverse relationships between Lin28 and miR-let-7g, and between miR-let-7g and PIK3IP1.
Conclusion
A conserved hypoxia-responsive Lin28-miR-let-7-PIK3IP1 regulatory axis is specific to cardiac myocytes and promotes apoptosis during myocardial ischemic injury.
Keywords: ischemia-reperfusion, apoptosis, let-7, Lin28, AKT, PIK3IP1
1. INTRODUCTION
A principal feature of myocardial infarction is the loss of cardiac myocytes through apoptosis and other modes of cell death; cell loss may be further exacerbated by oxidative stress during reperfusion [1, 2]. The resultant damage to the heart muscle compromises cardiac function and increases the likelihood of developing congestive heart failure. Preventing myocyte loss is therefore essential to minimizing the adverse effects of myocardial infarction.
The myocardium adapts to ischemia by switching from β-oxidative metabolism to glycolysis [3, 4]. Anaerobic glycolysis may permit short-term cell survival, but results in the production of acidic waste products and a fall in intracellular pH. Ischemia is also accompanied by increased generation of reactive oxygen species [5–7]. This acidic, oxidative environment leads to loss of cardiac myocytes and expansion of the area of myocardial damage[8]. At the same time, ischemia induces a wide variety of myocardial signaling pathways that can either mitigate or promote cell death [8, 9]. One protective pathway involves activation of phosphoinositide 3 kinase (PI3K), which activates the serine–threonine kinase Akt/PKB, an important regulator of cell growth and survival [10]. PI3K-dependent phosphorylation of AKT on serine 473 leads to its activation and phosphorylation of multiple AKT targets, including glycogen synthase kinase-β (GSKβ), reducing damage from myocardial ischemia and reperfusion in vitro and in vivo [11–13].
MicroRNAs (miRs) are short (~21 nt) evolutionarily conserved non-protein coding RNAs that inhibit gene expression by impeding translation from messenger RNA templates or by destabilizing mRNA in a sequence-specific manner. MicroRNAs are now understood to exert regulatory control over many important aspects of cardiovascular biology, including the response to ischemic stress, myocyte growth, differentiation and metabolism [14–17]. Among the first microRNAs to be identified and characterized were members of the miR-let-7 family[18]. The mammalian miR-let-7 family includes at least 14 members with a common seed sequence and minor sequence differences denoted by small letters (-7a, -7b etc.). Some let-7 species, e.g. let-7a, are encoded at multiple different genomic loci, denoted by numbers (e.g. let-7a-1, let-7a-2). All are probably functionally redundant [19]. Mammalian let-7 species are widely tissue-distributed and exhibit temporal regulation, increasing during development and differentiation.
The timing of miR-let-7 expression is determined both transcriptionally and post-transcriptionally. The mechanisms regulating let-7 differ depending on isoform-specific factors, on developmental stage and tissue type[20]. In developing brain and differentiating neurons, precursor let-7a, -c and -e transcripts were found to be abundant at all stages, but the mature transcripts were only measurable in mature cells[21]. Terminal loop structures in let-7 precursor forms are specifically bound by the RNA binding protein LIN28, blocking their processing by Drosha and Dicer and/or inducing their degradation [22–24]. Like let-7, Lin28 is strongly conserved through evolution from worms to humans, and its regulatory properties are similarly conserved. Through repression of let-7, LIN28 exerts control over multiple fundamental cell processes, including cell fate decisions, pluripotency, and tissue regeneration (Reviewed in [25, 26]). Thus, multiple redundant mechanisms help to achieve specific local and time-dependent regulation of let-7.
Although let-7 has been implicated in the regulation of various cardiovascular processes, it is not clear how let-7 functions nor how it is regulated in the heart. In keeping with the known role of let-7 in differentiation, upregulation of let-7c accompanies maturation of the developing heart, and repression of let-7 permits cardiomyocyte de-differentiation and regeneration [27, 28]. Some let-7 species may participate in the response to hypertrophic signals [29], and miR-let-7 is dysregulated in the failing human heart [30]. In other cell types, miR-let-7 has been shown to regulate genes involved in glucose metabolism and glycolysis [19, 28, 31, 32] and inflammation[33]. It is not known whether let-7 assists in the metabolic adaptation to hypoxia.
Here we show that hypoxia activates LIN28, resulting in the repression of multiple miR-let-7 species both in culture and in vivo. Notably, these responses to hypoxia are seen only in cardiac myocytes and not in cardiac fibroblasts. We show that the hypoxia-mediated loss of let-7 increases myocyte vulnerability to apoptosis during ischemia-reperfusion injury, and that restoring (“rescuing”) let-7g protects myocytes from apoptosis. Mechanistically, we identify a novel target of miR-let-7g, PIK3IP1, that acts as an inhibitor of Akt survival signaling and whose levels increase with loss of miR-let-7 both in mouse models and in human myocardium.
2. EXPERIMENTAL PROCEDURES
Primary culture of rat neonatal cardiomyocytes
Methods for primary culture of neonatal rat cardiac myocytes and fibroblasts have been previously described [34]. Protein kinases were inhibited using selective small molecule enzyme activity blockers at concentrations chosen to maximize specificity of action and minimize off-target effects, as reported [35, 36].
Lentiviral Transduction
Transduction of NRVMs was carried out using lentiviral pLemir-let-7g or a non-silencing control vector (Open Biosystems) as described in [37]. Lentiviral transduction of neonatal mouse pups was carried out exactly as in [37] using the same lentiviral vectors.
In vivo transduction and ischemia-reperfusion surgery
All experiments were performed on wild type C57BL/6 mice under protocols approved by Institutional Review Board at University of Miami. Lentiviral transduction of neonatal pups was carried out described in. Briefly, 2-day neonatal pups were placed on a transilluminator and 15x108 lentiviral vectors described earlier were injected using the external jugular vein. Mice were sacrificed 10 days and 9–10 weeks post injection. RNA and protein from whole heart was used to access the efficiency of injection. Ischemia-reperfusion was carried out as described in [38] with minor changes. Briefly, at 9–10 weeks of age, mice were anaesthetized with a cocktail of 40mg/kg ketamine and 5mg/kg xylocaine. Following thoracotomy, LAD artery was visualized and occluded using an 8-0 silk suture for 60 minute. Reperfusion was accomplished by releasing the suture and the chest wall was closed. After a reperfusion period of 24 hour, cardiac function was assessed using echocardiography.
PAR-CLIP
RNA immunoprecipitation was carried out as described in [39] with minor changes.
Human tissue samples
Anonymized left ventricular myocardial samples and associated clinical information were harvested within 4 hours post-demise or explantation and obtained through the Cooperative Human Tissue Network under a protocol approved by the UM Institutional Review Board. Samples were maintained at −80°C until use.
Statistical analysis
For all studies, analysis of variance (ANOVA) was used for comparison of multiple results within a single experiment, followed by Student's t-test using one- or two-tailed distributions as appropriate.
3. RESULTS
3.1. Ischemic stress initiates acute changes in miR-let-7 expression in the myocardium
In order to identify microRNAs regulated during acute myocardial ischemia, we compared ischemic and adjacent non-ischemic left ventricular myocardium in mice 2 hours after permanent coronary artery occlusion [40]. Induction of VEGF protein was used to confirm ischemia (not shown) [41, 42]. Among the relatively small number of differentially regulated microRNAs in this model were multiple miR-let-7 family members, including let-7c, let-7g and let-7a (Figure 1A), suggesting an important role for miR-let-7 in the early adaptive response to ischemia.
Figure 1. Cell type-specific repression of miR-let-7 by hypoxia in cardiac myocytes.

