

Summary
We have previously shown that Actinobacillus actinomycetemcomitans cytolethal-distending toxin (Cdt) is a potent immunosuppressive agent that induces G2/M arrest in human lymphocytes. In this study, we explored the possibility that Cdt-mediated immunotoxicity involves lipid membrane microdomains. We first determined that following treatment of Jurkat cells with Cdt holotoxin all three Cdt subunits localize to these microdomains. Laser confocal microscopy was employed to colocalize the subunits with GM1-enriched membrane regions which are characteristic of membrane rafts. Western blot analysis of isolated lipid rafts also demonstrated the presence of Cdt peptides. Cholesterol depletion, using methyl β-cyclodextrin, protected cells from the ability of the Cdt holotoxin to induce G2 arrest. Moreover, cholesterol depletion reduced the ability of the toxin to associate with Jurkat cells. Thus, lipid raft integrity is vital to the action of Cdt on host cells. The implications of our observations with respect to Cdt mode of action are discussed.
Introduction
The cytolethal-distending toxins (Cdts) are a family of heat-labile protein cytotoxins produced by several different bacterial species including diarrhoeal disease-causing enteropathogens such as Escherichia coli, Campylobacter jejuni, Shigella species, Haemophilus ducreyi, Salmonella typi and Actinobacillus actinomycetemcomitans (Pickett et al., 1994; Scott and Kaper, 1994; Okuda et al., 1995; 1997; Comayras et al., 1997; Mayer et al., 1999; Pickett and Whitehouse, 1999; Shenker et al., 2000; Haghjoo and Galan, 2004). We have previously shown that the Cdt derived from A. actinomycetemcomitans is a potent immunotoxin capable of inducing G2 arrest and ultimately apoptosis in human lymphocytes (Gelfanova et al., 1999; Shenker et al., 1999; 2001; Nalbant et al., 2003). There is now clear evidence that Cdt is encoded by three genes, designated cdtA, cdtB and cdtC, which encode polypeptides with apparent molecular masses of approximately 24–35 kDa (Lara-Tejero and Galan, 2001; Mao and DiRienzo, 2002; Deng and Hansen, 2003; Shenker et al., 2004). Moreover, we have recently demonstrated that the Cdt holotoxin consists of each of the Cdt subunits: CdtA, CdtB and CdtC. The Cdt holotoxin appears to function as an AB2 toxin where CdtB is the active (A) unit and the complex of CdtA and CdtC comprise the binding (B) unit (Elwell and Dreyfus, 2000; Lara-Tejero and Galan, 2001; Nesic et al., 2004). Furthermore, it is generally agreed that the active subunit, CdtB, enters the cell while CdtA and CdtC remain associated with the cell surface (Cortes-Bratti et al., 2000; McSweeney and Dreyfus, 2004; Nesic et al., 2004).
It is well documented that plasma membranes consist of microscopically and biochemically distinguishable lipid microdomains. Membrane bilayer organization into these microdomains known, colloquially as membrane rafts and super-rafts, provides the requisite temporal and spatial organization of protein and lipid components necessary for normal cellular function (Simons and Ikonen, 1997; Brown and London, 1998; Zajchowski and Robbins, 2002; Horejsi, 2004; Pike, 2004). A unique property of these membrane rafts is their resistance to solubilization at low temperatures by mild detergents such as Triton X-100, NP-40 and Brij; this insolubility is due, in part, to long saturated acyl chains of gangliosides that impart a high degree of order and also by stabilization of intercalating cholesterol molecules (Simons and Ikonen, 1997; Brown and London, 1998). In addition to their unique lipid profile, membrane rafts also contain a number of extracellular and cytoplasmic proteins involved in signal transduction. The organization of rafts restricts access so that only limited proteins can reside in rafts; these include those attached to the membrane by a lipid anchor, such as glycosylphosphatidylinoisitol (GPI)-anchored proteins, acylated cytosolic proteins and certain transmembrane proteins (Simons and Ikonen, 1997; Brown and London, 1998; Horejsi, 2004). Membrane rafts are involved in cellular processes as diverse as fertilization, viral infection, maintenance of cell polarity, chemotaxis, apoptosis and development (Simons and Ikonen, 1997; Brown and London, 1998; van der Goot and Harder, 2001; Zajchowski and Robbins, 2002; Horejsi, 2003; 2004). Thus, it has been proposed that rafts may play a pivotal role in the plasma membrane by serving as platforms that co-ordinate the induction of signalling pathways. Indeed, lipid rafts isolated from lymphocytes not only contain Src kinases but these membrane complexes also have been implicated in regulating cell activation following engagement of the T-cell receptor (TCR) as well as interleukin-2 receptor signalling (Xavier et al., 1998; Alonso and Millan, 2001; Marmor and Julius, 2001).
In addition to a role in cell signalling, it has become increasingly evident that several pathogens and microbial-derived toxins interact with their target cell via membrane rafts (van der Goot and Harder, 2001). For instance, rafts may provide a mechanism by which receptors are concentrated and thereby promote binding. One such example is cholera toxin which is pentameric and binds to targets cells via the ganglioside GM1. It is likely that cholera toxin simultaneously binds with high affinity to multiple receptors as result of receptor concentration within the lipid microdomain (Montecucco et al., 1994; van der Goot and Harder, 2001). Likewise, pore-forming toxins such as Aeromonas hydrophila, which binds to GPI-anchored proteins, utilize the concentrating properties of lipid rafts to facilitate oligomerization, a requisite for channel formation (Abrami and van der Goot, 1999; van der Goot and Harder, 2001). Membrane rafts may also facilitate signalling following the binding of bacterial toxins to raft-associated receptors. In this regard, bacterial lipopolysaccharride (LPS) interacts with rafts via CD14, a GPI-anchored receptor; LPS binding results in mitogen-activated protein (MAP) kinase activation and eventually cytokine production. Finally, membrane rafts may serve as entry sites for pathogens; in this regard several pathogens enter host cells in a cholesterol-dependent manner (van der Goot and Harder, 2001; Lencer, 2001). Furthermore, the uptake of E. coli strains which express FimH have been shown to involve cholesterol-rich microdomains. Similarly, Shigella invades cells via interaction between the invasin, IpaB, and the raft-associated receptor, CD44. Viruses such as SV40 also utilize rafts either for internalization or as a site for fusion with the plasma membrane (Lafont et al., 2002).
It is clear that lipid microdomains play a pivotal role in regulating T-cell activation and function. Moreover, there is growing evidence that several microbes specifically target raft domains in order to infect host cells and/or hijack these signalling platforms to promote pathogenic events. Therefore, we explored the possibility that Cdt-mediated immunotoxicity is cholesterol dependent. We now report that Cdt colocalizes to membrane rafts. The association of all three Cdt subunits with lipid rafts was evident from Western blot analysis of isolated raft fractions and confocal microscopy of toxin-treated cells. Cholesterol depletion of Jurkat cell membranes confers resistance to Cdt-induced G2 arrest indicating that the integrity of cholesterol-rich microdomains is crucial to toxin activity. Finally, cholesterol-depleted cells exhibited reduced ability to bind Cdt. These studies clearly indicate that lipid rafts are indeed critical to the mechanism by which Cdt induces cell cycle arrest in target cells.
Results
The Cdt holotoxin is composed of three truncated subunits: CdtA, CdtB and CdtC, and furthermore, a complex of CdtA and CdtC is required for interaction of the toxin with target cell membranes leading to toxin-induced G2 arrest. Moreover, following a 2 h exposure to Cdt holotoxin, the individual subunits could be detected associated with Jurkat cell membranes. We therefore wanted to determine whether any of the Cdt subunits associate with membrane lipid microdomains. In order to conduct these experiments, we first needed antibodies capable of recognizing the individual Cdt subunits when they are present in the holotoxin complex and associated with cells. Monoclonal antibodies (mAbs) were generated and characterized in terms of their peptide specificity by both Western blot analysis and enzyme-linked immunosorbent assay (ELISA) as well as for their ability to recognize the peptide in association with the holotoxin complex by surface plasmon resonance (data not sown). Figure 1 shows flow cytometric analysis of Jurkat cells after they have been treated with the Cdt holotoxin and then stained using three of these mAbs. The mAbs specifically recognize either CdtA (Fig. 1A), CdtB (Fig. 1B) or CdtC (Fig. 1C) when the toxin is associated with cells. These mAbs were used in this study to investigate the localization of Cdt subunits to membrane lipid microdomains.
Fig. 1.
