

Abstract
As a prerequisite for studies using mutant mice, we established a mouse model for investigating the molecular mechanisms by which testosterone (T) promotes muscle growth. Groups of six adult male mice (C57BL/6) received one of the following treatments: 1) vehicle (sterile distilled water; normal control) and 2) GnRH antagonist with empty (sham control) or 2 cm T-filled implant. Mice were killed 2, 6, and 8 weeks after treatment. T treatment for 8 weeks resulted in a significant (P<0.001) increase in fiber area of gastrocnemius muscles. T-induced fiber-hypertrophy was accompanied by up-regulation of the Notch ligand Delta 1 and activation of Notch signaling, as evidenced by increase in activated forms of Notch 1 and Notch 2. Consistent with this, we also observed an increase in the number of proliferating cell nuclear antigen (PCNA)-positive nuclei in muscles of T-treated mice, indicating that activation of Notch signaling enhanced cell proliferation. T supplementation not only triggered p38 mitogen-activated protein kinase (MAPK) activation but also concurrently inhibited c-Jun NH2-terminal kinase (JNK) activation within 2 weeks of treatment. Concomitant administration of SB203580, a p38 MAPK inhibitor, effectively blocked T-induced activation of Notch signaling and significantly (P<0.001) suppressed PCNA levels. Together, our results indicate that T induces muscle fiber hypertrophy through activation of Notch signaling and the inactivation of JNK together with the activation of p38 MAPK may be critical for T-induced activation of Notch signaling and, as a consequence, muscle fiber hypertrophy.
Introduction
A great deal of interest and effort has been focused on elucidating the underlying molecular mechanisms of skeletal muscle growth and development. A better understanding of this process may unveil novel targets for the development of anabolic therapies for the treatment of many diseases associated with muscle wasting. Systematic reviews of literature have concluded that testosterone (T) supplementation increases muscle mass in healthy young and old, hypogonadal, and men with chronic illnesses and with low testosterone levels (Bhasin et al. 2006). In earlier studies, we have shown that such T-induced increase in muscle size in both young and old men is associated with hypertrophy of muscle fibers and significant increases in myonuclear and satellite cell numbers (Sinha-Hikim et al. 2002, 2003, 2006). The mechanisms by which T increases satellite cell number and promotes muscle growth are not well understood.
An exciting advance in the understanding of the genetic modulation of skeletal muscle growth and development is the use of genetically altered mice either overexpressing or harboring null or loss-of-function mutations of specific genes (McPherron et al. 1997, Reisz-Porszasz et al. 2003, Kujoth et al. 2005, Vianzof et al. 2008). These mutant animals with additional manipulation (such as suppression of signal transduction pathways by the selective inhibitors during T-induced muscle growth) are invaluable tools not only for confirming or refuting a proposed function of a particular gene in an in vivo setting, but also for uncovering novel functions for a gene that are not anticipated in in vitro experiments. Thus, as a prerequisite for studies using mutant mice, we wish to establish a mouse model for investigating the molecular mechanisms by which T promotes muscle growth.
The regeneration of skeletal muscle is largely dependent on a small population of self-renewing committed stem cells, the satellite cells (Conboy & Rando 2002, Conboy et al. 2003, 2005, Morgan & Partridge 2003), which play an important role in mediating the anabolic response of T leading to muscle fiber hypertrophy (Sinha-Hikim et al. 2003, 2006). Activated satellite cells enter the cell cycle and give rise to muscle precursor cells, which undergo multiple rounds of proliferation prior to terminal differentiation and fusion with new or growing myofibers (Morgan & Partridge 2003). Notch signaling pathway is essential for the activation, proliferation, and myogenic progression of satellite cells necessary for muscle regeneration and repair (Conboy & Rando 2002, Conboy et al. 2003, 2005). Our recent studies on elderly men (Sinha-Hikim et al. 2006) also indicate the involvement of Notch signaling in T-mediated muscle fiber hypertrophy.
