

Abstract
Small molecule targeting of the DNA minor groove is a promising approach to modulate genomic processes necessary for normal cellular function. For instance, dicationic diamindines, a well-known class of minor groove binding compounds, have been shown to inhibit interactions of transcription factors binding to genomic DNA. The applications of these compounds could be significantly expanded if we understand sequence-specific recognition of DNA better and could use the information to design more sequence-specific compounds. Aside from polyamides, minor groove binders typically recognize DNA at A-tract or alternating AT base pair sites. Targeting sites with GC base pairs, referred to here as mixed base pair sequences, is much more difficult than those rich in AT base pairs. Compound 1 is the first dicationic diamidine reported to recognize a mixed base pair site. It binds in the minor groove of ATGA sequences as a dimer with positive cooperativity. Due to the well-characterized behavior of 1 with ATGA and AT rich sequences, it provides a paradigm for understanding the elements that are key for recognition of mixed sequence sites. Electrospray ionization mass spectrometry (ESI-MS) is a powerful method to screen DNA complexes formed by analogs of 1 for specific recognition. We also report a novel approach to determine patterns of recognition by 1 for cognate ATGA and ATGA-mutant sequences. We found that functional group modifications and mutating the DNA target site significantly affect binding and stacking, respectively. Both compound conformation and DNA sequence directionality are crucial for recognition.
Keywords: mass spectrometry, DNA recognition, dimerization, minor groove binder, mixed DNA sequence
Introduction
Genetic processes are regulated by transcription factors (TF) that target specific DNA sequences. Typically, conformational changes or other processes, such as hydration, that yield strong interactions with bases in the recognition site are involved in binding.[1] A major goal of fields from chemical biology to therapeutic development is control of gene expression through TF modulation by small molecules that target DNA.[2] Instead of targeting the major groove, like most TFs, a more effective approach involves using small molecules to form a complex in the minor groove of DNA and allosterically modulate transcription factor binding.[3] Both inhibition and enhancement of TF complexes are possible with this approach. Typical minor groove binding compounds are relatively planar, crescent-shaped structures which match the geometry of the minor groove. Reversible binders typically have positively charged groups and form non-covalent interactions with DNA sites through electrostatic, hydrogen bonds, and van der Waals contacts. Although most minor groove binding structural types have a high affinity for A-tract and multiple AT sequences, they do no distinguish well between various AT base pair sequences.[4]
Polyamides (PA), the paradigm minor groove binding compounds which can recognize mixed or AT and GC containing sequences pose difficulties with solubility, aggregation, and synthetic costs.[5] Dicationic diamidines are a class of minor groove binding drugs which have overcome many of the issues encountered by PAs, but lack the sequence-specific targeting characteristics of PAs. A breakthrough compound for dicationic diamidines is compound 1 (Figure 1) since it recognizes a target site with a GC base pair in addition to AT.[6] It is exceptional since it dimerizes in the minor groove of ATGA sequences with positive cooperativity in spite of being a dication. Earlier reports revealed two binding constants for the dimer, the second KA value considerably higher (>20-fold) than the first, demonstrating positive cooperativity in binding of 1.[6b] The first diamidine molecule is believed to insert itself in the minor groove followed by slight widening in the groove width to accommodate the second, energetically more favorable molecule. The second 1 molecule inserts itself into the groove and participates in π-π stacking with the first molecule in an antiparallel fashion. Surprisingly, the four positive charges, which would be expected to repel each other due to their close proximities, do not inhibit the cooperative binding.
Figure 1.
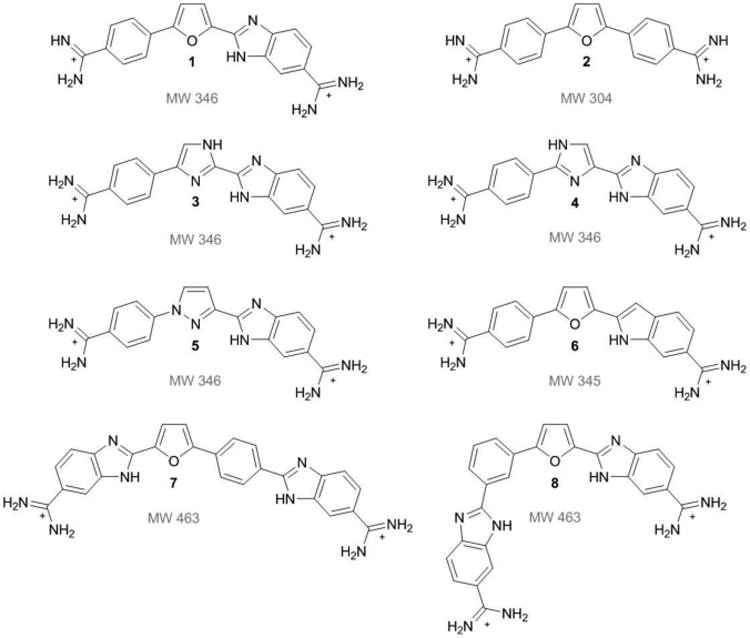
Structures of dicationic diamidine minor groove binding compounds used to investigate dimer formation in mixed sequence sites. Compound 1 is a reference compound known to dimerize in the mixed sequence site ATGA. Compound 2 is a classical minor groove binding compound known to recognize AT rich sites. Compound 3 to 8 are analogs of 1. Molecular weights are listed below the respective structures.
A similar and symmetric ligand, compound 2, is a well characterized dicationic diphenyl diamidine which behaves as a classical minor groove binding compound by recognizing AT rich sequences with 1:1 stoichiometry.[7] A single, asymmetric modification from benzimidazole to phenyl is the only difference between 1 and 2. Both compounds recognize AT sequences but 1 has a higher affinity for dimer formation with ATGA. Alternatively, variation in the flanking sequence of the target site has also been demonstrated to affect the binding affinity of small molecules in the minor groove.[6b] For instance, the binding affinity of 1 for ATGA can be affected by sequences flanking the target binding site. These examples provide important insights into structural and sequence-dependent effects of minor groove recognition.
A current research goal is to identify how variations in compound structure can affect the relative affinity for specific DNA sequences and how different sequences will be recognized by a single compound. Due to the large number of potential drug candidates and DNA sequences, a robust method to screen DNA and small molecule interactions is essential. Electrospray ionization mass spectrometry (ESI-MS) is a powerful method to investigate minor groove binder-DNA complexes.[8] It has been demonstrated that ESI-MS can be used for studying biological macromolecular systems such as DNA complexes because the soft ionization conditions used allow the non-covalent interactions that occur to remain essentially intact.[9] Necessary information such as stoichiometry and relative binding affinities can be determined directly, rapidly, and with little material. It is especially useful when examining interactions between DNA and small molecules. We recently reported a high-throughput method using ESI-MS to simultaneously screen multiple DNA-minor groove binder interactions.[10] This technique is advantageous over other screening methods because ESI-MS is gentle enough to detect complexes yet powerful enough to sort out similar complexes. The complexes detected are of minor groove binding compounds having relatively high binding affinities so that they can be detected at low concentrations. Previously, we reported the versatility of our ESI-MS method by demonstrating the cooperative dimer-forming nature of 1 with ATGA as well as monomer binding for AT rich sites.
Our goal in this report is to identify the features of 1 that make it ideal for dimerization with an ATGA sequence. Interactions of structurally similar compounds are compared with a mixed set of multiple DNA sequences since small modifications can affect minor groove recognition. The motifs chosen have the potential for dimer formation based on their similarities to the parent compound 1. Next, DNA-minor groove binder complexes are studied using the parent compound, 1, and mutated target sequences. This is a novel approach to gain insight into how 1 forms a sequence-specific dimer in the minor groove. It allows a better understanding of how sequence composition and directionality can affect the selectivity of 1 using cognate and variant ATGA sequences and the method can identify other compounds and sequences for mixed-site dimers.