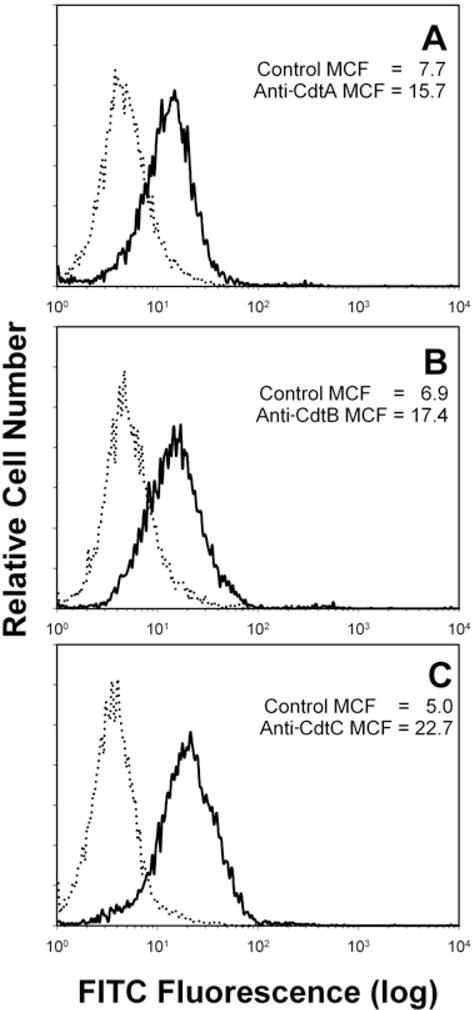
Detection of Cdt subunits associated with the Jurkat cell surface following treatment with Cdt holotoxin. Jurkat cells were exposed to Cdt holotoxin (2 μg ml−1) for 2 h and then treated with control murine IgG (dotted lines), anti-CdtA mAb [solid line in (A); mAb Cdt162]; anti-CdtB mAb [solid line in (B); mAb Cdt171] or anti-CdtC mAb [solid line in (C); mAb Cdt112]. Cells were then sequentially stained with goat anti-mouse Ig conjugated to biotin and streptavidin conjugated to FITC and analysed by flow cytometry. FITC fluorescence is plotted versus relative cell number. Numbers represent the mean channel fluorescence (MCF); at least 10 000 cells were analysed per sample. Results are representative of three experiments.
To demonstrate that the Cdt subunits not only associate with the cell membrane, but localize to lipid rafts, Jurkat cells were treated with the holotoxin and then dual stained to identify individual Cdt peptides and GM1, a well-established marker for membrane rafts. Cells were first exposed to cholera toxin B unit (CTB)-AlexaFluor 647 to identify GM1 and patches representing microdomains were induced by treatment with anti-CTB antisera (Figs 2B, E and H, 3B, E and H, 4B, E and H). Cells were then exposed to Cdt holotoxin for 2 h, washed and stained with mAb to either CdtA, CdtB or CdtC (Figs 2D and G, 3D and G, 4D and G). To control for non-specific staining with the anti-Cdt mAbs, isotype-matched control IgG was used instead (Figs 2A, 3A and 4A). Analysis of merged images indicates that both CdtA (Fig. 2F and I) and CdtC (Fig. 4F and I) colocalize with GM1 (CTB fluorescence); virtually all CdtA (> 96%) and CdtC (> 88%) fluorescence is found associated with the GM1 patches. In contrast, most, but not all, of CdtB fluorescence exhibited colocalization to GM1 (Fig. 3F and I). Analysis of the fluorescence distribution indicates that only 70% of anti-CdtB fluorescence is associated with GM1. While CdtB is known to enter cells, it should be noted that these experiments only assessed Cdt subunits associated with the cell surface; immunofluorescent staining was performed before permeabilization for DAPI staining. Wheat germ agglutinin was also employed to verify that the toxin did not alter cell permeability; no difference was noted in the immunofluorescent pattern for wheat germ agglutinin in control cells versus Cdt holotoxin-treated cells (data not shown).
Fig. 2.
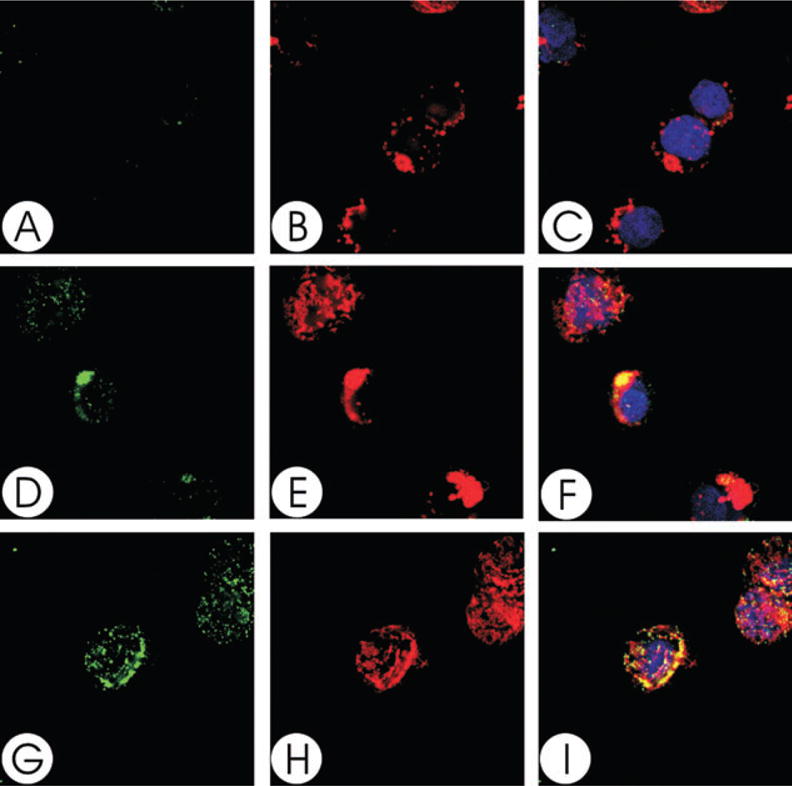
Colocalization of CdtA with GM1. Jurkat cells were treated with CTB conjugated to AlexaFluor 647 and patched with anti-CTB sera. The cells were then treated with Cdt holotoxin and stained with control IgG (A–C) or anti-CdtA (mAb162; D–I) as described in Experimental procedures and assessed using laser confocal microscopy. Images of FITC fluorescence (A, D and G) and AlexaFluor 647 fluorescence (B, E and H) are shown as well as merged images (C, F and I) showing both FITC (green) and AlexaFluor 647 (red) fluorescence along with DAPI-stained nuclei; colocalization is shown in yellow. Analysis of the images indicate that >96% of CdtA fluorescence colocalizes with CTB fluorescence. Results are representative of three experiments.
Fig. 3.
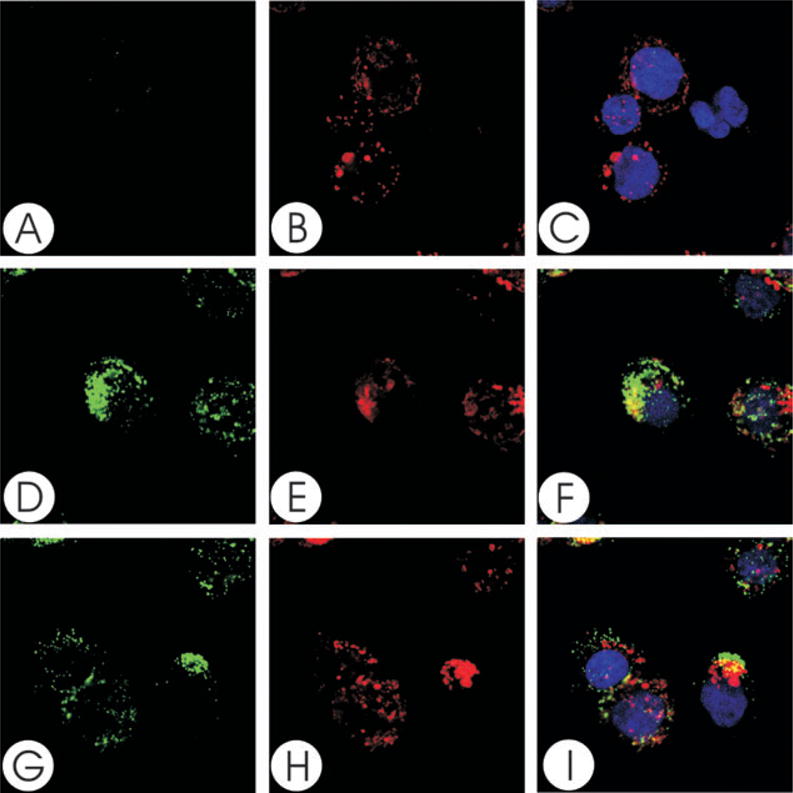
Colocalization of CdtB with GM1. Jurkat cells were treated with CTB conjugated to AlexaFluor 647 and patched with anti-CTB sera. The cells were then treated with Cdt holotoxin and stained with control IgG (A–C) or anti-CdtB (mAb171; D–I) as described in Experimental procedures and assessed using laser confocal microscopy. Images of FITC fluorescence (A, D and G) and AlexaFluor 647 fluorescence (B, E and H) are shown as well as merged images (C, F and I) showing both FITC (green) and AlexaFluor 647 (red) fluorescence along with DAPI-stained nuclei; colocalization is shown in yellow. Analysis of the images indicates that 70% of CdtB fluorescence colocalizes with CTB fluorescence. Results are representative of three experiments.