Mitogen-activated protein kinases (MAPKs) comprise a family of serine/threonine kinases that function as critical mediators of variety of extracellular signals (Johnson & Lapadat 2002, Wada & Penninger 2004). Members of the MAP kinase superfamily include the extracellular signal-regulated kineses (ERKs), the c-Jun NH2-terminal kinases (JNKs), also known as stress-activated protein kinases, and the p38 MAP kinases (p38 MAPKs). Available data from various cell systems suggest that ERK1 and ERK2 are activated in response to growth stimuli and promote cell growth, whereas both JNKs and p38 MAPKs are activated in response to a variety of environmental stresses and inflammatory signals and promote apoptosis and growth inhibition (Johnson & Lapadat 2002, Wada & Penninger 2004). However, the regulation of cellular homeostasis by MAPKs is more complex and varies depending on tissues, nature of the stimulus, and duration of their activation (Johnson & Lapadat 2002, Lin & Dibling 2002, Caughlan et al. 2004, Wada & Penninger 2004). The role of these kinases varies, at least in cardiac myocytes, between in vitro and in vivo (using genetically modified mouse models) settings (Liang & Mollkentin 2003). The p38 MAPK signaling has been implicated in the regulation of skeletal muscle gene expression at different stages of the myogenic process in cell culture (Lluis et al. 2006). A role of p38 MAPK in the activation of Notch signaling has also been suggested in some cell lines (Weijzen et al. 2002). Thus, a possible mechanism by which p38 MAPK can induce muscle growth is through the induction of Notch signaling.
The objectives of the present study were twofold. The first was to establish a mouse model for T-induced muscle fiber hypertrophy. The second was to examine the signal transduction pathways that mediate T-induced muscle fiber hypertrophy.
Materials and Methods
Animals
C57BL6J male mice of seven to eight-weeks of age were obtained from the Harlan Laboratories (Indianapolis, IN, USA). Animals were housed in a standard animal facility under controlled temperature (22 °C) and photoperiod (12 h of light:12 h of darkness) with access to food and water ad libitum. Groups of six adult male mice received a single s.c. injection of a long acting GnRH antagonist (GnRH-A), acyline (20 mg/kg BW) every 2 weeks for up to 8 weeks to suppress endogenous T production (Sinha Hikim et al. 2005, 2007). Acyline was kindly provided by Dr Richard P Blye (Contraceptive and Reproductive Health Branch, NICHHD, NIH). GnRH-A-treated mice were then implanted subdermally in the neck region, under ketamine-xylazine anesthesia, empty (sham control) or 2-cm T-filled SILASTIC implants and killed 2, 4, and 8 weeks after treatment. T-filled implants were prepared from polydimethylsilozane tubing (od, 1.96 mm; id, 1.47 mm; Dow-Corning, Midland, MI, USA). Implant lengths are based on the results of our previous study, which showed that a 2 cm T implant can provide the supraphysio-logical levels of T required for exerting its anabolic action on the muscle (Sinha-Hikim et al. 2007). An additional group of six mice received vehicle (distilled water) every 2 weeks for up to 8 weeks. This group was included to assess the efficacy of a high dose of T inducing muscle fiber hypertrophy when compared with mice with normal T levels (normal controls). A pilot study was performed to assess the changes in muscle fiber cross-sectional area (CSA) between normal controls and GnRH-A treated mice 8 weeks after vehicle or GnRH-A treatment. No significant differences (student's t-test) in the muscle fiber CSA was noted between normal controls and (1675±47 μm2) and GnRH-A treated mice (1379±47 μm2). Thus, normal controls were chosen for the main part of studies to compare the effects of Ton muscle growth.
To further explore the role of p38 MAPK signaling in T-induced muscle growth, we examined whether SB203580, a p38 MAPK inhibitor (Cuenda et al. 1995, Lahti et al. 2002), could prevent or attenuate T-mediated activation of Notch signaling, the key mediator of satellite cell activation and muscle growth (Conboy & Rando 2002, Conboy et al. 2003, 2005, Sinha-Hikim et al. 2006). This study was conducted after the results of the first study were known. Groups of six GnRHA treated mice received one of the following treatments for 2 weeks: 1) 2 cm T-filled implant; 2) 2 cm T-filled implant+ SB203580 (daily i.p. injection of 1 mg/kg BW; LC Laboratories, Woburn, MA, USA); and 3) SB203580+ empty capsule (inhibitor only). A group of six mice received a single s.c. injection of distilled water and served as normal controls.
Animal handling and experimentation were in accordance with the recommendation of the American Veterinary Medical Association and were approved by the Charles Drew University School of Medicine and Science animal care and use review committee.