Compound Design
Visually, the structural and conformational characteristics of 1 are typical for heterocyclic cations that bind specifically in the minor groove at AT sequence sites. With 1, however, two molecules can form a unique, antiparallel stacked tetracationic dimer in the minor groove and recognize an ATGA sequence with positive cooperativity.[6a] The phenyl-furan-benzimidazole system clearly has features that optimize stacking in sequences having a wider minor groove and altering these functional groups can modulate dimerization.[11] It is not clear that 1 is the optimum structure for this recognition mode or what other sequences could be recognized in a similar complex. To address these questions and better understand the molecular features that are required for the cooperative dimer complex, a number of analogs of 1 were prepared. The effects of structural and chemical changes on minor groove recognition of the ATGA target site and related sequences were then investigated with ESI-MS.
Modifications of the furan group give 3, 4, and 5 (Figure 1). Analog 4 contains an imidazole where one nitrogen is adjacent to the benzimidazole, while 3 is an isomer of 4 in which the nitrogen is positioned away from the benzimidazole and the third analog, 5, is a pyrrazole substituted system. For this class of compounds, modifying the furan group from a single hydrogen bond acceptor to a system containing both a donor and an acceptor should better define the stacking effects and hydrogen bonding found in ATGA recognition. The benzimidazole-amidine of 1 provides a strong minor groove recognition module. The indole analog 6 provides structural similarity to both 1 and DAPI[12] and the indole can preserve strong binding to the minor groove in AT sequences, but its effects on dimer formation are unknown. In functional groups with multiple nitrogens, this modification may affect the stacking and/or hydrogen bonding required for dimerization.
Lastly, two compounds have additions at the phenyl groups which lengthen the structure by including a second benzimidazole between the phenyl and amidine. Analogs 7 and 8 are isomers and differ in the benzimidazole-phenyl connectivity at the para- and meta-phenyl positions, respectively. This modification was chosen to determine how length, hydrogen bonding capability, and curvature of the moeity could affect DNA-ATGA interactions, and specifically dimer formation.
Results
Structural effects on selective recognition using analogs of 1
To begin our investigation of the effects of different functional groups and substitutions on minor groove recognition, a test was conducted using 1 as a reference with a mixed set of DNA sequences including ATATAT, AAATTT, and ATGA (Figure 2) since the binding affinities and modes of these sequences have been extensively studied with 1. A titration assay was performed with increasing concentrations of 1 to DNA and the spectra are shown in Figure 3. For each titration, the concentrations are expressed as a mole to mole ratio of 1 to a single DNA sequence to evaluate competition among the DNAs for ligand binding. This procedure allows lower concentrations of ligand to be used and enhances the preferred binding for one sequence over another. The spectrum in Figure 3A shows only DNA, where no 1 was included. Peaks are labeled as the “sequence name” over the corresponding molecular weight (m/z). In the following titrations (Figures 3B and 3C) where 1 is added, peaks begin to show for complexes formed between DNA and ligand. For example, a complex formed between 1 and AAATTT is shown at m/z 8,266 and labeled as AAATTT + (1) 1, where the integer in parentheses is the stoichiometric value for one molecule of 1 bound to AAATTT. In Figure 3B, the binding of two 1 compounds to ATGA is observed which indicates dimerization of the ligand with ATGA as expected. It is interesting to note that no 1:1 binding of compound 1 with ATGA is detected, which is reasonable since the affinity of the second molecule is more than 10-fold greater than binding of the first molecule.[6b] The monomeric binding of 1 binding with AAATTT and ATATAT sequences and dimeric binding to ATGA is in agreement with literature. The most distinctive characteristic of 1 is its ability to selectively bind as a cooperative dimer with ATGA while forming only monomer complexes with AT rich sequences. These results are clearly observed in Figure 3, further illustrating the positive cooperativity of 1 with ATGA by ESI-MS.
Figure 2.
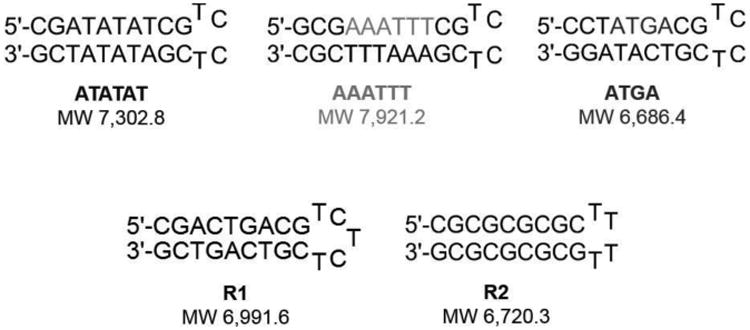
Mixed hairpin DNA sequences used to screen interactions for monomer and dimer-forming complex interactions with multiple sequences. Top row: ATATAT, AAATTT, and ATGA test sequences; bottom row: R1 and R2 as reference DNA sequences.
Figure 3.
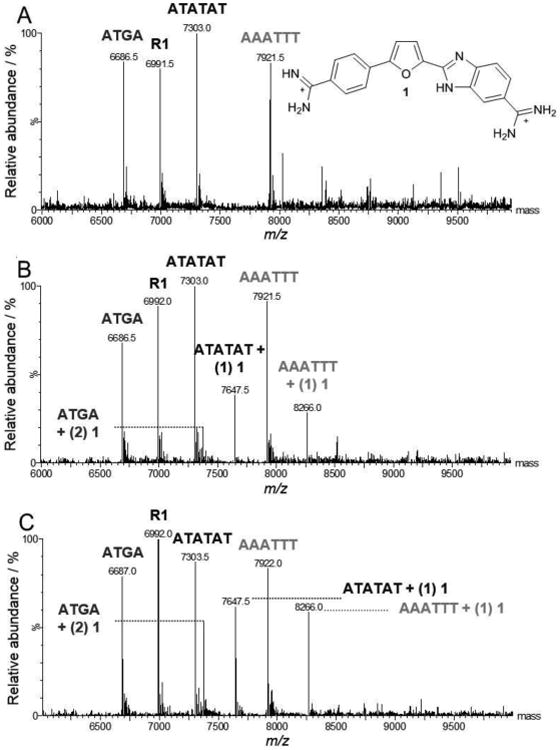
Example ESI-MS spectra of 1 titrated with multiple DNA sequences. Free DNA sequences are apparent by the sequence “name” above the corresponding peak (e.g. AAATTT m/z 7,921.5) and ligand-DNA complex as “name + (n ligands bound) ligand name” (e.g. ATGA + (2) 1, m/z 7,375). Concentrations of 1 are expressed as a mole to mole ratio for 1 to DNA and range [0:1] to [2:1]. Note that the positive cooperative nature of 1 binding to ATGA is indicated by increasing peak for the dimer species and no detectable 1:1 species. (A) [0:1], (B) [1:1], (C) [2:1].
To expand our understanding of how structural modifications of the 1 motif affects recognition in the DNA minor groove, the analogs of 1 were screened with the same mixed set of DNAs previously tested with 1. The structural conformation of each analog can vary considerably – depending on the modification made, and the intrinsic groove width for each DNA depends on the base pair sequence. The groove width of A-tract sequences are the most narrow of sequences compared to alternating AT sites, followed by mixed sequence sites, and GC rich sequences having the widest groove. Therefore, interaction between DNA and ligand is contingent on the inherent minor groove width matching the conformational space of the ligand(s). The AT rich sequences would be expected to bind the ligand as a monomer while the mixed sequence site, ATGA, should be able to recognize two ligands. The minor groove of the R1 sequence is too wide and has the steric effects of the GC hydrogen bond in the minor groove making it difficult to recognize small molecules such as our dicationic diamidines, by the R1 reference sequence. As aforementioned, complexes formed are labeled as “sequence name” + (n) ligand, where (n) is the stoichiometric value for one ligand molecule bound to DNA. Screening of the analogs first began with the compounds extended in length, 7 and 8. With 7 at a [1:1] ratio, only small peaks for AAATTT + (1) 7 and ATATAT + (1) 7 were detected (data not shown). A two-fold increase in ligand concentration showed higher peak intensities for AAATTT and ATATAT. In Figure 4A, a [4:1] titration of 7 with mixed sequences showed large peak intensities for 7 with the AT sequences, but no ATGA complex. Alternatively, no complexes with ATGA, ATATAT, or AAATTT sequences were detected with 8, an isomer of 7. Formation of complexes is contingent on the compound having a complementary shape to fit in the minor groove. For instance, based on the intrinsic helical nature of the DNA minor groove, and due to the extreme curvature of 8, one would expect to find a weakened interaction with any DNA (Figure 4B), as observed.