Fig. 4.
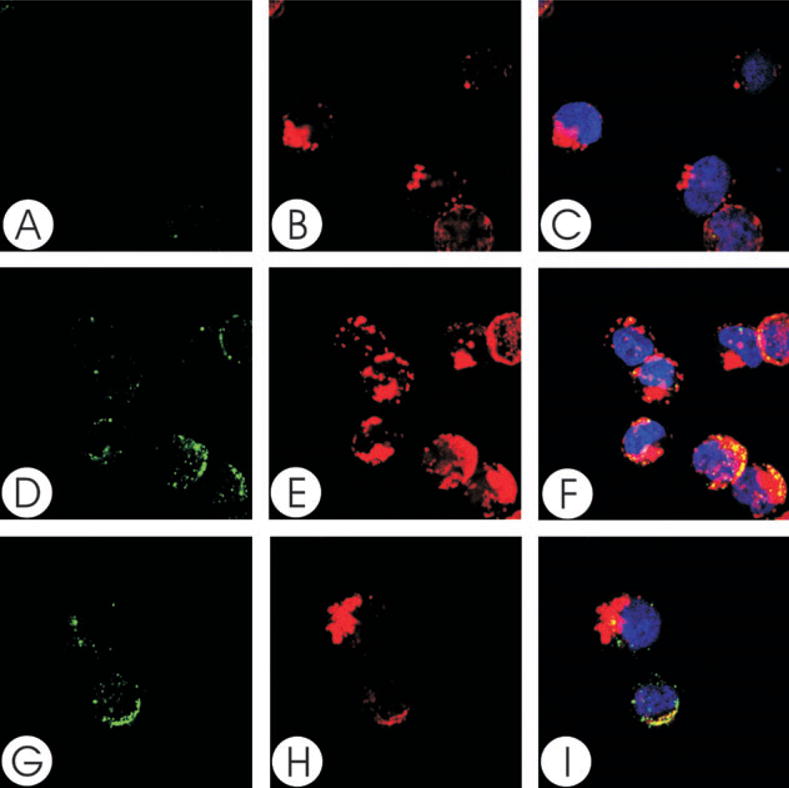
Colocalization of CdtC with GM1. Jurkat cells were treated with CTB conjugated to AlexaFluor 647 and patched with anti-CTB sera. The cells were then treated with Cdt holotoxin and stained with control IgG (A–C) or anti-CdtC (mAb112; D–I) as described in Experimental procedures and assessed using laser confocal microscopy. Images of FITC fluorescence (A, D and G) and AlexaFluor 647 fluorescence (B, E and H) are shown as well as merged images (C, F and I) showing both FITC (green) and AlexaFluor 647 (red) fluorescence along with DAPI-stained nuclei; colocalization is shown in yellow. Analysis of the images indicates that >88% of CdtC fluorescence colocalizes with CTB fluorescence. Results are representative of three experiments.
Further support for localization of Cdt peptides to cholesterol-rich microdomains was obtained by isolating detergent-resistant membranes (DRMs) from both untreated (no toxin) Jurkat cells and cells exposed to Cdt holotoxin. Following a 2 h incubation, Jurkat cells were disrupted, homogenized in ice-cold Triton X-100 and ultra-centrifuged on a sucrose gradient. Two distinct low-buoyant-density bands, designated DRM1 and DRM2, were obtained (Fig. 5). The lipid and protein composition of these bands was analysed to verify that they were indeed rafts. As shown in Fig. 5, both DRM1 and DRM2 were enriched in GM1 (dot blot) and in total cholesterol. Scanning densitometric analysis of the blots indicated that GM1 was approximately equal in DRM1 and DRM2 from both control and toxin-treated cells. DRM1 isolated from toxin treated cells had a cholesterol/phosphate ratio of 0.18; in contrast, the Triton X-100 soluble fraction had a ratio of 0.08. Similarly, DRM1 from untreated cells had a cholesterol/phosphate ratio of 0.14 and the ratio of the soluble fraction was 0.08. Moreover, the raft-associated protein, Lck, was enriched in these fractions as well. In contrast, the transferrin receptor (CD71), a non-raft-associated protein, was found only in the soluble fraction.
Fig. 5.
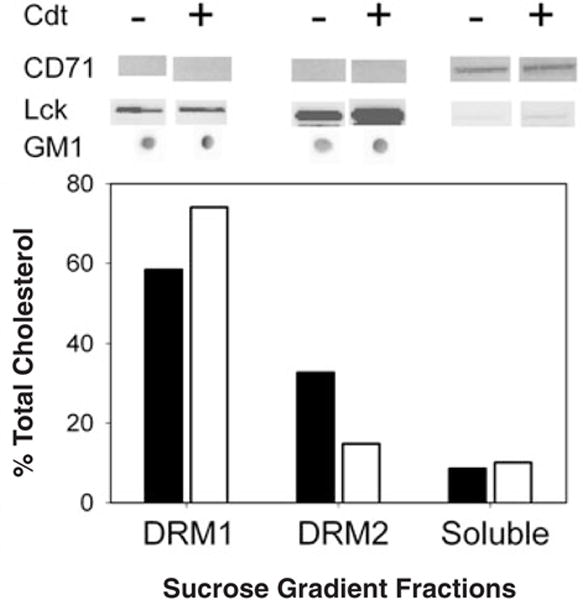
Isolation of Jurkat cell DRMs. Jurkat cells were treated with Cdt holotoxin for 2 h and DRMs were isolated as described in Experimental procedures. Two distinct low-buoyant-density bands, designated DRM1 and DRM2, were obtained. The composition of these bands was analysed to ascertain that they were indeed lipid rafts. DRM1, DRM2 and the soluble fraction were assessed for cholesterol (bottom) and GM1 (dot blot). The protein profile of the fractions was assessed by Western blot analysis for the presence of the raft-associated protein, Lck, which was enriched in these fractions. In contrast, the transferrin receptor (CD71), a non-raft-associated protein, was found in the soluble fraction. Results are representative of three experiments.
The DRMs were also analysed to determine the relative distribution of toxin subunits (Fig. 6). Following treatment of Jurkat cells with Cdt holotoxin, the DRMs were isolated and analysed by Western blot. All three subunits were found associated with DRM1; >95% of CdtA and CdtC were associated with this fraction. In contrast, 80% of CdtB was present in DRM1 and a significant amount of peptide was also observed in DRM2 (15.8%) as well as in the soluble fraction (3.5%). It should be noted that the amount of CdtB present in the soluble fraction was probably greater than 3.5% when total sample volume is taken into account. CdtA and CdtC were minimally detected in DRM2, 1.6% and 5.4%, respectively; neither of these subunits were found in the soluble fraction. Moreover, both CdtA and CdtC were found to be required for toxin localization to membrane lipid rafts. CdtB was not detected in DRMs when cells were exposed to this subunit alone. In contrast, both CdtA and CdtC were able to localize to DRM1 (> 98%) when Jurkat cells were treated with only these two subunits. These results were not surprising as the CdtA and CdtC subunits have been shown to be required for association of the holotoxin with cells.
Fig. 6.

Western blot analysis of Cdt peptides associated with DRMs. Jurkat cells were treated with Cdt holotoxin, CdtB peptide or both CdtA and CdtC for 2 h; DRMs were isolated as described in Experimental procedures. DRM1, DRM2 and the soluble fraction were analysed for the presence of each Cdt peptide by Western blot analysis using mAb specific for CdtA, CdtB and CdtC. The immunoblots were analysed by digitized scanning densitometry; the numbers represent the relative distribution (%) of each subunit within DRM1, DRM2 and the soluble (s) fractions respectively. Results are representative of three experiments.
In these studies we observed that the relative buoyancy of the DRM fractions differed upon toxin treatment; DRMs from toxin treated cells routinely floated at 13% (DRM1) and 22% (DRM2) sucrose while DRMs from untreated cells floated at 15% (DRM1) and 20% (DRM2) sucrose. The change in the banding pattern of the DRMs prompted us to further assess the lipid composition of membrane rafts from control and toxin-treated cells. As shown in Table 1, there was no significant difference between the major classes of phospholipids in control DRMs versus that of DRMs isolated from Cdt-treated cells. Also, the phospholipid content of DRM1 and DRM2 were similar.
Table 1.