Blood collection and tissue preparation
All mice were euthanized with a lethal injection of sodium pentobarbital (200 mg/kg BW). The gastrocnemius muscles were quickly removed and weighed. Portions of the tissue were quickly frozen in liquid N2 and stored frozen for subsequent analysis by western blotting and measurements of kinase activation by Enzyme Immunometric Assay (EIA). Additional portions in each group were fixed in 4% paraformaldehyde for histological and immunohistochemical observations. The rationale for using gastrocnemius muscles was based on the results of an earlier study that showed in general, muscle weights of adult (8 weeks) hypogonadal male mice was significantly less than normal males with the most notable difference seen in gastrocnemius muscle weights (Sciote et al. 2001). Of interest, these muscles also exhibited age-related decline in weight and muscle fiber CSA in mice (Braga et al. 2008). Blood samples were collected from each animal by cardiac puncture immediately after death, and plasma was separated and stored at −20 °C for subsequent T assay.
Hormone assay
Serum T levels were measured by a previously reported RIA (Sinha-Hikim et al. 2007). The minimal detection limit in the assay was 0.6 ng/dl. The intra-assay and inter-assay coefficient of variations were 8.2% and 13.2% respectively.
Muscle fiber CSA
Muscle fiber CSA was determined in 5 μm paraffin sections of gastrocnemius muscles using the ImagePro Plus, version 5.1 software (Media Cybernetics, Silver Spring, MD, USA) coupled to an Olympus BHS microscope equipped with a VCC video camera (Braga et al. 2008). For each animal at least 100 fibers were measured.
Measurements of kinase activation
Activation of p38 MAPK in muscle lysates was measured by TiterZyme EIA kit (Assay Designs Inc., Ann Arbor, MI, USA as described previously (Johnson et al. 2008).
Immunohistochemical and immunofluorescence analyses
Paraformaldehyde-fixed, paraffin-embedded muscle sections were immunostained as described previously (Sinha-Hikim et al. 2006, 2007, Braga et al. 2008). Primary antibodies included rabbit polyclonal Notch 1 and 2 (1:200), goat polyclonal Delta 1 (1:40), and mouse monoclonal phosphop38 MAPK, which detect p38 MAPK only when phosphorylated at tyrosine 182, (1:40) and myogenine (1:200) antibodies. All antibodies were obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Immunoreactivity was detected using biotinylated anti-rabbit, anti-mouse, or anti-goat IgG secondary antibody followed by avidinbiotinylated HRP complex, and visualized with diaminobenzidine tetrahydrochloride as per the manufacturer's instructions (VECTASTAIN Elile ABC Rabbit or Mouse IgG kit, Burlingame, CA, USA). Slides were counterstained with hematoxylin. Negative control was run for every assay and was processed in an identical manner, except the primary antibody was substituted by the rabbit, goat or mouse IgG.
Enumeration of phospho-p38 MAPK- and myogenin-positive nuclei was carried out using an American optical microscope with an ×40 objective and a pair of ×10 eyepieces. A square grid fitted with one eyepiece provided a reference area of 62 500 μm2. Cell count was expressed as the percentage of phospho-p38 MAPK- and myogenin-positive nuclei present within the reference area.
Colocalization of polyclonal paired box (PAX) 3/7 and PCNA was detected by confocal microscopy using double immunostaining as previously described (Sinha-Hikim et al. 2006, 2007, Johnson et al. 2008). In brief, after deparaffinization and rehydration, tissue sections were incubated with blocking serum for 20 min at room temperature. Sections were then incubated with a rabbit PAX3/7 antibody (1:50; Santa Cruz Biotechnology) at 4 °C overnight, followed by donkey anti-rabbit fluorescein isothiocynate secondary-antibody for 45 min at room temperature. The sections were then incubated with a mouse monoclonal PCNA (1:50; Santa Cruz Biotechnology) at 4 °C overnight, followed by goat-anti-mouse Texas Red-labeled secondary antibody for 45 min at room temperature. For controls, sections were treated only with secondary antibody, and no signals were detected. Immunofluorescence studies were also performed to assess the effect of SB203580 on the in vivo phospho-p38 MAPK, Delta 1, and Notch 1. The same primary antibodies used in immunohistochemistry were used. Confocal imaging was performed using a Leica TCS-SP-MP confocal microscope equipped with a 488 nm argon laser for excitation of green fluorophores such as FITC and a 543 nm helium-neon laser for excitation of red flurophores such as Texas Red.