Figure 4.
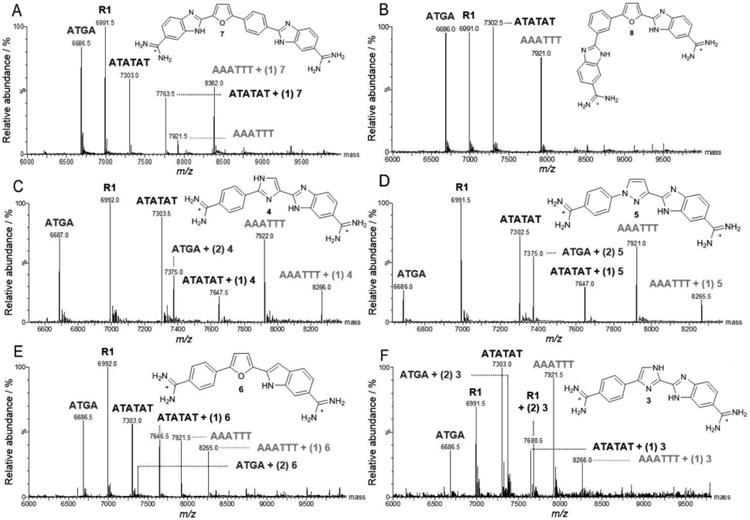
Spectra of DNA sequences titrated with compounds 3 to 8 analogs. Unbound DNAs are indicated by the sequence “name” above the respective peak (e.g. AAATTT, m/z 7,921.5) and ligand-DNA complex as “name + (n ligands bound) ligand name” (e.g. AAATTT + (1) 3, m/z 8,265.5). Molar ratios are expressed as [4:1] where ligand is to DNA. (A) 7, (B) 8, (C) 4, (D) 5, (E) 6, and (F) 3.
In our investigation of compound conformational space affecting recognition of ATGA, derivatives with the furan replaced by other five atom heterocycles were evaluated. A titration with the imidazole, 4, at [1:1] detected no complex interactions. On increasing concentrations of 4 to [2:1], 1:1 binding for AAATTT and ATATAT and 2:1 stoichiometry with ATGA were observed with similar intensities for both AT complexes (data not shown). After further increasing the concentration of 4 to [4:1], dimerization was enhanced and the peak intensity for ATGA increased relative to AAATTT + (1) 4 and ATATAT + (1) 4. In this case, the intensity for the AT sequences were comparable to one another (Figure 4C). A higher peak intensity of 4 with ATGA was observed with nearly equal intensities to AT complexes. It is important to note that upon increasing the concentration of 4, specifically from [1:1] to [2:1], only dimer complexes between 4 and ATGA were observed. The lack of any detectable 1:1 species illustrates the positive and cooperative binding behavior of 4 with ATGA.
Analog 5, which also has a central nitrogen heterocycle, was examined with the mixed DNA set. At lower concentrations of 5, a small peak for a complex formed with ATATAT was detected along with cooperative dimer binding with ATGA. Unlike its isomer 4, 5 showed a small peak at a molar ratio of [1:1] corresponding to ATATAT + (1) 5. At a [2:1] ratio, a complex with AAATTT was identified, but with less affinity than with ATGA and ATATAT. A 2:1 complex with 5 and ATGA was detectable with a higher intensity than AAATTT and ATATAT + (1) 5. By again doubling the concentration of 5 to [4:1], the intensity of ATGA + (2) 5 increased to more than double that of free ATGA, and increases in both ATATAT and AAATTT complexes were observed (Figure 4D). Based on the spectra shown in Figures 4C & 4D, ATGA recognition as a dimer appears stronger with the pyrrazole system found in 5 over the imidazole arrangement of 4. However, neither of these two systems is preferred over the furan found in 1 based on results obtained using ESI-MS with mixed sequences.[10]
Due to its structural similarity to 1, the indole-substituted analog, 6, would be expected to recognize ATGA as a dimer. At a mole:mole ratio of [1:1], a small complex peak for ATATAT + (1) 6 was detected. As the concentration was increased to [2:1], monomers with both AT sequences were observed with comparable intensities, but no complexes formed with ATGA. Finally, after again doubling the concentration of 6, a peak corresponding to ATGA + (2) 6 was detected with the intensity of nearly half that of free ATGA but with no 1:1 ATGA peak (Figure 4E). Analog 6 exhibits 1:1 stoichiometry with ATATAT and AAATTT; however, the highest complex peak corresponded to ATATAT + (1) 6 instead of the ATGA complex. Unlike 1 at lower concentrations, 6 formed monomeric complexes with near equal proportions from AT complexes. Increasing concentrations of 6 showed cooperative dimerization with ATGA, but the preference for AT sequences was greater than ATGA. This set of DNAs with 6 indicates that substituting the benzimidazole with an indole negatively affects the relative affinity for ATGA recognition as a stacked dimer versus monomer AT binding by a surprisingly large amount.
To investigate the relationship between ATGA recognition and the arrangement of hydrogen bond donors/acceptors, 3, an isomer of 4, was screened with the DNA set. The titration at [1:1] displayed a pattern similar to that found with 5 with only a small peak for ATATAT + (1) 3. Peak intensities from a [2:1] titration showed cooperative binding between ATGA and two 3s with a higher relative intensity than ATATAT + (1) 3. Doubling the concentration for 3 to [4:1] showed a dramatic increase in dimerization with ATGA (Figure 4F), an increase in ATATAT + (1) 3, and a new peak corresponding to AAATTT + (1) 3. The absence of any detectable 1:1 species again highlights the positive and cooperative behavior of 3 with ATGA. However, at [4:1] there was an additional – and relatively high – peak at m/z 7,680.5. Unexpectedly for this minor groove binding series, the new peak matches a 2:1 stoichiometry for a 3 complex with the reference DNA, R1 (Figure 5). This reference sequence has none of the usual diamidine minor groove binding sites and showed no interaction with the other compounds found in Figure 1.
Figure 5.
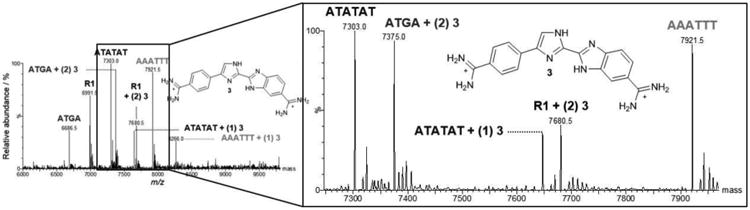
Mixed DNA sequence results with 3 are expanded between the range m/z 7,250 to 7,950 to highlight the unexpected dimerization of two molecules of 3 bound to R1. The molar ratio shown is [4:1].
As expected with 3 and DNA, monomer binding with the AT sequences was observed at lower concentrations. At higher concentrations, cooperative dimerization with ATGA was observed with a higher abundance relative to the AT sequences. The peak corresponding to two 3 molecules and R1, with higher relative intensity than complexes with the AT sequences was, however, unexpected and was not observed with the isomer 4. The dimeric binding of 3 to R1 is likely attributed to a common TGA, found in both ATGA and the CTGA in R1. The cooperativity of binding from 3 is comparable to 1 with ATGA. Structural simlarity would suggest similar interactions with the mixed set of DNAs since 4 and 3 are isomers of each other with only the inner imidazole reversed. Surprisingly, however, 3 showed a higher specificity for ATGA than 4 and 1, in addition to dimerization with R1. This rather significant difference in complex formation with 3 and 4 was certainly unexpected and illustrates the power of the ESI-MS mixed DNA sequences approach to discover new binding modes and sequences.