Lipid content of Jurkat DRMs (mole%).a
| Lipid | DRM1
|
DRM2
|
||
|---|---|---|---|---|
| Control | Cdt-treated | Control | Cdt-treated | |
| SM | 10.5 ± 2.3 | 8.2 ± 1.2 | 11.8 ± 6.2 | 9.7 ± 5.3 |
| PC | 54.3 ± 4.0 | 53.8 ± 5.7 | 39.7 ± 10.7 | 42.5 ± 7.3 |
| PS | 7.7 ± 1.7 | 7.5 ± 2.5 | 11.0 ± 5.3 | 7.4 ± 0.5 |
| PI | 6.2 ± 0.6 | 5.2 ± 2.0 | 6.1 ± 2.1 | 4.0 ± 1.3 |
| PE | 20.8 ± 2.6 | 23.4 ± 1.3 | 22.8 ± 5.0 | 30.2 ± 13.2 |
| PG | 0.2 ± 0.3 | 0.2 ± 0.3 | ND | ND |
| Lyso-PG | 2.7 ± 0.2 | 1.6 ± 1.6 | 0.7 ± 0.1 | ND |
| PA | 5.0 ± 5.0 | 0.7 ± 0.7 | ND | ND |
Results are mean ± SEM of three experiments each performed in duplicate.
SM, sphingomyelin; PC, phosphatidylcholine; PS, phosphatidylserine; PI, phosphatidylinositol; PE, phosphatidylethanolamine; PG, phosphatidylglycerol; PA, phosphatidic acid; ND, not detected.
To determine whether Cdt action is dependent on membrane microdomains, we assessed whether the integrity of these cholesterol enriched rafts was critical to Cdt-induced cell cycle arrest. Jurkat cell membranes were depleted of cholesterol with 0–10 mM methyl β-cyclodextrin (MβCD). After 30 min exposure, cells were washed, incubated for 18 h in the presence of Cdt holotoxin (40 pg ml−1) and analysed for cell cycle distribution. Jurkat cells exposed to only Cdt holotoxin exhibited 60% G2 cells (Fig. 7A); this compares with 15% G2 cells in untreated controls (data not shown). Pre-treatment of Jurkat cells with 2.5–10 mM MβCD resulted in a dose-dependent reduction in Cdt holotoxin-induced G2 accumulation: 46.9% G2/M (2.5 mM; Fig. 7B), 32.3% G2/M (5.0 mM; Fig. 7C) and 27.1% G2/M (10 mM; Fig. 7D). As reported by others, no cytotoxic effects of MβCD were noted at these concentrations (Nguyen and Taub, 2002; Pizzo et al., 2002; Rajendran et al., 2003). To control for MβCD effects unrelated to cholesterol depletion, Jurkat cells were also pre-treated with cholesterol-saturated MβCD. Cells were treated as described above with medium, 10 mM MβCD or 10 mM cholesterol-saturated MβCD, washed and then incubated for 18 h in the presence of Cdt holotoxin. Jurkat cells exposed to medium only exhibited 15.2% G2/M cells (Fig. 8A) while cells exposed to toxin alone exhibited more than threefold increase in the percentage of G2/M cells (49.3%; Fig. 8B). As shown in Fig. 8C, cells first treated with 10 mM MβCD and then exposed to Cdt holotoxin exhibited only a slight increase in G2/M cells (25%); this compares with 49% G2 cells when Jurkat cells were pre-treated with cholesterol-saturated MβCD (Fig. 8D).
Fig. 7.
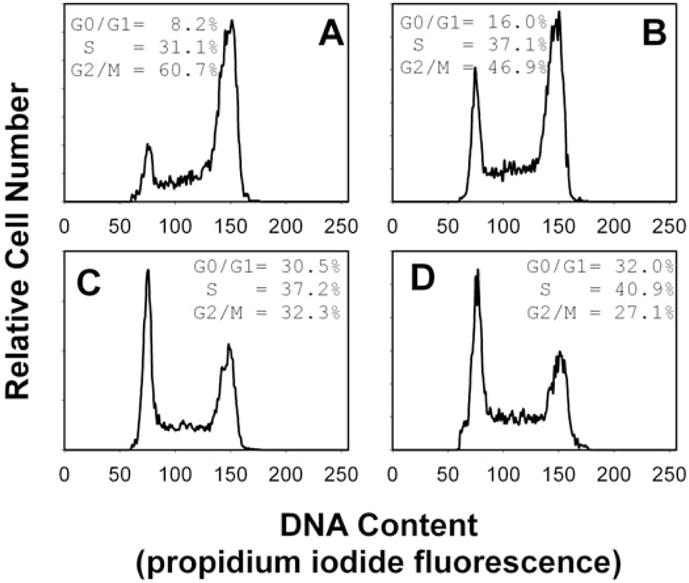
Effect of MβCD on Cdt-induced cell cycle arrest. Jurkat cells were pre-treated with medium (A), 2.5 mM (B), 5.0 mM (C) or 10 mM (D) MβCD for 30 min. The cells were washed, exposed to Cdt holotoxin (40 pg ml−1), incubated for 18 h, stained with propidium iodide and analysed for cell cycle distribution by flow cytometry. Cell cycle distribution is based on DNA content (propidium iodide fluorescence) which is plotted versus relative cell number. G0/G1 cells are found in the first peak (MCF of 75) and G2/M cells in the second peak (MCF of 150); the percentage of cells in S phase was determined based on computer modelling as noted in Experimental procedures. Numbers represent the percentage of cells in the G0/G1, S and G2/M phases of the cell cycle. Results are representative of four experiments; 15 000 cells were analysed for each sample. Parallel experiments verified that MβCD did not adversely affect Jurkat cell viability as determined by propidium iodide exclusion.
Fig. 8.
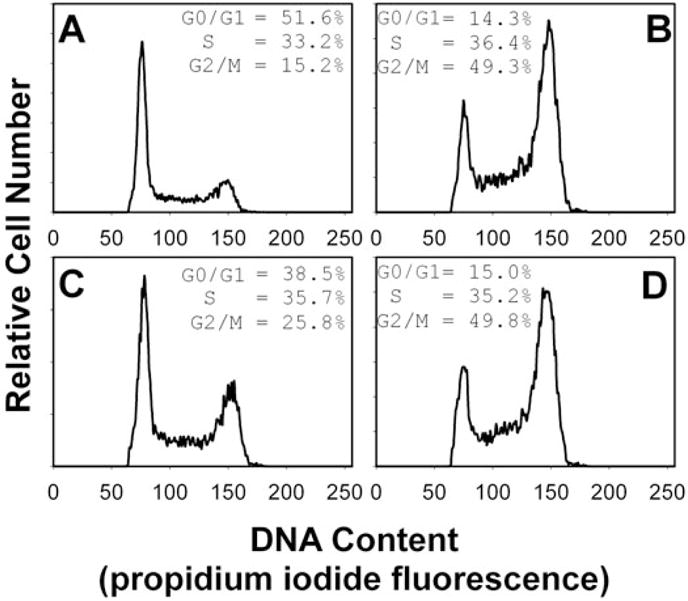
Effect of MβCD and cholesterol-saturated MβCD on Cdt-induced cell cycle arrest. Jurkat cells were pre-treated with medium (A and B), 10 mM MβCD (C) or cholesterol-saturated MβCD (D) for 30 min, washed, exposed to Cdt holotoxin (40 pg ml−1; B–D), incubated for 18 h, stained with propidium iodide and analysed for cell cycle distribution by flow cytometry as described. Numbers represent the percentage of cells in the G0/G1, S and G2/M phases of the cell cycle. Results are representative of four experiments; 15 000 cells were analysed for each sample.
In another series of experiments, we observed that cholesterol reconstitution of MβCD-treated cells restores susceptibility to Cdt holotoxin. As shown in Fig. 9, pretreatment of Jurkat cells with MβCD reduced cell susceptibility to Cdt-induced G2/M arrest. Control cells contained 13.4% G2/M cells (Fig. 9A); the percentage of cells in the G2/M phase increased in Cdt holotoxin-treated cells (62.9%; Fig. 9B) and cells pre-exposed to MβCD and then treated with Cdt holotoxin exhibited 30.2% G2/M (Fig. 9D). Cholesterol repletion of MβCD-treated cells with cholesterol-saturated MβCD restored susceptibility of Jurkat cells to Cdt holotoxin; these cells contained 57.3% G2/M (Fig. 9F). It should also be noted that Jurkat cells were not adversely affected by these agents; cell cycle profiles of cells treated with either MβCD (Fig. 9C) or MβCD plus cholesterol-saturated MβCD (Fig. 9E) in the absence of toxin were similar to control cells (Fig. 9A). Thus, it appears that intact membrane rafts are critical to the action of Cdt in lymphocytes.
Fig. 9.