Western blotting
Western blotting was performed using muscle lysates as described previously (Sinha-Hikim et al. 2007, Braga et al. 2008). In brief, proteins (50–80 μg) were separated on a 4–12% SDS-polyacrylamide gel with MES or MOPS buffer purchased from Invitrogen at 200 V. Gel was transferred on to immuno-blot PVDF membrane (Bio-Rad) overnight at 4 °C. Membranes were blocked in blocking solution (0.3% Tween 20 in Tris-buffered saline and 10% non-fat dry milk) for 1 h at room temperature then probed using mouse monoclonal PCNA (1:100; Santa Cruz Biotechnolgy), myogenin (1:300), phospho-JNK, which detects JNK only when phosphorylated at threonine 183 and tyrosine 185 (1:200; Santa Cruz Biotechnology), and total (1:200) and phospho-p38 MAPK (1:200; Santa Cruz Biotechnology) and rabbit polyclonal Delta 1 (1:200), Notch 1 and Notch 2 (1:300), and phospho-44/42 MAPK, which detects endogenous levels of ERK1 and ERK2 only when phosphorylated at threonine 202 and tyrosine 204 (1:300; Cell Signaling Technology, Inc., Beverly, MA, USA) antibodies for 1 h at room temperature or overnight at 4 °C with constant shaking. Following 3×10-min washes in TBS-T buffer, membranes were then incubated in anti-rabbit (Amersham Biosciences), anti-goat, or anti-mouse IgG-HRP (Santa Cruz Biotechnology) secondary antibodies at a 1:2000 dilution. All antibodies were diluted in blocking buffer. For immunodetection, membranes were washed three times in TBS-Twash buffer, incubated with ECL solutions per the manufacturer's specifications (Amersham Biosciences), and exposed to Hyper film ECL. The membranes were stripped and reprobed with a rabbit polyclonal GAPDH (1:2000) for normalization of the loading. Band intensities were determined using Quantity One software from Bio-Rad.
Statistical analysis
Statistical analyses were performed using the SigmaStat 2.0 Program (Jandel Cooperation, San Rafael, CA, USA). Results were tested for statistical significance using the Tukey or Student-Newman-Keuls test after one-way ANOVA. Differences were considered significant if P<0.05.
Results
Serum T levels and muscle fiber CSA
Temporal changes in serum T levels after T supplementation are summarized in Table 1. Serum T levels (mean±s.e.m.) were 0.6±0.1 ng/ml in normal mice (henceforth were referred as controls) and decreased significantly (P<0.001) to 10% of control values in mice treated with GnRH-A. Combined treatment with GnRH-A and 2-cm T-filled capsules resulted in supra-physiological levels of T over the values measured in controls throughout the treatment period. Muscle fiber CSA was determined 8 weeks after T-treatment. A significant (P<0.001) increase in the muscle fiber CSA by 1.4-fold was noted after T treatment in comparison with controls (Table 1).
Table 1. Serum T levels and muscle fiber cross-sectional area (CSA).
| Parameter | ||
|---|---|---|
|
|
||
| Serum T (ng/ml) | Muscle fiber CSA (μm2) | |
| Treatment | ||
| Control | 0.61±0.1a | 1675±47a |
| GnRH-A | 0.06±0.002b | 1379±47a |
| T-treated | ||
| 2 weeks | 21.5±3.8c | |
| 4 weeks | 17.4±3.0c | |
| 8 weeks | 14.4±2.4c | 2414±104b |
|
|
||
Values are given as mean±s.e.m. Means with unlike superscripts are significantly different.
MAPKs activation
To characterize the molecular mechanisms by which T promotes muscle growth, we examined the potential role of p38 MAPK, JNK, and ERK in this process. Western blot analyses revealed a significant increase in phospho-p38 MAPK levels in muscle lysates at 2, 4, and 8 weeks after T treatment (Fig. 1A). This was further corroborated by densitometric evaluation (Fig. 1B). Activation of p38 MAPK was also ascertained by immunohistochemistry. Compared with control, where no staining was detected, strong phosphop38 MAPK immunoreactivity was noted in the nuclei of muscle cells as early as 2 weeks after T treatment (Fig. 1C). To determine if JNK is also stimulated after T supplementation, we assessed activation of JNK by western blotting. Unlike p38 MAPK, we found inhibition of JNK, as evidenced by a substantial decrease in phospho-JNK levels at 2 and 4 weeks after T treatment (Fig. 2A). By 8 weeks, phospho-JNK levels, however, returned to normal levels (Fig. 2A). The observed decrease in phospho-JNK levels was statistically significant (P<0.05) as revealed by densitometric analysis (Fig. 2B). Western blot analyses also revealed that T supplementation had no effect on ERK activation (data not shown). Thus, T supplementation resulted in p38 MAPK activation but inhibition of JNK signaling within 2 weeks of treatment.