Molecular modeling of the compounds illustrates the effect of structural conformation on minor groove recogniton
In an effort to explain the compound differences in recognition of ATGA, ab initio calculations and molecular modeling were performed to better understand the conformation of 1 and its analogs and understand how slight differences in composition can affect overall conformation (Figure 6, see Electronic Supporting Information color image). The conformation and curvature of 3 are very similar to 1 and yet, interestingly, the behavior of 3 is different from its 4 isomer. The planarity of 3 matches that of 1 as does the electrostatic potential map. These qualities of 3 innately enhance its ability to stack as a dimer with mixed sequences including sequences with multiple GC base pairs (i.e. CTGA of R1). Structural information of 4 and 5 were also compared to 1. Surprisingly, the electrostatic potential map for 5 is very similar to 1 but the overall structures do not match. A model of 5 shows a twist in the dihedral angle between the phenyl and pyrrazole ring systems. This twist likely arises from the hydrogens of the phenyl and pyrrazole groups in close enough proximity to clash which is relieved by a 20° rotation. A twist of the same degree is also observed in 4 between its phenyl and imidazole groups. As with 5, 4 likely experiences clashing between the phenyl and imidazole hydrogens. It appears that a mostly planar conformation, such as that found in 1 and 3, is necessary for strong dimerization in the minor groove of ATGA. Compromising this planarity appears to hinder the ability of 5 and 4 to recognize ATGA presumably due to the conformational changes required to fit the minor groove, particularly as a stacked system. For instance, modifying the core imidazole system in 3 to 4 results in a decreased curvature for 4 compared to 3 which has a more crescent shape.
Figure 6.
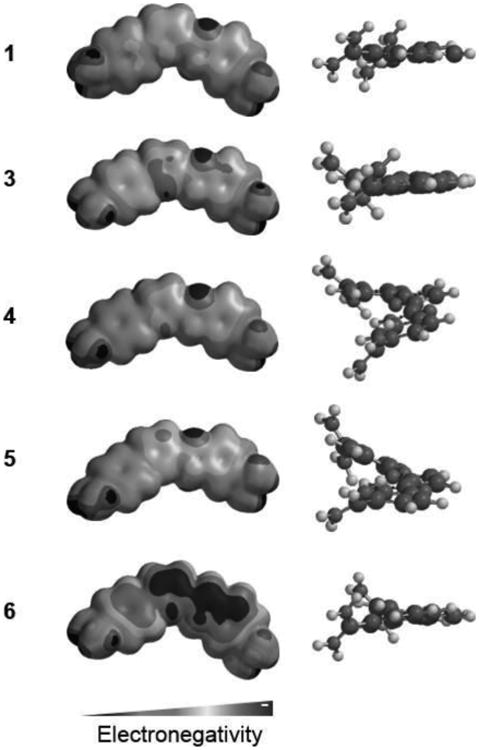
Comparison of 1 and analogs 3, 4, 5, and 6. Left illustrates the electrostatic potential map for the compounds. The right column shows a side view of the twists experienced in the overall structures. Molecules were minimized and electrostatic potential maps calculated using Spartan.
A model of two 3 molecules bound in the minor groove of ATGA is illustrated in Figure 7 and is based on our current understanding of the interactions between 1 and ATGA.[13] Figure 7A is a model portraying the stacked recognition of two 3 molecules in the ATGA minor groove. The 5′-ATGA-3′ is represented in cyan while the complementary 5′-TCAT-3′ is purple. A side view (Figure 7B) of the two stacked molecules illustrates the antiparallel, stacked nature of the compounds. The bottom ligand, in orange, binds in an orientation in which the benizimidaole-amidine motif is at the 3′ end of 5′-ATGA-3′. The N-H group of the central imidazole is solvent accessible while the nitrogen faces the floor of the minor groove to act as a hydrogen bond acceptor with the amino group of G (Figure 7C). The N-H of the benzimidazole faces the floor of the minor groove while the attached amidine can hydrogen bond with the keto oxygen of T (adjacent to C) on the complementary strand. The top ligand, shown as green, is orientated with the phenyl-amidine group at the 5′ end of 5′-TCAT-3′ with the curvature facing away from the ATGA minor groove. This arrangement moves the amidine groups apart and helps prevent electrostatic repulsion. The adjacent amidine is also capable of forming a hydrogen bond with the carbonyl oxygen of T of the 3′ end of 3′-TACT-5′. This indicates that rearrangement of nitrogens in the central ring system clearly has an overall effect on binding with ATGA. Reversing the central imidazole ring in 3 so that the two nitrogens are facing the benzimidazole-amidine system increases its curvature to more closely match the contour of the minor groove and improves its affinity for mixed DNA sequences.
Figure 7.
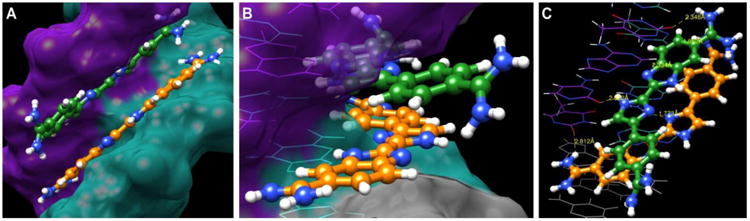
Models of compound 3 recognizing the mixed sequences ATGA as a dimer. (A) The spaced-filled model illustrates the stacked dimer formation of 3 in the minor groove of ATGA. (B) Side view of the stacked compounds. The curvature of the bottom molecule (orange) turns in towards the floor of the minor groove whereas the top molecule (green) faces out toward the solvent. (C) The stacked 3 dimer interactions with the base pairs 5′-ATGA-3′ and 3′-TCAT-5′. H-bond interactions between the base pairs are shown having dashed lines with distances in Å.
DNA sequence and directionality influence selective recognition
An alternative approach to investigate minor groove binding is with the modification of a known target sequence to understand sequence-specificity of a single compound and was inspired by the surprising interaction of 3 with R1 (CTGA). Figure 8 shows a scheme of the cognate ATGA and ATGA-mutant sequences studied simultaneously with 1. Strong and separated peak intensities were observed for complexes of 1 with ATGA and ATGA-mutant sequences at lower concentrations. Complex peaks had lower intensities compared with peaks of free DNA, but dimerization was observed with ATGA, TTGA, and ATAA. Additional peaks were present for ATAA + (1) 1 and ATGT + (1) 1. Peak intensities for ATAA + (1) 1 and TTGA + (2) 1 were comparable to ATGA + (2) 1. At a [4:1] ratio, the peak for ATGA + (2) 1 showed the highest abundance of the dimer complexes (Figure 9). It was followed next, in decreasing order, by TTGA, ATAA, and ATGT dimer complexes. Monomer complexes were also detected for ATAA, ATGT, and TTGA; however, peak intensities for ATGT + (1) 1 and TTGA + (1) 1 were difficult to distinguish from background noise. The cooperative binding of ATGA is evident by 2:1 complexes and no 1:1 interactions detected. Of the DNA sequences which have both monomer and dimer complexes, the intensity for ATAA + (1) 1 was greater than ATAA + (2) 1. This differs from TTGA where the peak for 2:1 was greater than 1:1. Intensities for ATGT + (1) 1 and ATGT + (2) 1 were nearly equal to each other and no complex between AGTA and 1 was observed.
Figure 8.