A. Acute myocardial ischemia in vivo represses multiple miR-let-7 species. MiR-let-7c,-7g and -7a were quantitated in mouse myocardium 2hr after LAD ligation (black bars) or a sham procedure (white bars) using a Taqman low-density microfluidic array (n=3). B. Myocyte-specific repression of miR-let-7c is mediated by hypoxia. Control neonatal rat ventricular myocytes were cultured under normoxia with 5 mM glucose. Experimental groups included normoxia without glucose (LG, diamonds), hypoxia with glucose (circles) and hypoxia without glucose (triangles). Cardiac fibroblasts (squares) were cultured under normoxia with glucose. MiR-let-7c was quantitated by RT-PCR at 4, 8 and 24 hours. C. Hypoxia represses miR-let-7g and -7a. CMs were cultured as in (B) and miRNAs were quantitated at 24 hour. For (B) and (C), graphs display miRNA expression levels in experimental conditions relative to normoxic cells (n=5). D. Hypoxia downregulates miR-let-7 at post-transcriptional level. Primary and precursor miR-let-7c transcripts were quantitated by qPCR in CM cultured as in (B) and expressed relative to those in normoxic CMs (n=6) E. Chemical induction of HIF1 is insufficient to downregulate let-7. Cardiac myocytes were subjected to treatment with 50mM cobalt chloride (CoCl2) and vehicle (NoCo), and expression of HO-1 protein and miR-let-7c were assayed by immunoblot and/or RT-PCR respectively. (top) Representative Western blot. (bottom) HO-1 protein/β-actin and miR-let-7c transcript levels (n=6). Hypoxia treated CM serves as positive control. F. Hypoxic downregulation of let-7 requires MAPK/ERK and class I-II histone deacetylase activity. MiR-let-7c levels under hypoxic NRVMs in the presence and absence of the non-selective HDAC inhibitor trichostatin A (TSA) (10nM) and the MEK1/2 inhibitor U0126 (U0, (10μM)) (n=4). Norm= normoxic culture.
Because miR-let-7 is ubiquitously expressed, we asked whether ischemic let-7 downregulation occurred equally in myocytes and non-myocytes (mostly fibroblasts) within the myocardium. Neonatal rat ventricular myocytes (NRVMs) and cardiac fibroblasts (CFs) derived from the pre-plating step in the same preparation were placed under normoxic or hypoxic (0.5% O2, 5% CO2, balance nitrogen) as previously described[34]. In myocytes, miR-let-7c levels declined in a time-dependent manner between 4 and 24 hours of hypoxia (Figure 1B). This decline was not seen in parallel cultures of fibroblasts (Figure 1B). Reducing media glucose (0.5mM) had no additional effect, and low glucose alone was not sufficient to downregulate let-7 (Figure 1B), indicating that miR-let-7 downregulation was dependent on hypoxia alone. Similar reductions were seen with miR-let-7g and miR-let-7a (Figure 1C).
In looking for mechanisms of miR-let-7 downregulation, we found that precursor forms of let-7c were differentially affected. The processed precursor pre-miR-let-7c decreased, but levels of the primary transcript pri-miR-let-7c were not changed by hypoxia (Figure 1D). To determine whether miR-let-7 was repressed by a hypoxia-inducible-factor-1 (HIF-1)-dependent mechanism, we used cobalt chloride (CoCl2) to achieve non-hypoxic induction of HIF-1[43, 44]. HIF-1 activation was confirmed by upregulation of the known HIF-1 target heme oxygenase 1 (HO-1) (Figure 1E). However, mature miR-let-7c levels were not reduced by CoCl2, (Figure 1E, bottom), suggesting that HIF-1 is unlikely to be directly involved.
In other systems, miR-let-7 has been reported to be suppressed by MEK1/2 activation [45], and by histone deacetylase activity[46]. Congruent with these reports, we found that treatment with either the MEK1/2 inhibitor UO126 or the class I, II HDAC inhibitor trichostatin A (TSA) prevented hypoxic downregulation of miR-let-7c (Figure 1F).
3.2. Ischemic stress induces Lin28 expression in the myocardium
Mature microRNAs are generated through sequential transcriptional and enzymatic processing steps, each of which may be subject to regulation (reviewed in [47, 48]). The lack of impact on pri-miR-let-7 indicated a likely post-transcriptional regulatory mechanism. The RNA binding protein Lin28 regulates levels of mature miR-let-7 species in many tissues and organism [32, 49–51]. We observed that Lin28 protein was induced in ischemic myocardium (Figure 2A), and in hypoxic NRVMs (Figure 2B) in a time-dependent manner, along with increased Lin28 mRNA (Figure 2C). Hypoxia failed to induce Lin28 in cardiac fibroblasts (Figure 2D). These findings suggest that hypoxia represses miR-let-7 through myocyte-specific induction of Lin28 Of note, hypoxic induction of Lin28 was prevented by the HDAC inhibitor TSA (Figure 2E) indicating that hypoxia may regulate the myocyte Lin28/let7 axis through elevated HDAC activity.
Figure 2. Myocyte-specific induction of Lin28 by hypoxia.

A. Acute myocardial ischemia in vivo induces Lin28. Lin28 was assayed by immunoblot in non-ischemic (white bar) and ischemic (black bar) zones of murine left ventricle following a 2-hour LAD occlusion. (Top) Representative Western blot. (Bottom) Graph summarizing 3 experiments. B. Myocyte-specific induction of Lin28 is mediated by hypoxia. NRVMs were cultured under normoxic and hypoxic conditions for the indicated times and assayed for Lin28 by immunoblot. (Top) Representative Western blot. (Bottom) Graph summarizing 3 experiments. C. Hypoxia induces Lin28 mRNA in NRVMs. NRVMs were cultured in normoxic and hypoxic conditions and mRNA levels measured using RT-PCR (n=6). D. Hypoxia does not induce Lin28 in cardiac fibroblasts. Fibroblasts (CFs) were exposed to normoxic and hypoxic cultures for 24-hour and protein samples were assayed using immunoblot. (Top) Representative Western blot. (Bottom) Graph summarizing 4 experiments. For B, C and D, Lin28 levels are expressed relative to levels in normoxic cells. E. Lin28 induction in hypoxia requires class I-II histone deacetylase activity. Lin28 was assayed by immunoblot in normoxic and hypoxic myocytes cultured in the presence and absence of TSA (10nM). (Top) Representative Western blot. (Bottom) Graph summarizing 7 experiments. Lin28 protein was normalized to β-actin. Norm=normoxic cultures.
3.3. MiR-let-7 regulates cardiac myocyte cell death under hypoxic stress
Our results suggest that hypoxia induces a gradual, time-dependent downregulation of let-7 in cardiac myocytes that continues over at least 24 hours. To assess the consequences of miR-let-7 loss in sustained hypoxia, we adopted a rescue strategy to restore miR-let-7g levels to normoxic levels in hypoxic myocytes, using lentiviral transduction. As expected, exogenous miR-let-7g was also subject to negative post-transcriptional control. However, we were able to achieve comparable levels of mature miR-let-7g in normoxic and hypoxic NRVM (Figure 3A).
Figure 3. MiR-let-7 rescue prevents hypoxia-induced myocyte death by sustaining cardio-protective signaling.

A. Lentiviral miR-let-7g transduction rescues miR-let-7g expression levels in hypoxic NRVMS. NRVMs were cultured as described above. 48 hours after transduction with lentiviral vectors expressing NT (non-targeting) and miR-let-7g, myocyte levels of miR-let-7g were measured using RT-PCR (n=5). Mature sequences for let-7g and let-7g are provided for comparison. B. miR-let-7g rescue prevents myocyte apoptosis during hypoxia. Protein levels of caspase3 and its cleavage products were assayed using immunoblot. LDH release was measured by colorimetric assay as described in Methods, after 24 hours of hypoxia. (Top and bottom left) Representative Western blot and quantitation. (Bottom right) Quantitation of LDH release (n=6-9). C. miR-let-7g rescue promotes Akt activation in hypoxia. NRVMs transduced with lenti-NT and lenti-let-7g were assayed after 24h for pSer473-AKT, pSer9-GSKβ, total GSKβ and total AKT by immunoblot. (Left) Representative Western blots. (Center and right) Quantitation of pAKT and pGSK (n=6-9 per treatment group). All phosphoprotein levels are normalized to their respective total proteins.
We initially hypothesized that loss of miR-let-7 might affect cell fate decisions in NRVMs during hypoxia. Overexpression of miR-let-7g in normoxic cells did not affect basal rates of cell death or apoptosis as determined by LDH release and caspase-3 cleavage, respectively (Figure 3B). In contrast, both cell death and apoptosis increased in hypoxic cells transduced with a non-targeting (NT) vector, but not in cells rescued with the miR-let-7g vector (Figure 3B), implying that apoptotic signaling in hypoxic myocytes is due in part to loss of miR-let-7.