Figure 1.
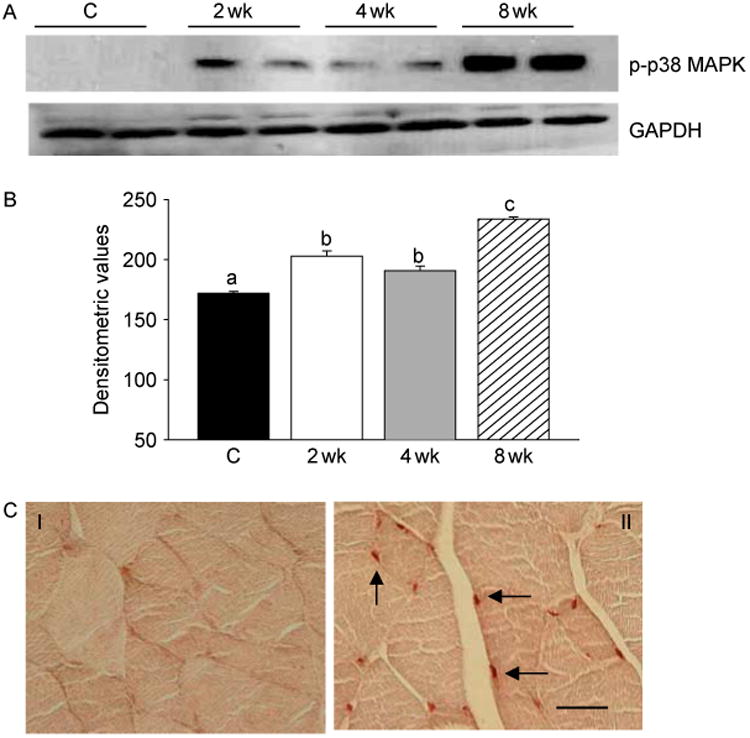
T supplementation results in activation of p38 MAPK. (A) Western blots of muscle lysates from control and T-treated mice show increased phospho-p38 MAPK (p-p38 MAPK) levels at 2, 4, and 8 weeks after treatment. The gels are representative of two animals at each time point from one of three separate experiments. GAPDH in the immunoblot is shown as a loading control. (B) Densitometric analysis shows a significant increase in phospho-p38 MAPK levels at 2, 4, and 8 weeks after T treatment. Values are mean±s.e.m. Means with unlike superscripts are significantly (P<0.05) different. (C) p38 MAPK activation visualized by immunohistochemistry using a phosphospecific antibody that detects p38 MAPK only when phosphorylated at tyrosine 182. Compare with control (panel I), where no staining is detected, strong phospho-p38 MAPK immunoreactivity is detected in the nuclei of muscle cells (arrow) within 2 weeks of T treatment (panel II). Scale bar=50 μm.
Figure 2.
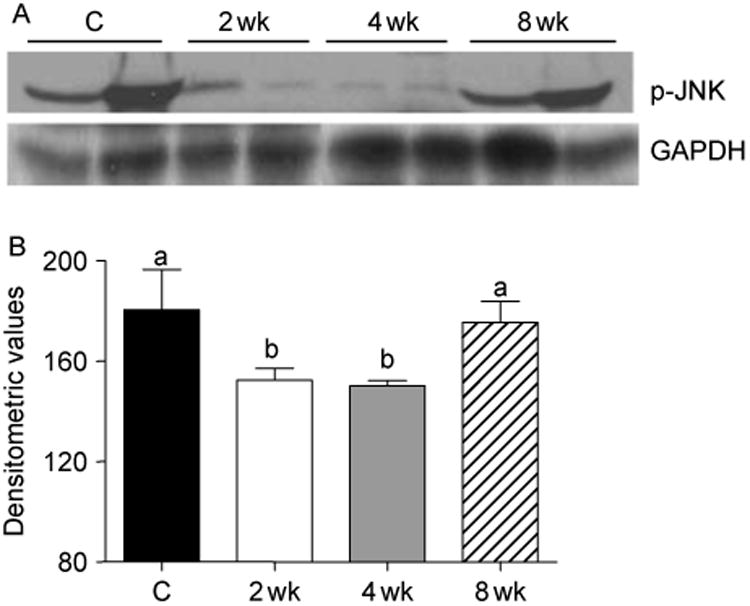
(A) Western blots of muscle lysates from control and T-treated mice show a transient decrease in phospho-JNK levels at 2 and 4 weeks of treatment. The gels are representative of two animals at each time point from one of three separate experiments. GAPDH in the immunoblot is shown as a loading control. (B) Densitometric analysis shows a significant decrease in phospho-JNK levels at 2 and 4 weeks after T treatment. Values are mean±s.e.m. Means with unlike superscripts are significantly (P<0.05) different.