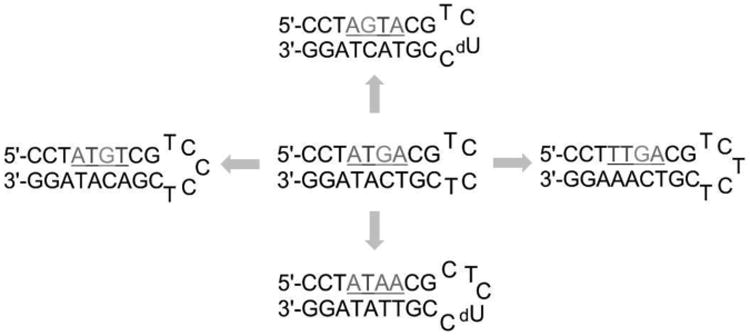
ATGA cognate and ATGA sequence variants used to examine the sequence specificity of 1. Base pairs flanking the target sites were maintained to allow similar response. Loops were modified for distinguishability using ESI-MS.
Figure 9.
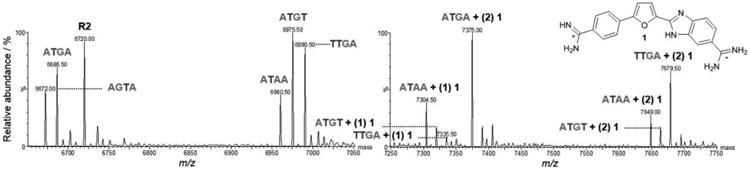
Spectra of ATGA cognate and ATGA mutant sequences with 1. Free DNA sequences range m/z 6,650-7,750 (left) and compound 1-DNA complexes m/z 7,250-7,975 (right). Both spectra belong to the same titration sample having a molar ratio of [4:1]. Peak intensities for the complexes are relative to the peak for ATGA + (2) 1.
The strong, cooperative binding of 1 with ATGA indicates a preference over all other similar sequence variants and is clearly optimized for dimer formation. As evident in Figure 9, smaller peaks occurred between 1 and ATAA in 1:1 and 2:1 stoichiometries, with a slight preference for the monomer complex, indicating low cooperativity for dimer formation. Dimerization was shown with TTGA at a higher relative abundance. A 1:1 complex with TTGA is also detected, but with lower intensity and only at higher concentrations. Peaks were visible for both monomer and dimer-ATGT complexes at m/z 7,322 and 7,668.5, respectively. However, the intensities were low and signals nearly merged with the background. The strongest dimer-forming complexes were ATGA and TTGA in which peak intensities continued to increase as the concentration of 1 increased.
Additional evidence for DNA complex formation with 1 and analogs by thermal melting and circular dichroism
Thermal melting is a robust method to qualitatively measure the stability of DNA and DNA complexes and offers valuable insight on single complexes for comparison with our ESI-MS competition experiments. The ΔTm values (Electronic Supporting Information, Table S1) suggest ATGA has a higher affinity for 6 over 4 and 5. The preference of 3 for ATGA over R1 (i.e. CTGA) is in agreement with the results shown in Figures 4F and 5. This is expected since the hydrogen bonding pattern necessary for recognition will be disrupted when substituting adenosine to cytidine (ATGA → CTGA). The peak intensities and ΔTm values for 1 and its analogs with initial mixed sequences are compared in Figure 10A with a superimposed model of the compounds (Figure 10B) to illustrate differences in structural conformations. The values for 1 and ATGA were taken from previously published results using ESI-MS.[10] A comparison of ΔTm values for 1 with ATGA (Table S2) shows that 1 prefers TTGA and ATGA over ATGT and AGTA. This is consistent with the results obtained using ESI-MS, as shown in Figure 9, which indicates that the choice and arrangement of base pairs in the target site plays a key role in forming complexes between the parent compound 1 and DNA.
Figure 10.
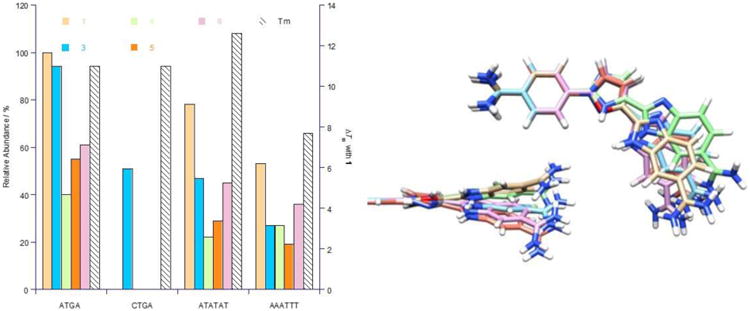
(A) Comparison of the relative peak intensities (± 3%) for complexes and ΔTm values (± 0.5 °C) for mixed DNA sequences with 1 and the dimer-forming analogs 3 – 6. ΔTm values (secondary y-axis) are for dimer-complexes formed between ligands and ATGA at a [4:1] molar ratio. (B) Structural variability and spatial arrangement for dimer-forming compounds are shown by superimposing the molecules over their mutual phenyl-amidines. 1 (tan), 3 (blue), 4 (green), 5 (orange), and 6 (pink).
It is interesting to see that the DNA complexes formed are well-defined. The binding behavior established by 1 translates well for some of its analogs with ATGA recognition. Specifically, cooperative binding is observed with increasing concentrations of ligand where the complexes formed between ATGA and 1 or its analogs show only 2:1 dimer ligand to DNA complexation. Any 1:1 complexes formed are at very low concentrations and, therefore, not detected. This illustrates that as concentrations of ligand are increased, so does the amount of complexes detected. The results correlate well with the thermal melting studies which allows the relative affinities of DNA and DNA-ligand complexes to be directly compared.
The overall structure of DNA and DNA complexes can be evaluated using circular dichroism spectroscopy (CD). CD studies are useful in examining the global conformation of DNA and other biomolecules. Studies were performed to compare the conformation of ATGA to its mutated sequences (Electronic Supporting Information, Figure S2). Curves were normalized and superimposed to facilitate the comparison. No change in CD signal was detected when comparing ATGT or AGTA to our cognate sequence, and only minute differences in the normalized signals of ATAA and TTGA were observed when compared to ATGA. The spectra showed that ATGA and its mutant sequences were very similar in their overall DNA conformations. Based on the structural similarities of ATGT and AGTA when compared to ATGA, it is becomes more clear that it is sequence which plays a direct role for recognition by 1. The slight variation in the CD curves are likely attributed to the individual base pair stacking which will affect the DNA microstructure but not the overall conformation. This may offer an explanation as to why interaction of 1 with ATAA can occur by both monomerization and dimerization since the microstructures between ATAA and mixed sites will be different.
Discussion
This report indicates that several features of the DNA complexes of 1 and analogs with DNA are critical in whether the compounds bind as a cooperative dimer or monomer. For DNA, the groove width and sequences of hydrogen bond donors and acceptors on the base pair edges at the floor of the minor groove are crucial. For the compounds, the stacking ability in the correct conformation to place hydrogen bond donors and acceptors to match the groups on DNA are necessary. These features are illustrated in Figure 7 and are based on the known binding mode of 1 with ATGA.[6a, 6b] Because of the complexity of these factors, experimental methods are required to determine whether the compounds bind as cooperative dimers, monomers, or if binding occurs at all. To do this in a competitive format for evaluating a large number of DNA sequences and compounds as rapidly as possible while using little sample, we have established the competitive ESI-MS method. Our method provides detailed insight into the features necessary to form a stacked cooperative dimer with DNA. We have discovered an entirely new and unexpected binding mode for 3. This is the first report of recognition by a synthetic, non-polyamide compound for a multiple GC sequence such as R1 with positive cooperativity. The analog, 3, forms not only a strong cooperative dimer complex with ATGA, but also forms a strong cooperative dimer with CTGA in R1. The core site, CTGA, has none of the traditional sites found in sequences recognized by 1 or similarly designed compounds. Steric hindrance from the hydrogen bond between the carbonyl group of cytidine and the amino group of guanosine in the minor groove likely affects the stacking ability of 3 and yet the compound is still capable of forming a strong positive – cooperativity dimer complex with CTGA. It is also interesting to see that the isomer of 3, 4, is unable to bind CTGA and binds weakly with ATGA. A seemingly subtle reorientation of the central imidazole places it in a way so that the compound is unable to form hydrogen bonds with the bases in the minor groove. The strategic placement of groups that act as hydrogen bond donors or acceptors is key for stacked binding in the minor groove and these complexes are detected using a competitive ESI-MS method. This important discovery provides a new paradigm for rationally designed, synthetic compounds to recognize mixed and/or GC rich sequences.