The serine/threonine kinase Akt plays a critical role in cardiomyocyte survival under stress [11–13]. AKT is initially activated under hypoxia and then progressively inhibited through a negative feedback mechanism involving the upstream adaptor IRS-1 [52, 53]. Consistent with this, we observed a decline in the activating phosphorylation of Akt at serine 473, as well as reduced phosphorylation on Ser9 of GSKβ, an Akt substrate (Figure 3C), during hypoxia (Figure 3C). Transduction of miR-let-7g, but not the NT vector, prevented the hypoxic decline of both pSer473-Akt and pSer9-GSKβ. These findings suggest that restoring miR-let-7 during hypoxia protects against myocyte apoptosis by permitting AKT activation.
We next asked whether miR-let-7g-mediated cytoprotection was dependent on PI3K, an important upstream activator of Akt. The miR-let-7g-dependent increases in pSer473-Akt and pSer9-GSKβ were abrogated by the selective PI3K inhibitor LY-294-002, but not by its vehicle. Similarly, the protective effect of miR-let-7g rescue against caspase 3 cleavage was abrogated by LY-294-002 (Figure 4). These findings demonstrate that loss of miR-let-7g in hypoxia leads to loss of survival signaling through PI3K, upstream of AKT.
Figure 4. MiR-let-7g regulates AKT activation and cell survival through the PI3K pathway.

NRVMs were cultured and transduced as before and treated with 10μM PI3K inhibitor LY 294-002 (LY) or vehicle control. Protein samples were assayed for phosphorylated GSKβ at serine9, cleaved caspase3 and total caspase3 using immunoblot. A. PI3K inhibitor LY-294-002 (LY) reverses cytoprotective effects of let-7 rescue. (Above) Representative Western blot. (Below) Graphs summarize results from 3 experiments. B, C, D. PI3K inhibition reverses effect of let-7 rescue on Akt Ser473 phosphorylation. B. Representative Western blots. C. Quantitation of pAkt. D. Quantitation of pGSK. All phosphoproteins were normalized to their respective total proteins. N = 6 per condition.
3.4. MiR-let-7 regulates cell fate decisions in the ischemic myocardium in vivo
The effects of miR-let-7 on Akt and apoptosis were confirmed in vivo using the same lentiviral vectors in a neonatal mouse transduction protocol [37, 54]. Transduction achieved a functionally significant increase in miR-let-7g, together with downregulation of the established let-7 target INSRβ (Figure 5A, left and right). Elevated levels of miR-let-7g were sustained to at least 9–10 weeks of age (Figure 5B). Although miR-let-7 transgenic mice have been reported to have smaller body weights [19], we did not any observe difference in weight between NT- and miR-let-7g-overexpressing mice.
Figure 5. miR-let-7g protects myocardium from acute ischemia-reperfusion injury by increasing AKT activation.

A. Lentiviral transduction of miR-let-7g in myocardium in vivo. Transcripts levels of miR-let-7 family members and protein levels of INSRβ, a validated miR-let-7 target, were assayed in whole heart tissue using RT-PCR and immunoblot 10 days after lentiviral vector injection. (Left) Transcript levels relative to lenti-NT-transduced animals. (Above) Representative Western blot. B. miR-let-7g expression is sustained to adulthood. MiR-let-7g transcript levels were measured in whole hearts 9–10 weeks after transduction (n=3-5). C. Increasing miR-let-7g reduces apoptosis in ischemia-reperfusion injury. Sections from sham-operated hearts, and from non-ischemic and ischemic zones of NT- and miR-let-7g-transduced mice were examined for apoptotic cells following 60 minutes of ischemia and 24 hours of reperfusion as described in Experimental Procedures. (Left) Representative sections and (top) enlargements. Sham = sham-operated. I/R NI = Ischemia-reperfusion, non-ischemic zone. I/R-NT, I/R-let-7g = I/R + non-targeting and let-7g vectors respectively. (Bottom right) Quantitation of apoptotic nuclei (n=2500–4000 nuclei) in n = 4 (NT) or 5 (let-7g) individual mice. D. AKT is activated in vivo with miR-let-7g overexpression. Non-ischemic and ischemic zones of hearts from NT and miR-let-7g-transduced animals were assayed for pSer473-Akt, total Akt and β-actin. (Left) Representative Western blots. (Right) Graph summarizes data from n=4 (NT) and n=5 (let-7g) mice.
Lentivirus-transduced mice were then subjected to an ischemia-reperfusion injury protocol [40] at 9–10 weeks of age. NT-expressing myocardium contained large numbers of apoptotic cells in the ischemic zone, while miR-let-7g-expressing mice had low rates of apoptosis that were indistinguishable between ischemic and non-ischemic zones (Figure 5C). While we did not specifically differentiate between apoptotic myocytes and fibroblasts, myocytes constitute the vast majority of apoptotic cells following ischemia-reperfusion injury [2, 55], whereas fibroblasts are relatively resistant to apoptosis under the same conditions [56]. Transduction of miR-let-7g, but not the NT sequence, was associated with increased pSer473 AKT in the ischemic myocardium (Figure 5D). Although let-7 “rescue” did not significantly affect calculated ejection fraction, both diastolic and systolic volumes were improved in these mice compared with mice receiving the non-targeting control vector (LViVd 71.9 ± 1.1 vs. 66.5 ± 2.4, p= 0.039; LViVs 22.8 ± 0.4 vs. 20.6 ± 0.91, p = 0.05, NT vs. Let7 n = 4,5) (Supplemental Table 1), consistent with reduced post-infarct remodeling.
3.5. MiR-let-7 regulates PI3K signaling by direct targeting of PIK3IP1
Phosphatidylinositol 3’ kinase (PI3K) transmits an activating signal to Akt, and PI3K inhibition by phosphatidylinositol 3’ kinase interacting protein 1 (PIK3IP1) results in decreased AKT phosphorylation [57–59]. The PIK3IP1 3’UTR contains a predicted binding site for miR-let-7, and miR-let-7-induced degradation of PIK3IP1 would be predicted to increase AKT phosphorylation. Concomitant with loss of miR-let-7c/g, PIK3IP1 expression was increased in hypoxic NRVMs (Figure 6A). CMs expressing lenti-miR-let-7g had significantly decreased mRNA levels of Lin28, a previously validated target of miR-let-7 and PIK3IP1 compared to CMs expressing the NT sequence (Figure 6B). To validate PIK3IP1 as a direct miR-let-7g target in cardiac myocytes, we used a direct biochemical approach based on Photoactivable Ribonucleoside Crosslinking and Immunoprecipitation (PAR-CLIP) [39], a robust method for validating miR and target interaction in the cardiac RISC[60]. Myocyte RISC complexes were programmed with miR-let-7g or a non-targeting sequence using lentiviral vectors, and Argonaute 2 (Ago2), a major component of the RISC, was immunoprecipitated from NRVM lysates 48 hours later. Co-precipitated RNA was extracted and analyzed by RT-PCR for the presence of potential miR-let-7 targets. In myocytes programmed with the miR-let-7g vector, miR-let-7 was enriched in Ago2-associated immunoprecipitates relative to NT-transduced cells; unrelated miR-142-5p and snoRNA species were not (Figure 6C, top). PIK3IP1 mRNA was also significantly enriched in the miR-let-7g-programmed RISC, as was Lin28, while control β-actin and 18S RNAs were not (Figure 6C, bottom). The wild type 3’UTR of PIK3IP1 was also functionally targeted by miR-let-7g but not NT sequence (Figure 6D). Thus, in cardiac myocytes, miR-let-7g promotes Akt activation and cell survival by directly targeting PIK3IP1 to the RISC.
Figure 6. MiR-let-7g regulates PI3K signaling to modulate AKT activation by targeting PIK3IP1.

A. PIK3IP1 is induced by hypoxia in NRVMs. NRVMs were cultured in normoxia and hypoxia and mRNA levels of PIK3IP1 were measured using RT-PCR (n=4). B. miR-let-7g modulates mRNA levels of PIK3IP1. NRVMs were cultured in normoxia and transduced with lenti-let-7g and NT vector and mRNA levels of Lin28 and PIK3IP1 were measured using RT-PCR (n=4). C. miR-let-7g targets PIK3IP1 to cardiac myocyte RISC. NRVMs were cultured in normoxia and transduced with lenti-let-7g and NT vector so as to program the myocyte RISC. Following immunoprecipitation with Ago2 antibody as described in Methods levels of various miRs and genes were quantified in the immunoprecipitated RNA fraction using RT-PCR. (top) Transcript levels of miR-let-7g, miR-142 and snoRNA. (bottom) mRNA levels of PIK3IP1, Lin28, β-actin and 18S. Transcript levels were normalized to immunoprecipitated AGO2 protein (n=3). D. miR-let-7g functionally targets WT PIK3IP1 3′fUTR. 293Ts expressing miR-let-7g and NT were transfected with pGLO dual luciferase vector. Renilla luciferase units were normalized to firefly luciferase units (n=5).