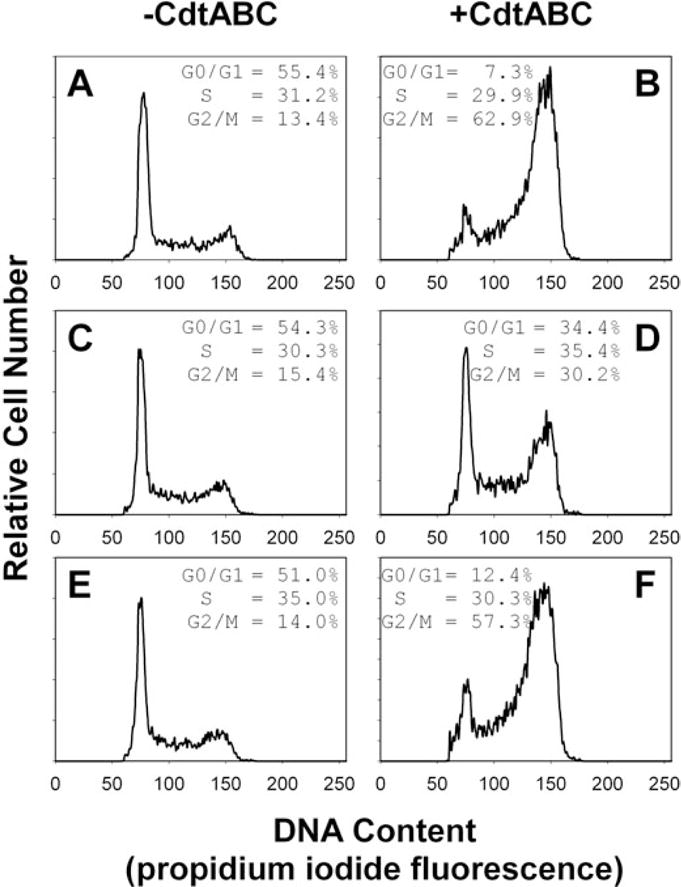
Cholesterol repletion in MβCD-treated cells restores susceptibility to Cdt. Jurkat cells were first exposed to medium (A and B) or 5 mM MβCD (C–F) for 30 min. The cells were washed and incubated for 30 min in the presence of medium (A–D) or cholesterol-saturated MβCD (E and F; 0.5 mM). Cells were then incubated for 18 h in the presence of medium (A, C and E) or Cdt holotoxin (40 pg ml−1; B, D and F), stained with propidium iodide and analysed for cell cycle distribution by flow cytometry. Numbers represent the percentage of cells in the G0/G1, S and G2/M phases of the cell cycle. Results are representative of three experiments; 15 000 cells were analysed for each sample.
In a final series of experiments, we assessed whether cholesterol depletion also altered the ability of the toxin to associate with cells. Jurkat cells were pre-treated with 10 mM MβCD for 30 min, exposed to Cdt holotoxin for 1 h and then the cells were analysed by flow cytometry for the presence of toxin subunits on the surface following staining with anti-Cdt mAb. As shown in Fig. 10, all three Cdt subunits could be detected on cells pre-incubated in medium alone; mean channel fluorescence (MCF) was 34.9, 20.4 and 17.8 for anti-CdtA (Fig. 10C), anti-CdtB (Fig. 10E) and anti-CdtC (Fig. 10G) respectively. Pretreatment of cells with MβCD reduced toxin binding; the MCF was reduced for all three subunits: anti-CdtA (9.0; Fig. 10D), anti-CdtB (13.0; Fig. 10F) and anti-CdtC (10.2; Fig. 10G).
Fig. 10.
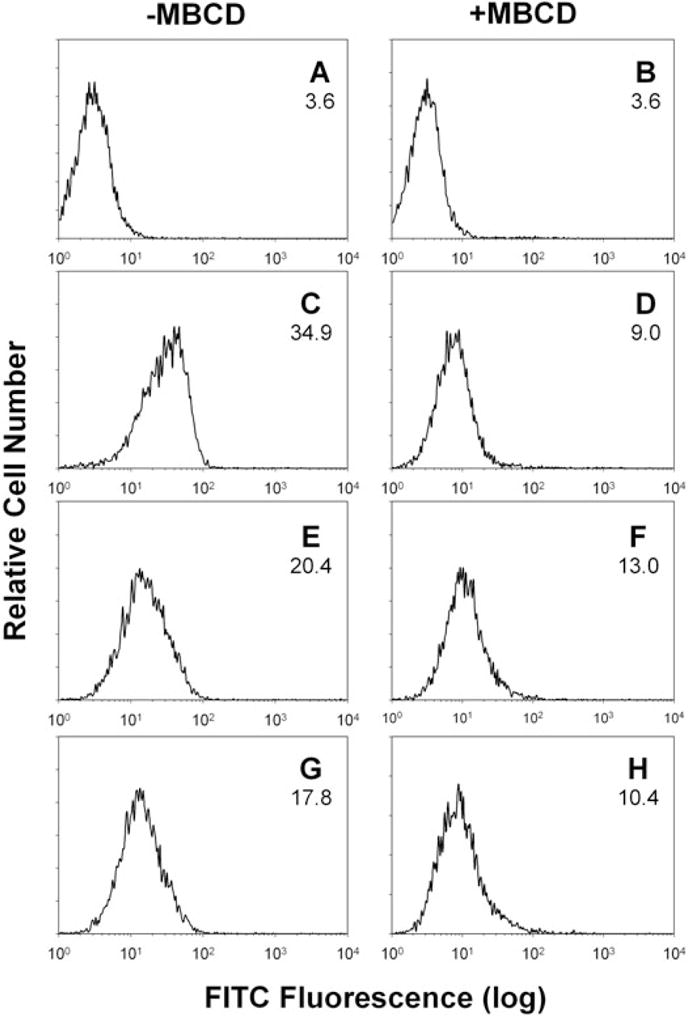
Effect of cholesterol depletion on Cdt binding to Jurkat cells. Jurkat cells were pretreated with medium (A, C, E and G) or 10 mM MβCD (B, D, F and H) for 30 min. The cells were washed and treated with Cdt for 1 h. Jurkat cells were then washed and stained with control IgG (A and B), anti-CdtA mAb (mAb162; C and D), anti-CdtB mAb (mAb171; E and F) or anti-CdtC mAb (mAb112; G and H); cells were then exposed to goat anti-mouse IgG conjugated to FITC as described in Experimental procedures. Cells were analysed by flow cytometry for FITC fluorescence; numbers in each panel represent the MCF. Results are representative of three experiments; at least 15 000 cells were analysed per sample.
Discussion
In this study, we provide evidence that Cdt preferentially associates with cholesterol-rich membrane rafts. The association of Cdt subunits with lipid rafts was corroborated utilizing two independent approaches. It is well established that lipid rafts are enriched in glycosphingolipids, including the ganglioside GM1. GM1 can be detected within lipid microdomains with CTB, the membrane-binding cholera toxin subunit. Once bound to GM1, CTB is readily observed microscopically following patch formation induced by exposing cells to anti-CTB. Utilizing this approach, we were able to demonstrate colocalization of all three Cdt subunits with CTB bound to GM1. It is noteworthy that essentially all of the CdtA subunit and most of the CdtC subunit co-patched with GM1. In contrast, approximately 30% of CdtB was not associated with GM1. In addition to microscopic colocalization, we also employed a biochemical approach to ascertain that Cdt peptides associate with membrane rafts. Following treatment of Jurkat cells with Cdt holotoxin, detergent-resistant cholesterol rich microdomains were isolated and assessed by Western blot for the presence of toxin subunits. All three subunits were found within the isolated rafts. Both CdtA and CdtC were restricted to the raft, whereas a portion of CdtB was also observed to be in the soluble fraction. The Western blot is consistent with the notion that CdtB enters the cell and that the other two subunits most likely remain membrane associated. These findings are consistent with observations made by several laboratories suggesting that CdtB, the active Cdt subunit, must enter the cell to induce cell cycle arrest (Shenker et al., 1999; 2004; Cortes-Bratti et al., 2000; Lara-Tejero and Galan, 2000; McSweeney and Dreyfus, 2004; Guerra et al., 2005). Moreover, our studies clearly demonstrate that CdtA and CdtC are required for localization to these membrane micodomains; these results are consistent with the notion that CdtA and CdtC represent the binding components of the holotoxin.
The microscopic and biochemical analyses provide strong support that the Cdt subunits associate with lipid rafts. However, these data do not provide information as to whether the microdomains actually participate in the sequence of toxin-induced events leading to cell cycle arrest. A frequently employed approach to address this possibility consists of disrupting rafts by extracting cholesterol with sequestering agents, such as MβCD. This drug is efficient in extracting membrane cholesterol resulting in dispersal of the rafts into the plasma membrane; as a consequence the rafts are not only disrupted but also lose their detergent-resistant properties. Thus, Jurkat cells were pre-treated with 2.5–10 mM MβCD for 30 min, exposed to Cdt and then the cells were monitored for cell cycle progression. Cells treated with MβCD were clearly resistant to Cdt-induced G2 arrest. These effects were dependent on the dose of MβCD employed. MβCD has been shown to have a number of other effects on cells besides depletion of cholesterol; these include plasma membrane depolarization and inhibition of calcium mobilization among others (Pizzo and Viola, 2003). To control for these effects, we also employed cholesterol-saturated MβCD which is not able to extract cholesterol but retains the capacity to induce these other effects. Indeed, cholesterol-saturated MβCD failed to inhibit Cdt-induced G2 arrest. In other experiments, cholesterol reconstitution of MβCD depleted cells restored susceptibility to Cdt indicating that MβCD was not toxic under these conditions. Collectively, these results clearly demonstrate that the integrity of membrane lipid rafts is crucial to the action of Cdt.