Parent compound 1 binds in the minor groove of ATGA as a dimer and recognizes AT rich sequences as a monomer. In contrast to this generalization, detection of ATAA as both monomer and dimer 1 expands our understanding of earlier evidence of dimerization of 1 at an ATAA site.[6b] For AT rich sequences, the minor groove of A-tracts is distinguished by a narrower groove width while alternating AT sequences, including those with the TA step, are wider.[4a, 15] Monomer and dimer binding should be possible for an ATAA sequence due to the TA step and wider minor groove. Based on the dual recognition of ATAA by 1 as a monomer and dimer and with nearly equal intensities, the ATAA minor groove is more closely related to alternating AT sequences than A-tracts. Binding of 1 to ATAA as a monomer can slightly narrow the groove width while binding as a dimer can slightly widen the groove.
For our mixed sequence mutants, the melting temperature of the free DNAs fall within ± 1.0 °C of ATGA and so the particular arrangement of the base pairs in these sequences does not have a large effect on the thermal stability of free DNA. The sequence arrangement, however, has an effect on binding of 1. For instance, mutation of the cognate sequence to read GT in the 5′ to 3′ direction within AGTA or ATGT, results in a significant decrease in binding of 1 compared to ATGA. Footprinting studies with 1 and a single mutation from ATGA to AGTA have shown similar results with no AGTA recognition.[6c] Additionally, as evident with TTGA at the 5′ end when A is replaced by T, cooperative binding of 1 is present, but decreased. These results suggest that because the base pair composition is very well maintained (GC and AT content), it is the stacking of the base pairs AA∙TT vs. AT∙AT that influences changes in minor groove microstructure and affects the affinity and binding mode of 1. Further investigations are necessary to identify minor groove microstructures for sequences with similar structures to ATGA.
Based on the ESI-MS studies of 1 with several DNAs, we can now see that it binds as a highly cooperative dimer to ATGA-like sequences but as a monomer to A-tract sequences. Based on the structural similarity of benzimidazole and indole groups, we expected the indole analog of 1, 6, to bind as a similar cooperative dimer. With a few exceptions, however, dimerization among minor groove binders containing an indole system is rare.[11b, 14] Most indole-containing minor groove binders recognize AT sequences strictly as a monomer. For instance, DAPI, the most thoroughly studied indole-containing compound, binds AT sequences as a monomer only.[12] More interestingly, however, is the higher affinity of 6 over 1 for ATGA which is unexpected since the curvature and conformation of the benzimidazole and indole systems are essentially the same (Figure 6). Biosensor-SPR studies (not published) have shown that 6 binds as a strong dimer to ATGA with a higher affinity over 1 which is in agreement with the thermal melting studies, however, the results from ESI-MS are not completely consistent. In the mass spectra, the 6-ATGA relative peak abundances are not as high as one would anticipate based on the results with 1. At this time, it is not completely understood why the 6-DNA peaks, which includes 6 with ATGA and both AT sequences, are less than expected. This is especially surprising since there has been excellent correlation between ESI-MS and thermal melting with 1 and the other analogs. One possible explanation may be technique-related in which the compound interacts with the injection tubing so that the total concentration of 6 in the sample solution decreases below the expected amount. A lower concentration of 6 would then result in less 6 complex formed and lower abundances of 6-DNA complexes detected.
To examine competition for DNA sites by 1 and analogs using ESI-MS, proper care must be taken to ensure that the molecular weights of the small molecules and their complexes, and all possible stoichiometries, are distinguishable. On the other hand, another approach is to examine the binding of a single compound with an array of target sequences and their mutations. Different DNA sequences can be examined simultaneously in this way as long as the molecular weights of the DNAs and complexes are distinguishable. A combination of an ATGA cognate sequence, ATGA-mutant sequences, and a reference DNA (R2) were screened with 1. To obtain different molecular weights for the variants, such as ATGA and AGTA which have the same stem molecular weights, the hairpin loops of the DNAs were altered with different numbers of thymidine and cytidine or by incorporation of a deoxyuridine so that the flanking base pairs were preserved.
In the spectra shown, peaks of the systems correspond well to their expected molecular weights (i.e. m/z) for free DNA and DNA-ligand complexes. The ionization process of ESI-MS results in multiply charged species and for the raw data, every system shows multiple, charge states (Electronic Supporting Information, Figure S4). Due to the nature of the analyte and negative mode analysis, the most abundant charge states range between -3 and -6. These lower net charges indicate the DNA backbone becomes partially neutralized during the electrospray process during which ammonium ions transfer a proton to the phosphate backbone and the ammonia ions evaporate. The amount of neutralization occurred depends on the size of the DNA, concentration of ammonium ions, and instrument parameters used.[8a, 19] Positively charged dicationic diamidines help in neutralizing the backbone, however, the presence of ligand does not affect the overall charge after forming a complex. For instance, peaks remain the most abundant in -4 and -5 charge states for both free AAATTT and AAATTT + (1) 1 complexes. The spectral peaks are transformed via deconvolution – the ability to transform multiple charge peaks into the single peak, zero charge molecular ion species. Deconvolution greatly simplifies the spectra for optimum visualization and is achieved by multiplying the charge of the species by its respective m/z.
Lower DNA concentrations such as 2.5 μm have been tested and not surprisingly, there is little difference in the peak intensities when comparing 2.5 μm of DNA versus 5 μm of DNA. The level of cooperativity is still observed, and is in agreement with earlier reports from our group demonstrating the cooperative binding of 1 to ATGA by ESI-MS using 5 μm of DNA.[10] For our systems, there is a general preference for using 5 μm of DNA since it results in a larger signal for the DNA and/or complexes over using 2.5 μm. A spectrum using 2.5 μm concentrations of DNA with compound 1 can be found in the Electronic Supporting Information (Figure S5). Due to the nature of compounds 1 – 8 and other dicationic diamidines, an unknown amount of ligand is often lost during the injection process. At times, the ligand will presumably become stuck and remain fixed to the inside of the injection tubing, therefore reducing the total ligand concentration. This phenomenon has been experienced on multiple occasions and requires thorough cleanings of the instrument between different samples. Samples containing DNA only (no compound) are routinely injected before beginning any new analysis to check for and remove residual ligand through binding of free DNA. Results can be successfully quantified using ESI-MS, as long as the specific response sensitivity and the concentrations are accurately known. It is possible to determine an equilibrium binding constant for DNA and small molecule systems and there are examples in literature demonstrating this.[18, 20] The ability to determine binding constants for dicationic diamidines is primarily limited to the loss of ligand during injection and response factors for the DNA and complexes, and these limitations influence our preference to use ESI-MS for qualitative purposes only.
Other methods can also be used, with or without ESI-MS, to efficiently screen for DNA binding compounds. For instance, thermal melting studies are commonly used to screen for binding of ligand to DNA. Additional techniques can include fluorescence assays, competitive dialysis experiments,[16] and separation techniques such as gel electrophoresis.[17] While these methods can provide important information, they can often demand more time and sample than ESI-MS. The ESI-MS technique reported here is rapid and convenient, requires little sample, and can provide quantitative information.[18] However, the most important feature from this method is that it can offer quick insight into the preferential binding of ligands based on compound structure and/or DNA sequence. With this, one can determine the stoichiometry, relative affinity, the binding mode (cooperative vs. non-specific) and it can even be used to determine heterodimeric binding.