3.6. MiR-let-7 modulates its target genes in human heart failure
Previous studies have separately shown that that miR-let-7 family members are dysregulated in human heart failure [30] and that Lin28 expression is upregulated in the ischemic human heart [61]. To obtain evidence for a functional LIN28-let-7 regulatory axis in human heart failure, we determined the protein levels of Lin28 and mature transcript levels of miR-let-7g and miR-let-7a in a series of failing and non-failing human hearts. Clinical characteristics of samples are provided in Table 1. A significant inverse relationship was found between Lin28 expression and levels of both let-7 family members (Figure 7A, left and right, respectively). Likewise, PIK3IP1 protein content varied inversely with miR-let-7 family members, miR-let-7g and miR-let-7a in the same human heart failure samples (Figure 7B, left, and right, respectively). The inverse relationship between let-7g and both Lin28 and PIK3IP1 was observed across all samples and was particularly evident in the failing heart group. In contrast, there was no relationship between Lin28 or PIK3IP1 and levels of the unrelated miR-141 (Figure 7C left and right, respectively).
Table 1.
Characteristics of human subjects analyzed for this study.
| ID | Gender | Condition | Heart Weight (g) | Age | Ethnicity | Notes |
|---|---|---|---|---|---|---|
| 39306A1D | Male | non-failing | 414 | 55 | White | Normal anatomy |
| 322A20D | Male | non-failing | N/A | 46 | Black | Stent in LAD, apical scarring |
| 51565T_004 | Male | non-failing | 472 | 40 | White | Triple vessel CAD, septal infarct |
| 1030528A2 | Male | ICM | 518 | 42 | Black | Biventricular dilatation and hypertrophy.interstitial fibrosis |
| 1050968B3 | Male | Hypertrophic,failing | 840 | 51 | Unknown | |
| 1040221A2 | Female | Hypertrophic,failing | 280 | 33 | White | Stent in LAD, aortic valve prosthesis |
| 1050508A3 | Female | CHF | unknown | 59 | White | Posterior infarction, fibrosis |
| 1040319A3 | Male | ICM, aortic valve disease | 548 | 67 | White | Concentric hypertrophy, patchy fibrosis, ICD in place |
| 1030952A3 | Male | ICM | 547 | 62 | White | Triple vessel CAD,septal infarct |
Figure 7. miR-let-7 microRNA family members modulate levels of Lin28 and PIK3IP1 in human myocardium.

A. miR-let-7 family members inversely correlate with Lin28. Scatter plot showing the protein levels of Lin28 and transcript levels of miR-let-7g (left) and miR-let-7a (right) in corresponding human myocardial samples. B. miR-let-7 family members inversely correlate with PIK3IP1. Scatter plot showing the protein levels of PIK3IP1 and transcript levels of miR-let-7g (left) and miR-let-7a (right) in corresponding human myocardial samples. C. No inverse correlation between Lin28 or PIK3IP1 and miR-141. Scatter plot showing the protein levels of Lin28 (left) and PIK3IP1 (right) and transcript levels of miR-141 in corresponding human myocardial samples. (n=3 non-failing, n=6 failing, p 0.05 αat 5%).
4. DISCUSSION
MiR-let-7 expression is dysregulated in human heart failure [30], and several miR-let-7 species decline after myocardial injury in zebrafish and rat [28, 62, 63]. The biological significance of these changes is unknown, although loss of let-7 has been proposed to initiate a regenerative process [28]. The regulatory mechanisms and key targets of miR-let-7 in the diseased heart are also unknown. Here we show that miR-let-7 is controlled in the postnatal heart through myocyte-restricted, hypoxia-dependent induction of the pluripotency factor Lin28. Activation of this pathway results in suppression of Akt through a direct let-7 target, PIK3IP1, increasing myocyte vulnerability to apoptosis that can be reversed by restoring let-7g levels. Finally, we show that Lin28, let-7, and PIK3IP1 are specifically co-regulated across a spectrum of normal and diseased human hearts. Whether hypoxic activation of this pathway potentiates metabolic adaptation (see below) or regenerative mechanisms, it clearly does so at the expense of PI3K/Akt-mediated survival signaling.
4.1. Loss of miR-let-7 is potentially adaptive to hypoxia
The fall in miR-let-7 and rise in Lin28 coincide with a period of rapid adaptation to ischemic stress in which the myocyte is required to generate ATP through glycolysis. MiR-let-7 is well-established as a repressor of glucose utilization by targeting multiple genes in the PI3K-mTOR pathway regulating insulin signaling and glucose metabolism, including INSRβ, IGF1R and IRS2 [19, 32]. Global let-7 transgenic animals are glucose intolerant and insulin-resistant, and let-7 inhibitors prevent and treat high-fat induced glucose intolerance in vivo [19, 32]. In cancer cells, loss of miR-let-7 promotes high rates of glycolysis even when oxygen is abundant, a phenomenon known as the Warburg effect [64] through de-repression of pyruvate dehydrogenase kinase (PDK), a key glycolytic switch [65]. The observed loss of let-7 in hypoxic cardiac myocytes would thus act to facilitate the adaptive switch from β oxidation to anaerobic glycolysis [66, 67], and to increase glucose uptake through insulin signaling.
4.2. A myocyte-specific mechanism for let-7 downregulation in ischemia
Multiple pre- and post-transcriptional mechanisms exist to permit close control of the timing and quantity of miR-let-7. Our results show that hypoxia acts post-transcriptionally to repress let-7, but only in cardiac myocytes and not in fibroblasts from the same tissue. Processing of mature miR-let-7 from its precursor forms is potentially sensitive to extracellular signals and cellular context. One potential control point for this process is binding of the KH-type splicing regulatory protein (KSRP) to the terminal loop of precursor species, which is required for accumulation of mature let-7 in multiple cell types, and for promotion of myogenic differentiation of C2C12 cells[23, 68]. This interaction is regulated in part by PI3K/Akt signaling and thus potentially by other extracellular signals [23]. The same terminal loop is targeted by Lin28a and heteronuclear ribonucleoprotein A1 (hnRNP A1) to inhibit production of miR-let-7 and maintain cell stemness.[22, 45, 69–71]. Functioning of mature let-7 is directly inhibited by the lncRNA H19, which contains multiple let-7 binding sites and thus acts as an endogenous sponge [72]. Our findings show that the Lin28/let-7 regulatory axis can be invoked in a cell type-specific and stimulus-specific manner to regulate the adaptation to myocardial hypoxia, and may be important in regulating miR-let-7 levels in the failing human heart. This axis is evolutionarily ancient, being conserved from worms to mammals, and appears to be preserved in adult mammalian heart as a means of responding to extracellular conditions.
Lin28 itself is subject to repression by miR-let-7, setting up a mutual negative feedback loop that can facilitate rapid adaptations. In our system, upregulation of Lin28 protein and downregulation of miR-let-7 were immediate-early responses to myocardial ischemia, occurring within 2 hours of coronary occlusion in vivo and beginning at 8 hours in hypoxic myocytes in vitro. Although we did not see any effect of hypoxia on Lin28 acetylation, hypoxic modulation of both Lin28 and miR-let-7 was sensitive to broad-spectrum HDAC inhibition, suggesting that other post-transcriptional modifications such as acetylation could regulate let-7 directly or indirectly[46]. MEK1/2 and/or its upstream activators, which also respond rapidly to ischemia, further facilitate this fast response [73].
MEK/ERK activity is known to repress let-7, despite generally increasing microRNA expression through effects on Dicer [45]. Using the MEK1/2 inhibitor U0126, Paroo et.al [45] showed that active ERK, through a DICER-dependent mechanism, downregulates multiple let-7 family members. Our experiments provide evidence that a similar pathway may be activated in myocytes by hypoxia. ERK activation has been observed in myocytes under hypoxic and oxidative stress [73, 74]. However, ERK activation is likely cardioprotective, thus the enhanced vulnerability of myocytes following downregulation of let-7 must result from a mechanism downstream of let-7 itself. We propose that myocyte-selective inhibition of survival signaling through PI3 kinase is a major contributor to the selective apoptosis of myocytes when let-7 levels are reduced by hypoxia (Figure 8).
Figure 8. Summary diagram.