An unanticipated effect of Cdt treatment was that the relative buoyancy of the DRMs from toxin-treated cells changed suggesting that the lipid composition of membrane rafts had been altered. Therefore, we analysed the lipid content of both DRM 1 and DRM2. Cdt treatment did not induce any significant change in content of the major classes of phospholipid in either DRM1 or DRM2. It is possible that the change in DRM buoyance is the result of altered protein content; this possibility is the focus of ongoing studies.
We have previously shown that the CdtA and CdtC subunits are responsible for the ability of the Cdt holotoxin to interact with the cell surface. Furthermore, analysis of the crystal structure of the holotoxin suggests that CdtA and CdtC together contribute towards the formation of two notable surface elements; these include an aromatic cluster and a deep groove both of which may be critical to the interaction of Cdt with the cell surface (Nesic et al., 2004). Indeed, mutagenesis studies suggest that these structures are vital for toxin activity. To date, no definitive receptor has been identified for Cdt although recent evidence suggests that carbohydrate residues might be important (McSweeney and Dreyfus, 2005). However, our studies do indicate that these membrane microdomains must be intact not only for Cdt to intoxicate cells, but also to associate with cells as well. In conclusion, we demonstrate that the Cdt holotoxin interacts with lipid rafts and that the integrity of these microdomains is vital to both the binding and action of the toxin. This association between Cdt subunits and lipid rafts significantly advances our understanding of how the holotoxin interacts with host cells and raises several possibilities. Lipid microdomains have been shown to serve as a site of attachment for pathogens and provide a means to concentrate receptors for toxins and thereby increase binding affinity. These microdomains also play a key role in triggering internalization and may provide a means of entry for the CdtB subunit leading to retrograde transport through the trans-golgi network. Finally, rafts may also serve as signalling platforms; this raises the possibility that a component of the mechanism of Cdt action may indeed involve signal transduction. Studies are currently in progress to further define the role of lipid rafts in action of this toxin.
Experimental procedures
Preparation of Cdt holotoxin and Cdt peptides
Recombinant Cdt holotoxin was used for most of the experiments described in this study. Construction and characterization of the plasmid pUCAacdtABChis which expresses the Cdt holotoxin containing a His tag on the CdtC peptide has previously been reported (Shenker et al., 2004). The plasmid was constructed so that the cdt genes were under control of the lac promoter; all ligation mixtures were transformed into E. coli DH5α. Cultures of transformed E. coli were grown in 1 l of LB broth and induced with 0.1 mM IPTG for 2 h; bacterial cells were harvested, washed and resuspended in 50 mM Tris (pH 8.0). The cells were frozen overnight, thawed and sonicated. The His-tagged holotoxin was isolated by nickel affinity chromatograpy as previously described (Shenker et al., 2000). Briefly, the sonicated bacterial extracts were applied to a histidine-binding column (HiTrap Chelating HP; Amersham Biosciences, Uppsala, Sweden). The column was washed and His-tagged proteins were eluted with 500 mM imidazole; previous studies have demonstrated that the isolated holotoxin, designated CdtABC, consists of a heterotrimeric complex of CdtA, CdtB and CdtC (Shenker et al., 2004).
Individual Cdt peptides were expressed using an in vitro expression system; construction of the plasmids pGEMCdtA, pGEMCdtB and pGEMCdtC was previously described (Shenker et al., 2005). We employed the Rapid Translation System to express recombinant peptides (RTS 500 ProteoMaster; Roche Applied Science); reactions were run according to the manufacturer’s specification (Roche Applied Science) using 10–15 μg of template DNA. After 20 h at 30°C, the reaction mix was removed and the expressed Cdt peptides were purified by nickel affinity chromatography as previously described.
Cell culture and analysis of cell cycle
The T cell leukaemia cell line Jurkat (E6-1; American Type Tissue Culture Collection, Manassas, VA) was maintained as previously described (Shenker et al., 2004). Briefly, cells were maintained in RPMI 1640 supplemented with 10% FCS, 2 mM glutamine, 10 mM Hepes, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin. Cells were harvested in mid-log growth phase and plated at 5 × 105 cells ml−1 in 24-well tissue culture plates or as described below. The cells were exposed to medium, 2.5–10 mM MβCD (Sigma Chemical, St Louis, MO) or 10 mM cholesterol-saturated MβCD; 0.5 mM cholesterol-saturated MβCD was employed for repletion experiments. Cholesterol-saturated MβCD was prepared as described by Christian et al. (1997). Following a 30 min incubation, cells were treated with Cdt holotoxin or medium (control) and incubated for 18 h. To measure Cdt-induced cell cycle arrest, Jurkat cells were washed and fixed for 60 min with cold 80% ethanol. After washing, the cells were stained with 10 μg ml−1 propidium iodide containing 1 mg ml−1 RNase (Sigma Chemical) for 30 min. Samples were analysed on a Becton–Dickinson FacstarPLUS flow cytometer (BD Biosciences, San Jose, CA). Propidium iodide fluorescence was excited by an argon laser operating at 488 nm and fluorescence measured with a 630/22 nm bandpass filter using linear amplification. A minimum of 15 000 events were collected on each sample; cell cycle analysis was performed using Modfit (Verity Software House, Topsham, ME).
Isolation of Triton X-100-resistant membrane rafts
Triton X-100-resistant membrane rafts were prepared from the T-cell leukaemia cell line Jurkat (E6-1); the cells (2.0 × 106 cells ml−1) were incubated in T-75 flasks in the presence of medium or Cdt holotoxin for 2 h. Following the incubation period, cells were washed in MOPS buffer and detergent-resistant fractions were isolated as previously described (Boesze-Battaglia et al., 2002). Briefly, the cell pellet (4–5 × 108 cells) was resuspended in 1.0 ml of MOPS buffer; Triton X-100 (2% w/v) was added to a final concentration of 1% Triton X-100. To distinguish between Triton X-100-resistant membranes and simply a partial detergent-dependent non-raft-specific solubilization, control cells were homogenized in 1% OG (final) in parallel with the Triton X-100-treated samples. Both Triton X-100- and OG-treated samples were homogenized with three passes of a glass pestle through a glass Tenboeck tissue grinder. Sucrose was added to the homogenate (final concentration of 0.9 M) and overlayed with 1.0 ml of 0.8, 0.70, 0.65, 0.60, 0.55 and 0.50 M sucrose. The homogenates were centrifuged at 40 000 r.p.m. for 20 h at 4°C. Two prominent bands were recovered, designated DRM1 and DRM2; these were washed and resuspended in 0.2 ml of Hepes buffer.
Western blot and GM1 analyses
Samples (20 μl) were separated on 12% SDS-PAGE and then transferred to nitrocellulose. The membrane was blocked with BLOTTO and then incubated with one of the following primary antibodies for 18 h at 4°C (Shenker et al., 1999): anti-CdtA mAb (CdtA21), anti-CdtB mAb (CdtB194), anti-CdtC mAb (CdtC63), anti-CD71 mAb (Santa Cruz Biotechnology, Santa Cruz, CA) or anti-Lck mAb (Upstate Cell Signaling, Charlottesville, VA). Membranes were washed, incubated with goat anti-mouse immunoglobulin (1:1000 dilution; Southern Biotechnology, Birmingham, AL) conjugated to horseradish peroxidase. The Western blots were developed using chemiluminescence (ECL; Amersham Biosciences) and analysed by digital densitometry (Kodak Image Systems). GM1 was detected by spotting 1 μl directly on nitrocellulose membranes; samples were adjusted for phosphate content. The membranes were blocked in BLOTTO and then incubated with CTB conjugated to horseradish peroxidase (85 ng ml−1; Sigma Chemical) for 1 h. The membranes were washed and analysed as described above.