Activation of p38 MAPK is associated with stimulation of Notch signaling and increased expression of PCNA and myogenin
Because Notch signaling is essential for activation, proliferation, and myogenic progression of satellite cells necessary for muscle growth (Conboy & Rando 2002, Conboy et al. 2003, 2005), and this can be stimulated by p38 MAPK (Weijzen et al. 2002), we examined the participation of Notch signaling in T-induced muscle fiber hypertrophy. We found increased expression of Notch 1 and 2 and their ligand Delta 1 in muscle lysates by immunoblotting, as early as 2 weeks of T treatment (Fig. 3A). The increased levels of Delta 1 (Fig. 3B) were significant (P<0.001) at 2 weeks after treatment, which declined to near normal levels by 4 weeks and subsequently elevated again by 8 weeks of T treatment. Increased expression of Notch 1 was also noted at 2, 4, and 8 weeks after T treatment, but the difference was only significant at 8 weeks after treatment (Fig. 3C). The increased levels of Notch 2 (Fig. 3D) were significant (P<0.01) only at 2 and 8 weeks after treatment.
Figure 3.
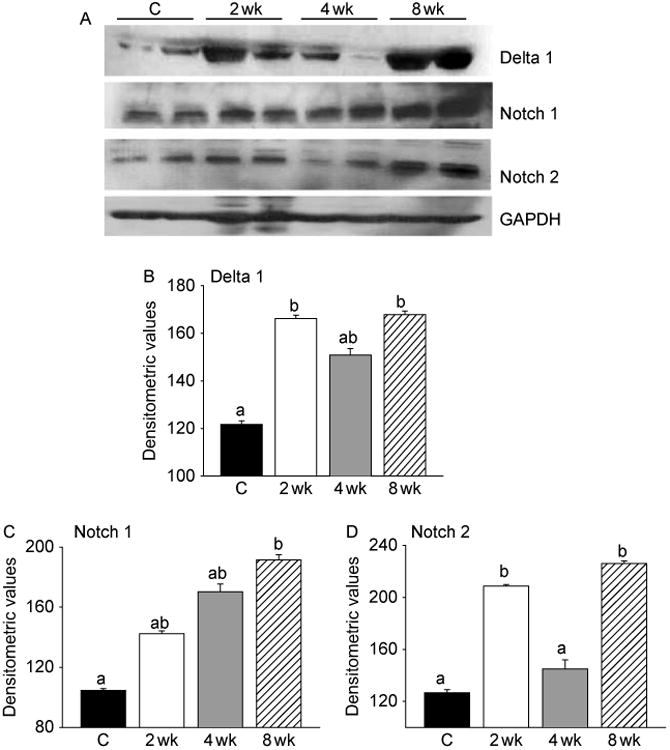
Activation of p38 MAPK is associated with stimulation of Notch signaling. (A) Western blots of muscle lysates from control and T-treated mice show increased levels of Delta 1, Notch 1, and Notch 2 within 2 weeks of treatment. The gels are representative of two animals at each time point from one of three separate experiments. GAPDH in the immunoblot is shown as a loading control. (B) Densitometric analysis shows a significant increase in Delta 1 and Notch 2 at 2 and 8 weeks and Notch 1 at 8 weeks after treatment. Values are mean±s.e.m. of six animals. Means with unlike superscripts are significantly (P<0.001 for Delta 1 and P<0.01 for Notch 1 and 2) different.