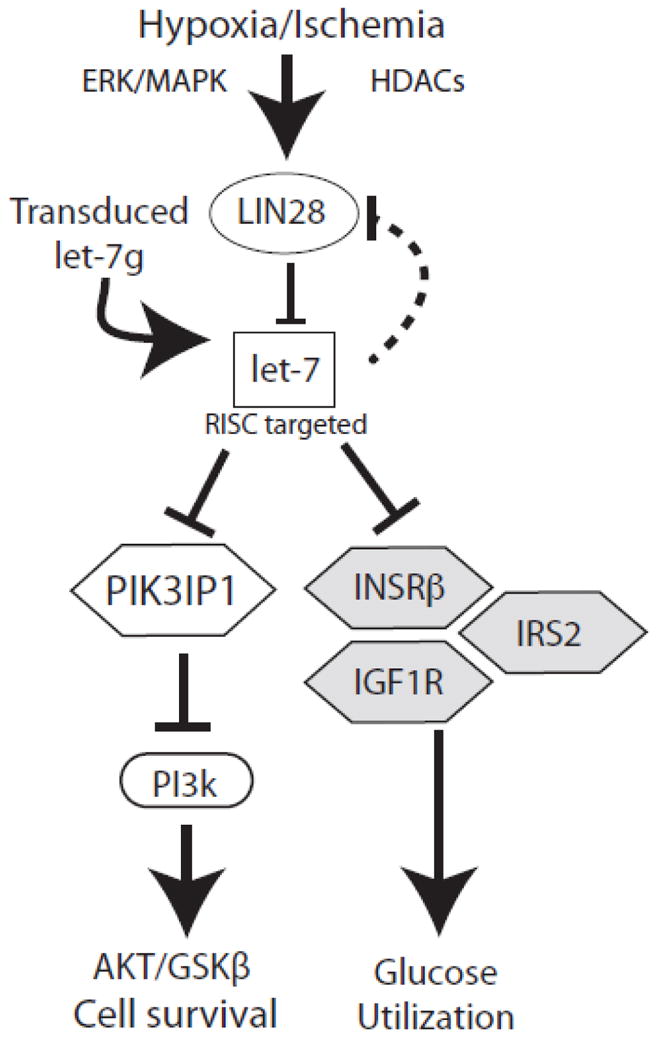
Hypoxia induces expression of the RNA binding protein Lin28 by a mechanism that is sensitive to ERK signaling and class 1/2 HDAC activity. Lin28 destabilizes members of the miR-let-7 family, potent repressors of genes (Insrβ, Irs2, Igf1r) involved in glucose utilization, promoting an adaptive shift to glycolysis. However, let-7 also represses PIK3IP1, an inhibitor of Akt, thus cell survival signaling is impaired by let-7 loss. Restoration of let-7g during hypoxia boosts Akt signaling and improves cell survival.
4.3. Preventing let-7 downregulation reduces myocyte apoptosis
As noted above, the immediate loss of let-7 in hypoxic cardiac myocytes could have short-term value in the ischemic myocardium by facilitating glucose uptake and glycolysis. However, at later time points, preventing the fall in miR-let-7g reduced, rather than increased, cardiac myocyte death both in hypoxia in vitro, and following ischemia-reperfusion injury in vivo. This observation is consistent with a recent study in which overexpression of let-7b protected against cell death in an in vitro model of simulated ischemia-reperfusion [75]. Similarly, overexpression of the let-7 family member miR-98 conferred survival benefit in a model of angiotensin II-induced myocardial hypertrophy [29]. Efforts to downregulate let-7 have produced discordant results. Aguirre et al described improved systolic function with combined intracardiac injection of anti-miR-let-7a/c and anti-miR-99/100 late in the course of experimental myocardial infarction, associated with evidence of increased cardiomyocyte mitosis [28]; Tolonen reported that intravenous infusion of a miR-let-7c antagomiR 4 weeks after coronary occlusion prevented post-MI remodeling, associated with reduced apoptosis and fibrosis[76]. These conflicting results could be due to the significant difference in timing of intervention in these latter studies, and possibly also in model systems and microRNA manipulation strategies. The balance of competing signals and outcomes following myocardial infarction in vivo depends greatly on the intensity and duration of hypoxia. Moreover, the targets and functions of let-7 are strongly influenced by concentration and by other factors in the cellular milieu [20], and potentially involve other cell types present at lower abundance in the myocardium, such as endothelial cells[33, 77].
Our data show that transduction of miR-let-7 markedly reduces apoptosis rates. Although we did not identify significant improvement in ejection fraction, both systolic and diastolic dimensions were significantly reduced, indicative of an anti-remodeling effect after infarction. It is important to note that end-systolic and end-diastolic volumes are also excellent predictors of adverse cardiovascular events after infarction[78]. As a technical point, it has been reported that treatments targeting myocardial infarctions directly (e.g. stem cell injections) result in local improvement in contractility without affecting measurements of global ejection fraction [79]. Hence it is possible that beneficial effects of let-7 were exerted locally within the infarcted area, a measurement technically inaccessible to our probe. It is also possible that the effect size on global contractility was below the threshold of measurement and would have been increased by greater let-7 transduction, or by co-transduction of let-7 and other ischemia-regulated microRNAs, including miR-99 and miR-100, as suggested by the work of Aguirre et al. [28]. Understanding the targets and regulation of the Lin28-let-7 axis may shed light on how the complex myocardial injury response is regulated in vivo.
4.4. MiR-let-7 reduces apoptosis by promoting Akt signaling via targeting PIK3IP1
One novel finding of this study is that miR-let-7 can act to promote Akt signaling in the heart. Although miR-let-7 negatively regulates the INSRβ-PI3K pathway in many cell types, in cortical neurons, let-7 “mimics” failed to decrease AKT phosphorylation [31], suggesting additional cell-type specific or context-dependent factors. A protein, PIK3IP1 that shares significant homology with the PI3K p85 regulatory subunit, negatively regulates PI3K. PIK3IP1 binds to the catalytic p110 subunit to prevent PI3K activation and phosphorylation of Akt in multiple cell types, including cardiac myocytes [57–59, 80]. Our study provides substantial evidence for PIK3IP1 as a novel target for let-7 in the myocardium. The PIK3IP1 3’ UTR contains a functional miR-let-7 binding site; overexpression of let-7g represses expression of PIK3IP1 in cardiac myocytes; PIK3IP1 and miR-let-7g are reciprocally expressed in human myocardium; gain of miR-let-7g induces AKT phosphorylation both in cultured myocytes and in vivo. Finally, PIK3IP1 is specifically enriched in the RISC of cardiac myocytes programmed with miR-let-7g. We propose that among other mechanisms targeting of PIK3IP1 by miR-let-7g enhances Akt activation and reduces loss of myocytes through apoptosis.
5. CONCLUSION
Important work to date has focused on the role of let-7 in differentiation, de-differentiation and maturation, of the cardiac myocyte [28, 81]; however, it is not clear that these processes contribute meaningfully to repair of the adult mammalian heart, so that the physiologic significance of altered miR-let-7 levels in conditions such as ischemic damage and heart failure remains obscure. As in the developing organism, the timing and amplitude of miR-let-7 changes may be critical to its functions after birth. Our study shows that in the postnatal cardiac myocyte, hypoxia induces Lin28 to repress miR-let-7, which normally promotes cell survival signaling by repressing the PI3K inhibitor PIK3IP1, but also represses glucose utilization. We propose that this myocyte-specific mechanism serves to promote acute glycolytic adaptation to hypoxia at the expense of increased vulnerability to apoptosis.
Supplementary Material
HIGHLIGHTS.
Hypoxia induces Lin28 expression in a cardiac myocyte-specific manner to downregulate members of the miR-let-7 family.
Downregulation of let-7 promotes cardiac myocyte cell death during hypoxia-reoxygenation.
Mir-let-7g targets a negative regulator of PI3K, PIK3IP1 to the RISC.
Hypoxia represses PI3K-Akt signaling in cardiac myocytes through a fall in let-7, and exogenous restoration of miR-let-7g restores PI3K signalling.
A similar Lin28-let-7 axis appears to control PIK3IP1 in human myocardium.
Acknowledgments
We thank Svetlana Speransky for her excellent technical assistance, and Drs. Sumit Jain and Salil Sharma for their helpful advice and suggestions.