Analysis of lipid composition
Cholesterol was determined as described by Allain et al. (1974) and total phosphate as described by Bartlett (1959). Lipids were extracted from the low-buoyant-density fractions as previously described (Bligh and Dyer, 1959). Before the extraction of lipids from the detergent solubilized fractions, samples were dialysed for 48 h against 10 mM Hepes, 0.5 M NaCl, pH 7.4 to remove detergent. The extraction yield of total phospholipid was 90 ± 3.6% indicating that the presence of residual detergent (Triton X-100 or OG) in the solubilized fractions did not interfere with the extraction. Chloroform extracts were evaporated under nitrogen and diluted into chloroform immediately before lipid analysis by gradient elution using normal phase HPLC/ELSD (Avanti-Polar Lipids, Alabaster, AL). Standards 1 mg ml−1 included: triolein, cholesterol, ceramide, oleic acid, phosphatidylglycerol (PG), phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), phosphatidic acid (PA), soy-phosphatidylinositol (PI), and 0.5 mg ml−1 lyso-phosphatidylcholine (lyso-PC), lyso-phosphatidylethanolamine (lyso-PE), lyso-phosphatidylglycerol (lyso-PG) and lyso-phosphatidylserine (lyso-PS). Standards and samples were injected on a normal phase HPLC column and analysed by an evaporative light scattering detector (ELSD). A five-level calibration curve was used to calculate the amount of lipid species in each sample.
Laser confocal microscopy
Jurkat cells were incubated (2 × 106 ml−1) with CTB conjugated to AlexaFluor 647 (1 μg ml−1; Molecular Probes) for 30 min at 4°C; after washing twice with medium, CTB was cross-linked with goat anti-CTB sera (1:25 dilution; Calbiochem) for 30 min at 4°C followed by an additional 45 min incubation at 37°C. Cells were washed and incubated with Cdt holotoxin (2 μg ml−1) for 2 h at 37°C; after two washes the cells were blocked with normal goat IgG (Southern Biotechnology), treated with anti-Cdt mAb for 30 min at 4°C, washed, stained with goat anti-mouse Ig conjugated to biotin (1 μg; Southern Biotechnology) for 30 min at 4°C, washed and exposed to streptavidin conjugated to FITC (0.2 μg; Molecular Probes). The cells were then fixed in 2% paraformaldehyde, washed, permeabilized with 0.1% sodium citrate and 0.1% Triton X-100 for 2 min, washed and stained with DAPI (0.75 μM); stained cells were centrifuged and mounted onto microscope slides using Fluoromount-G (Southern Biotechnology). Images of cells were taken on a Bio-Rad Radiance 2100 confocal Microscope (Bio-Rad, Hercules, CA). DAPI, FITC and AlexaFluor 647 fluorescence were excited by blue diode, argon (488 nm) and red diode lasers respectively. Fluorescence emission was captured using the following filters: 376 ± 24 nm (DAPI); 515 ± 30 nm (FITC) and 660 nm and above (AlexaFluor 647). All images were captured with the same laser settings. Analysis of fluorescent probe colocalization was performed using image analysis software (Metamorph; Universal Imaging Corporation, Downingtown, PA). Regions of interest were defined to include cells that did not overlap. The region was segmented to select pixels above a constant threshold value (> 60% above background) which represent true fluorescence. As both spatial location and intensity of pixels contribute to colocalization, the values represent the integrated intensity; pixels in both FITC and AlexaFluor 647 images had similar brightness values and spatial location. Colocalization analysis was performed on all cells present in Figs 2–4.
Generation of anti-Cdt mAbs
Recombinant Cdt holotoxin was expressed in E. coli using a plasmid containing all three cdt genes (pUCAacdtABChis) as previously described; the His-tagged complex was purified by nickel affinity chromatography (Shenker et al., 2004). Anti-Cdt mAbs were generated as previously described (Lally et al., 1997). Briefly, BALB/c mice (Charles River Laboratories, Wilmington, MA) 10–12 weeks old were immunized by IP injection with 10–20 μg Cdt holotoxin on days 0, 10, 20 and 30 and allowed to rest for 30 days. Three days before fusion, the animals received 10 μg of peptide intravenously. Splenocytes were fused to Sp2/0-Ag14 myeloma cells in the presence of 50% polyethylene glycol (Kodak 1450). The cells were then dispersed in Kennett’s HY medium containing 20% FBS, glutamine, oxaloacetate, pyruvate, hypoxanthine and azaserine and fed 7 days later in medium lacking azaserine. Clones were visible 7–9 days after fusion and were first screened by Cdt ELISA. All mAbs were further screened by ELISA to identify the specific subunit with which they react as well as by immunofluorescence to determine whether the antibody was able to recognize an available epitope once the toxin had reacted with cells. Briefly, Jurkat cells were treated with Cdt holotoxin (2 μg ml−1) for 2 h at 37°C; cells were then washed and treated with varying amounts of anti-Cdt mAb for 30 min After washing the cells were first blocked with normal goat IgG (Southern Biotechnology) and exposed to goat anti-mouse Ig conjugated to biotin, washed, and treated with streptavidin-FITC (0.2 μg; Molecular Probes) for 30 min. FITC fluorescence was assessed by flow cytometry as previously described (Shenker et al., 2004).
Acknowledgments
The authors wish to acknowledge the School of Dental Medicine Flow Cytometry and Imaging Facility for their support of these studies as well as Lisa Pankoski, Rose Espiritu and Cheryl Gretzula for their technical expertise. This work was supported by USPHS Grant DE06014 from the National Instititute of Dental and Craniofacial Research.
References
- Abrami L, van der Goot FG. Plasma membrane microdomains act as concentration platforms to facilitate intoxication by aerolysin. J Cell Biol. 1999;147:175–184. doi: 10.1083/jcb.147.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allain CC, Poon LS, Chan CS, Richmond W, Fu PC. Enzymatic determination of total serum cholesterol. Clin Chem. 1974;20:470–475. [PubMed] [Google Scholar]
- Alonso M, Millan J. The role of lipid rafts in signaling and membrane trafficking in T lymphocytes. J Cell Science. 2001;114:3957–3965. doi: 10.1242/jcs.114.22.3957. [DOI] [PubMed] [Google Scholar]
- Bartlett GR. Phosphorous assay in column chromatography. J Biol Chem. 1959;234:466–473. [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Med Sci. 1959;8:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Boesze-Battaglia K, Dispoto J, Kahoe MA. Association of a photoreceptor-specific tetraspanin protein, ROM-1, with triton X-100-resistant membrane rafts from rod outer segment disk membranes. J Biol Chem. 2002;277:41843–41849. doi: 10.1074/jbc.M207111200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, London E. Functions of lipid rafts in biological membranes. Annu Rev Cell Dev Biol. 1998;14:111–136. doi: 10.1146/annurev.cellbio.14.1.111. [DOI] [PubMed] [Google Scholar]
- Christian A, Haynes M, Phillips M, Rothblat G. Use of cyclodextrins for manipulating cellular cholesterol content. J Lipid Res. 1997;38:2264–2272. [PubMed] [Google Scholar]
- Comayras C, Tasca C, Peres SY, Ducommun B, Oswald E, De Rycke J. Escherichia coli cytolethal distending toxin blocks the HeLa cell cycle at the G2/M transition by preventing cdc2 protein kinase dephosphorylation and activation. Infect Immun. 1997;65:5088–5095. doi: 10.1128/iai.65.12.5088-5095.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes-Bratti X, Chaves-Olarte E, Lagergard T, Thelastam M. Cellular internalization of cytolethal distending toxin from Haemophilus ducreyi. Infect Immun. 2000;68:6903–6911. doi: 10.1128/iai.68.12.6903-6911.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng K, Hansen JC. A CdtA–CdtC complex can block killing of HeLa cells by Haemophilus ducreyi cytolethal distending toxin. Infect Immun. 2003;71:6633–6640. doi: 10.1128/IAI.71.11.6633-6640.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elwell CA, Dreyfus LA. DNase I homologous residues in CdtB are critical for cytolethal distending toxin-mediated cell cycle arrest. Mol Microbiol. 2000;37:952–963. doi: 10.1046/j.1365-2958.2000.02070.x. [DOI] [PubMed] [Google Scholar]
- Gelfanova V, Hansen E, Spinola S. Cytolethal distending toxin of Haemophilus ducreyi induces apoptotic death of Jurkat T cells. Infect Immun. 1999;67:6394–6402. doi: 10.1128/iai.67.12.6394-6402.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Goot FG, Harder T. Raft membrane domains: from a liquid-ordered membrane phase to site of pathogen attack. Semin Immunol. 2001;13:89–97. doi: 10.1006/smim.2000.0300. [DOI] [PubMed] [Google Scholar]