We also analyzed the expression of PCNA and myogenin by immunoblotting. Activation of Notch signaling was associated with cell proliferation as demonstrated by increased expression of PCNA in muscle lysates (Fig. 4A). A significant (P<0.001) increase in PCNA levels could be detected as early as within 2 weeks of T treatment and became more pronounced by 8 weeks (Fig. 4B). Myogenin expression also increased at 4 and 8 weeks after T treatment (Fig. 4A). This was further corroborated by densitometric evaluation (Fig. 4C). Co-staining for PCNA and PAX3/7 further confirmed increased expression of PCNA after T treatment in satellite cells (Fig. 5). The number of myogenin-positive nuclei was significantly (P<0.001) higher after T treatment when compared with controls (Fig. 6).
Figure 4.
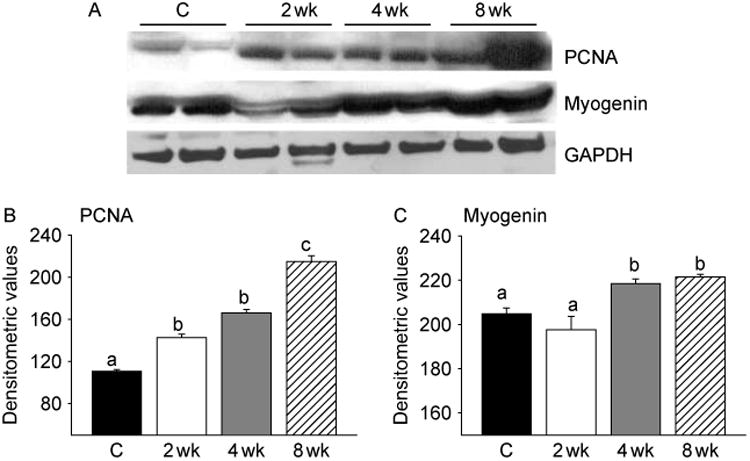
T-induced activation of Notch signaling is associated with increased expression of PCNA and myogenin. (A) Western blot analysis of muscle lysates from control and T-treated mice shows increased expression of PCNA through the treatment period and myogenin at 4 and 8 weeks of treatment. The gels are representative of two animals at each time point from one of three separate experiments. GAPDH in the immunoblot is shown as a loading control. (B) Densitometric analysis shows a significant increase in PCNA levels during the entire treatment duration and myogenin at 4 and 8 weeks after treatment. Values are mean±s.e.m. of six mice. Means with unlike superscripts are significantly (P<0.001) different.
Figure 5.
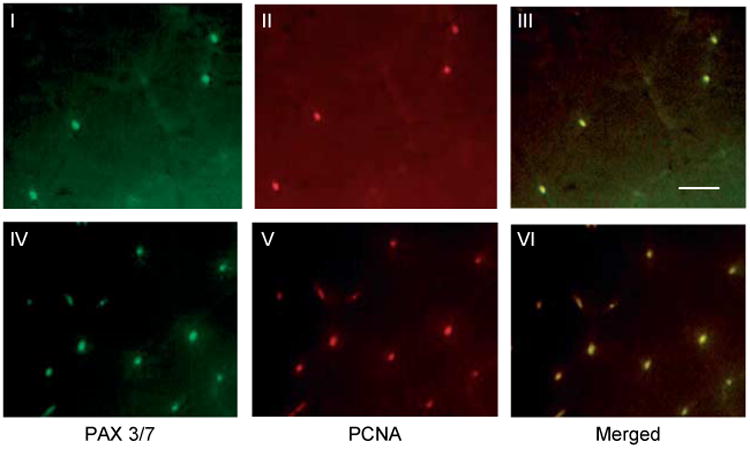
Double immunofluorescence staining for PAX3/7(green) and PCNA (red) from control (panels I–III) and 2 week T-treated gastrocnemius muscles (panels III–V) shows colocalization of PAX3/7 and PCNA (yellow). Scale bar=50 μm.
Figure 6.