SOURCES OF FUNDING
This study was funded by grants from the National Institutes of Health (NHLBI R-01-HL71094) and the Florida Heart Research Institute (to N.H.B), and by an American Heart Association Greater Southeastern Affiliate predoctoral fellowship (12PRE12080052, to S.J.)
Non-standard abbreviation and acronyms
- AGO2
Argonaute2
- let-7
lethal-7
- miR
microRNA
- NT
Non-targeting
- PAR-CLIP
Photoactivable Ribonucleoside Crosslinking and Immunoprecipitation
- PIK3IP1
Phosphotidyl-inositol kinase 3 interacting protein 1
- RISC
RNA-induced silencing complex
- TSA
Trichostatin A
Footnotes
DISCLOSURES
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Anversa P, Olivetti G, Capasso JM. Cellular basis of ventricular remodeling after myocardial infarction. Am J Cardiol. 1991;68:7D–16D. doi: 10.1016/0002-9149(91)90256-k. [DOI] [PubMed] [Google Scholar]
- 2.Gottlieb RA, Burleson KO, Kloner RA, Babior BM, Engler RL. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest. 1994;94:1621–1628. doi: 10.1172/JCI117504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bishop SP, Altschuld RA. Increased glycolytic metabolism in cardiac hypertrophy and congestive failure. Am J Physiol. 1970;218:153–159. doi: 10.1152/ajplegacy.1970.218.1.153. [DOI] [PubMed] [Google Scholar]
- 4.Taegtmeyer H. Energy metabolism of the heart: from basic concepts to clinical applications. Curr Probl Cardiol. 1994;19:59–113. doi: 10.1016/0146-2806(94)90008-6. [DOI] [PubMed] [Google Scholar]
- 5.Guzy RD, Schumacker PT. Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Exp Physiol. 2006;91:807–819. doi: 10.1113/expphysiol.2006.033506. [DOI] [PubMed] [Google Scholar]
- 6.Taylor CT, Moncada S. Nitric oxide, cytochrome C oxidase, and the cellular response to hypoxia. Arterioscler Thromb Vasc Biol. 2010;30:643–647. doi: 10.1161/ATVBAHA.108.181628. [DOI] [PubMed] [Google Scholar]
- 7.Chouchani ET, Methner C, Nadtochiy SM, Logan A, Pell VR, Ding S, James AM, Cocheme HM, Reinhold J, Lilley KS, Partridge L, Fearnley IM, Robinson AJ, Hartley RC, Smith RA, Krieg T, Brookes PS, Murphy MP. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat Med. 2013;19:753–759. doi: 10.1038/nm.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frohlich GM, Meier P, White SK, Yellon DM, Hausenloy DJ. Myocardial reperfusion injury: looking beyond primary PCI. Eur Heart J. 2013;34:1714–1722. doi: 10.1093/eurheartj/eht090. [DOI] [PubMed] [Google Scholar]
- 9.Heusch G. Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ Res. 2015;116:674–699. doi: 10.1161/CIRCRESAHA.116.305348. [DOI] [PubMed] [Google Scholar]
- 10.Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the akt proto-oncogene product by phosphatidyl-3,4-bis phosphate. Science. 1997;275:665–667. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- 11.Matsui T, Li L, del M, Fukui Y, Franke TF, Hajjar RJ, Rosenzweig A. Adenoviral gene transfer of activated phosphatidylinositol 3'-kinase and Akt inhibits apoptosis of hypoxic cardiomyocytes in vitro. Circulation. 1999;100:2373–2379. doi: 10.1161/01.cir.100.23.2373. [DOI] [PubMed] [Google Scholar]
- 12.Fujio Y, Nguyen T, Wencker D, Kitsis RN, Walsh K. Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation. 2000;101:660–667. doi: 10.1161/01.cir.101.6.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Juhaszova M, Zorov DB, Yaniv Y, Nuss HB, Wang S, Sollott SJ. Role of glycogen synthase kinase-3beta in cardioprotection. Circ Res. 2009;104:1240–1252. doi: 10.1161/CIRCRESAHA.109.197996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- 15.Small EM, Frost RJ, Olson EN. MicroRNAs add a new dimension to cardiovascular disease. Circulation. 2010;121:1022–1032. doi: 10.1161/CIRCULATIONAHA.109.889048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boon RA, Dimmeler S. MicroRNAs in myocardial infarction. Nat Rev Cardiol. 2015;12:135–142. doi: 10.1038/nrcardio.2014.207. [DOI] [PubMed] [Google Scholar]
- 17.Fiedler J, Thum T. MicroRNAs in myocardial infarction. Arterioscler Thromb Vasc Biol. 2013;33:201–205. doi: 10.1161/ATVBAHA.112.300137. [DOI] [PubMed] [Google Scholar]
- 18.Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901–906. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- 19.Frost RJ, Olson EN. Control of glucose homeostasis and insulin sensitivity by the Let-7 family of microRNAs. Proc Natl Acad Sci U S A. 2011;108:21075–21080. doi: 10.1073/pnas.1118922109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Su JL, Chen PS, Johansson G, Kuo ML. Function and regulation of let-7 family microRNAs. Microrna. 2012;1:34–39. doi: 10.2174/2211536611201010034. [DOI] [PubMed] [Google Scholar]
- 21.Wulczyn FG, Smirnova L, Rybak A, Brandt C, Kwidzinski E, Ninnemann O, Strehle M, Seiler A, Schumacher S, Nitsch R. Post-transcriptional regulation of the let-7 microRNA during neural cell specification. FASEB J. 2007;21:415–426. doi: 10.1096/fj.06-6130com. [DOI] [PubMed] [Google Scholar]
- 22.Heo I, Joo C, Cho J, Ha M, Han J, Kim VN. Lin28 mediates the terminal uridylation of let-7 precursor MicroRNA. Mol Cell. 2008;32:276–284. doi: 10.1016/j.molcel.2008.09.014. [DOI] [PubMed] [Google Scholar]
- 23.Briata P, Lin WJ, Giovarelli M, Pasero M, Chou CF, Trabucchi M, Rosenfeld MG, Chen CY, Gherzi R. PI3K/AKT signaling determines a dynamic switch between distinct KSRP functions favoring skeletal myogenesis. Cell Death Differ. 2012;19:478–487. doi: 10.1038/cdd.2011.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Viswanathan SR, Daley GQ, Gregory RI. Selective blockade of microRNA processing by Lin28. Science. 2008;320:97–100. doi: 10.1126/science.1154040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rehfeld F, Rohde AM, Nguyen DT, Wulczyn FG. Lin28 and let-7: ancient milestones on the road from pluripotency to neurogenesis. Cell Tissue Res. 2015;359:145–160. doi: 10.1007/s00441-014-1872-2. [DOI] [PubMed] [Google Scholar]
- 26.Thornton JE, Gregory RI. How does Lin28 let-7 control development and disease? Trends Cell Biol. 2012;22:474–482. doi: 10.1016/j.tcb.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coppola A, Romito A, Borel C, Gehrig C, Gagnebin M, Falconnet E, Izzo A, Altucci L, Banfi S, Antonarakis SE, Minchiotti G, Cobellis G. Cardiomyogenesis is controlled by the miR-99a/let–7c cluster and epigenetic modifications. Stem Cell Res. 2014;12:323–337. doi: 10.1016/j.scr.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 28.Aguirre A, Montserrat N, Zacchigna S, Nivet E, Hishida T, Krause MN, Kurian L, Ocampo A, Vazquez-Ferrer E, Rodriguez-Esteban C, Kumar S, Moresco JJ, Yates JR, 3rd, Campistol JM, Sancho-Martinez I, Giacca M, Izpisua Belmonte JC. In vivo activation of a conserved microRNA program induces mammalian heart regeneration. Cell Stem Cell. 2014;15:589–604. doi: 10.1016/j.stem.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang Y, Ago T, Zhai P, Abdellatif M, Sadoshima J. Thioredoxin 1 negatively regulates angiotensin II-induced cardiac hypertrophy through upregulation of miR-98/let-7. Circ Res. 2011;108:305–313. doi: 10.1161/CIRCRESAHA.110.228437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ikeda S, Kong SW, Lu J, Bisping E, Zhang H, Allen PD, Golub TR, Pieske B, Pu WT. Altered microRNA expression in human heart disease. Physiol Genomics. 2007;31:367–373. doi: 10.1152/physiolgenomics.00144.2007. [DOI] [PubMed] [Google Scholar]