Conclusion
Mixed DNA sequence investigations using ESI-MS has allowed the discovery of important features of 1 and analogs with ATGA and mutant sequences. For specificity and cooperative binding affinity to ATGA, these results show that at this point, 1 is the optimum compound. The results also show that 3 binds very well to ATGA but has many other strong interactions. A surprising result is that 3 binds quite well as a 2:1 dimer species to the GC sequence, R1, which was selected because heterocyclic dicationic diamidines have not been observed to bind to such GC rich sequences. Analog 4, the imidazole isomer of 3, does not bind as well to ATGA and does not bind at all to R1. The surprising binding of 3 needs additional investigation.
For sequence-specificity, the sequence ATAA is capable of binding 1 as both a monomer and dimer, despite containing no G or C bases in the target site. In sequences containing a GC base pair, the order of base pairs played a strong role in recognition by 1 such that the GT and TG steps had surprisingly different binding modes. Sites with TG have a preference for 1, whereas, GT sites tend to avoid forming complexes. Overall, ATGA remains the preferred site for cooperative 2:1 binding of 1 and these results further illustrate that sequence is crucial for minor groove recognition.
Experimental Procedures
Materials
Compounds 1[11a], 2[21], and 3[22] were synthesized using previously reported methods, and syntheses for the new analogs 4 – 8 are available in the Electronic Supporting Information. All compound stock solutions were prepared in doubly distilled water at a concentration of 1 mm. DNA sequences were purchased from Integrated DNA Technologies (IDT, Coralville, IA). Based on the predicted amount of DNA provided by IDT, DNAs were dissolved in doubly distilled water (1 mm). All sequences were converted to ammonium acetate salts by three steps of dialysis in 0.15 m ammonium acetate vacuum-filtered buffer (0.22 μM Millipore filter, pH 6.7) using a 1000 Da cut-off membrane (Spectrum Laboratories, Rancho Dominguez, CA). Following dialysis, concentrations of DNA were spectroscopically determined at 260 nm with extinction coefficients calculated using the nearest-neighbor method.[23] Sequences were denatured at 95 °C and immediately quenched on ice to initiate hairpin formation. Ligand stock solutions and dialyzed DNAs were stored at 4 °C.
Titration experiments were performed with a mixed set of DNAs in a single Eppendorf tube (100 μL, total volume). Ammonium acetate buffer was used due to its volatility under mass spectrometric conditions.[8a, 9b] DNAs were diluted (5 μm, 0.15 m ammonium acetate buffer, pH 6.7) with the appropriate concentration of ligand, vortexed, and stored at 4 °C until injection. Ratios with no surrounding punctuations refer to stoichiometry (i.e. 1:1 is ligand:DNA) whereas titration ratios are enveloped by brackets. For example, titration ratios are written as [n:m], where n and m are empirical concentrations of ligand and DNA, respectively. Titration ratios were prepared as compound-to-single-DNA. The ligand-to-single-DNA approach is more desirable for competitive binding analyses using multiple DNA sequences and avoids higher ratios being prepared. Two distinct hairpin DNA sequences were used to compare the formation and relative abundances of free DNA and DNA-complexes to a reference peak. For experiments with 1 and its analogs, R1 was used because it contained no known target sequence. R2 was later used as a reference to compare 1 with mutant DNA sequences due to the reference base pair composition which consisted of GC base pairs only in the DNA stem.
Electrospray ionization mass spectrometry
ESI-MS experiments were performed using a Waters Micromass Q-TOF (Waters, Milford, MA) in negative ion mode and MassLynx 4.1 software. Capillary voltage was set to 2500 V, sample cone voltage to 30 V, and extraction cone voltage at 3 V. Source block temperature was set to 70 °C and desolvation temperature at 100 °C. Prior to injection, the instrument was flushed with ammonium acetate buffer (0.15 m). Samples were injected at a rate of 5 μL∙min-1 and run for several minutes until the MassLynx chromatogram reached stabilization. Scanned peaks ranged m/z 300-3000 and the most abundant peaks observed belonged to -3 to -6 charge states. Scans were averaged over the last 2 min of analysis. Spectra were deconvoluted for comparative purposes. Deconvolution was acheived through multiplying peak intensities (m/z) by the charge (z) using the Maximum Entropy 1 Function (MassLynx 4.1).
Thermal Melting
Thermal melting studies were performed in cacodylate buffer (0.01 m cacodylic acid, 1 mm EDTA, 0.1 m NaCl, pH 7.1) using a Cary 300 UV-Vis spectrophotometer (Varian, Walnut Creek, CA) and a 1 cm quartz cuvette. Compound concentrations were chosen to give the desired ratio of compounds to hairpin DNA (3 μm). Scans were run from 25 °C to 95 °C at a rate of 0.5 °C∙min-1.
Circular Dichroism
Circular dichroism studies were performed using DNA prepared in cacodylate buffer (5 μm, 0.01 m cacodylic acid, 1 mm EDTA, 0.1 m NaCl, pH 7.1) using a Jasco J-810 Spectropolarimeter (Jasco Analytical Instruments Inc., Easton, MD) and a 1 cm quartz cuvette. Scans were performed at a rate of 50 nm∙min-1 from 320 nm to 220 nm, acquired in triplicate, and averaged.
Molecular Modeling
Ab initio calculations were performed in Spartan 10. Structures were minimized in the equilibrium geometry setting using a Hartree-Fock wavefunction and 6-31G* basis set. Molecules were set to dications in a vacuum environment. Canonical B-form doubled stranded DNA was built using the Sybyl software and coordinates saved as .pdb file. Hydrogen atoms were added to DNA using xLeap, solvated within a 10.0 Å TIP3PBOX waterbox, and neutralized by sodium ions. DNA minimization was achieved using AMBER99 force fields. DNA was visualized in VMD and coordinates were saved. The DNA sequence was then visualized and modeled with compound 3 using Chimera 1.8.1.
Supplementary Material
Figure 11.
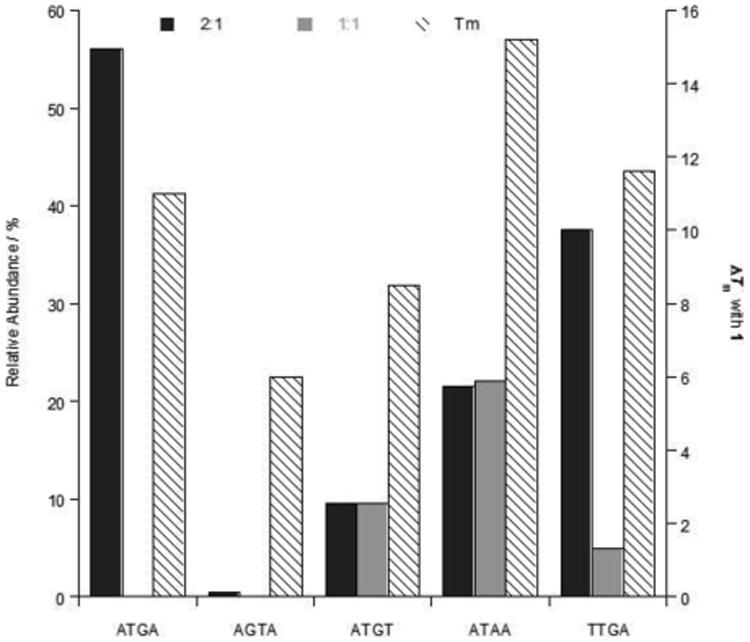
Comparison of the relative peak intensities (± 3%) of complexes and ΔTm values (± 0.5 °C) for ATGA cognate and ATGA mutant sequences with 1. Relative abundances (primary y-axis) display the peak intensities for both 1:1 and 2:1 binding of 1 with DNA as monomer and dimer complexes, respectively. ΔTm values (secondary y-axis) from studies performed using a [4:1] molar ratio of compound 1 to DNA sequence.
Acknowledgments
National Institutes of Health (NIH) GM111749 grant awarded to WDW and DWB, the Molecular Basis of Diseases Area of Focus (MBDAF) Fellowship to SRL, to Professor Donald Hamelberg for helpful discussions, and to Carol Wilson for help in manuscript preparation.