- Guerra L, Teter K, Lilley B, Stenerlow B, Holmes R, Ploegh H, et al. Cellular internalization of cytolethal distending toxin: a new end to a known pathway. Cell Microbiol. 2005;7:921–934. doi: 10.1111/j.1462-5822.2005.00520.x. [DOI] [PubMed] [Google Scholar]
- Haghjoo E, Galan JE. Salmonella typhi encodes a functional cytolethal distending toxin that is delivered into host cells by a bacterial-internalization pathway. Proc Natl Acad Sci USA. 2004;101:4614–4619. doi: 10.1073/pnas.0400932101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horejsi V. The roles of membrane microdomains (rafts) in T cell activation. Immunol Rev. 2003;191:148–164. doi: 10.1034/j.1600-065x.2003.00001.x. [DOI] [PubMed] [Google Scholar]
- Horejsi V. Transmembrane adaptor proteins in membrane microdomains: important regulators of immunoreceptor signaling. Immunol Lett. 2004;92:43–49. doi: 10.1016/j.imlet.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Lafont F, Tran Van Nhieu G, Hanada K, Sansonetti P, van der Goot FG. Initial steps of Shigella infection depend on the cholesterol/sphingolipid raft-mediated CD44–IpaB interaction. EMBO J. 2002;21:4449–4457. doi: 10.1093/emboj/cdf457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lally ET, Kieba I, Sato A, Green C, Rosenbloom J, Korostoff J, et al. RTX toxins recognize a β2 integrin on the surface of human target cells. J Biol Chem. 1997;272:30463–30469. doi: 10.1074/jbc.272.48.30463. [DOI] [PubMed] [Google Scholar]
- Lara-Tejero M, Galan JE. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science. 2000;290:354–357. doi: 10.1126/science.290.5490.354. [DOI] [PubMed] [Google Scholar]
- Lara-Tejero M, Galan JE. CdtA, CdtB, and CdtC form a tripartite complex that is required for cytolethal distending toxin activity. Infect Immun. 2001;69:4358–4365. doi: 10.1128/IAI.69.7.4358-4365.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lencer W. Microbes and microbial toxins: paradigms for microbial–mucosal interactions V. cholera: invasion of the intestinal epithelial barrier by a stably folded protein toxin. Am J Physiol Gastrointest Liver Physiol. 2001;280:G781–G786. doi: 10.1152/ajpgi.2001.280.5.G781. [DOI] [PubMed] [Google Scholar]
- McSweeney L, Dreyfus LA. Nuclear localizaton of the Escherichia coli cytolethal distending toxin CdtB subunit. Cell Microbiol. 2004;6:447–458. doi: 10.1111/j.1462-5822.2004.00373.x. [DOI] [PubMed] [Google Scholar]
- McSweeney L, Dreyfus LA. Carbohydrate-binding specificity of the Escherichia coli cytolethal distending toxin CdtA-II and CdtC-II subunits. Infect Immun. 2005;73:2051–2060. doi: 10.1128/IAI.73.4.2051-2060.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, DiRienzo JM. Functional studies of the recombinant subunits of a cytolethal distending toxin. Cell Microbiol. 2002;4:245–255. doi: 10.1046/j.1462-5822.2002.00186.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmor M, Julius M. Role for lipid rafts in regulating interleukin-2 receptor signaling. Immunobiology. 2001;98:1489–1497. doi: 10.1182/blood.v98.5.1489. [DOI] [PubMed] [Google Scholar]
- Mayer M, Bueno L, Hansen E, DiRienzo JM. Identification of a cytolethal distending toxin gene locus and features of a virulence-associated region in Actinobacillus actinomycetemcomitans. Infect Immun. 1999;67:1227–1237. doi: 10.1128/iai.67.3.1227-1237.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montecucco C, Papini E, Schiavo G. Bacterial protein toxins penetrate cells via a four-step mechanism. FEBS Lett. 1994;3466:92–98. doi: 10.1016/0014-5793(94)00449-8. [DOI] [PubMed] [Google Scholar]
- Nalbant A, Chen C, Wang Y, Zadeh HH. Induction of T-cell apoptosis by Actinobacillus actinomycetemcomitans mutants with deletion of ltxA and cdtABC genes: possible activity of GroEllike molecule. Oral Micro-biol Immunol. 2003;18:339–349. doi: 10.1046/j.0902-0055.2003.00082.x. [DOI] [PubMed] [Google Scholar]
- Nesic D, Hsu Y, Stebbins CE. Assembly and function of a bacterial genotoxin. Nature. 2004;429:429–433. doi: 10.1038/nature02532. [DOI] [PubMed] [Google Scholar]
- Nguyen DH, Taub D. CXCR4 function requires membrane cholesterol: implications for HIV infection. J Immunol. 2002;168:4121–4126. doi: 10.4049/jimmunol.168.8.4121. [DOI] [PubMed] [Google Scholar]
- Okuda J, Kurazono H, Takeda Y. Distribution of the cytolethal distending toxin A gene (cdtA) among species of Shigella and Vibrio, and cloning and sequencing of the cdt gene from Shigella dysenteriae. Microb Pathog. 1995;18:167–172. doi: 10.1016/s0882-4010(95)90022-5. [DOI] [PubMed] [Google Scholar]
- Okuda J, Fukumoto M, Takeda Y, Nishibuchi M. Examination of diarrheagenicity of cytolethal distending toxin: suckling mouse response to the products of the cdtABC genes of Shigella dysenteriae. Infect Immun. 1997;65:428–433. doi: 10.1128/iai.65.2.428-433.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickett CL, Whitehouse CA. The cytolethal distending toxin family. Trends Microbiol. 1999;7:292–297. doi: 10.1016/s0966-842x(99)01537-1. [DOI] [PubMed] [Google Scholar]
- Pickett CL, Cottle DL, Pesci EC, Bikah G. Cloning, sequencing, and expression of the Escherichia coli cytolethal distending toxin genes. Infect Immun. 1994;62:1046–1051. doi: 10.1128/iai.62.3.1046-1051.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike LJ. Lipid rafts; heterogeneity on the high seas. Biochem J. 2004;378:281–292. doi: 10.1042/BJ20031672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzo P, Viola A. Lymphocyte lipid rafts: structure and function. Curr Opin Immunol. 2003;15:255–260. doi: 10.1016/s0952-7915(03)00038-4. [DOI] [PubMed] [Google Scholar]
- Pizzo P, Giurisato E, Tassi M, Benedetti A, Pozzan T, Viola A. Lipid rafts and T cell receptor signaling: a critical re-evaluation. Eur J Immunol. 2002;32:3082–3091. doi: 10.1002/1521-4141(200211)32:11<3082::AID-IMMU3082>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Rajendran L, Masilamani M, Solomon S, Tikkanen R, Stuermer C, Plattner H, et al. Asymmetric localization of flotillins/reggies in preassembled platforms confers inherent polarity to hematopoietic cells. Proc Natl Acad Sci USA. 2003;100:8241–8246. doi: 10.1073/pnas.1331629100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DA, Kaper JB. Cloning and sequencing of the genes encoding Escherichia coli cytolethal distending toxin. Infect Immun. 1994;62:244–251. doi: 10.1128/iai.62.1.244-251.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenker BJ, McKay TL, Datar S, Miller M, Chowhan R, Demuth DR. Actinobacillus actinomycetemcomitans immunosuppressive protein is a member of the family of cytolethal distending toxins capable of causing a G2 arrest in human T cells. J Immunol. 1999;162:4773–4780. [PubMed] [Google Scholar]
- Shenker BJ, Hoffmaster RH, McKay TL, Demuth DR. Expression of the cytolethal distending toxin (Cdt) operon in Actinobacillus actinomycetemcomitans: evidence that the CdtB protein is responsible for G2 arrest of the cell cycle in human T-cells. J Immunol. 2000;165:2612–2618. doi: 10.4049/jimmunol.165.5.2612. [DOI] [PubMed] [Google Scholar]
- Shenker BJ, Hoffmaster RH, Zekavat A, Yamguchi N, Lally ET, Demuth DR. Induction of apoptosis in human T cells by Actinobacillus actinomycetemcomitans cytolethal distending toxin is a consequence of G2 arrest of the cell cycle. J Immunol. 2001;167:435–441. doi: 10.4049/jimmunol.167.1.435. [DOI] [PubMed] [Google Scholar]
- Shenker BJ, Besack D, McKay TL, Pankoski L, Zekavat A, Demuth DR. Actinobacillus actinomycetemcomitans cytolethal distending toxin (Cdt): evidence that the holotoxin is composed of three subunits: CdtA, CdtB, and CdtC. J Immunol. 2004;172:410–417. doi: 10.4049/jimmunol.172.1.410. [DOI] [PubMed] [Google Scholar]
- Shenker BJ, Besack D, McKay T, Pankoski L, Zekavat A, Demuth DR. Induction of cell cycle arrest in lymphocytes by Actinobacillus actinomycetemcomitans cytolethal distending toxin requires three subunits for maximum activity. J Immunol. 2005;174:2228–2234. doi: 10.4049/jimmunol.174.4.2228. [DOI] [PubMed] [Google Scholar]
- Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- Xavier R, Brennan T, Li Q, McCormack C, Seed B. Membrane compartmentation is required for efficient T cell activation. Immunity. 1998;8:723–732. doi: 10.1016/s1074-7613(00)80577-4. [DOI] [PubMed] [Google Scholar]
- Zajchowski LD, Robbins SM. Lipid rafts and little caves: compartmentalized signaling in membrane microdomains. Eur J Immunol. 2002;269:737–752. doi: 10.1046/j.0014-2956.2001.02715.x. [DOI] [PubMed] [Google Scholar]