- 31.Dubinsky AN, Dastidar SG, Hsu CL, Zahra R, Djakovic SN, Duarte S, Esau CC, Spencer B, Ashe TD, Fischer KM, MacKenna DA, Sopher BL, Masliah E, Gaasterland T, Chau BN, Pereira de Almeida L, Morrison BE, La Spada AR. Let-7 coordinately suppresses components of the amino acid sensing pathway to repress mTORC1 and induce autophagy. Cell Metab. 2014;20:626–638. doi: 10.1016/j.cmet.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu H, Shyh-Chang N, Segre AV, Shinoda G, Shah SP, Einhorn WS, Takeuchi A, Engreitz JM, Hagan JP, Kharas MG, Urbach A, Thornton JE, Triboulet R, Gregory RI, Altshuler D, Daley GQ. The Lin28/let-7 axis regulates glucose metabolism. Cell. 2011;147:81–94. doi: 10.1016/j.cell.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liao YC, Wang YS, Guo YC, Lin WL, Chang MH, Juo SH. Let-7g improves multiple endothelial functions through targeting transforming growth factor-beta and SIRT-1 signaling. J Am Coll Cardiol. 2014;63:1685–1694. doi: 10.1016/j.jacc.2013.09.069. [DOI] [PubMed] [Google Scholar]
- 34.Bishopric NH, Kedes L. Adrenergic regulation of the skeletal alpha-actin gene promoter during myocardial cell hypertrophy. Proc Natl Acad Sci USA. 1991;88:2132–2136. doi: 10.1073/pnas.88.6.2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turner MS, Haywood GA, Andreka P, You L, Martin PE, Evans WH, Webster KA, Bishopric NH. Reversible connexin 43 dephosphorylation during hypoxia and reoxygenation is linked to cellular ATP levels. Circ Res. 2004;95:726–733. doi: 10.1161/01.RES.0000144805.11519.1e. [DOI] [PubMed] [Google Scholar]
- 36.Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharma S, Liu J, Wei J, Yuan H, Zhang T, Bishopric NH. Repression of miR-142 by p300 and MAPK is required for survival signalling via gp130 during adaptive hypertrophy. EMBO Mol Med. 2012;4:617–632. doi: 10.1002/emmm.201200234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wei J, Wang W, Chopra I, Li HF, Dougherty CJ, Adi J, Adi N, Wang H, Webster KA. c-Jun N-terminal kinase (JNK-1) confers protection against brief but not extended ischemia during acute myocardial infarction. J Biol Chem. 2011;286:13995–14006. doi: 10.1074/jbc.M110.211334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jr, Jungkamp AC, Munschauer M, Ulrich A, Wardle GS, Dewell S, Zavolan M, Tuschl T. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Michael LH, Entman ML, Hartley CJ, Youker KA, Zhu J, Hall SR, Hawkins HK, Berens K, Ballantyne CM. Myocardial ischemia and reperfusion: a murine model. Am J Physiol. 1995;269:H2147–2154. doi: 10.1152/ajpheart.1995.269.6.H2147. [DOI] [PubMed] [Google Scholar]
- 41.Hashimoto E, Ogita T, Nakaoka T, Matsuoka R, Takao A, Kira Y. Rapid induction of vascular endothelial growth factor expression by transient ischemia in rat heart. Am J Physiol. 1994;267:H1948–1954. doi: 10.1152/ajpheart.1994.267.5.H1948. [DOI] [PubMed] [Google Scholar]
- 42.Ladoux A, Frelin C. Hypoxia is a strong inducer of vascular endothelial growth factor mRNA expression in the heart. Biochem Biophys Res Commun. 1993;195:1005–1010. doi: 10.1006/bbrc.1993.2144. [DOI] [PubMed] [Google Scholar]
- 43.An WG, Kanekal M, Simon MC, Maltepe E, Blagosklonny MV, Neckers LM. Stabilization of wild-type p53 by hypoxia-inducible factor 1alpha. Nature. 1998;392:405–408. doi: 10.1038/32925. [DOI] [PubMed] [Google Scholar]
- 44.Xi L, Taher M, Yin C, Salloum F, Kukreja RC. Cobalt chloride induces delayed cardiac preconditioning in mice through selective activation of HIF-1alpha and AP-1 and iNOS signaling. Am J Physiol Heart Circ Physiol. 2004;287:H2369–2375. doi: 10.1152/ajpheart.00422.2004. [DOI] [PubMed] [Google Scholar]
- 45.Paroo Z, Ye X, Chen S, Liu Q. Phosphorylation of the human microRNA-generating complex mediates MAPK/Erk signaling. Cell. 2009;139:112–122. doi: 10.1016/j.cell.2009.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang LX, Wang J, Qu TT, Zhang Y, Shen YF. Reversible acetylation of Lin28 mediated by PCAF and SIRT1. Biochim Biophys Acta. 2014;1843:1188–1195. doi: 10.1016/j.bbamcr.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 47.Cullen BR. Transcription and processing of human microRNA precursors. Mol Cell. 2004;16:861–865. doi: 10.1016/j.molcel.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 48.Siomi H, Siomi MC. Posttranscriptional regulation of microRNA biogenesis in animals. Mol Cell. 2010;38:323–332. doi: 10.1016/j.molcel.2010.03.013. [DOI] [PubMed] [Google Scholar]
- 49.Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139:693–706. doi: 10.1016/j.cell.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kawahara H, Okada Y, Imai T, Iwanami A, Mischel PS, Okano H. Musashi1 cooperates in abnormal cell lineage protein 28 (Lin28)-mediated let-7 family microRNA biogenesis in early neural differentiation. J Biol Chem. 2011;286:16121–16130. doi: 10.1074/jbc.M110.199166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang X, Lin X, Zhong X, Kaur S, Li N, Liang S, Lassus H, Wang L, Katsaros D, Montone K, Zhao X, Zhang Y, Butzow R, Coukos G, Zhang L. Double-negative feedback loop between reprogramming factor LIN28 and microRNA let-7 regulates aldehyde dehydrogenase 1-positive cancer stem cells. Cancer Res. 2010;70:9463–9472. doi: 10.1158/0008-5472.CAN-10-2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gual P, Le Marchand-Brustel Y, Tanti JF. Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie. 2005;87:99–109. doi: 10.1016/j.biochi.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 53.Weigert C, Kron M, Kalbacher H, Pohl AK, Runge H, Haring HU, Schleicher E, Lehmann R. Interplay and effects of temporal changes in the phosphorylation state of serine-302, -307, and -318 of insulin receptor substrate-1 on insulin action in skeletal muscle cells. Mol Endocrinol. 2008;22:2729–2740. doi: 10.1210/me.2008-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kienstra KA, Freysdottir D, Gonzales NM, Hirschi KK. Murine neonatal intravascular injections: modeling newborn disease. J Am Assoc Lab Anim Sci. 2007;46:50–54. [PubMed] [Google Scholar]
- 55.Bialik S, Geenen DL, Sasson IE, Cheng R, Horner JW, Evans SM, Lord EM, Koch CJ, Kitsis RN. Myocyte apoptosis during acute myocardial infarction in the mouse localizes to hypoxic regions but occurs independently of p53. J Clin Invest. 1997;100:1363–1372. doi: 10.1172/JCI119656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Philip L, Shivakumar K. cIAP-2 protects cardiac fibroblasts from oxidative damage: an obligate regulatory role for ERK1/2 MAPK and NF-kappaB. J Mol Cell Cardiol. 2013;62:217–226. doi: 10.1016/j.yjmcc.2013.06.009. [DOI] [PubMed] [Google Scholar]