Abbreviations
- DNA
deoxyribonucleic acid
- ESI-MS
electrospray ionization mass spectrometry
- Ka
association constant
- MGB
minor groove binder
- m/z
mass-over-charge
- SPR
surface plasmon resonance
References
- 1.a) Wang S, Linde MH, Munde M, Carvalho VD, Wilson WD, Poon GM. J Biol Chem. 2014;289:21605–21616. doi: 10.1074/jbc.M114.575340. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lefstin JA, Yamamoto KR. Nature. 1998;392:885–888. doi: 10.1038/31860. [DOI] [PubMed] [Google Scholar]; c) Sharma H, Yu S, Kong J, Wang J, Steitz TA. Proc Natl Acad Sci USA. 2009;106:16604–16609. doi: 10.1073/pnas.0908380106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Darnell JE. Nat Rev Cancer. 2002;2:740–749. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]; b) Koehler AN. Curr Opin Chem Biol. 2010;14:331–340. doi: 10.1016/j.cbpa.2010.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Peixoto P, Liu Y, Depauw S, Hildebrand MP, Boykin DW, Bailly C, Wilson WD, David-Cordonnier MH. Nucleic Acids Res. 2008;36:3341–3353. doi: 10.1093/nar/gkn208. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mapp AK, Ansari AZ, Ptashne M, Dervan PB. Proc Natl Acad Sci USA. 2000;97:3930–3935. doi: 10.1073/pnas.97.8.3930. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Dervan PB, Burli RW. Curr Opin Chem Biol. 1999;3:688–693. doi: 10.1016/s1367-5931(99)00027-7. [DOI] [PubMed] [Google Scholar]; d) Munde M, Poon GMK, Wilson WD. J Mol Biol. 2013;425:1655–1669. doi: 10.1016/j.jmb.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Abudaya A, Brown PM, Fox KR. Nucleic Acids Res. 1995;23:3385–3392. doi: 10.1093/nar/23.17.3385. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu Y, Kumar A, Boykin DW, Wilson WD. Biophys Chem. 2007;131:1–14. doi: 10.1016/j.bpc.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Kielkopf CL, Baird EE, Dervan PD, Rees DC. Nat Struct Biol. 1998;5:104–109. doi: 10.1038/nsb0298-104. [DOI] [PubMed] [Google Scholar]; b) Satam V, Patil P, Babu B, Gregory M, Bowerman M, Savagian M, Lee M, Tzou S, Olson K, Liu Y, Ramos J, Wilson WD, Bingham JP, Kiakos K, Hartley JA, Lee M. Bioorg Med Chem Lett. 2013;23:1699–1702. doi: 10.1016/j.bmcl.2013.01.075. [DOI] [PubMed] [Google Scholar]
- 6.a) Wang L, Carrasco C, Kumar A, Stephens CE, Bailly C, Boykin DW, Wilson WD. Biochemistry. 2001;40:2511–2521. doi: 10.1021/bi002301r. [DOI] [PubMed] [Google Scholar]; b) Tanious F, Wilson WD, Wang L, Kumar A, Boykin DW, Marty C, Baldeyrou B, Bailly C. Biochemistry. 2003;42:13576–13586. doi: 10.1021/bi034852y. [DOI] [PubMed] [Google Scholar]; c) Bailly C, Tardy C, Wang L, Armitage B, Hopkins K, Kumar A, Schuster GB, Boykin DW, Wilson WD. Biochemistry. 2001;40:9770–9779. doi: 10.1021/bi0108453. [DOI] [PubMed] [Google Scholar]
- 7.Laughton CA, Tanious F, Nunn CM, Boykin DW, Wilson WD, Neidle S. Biochemistry. 1996;35:5655–5661. doi: 10.1021/bi952162r. [DOI] [PubMed] [Google Scholar]
- 8.a) Rosu F, De Pauw E, Gabelica V. Biochimie. 2008;90:1074–1087. doi: 10.1016/j.biochi.2008.01.005. [DOI] [PubMed] [Google Scholar]; b) Rosu F, Gabelica V, Houssier C, De Pauw E. Nucleic Acids Res. 2002;30:e82. doi: 10.1093/nar/gnf081. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Brodbelt JS. Annu Rev Anal Chem. 2010;3:67–87. doi: 10.1146/annurev.anchem.111808.073627. [DOI] [PubMed] [Google Scholar]; d) Buchmann W, Boutorine A, Halby L, Tortajada J, De Pauw E. J Mass Spectrom. 2009;44:1171–1181. doi: 10.1002/jms.1592. [DOI] [PubMed] [Google Scholar]
- 9.a) Dass C. Fundamentals of contemporary mass spectrometry. Wiley-Interscience; Hoboken, N.J.: 2007. [Google Scholar]; b) Loo JA. Int J Mass Spectrom. 2000;200:175–186. [Google Scholar]; c) Gale DC, Goodlett DR, Lightwahl KJ, Smith RD. J Am Chem Soc. 1994;116:6027–6028. [Google Scholar]; d) Gale DC, Smith RD. J Am Soc Mass Spectr. 1995;6:1154–1164. [Google Scholar]; e) Ganem B, Li YT, Henion JD. Tetrahedron Lett. 1993;34:1445–1448. [Google Scholar]
- 10.Laughlin S, Wang S, Kumar A, Boykin DW, Wilson WD. Anal Bioanal Chem. 2014;406:6441–6445. doi: 10.1007/s00216-014-8044-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Hopkins KT, Wilson WD, Bender BC, McCurdy DR, Hall JE, Tidwell RR, Kumar A, Bajic M, Boykin DW. J Med Chem. 1998;41:3872–3878. doi: 10.1021/jm980230c. [DOI] [PubMed] [Google Scholar]; b) Munde M, Kumar A, Nhili R, Depauw S, David-Cordonnier MH, Ismail MA, Stephens CE, Farahat AA, Batista-Parra A, Boykin DW, Wilson WD. J Mol Biol. 2010;402:847–864. doi: 10.1016/j.jmb.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Williamson DH, Fennell DJ. Methods Cell Biol. 1975;12:335–351. doi: 10.1016/s0091-679x(08)60963-2. [DOI] [PubMed] [Google Scholar]
- 13.Wang L, Bailly C, Kumar A, Ding D, Bajic M, Boykin DW, Wilson WD. Proc Natl Acad Sci USA. 2000;97:12–16. doi: 10.1073/pnas.97.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kwok Y, Zhang WT, Schroth GP, Liang CH, Alexi N, Bruice TW. Biochemistry. 2001;40:12628–12638. doi: 10.1021/bi0109865. [DOI] [PubMed] [Google Scholar]
- 15.Fox KR. Nucleic Acids Res. 1992;20:6487–6493. doi: 10.1093/nar/20.24.6487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ren J, Chaires JB. Biochemistry. 1999;38:16067–16075. doi: 10.1021/bi992070s. [DOI] [PubMed] [Google Scholar]
- 17.Wang S, Munde M, Wang SM, Wilson WD. Biochemistry. 2011;50:7674–7683. doi: 10.1021/bi201010g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gabelica V, Galic N, Rosu F, Houssier C, De Pauw E. J Mass Spectrom. 2003;38:491–501. doi: 10.1002/jms.459. [DOI] [PubMed] [Google Scholar]
- 19.Gabelica V, De Pauw E, Rosu F. J Mass Spectrom. 1999;34:1328–1337. doi: 10.1002/(SICI)1096-9888(199912)34:12<1328::AID-JMS889>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 20.Sakakibara Y, Abeysirigunawardena SC, Duc AC, Dremann DN, Chow CS. Angew Chem Int Ed Engl. 2012;51:12095–12098. doi: 10.1002/anie.201206000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Das BP, Boykin DW. J Med Chem. 1977;20:531–536. doi: 10.1021/jm00214a014. [DOI] [PubMed] [Google Scholar]
- 22.Batista-Parra A. PhD thesis. Georgia State University (USA); 2003. [Google Scholar]
- 23.Fasman GD. Handbook of biochemistry and molecular biology. 3d. CRC Press; Cleveland: 1975. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.