- 57.DeFrances MC, Debelius DR, Cheng J, Kane LP. Inhibition of T-cell activation by PIK3IP1. European journal of immunology. 2012;42:2754–2759. doi: 10.1002/eji.201141653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.He X, Zhu Z, Johnson C, Stoops J, Eaker AE, Bowen W, DeFrances MC. PIK3IP1, a negative regulator of PI3K, suppresses the development of hepatocellular carcinoma. Cancer Res. 2008;68:5591–5598. doi: 10.1158/0008-5472.CAN-08-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhu Z, He X, Johnson C, Stoops J, Eaker AE, Stoffer DS, Bell A, Zarnegar R, DeFrances MC. PI3K is negatively regulated by PIK3IP1, a novel p110 interacting protein. Biochem Biophys Res Commun. 2007;358:66–72. doi: 10.1016/j.bbrc.2007.04.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Matkovich SJ, Van Booven DJ, Eschenbacher WH, Dorn GW., 2nd RISC RNA sequencing for context-specific identification of in vivo microRNA targets. Circ Res. 2011;108:18–26. doi: 10.1161/CIRCRESAHA.110.233528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Al-Lamki RS, Lu W, Wang J, Yang J, Sargeant TJ, Wells R, Suo C, Wright P, Goddard M, Huang Q, Lebastchi AH, Tellides G, Huang Y, Min W, Pober JS, Bradley JR. TNF, acting through inducibly expressed TNFR2, drives activation and cell cycle entry of c-Kit+ cardiac stem cells in ischemic heart disease. Stem Cells. 2013;31:1881–1892. doi: 10.1002/stem.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gray C, Li M, Patel R, Reynolds CM, Vickers MH. Let-7 miRNA profiles are associated with the reversal of left ventricular hypertrophy and hypertension in adult male offspring from mothers undernourished during pregnancy after preweaning growth hormone treatment. Endocrinology. 2014;155:4808–4817. doi: 10.1210/en.2014-1567. [DOI] [PubMed] [Google Scholar]
- 63.Ahn MY, Chung HY, Choi WS, Lee BM, Yoon S, Kim HS. Anti-tumor effect of apicidin on Ishikawa human endometrial cancer cells both in vitro and in vivo by blocking histone deacetylase 3 and 4. Int J Oncol. 2010;36:125–131. [PubMed] [Google Scholar]
- 64.Courtnay R, Ngo DC, Malik N, Ververis K, Tortorella SM, Karagiannis TC. Cancer metabolism and the Warburg effect: the role of HIF-1 and PI3K. Mol Biol Rep. 2015;42:841–851. doi: 10.1007/s11033-015-3858-x. [DOI] [PubMed] [Google Scholar]
- 65.Ma X, Li C, Sun L, Huang D, Li T, He X, Wu G, Yang Z, Zhong X, Song L, Gao P, Zhang H. Lin28/let-7 axis regulates aerobic glycolysis and cancer progression via PDK1. Nat Commun. 2014;5:5212. doi: 10.1038/ncomms6212. [DOI] [PubMed] [Google Scholar]
- 66.Eberli FR, Weinberg EO, Grice WN, Horowitz GL, Apstein CS. Protective effect of increased glycolytic substrate against systolic and diastolic dysfunction and increased coronary resistance from prolonged global underperfusion and reperfusion in isolated rabbit hearts perfused with erythrocyte suspensions. Circ Res. 1991;68:466–481. doi: 10.1161/01.res.68.2.466. [DOI] [PubMed] [Google Scholar]
- 67.Malhotra R, Sadoshima J, Brosius FC, Izumo S. Mechanical stretch and angiotensin II differentially upregulate the renin-angiotensin system in cardiac myocytes In vitro. Circ Res. 1999;85:137–146. doi: 10.1161/01.res.85.2.137. [DOI] [PubMed] [Google Scholar]
- 68.Trabucchi M, Briata P, Garcia-Mayoral M, Haase AD, Filipowicz W, Ramos A, Gherzi R, Rosenfeld MG. The RNA-binding protein KSRP promotes the biogenesis of a subset of microRNAs. Nature. 2009;459:1010–1014. doi: 10.1038/nature08025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rybak A, Fuchs H, Smirnova L, Brandt C, Pohl EE, Nitsch R, Wulczyn FG. A feedback loop comprising lin-28 and let-7 controls pre-let-7 maturation during neural stem-cell commitment. Nat Cell Biol. 2008;10:987–993. doi: 10.1038/ncb1759. [DOI] [PubMed] [Google Scholar]
- 70.Michlewski G, Caceres JF. Antagonistic role of hnRNP A1 and KSRP in the regulation of let-7a biogenesis. Nat Struct Mol Biol. 2010;17:1011–1018. doi: 10.1038/nsmb.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Towbin H, Wenter P, Guennewig B, Imig J, Zagalak JA, Gerber AP, Hall J. Systematic screens of proteins binding to synthetic microRNA precursors. Nucleic Acids Res. 2013;41:e47. doi: 10.1093/nar/gks1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kallen AN, Zhou XB, Xu J, Qiao C, Ma J, Yan L, Lu L, Liu C, Yi JS, Zhang H, Min W, Bennett AM, Gregory RI, Ding Y, Huang Y. The imprinted H19 lncRNA antagonizes let-7 microRNAs. Mol Cell. 2013;52:101–112. doi: 10.1016/j.molcel.2013.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yue TL, Wang C, Gu JL, Ma XL, Kumar S, Lee JC, Feuerstein GZ, Thomas H, Maleeff B, Ohlstein EH. Inhibition of extracellular signal-regulated kinase enhances ischemia/reoxygenation-induced apoptosis in cultured cardiac myocytes and exaggerates reperfusion injury in isolated perfused heart. Circ Res. 2000;86:692–699. doi: 10.1161/01.res.86.6.692. [DOI] [PubMed] [Google Scholar]
- 74.Clerk A, Michael A, Sugden PH. Stimulation of multiple mitogen-activated protein kinase sub-families by oxidative stress and phosphorylation of the small heat shock protein, HSP25/27, in neonatal ventricular myocytes. Biochem J. 1998;333(Pt 3):581–589. doi: 10.1042/bj3330581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Varga ZV, Zvara A, Farago N, Kocsis GF, Pipicz M, Gaspar R, Bencsik P, Gorbe A, Csonka C, Puskas LG, Thum T, Csont T, Ferdinandy P. MicroRNAs associated with ischemia-reperfusion injury and cardioprotection by ischemic pre- and postconditioning: protectomiRs. Am J Physiol Heart Circ Physiol. 2014;307:H216–227. doi: 10.1152/ajpheart.00812.2013. [DOI] [PubMed] [Google Scholar]
- 76.Tolonen AM, Magga J, Szabo Z, Viitala P, Gao E, Moilanen AM, Ohukainen P, Vainio L, Koch WJ, Kerkela R, Ruskoaho H, Serpi R. Inhibition of Let-7 microRNA attenuates myocardial remodeling and improves cardiac function postinfarction in mice. Pharmacol Res Perspect. 2014;2:e00056. doi: 10.1002/prp2.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Frangogiannis NG. MicroRNAs and endothelial function: many challenges and early hopes for clinical applications. J Am Coll Cardiol. 2014;63:1695–1696. doi: 10.1016/j.jacc.2013.10.056. [DOI] [PubMed] [Google Scholar]
- 78.Dart AM, Otterstad JE, Kirwan BA, Parker JD, de Brouwer S, Poole-Wilson PA, Lubsen J. Predictive value of local and core laboratory echocardiographic assessment of cardiac function in patients with chronic stable angina: The ACTION study. European journal of echocardiography : the journal of the Working Group on Echocardiography of the European Society of Cardiology. 2007;8:275–283. doi: 10.1016/j.euje.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 79.Suncion VY, Ghersin E, Fishman JE, Zambrano JP, Karantalis V, Mandel N, Nelson KH, Gerstenblith G, DiFede Velazquez DL, Breton E, Sitammagari K, Schulman IH, Taldone SN, Williams AR, Sanina C, Johnston PV, Brinker J, Altman P, Mushtaq M, Trachtenberg B, Mendizabal AM, Tracy M, Da Silva J, McNiece IK, Lardo AC, George RT, Hare JM, Heldman AW. Does transendocardial injection of mesenchymal stem cells improve myocardial function locally or globally?: An analysis from the Percutaneous Stem Cell Injection Delivery Effects on Neomyogenesis (POSEIDON) randomized trial. Circ Res. 2014;114:1292–1301. doi: 10.1161/CIRCRESAHA.114.302854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Song HK, Kim J, Lee JS, Nho KJ, Jeong HC, Ahn Y, Park WJ, Kim do H. Pik3ip1 modulates cardiac hypertrophy by inhibiting PI3K pathway. PLoS One. 2015;10:e0122251. doi: 10.1371/journal.pone.0122251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kuppusamy KT, Jones DC, Sperber H, Madan A, Fischer KA, Rodriguez ML, Pabon L, Zhu WZ, Tulloch NL, Yang X, Sniadecki NJ, Laflamme MA, Ruzzo WL, Murry CE, Ruohola-Baker H. Let-7 family of microRNA is required for maturation and adult-like metabolism in stem cell-derived cardiomyocytes. Proc Natl Acad Sci U S A. 2015 doi: 10.1073/pnas.1424042112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.