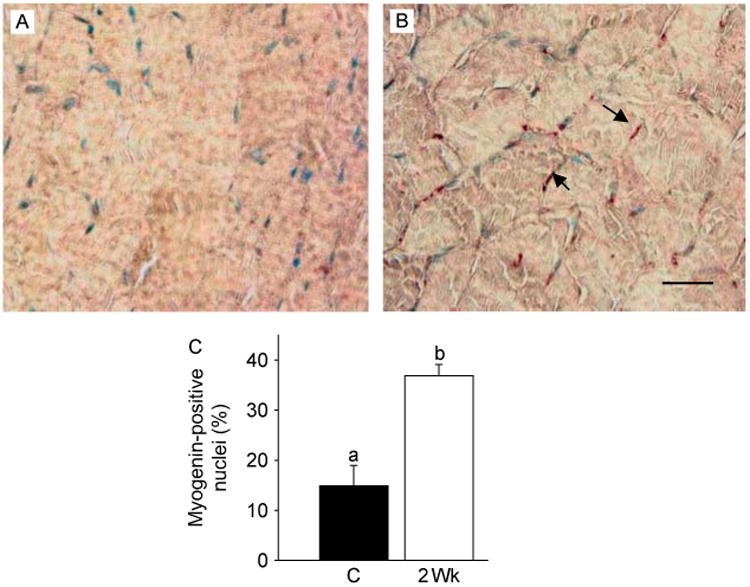
T treatment increases the number of myogenin positive cells. Gastrocnemius muscles from normal control (A) and T-treated (B) mice show a substantial increase in the number of myogenin-positive nuclei (arrow) 2 weeks after T treatment. Scale bar=50 μm. (C) Quantitative analysis shows a significant increase in the number of myogenin-positive nuclei 2 weeks after T treatment compared with control. Values are mean±s.e.m. Means with unlike superscripts are significantly (P<0.001) different.
SB203580 prevents T-induced activation of Notch signaling and muscle cell proliferation
To further explore the role of p38 MAPK signaling in T-induced activation of Notch signaling and muscle growth, we examined whether SB203580, a p38 MAPK inhibitor (Cuenda et al. 1995, Lahti et al. 2002), could prevent or attenuate T-mediated activation of Notch signaling. Concomitant administration of SB203580 significantly (P<0.001) prevented T-induced activation of p38 MAPK, as evidenced by an EIA assay (Fig. 7A), immunoblotting (Fig. 7B), as well as by immunohistochemistry (Fig. 7C). Morphometric analysis further revealed a significant (P<0.01) increase in the number of phospho-p38 MAPK-positive nuclei after T treatment when compared with controls and that could be fully prevented by SB203580 treatment (Fig. 7D). We then looked at the expression profiles of Delta 1, Notch 1, Notch 2, and PCNA after combined treatment with T and SB203580. Western blot analysis revealed that addition of SB203580 to T treatment effectively blocked Notch signaling, as demonstrated by decreased levels of Delta 1, Notch 1, and Notch 2 (Fig. 8A). Addition of SB203580 to T treatment also resulted in a significant (P<0.001) suppression of PCNA levels (Fig. 8A). These findings were further corroborated by densitometry (Fig. 8B–E).
Figure 7.
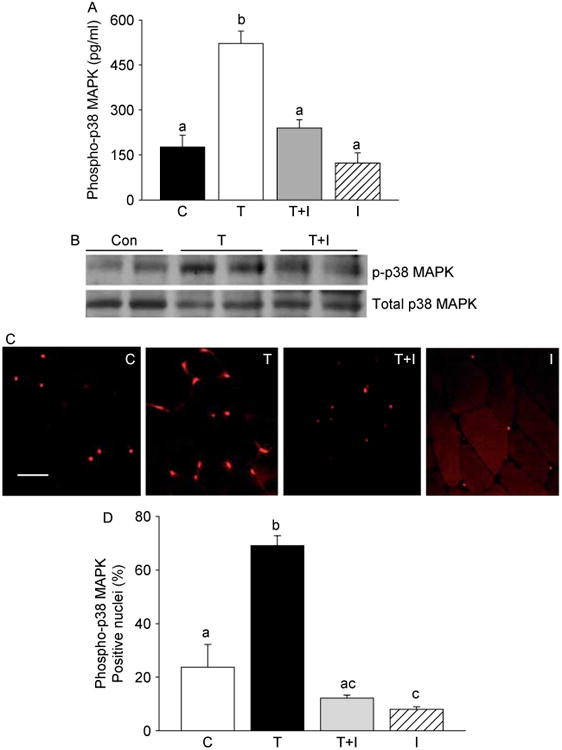
Concomitant administration of SB203580, a p38 MAPK inhibitor (I), prevents T-induced activation of p38 MAPK. (A) EIA assay shows a significant increase in phosphop38 MAPK levels 2 week after T treatment, which can be effectively prevented by SB203580. Values are mean±s.e.m. Means with unlike superscripts are significantly (P<0.001) different. (B) Western blots of muscle lysates from control, testosterone (T), and T+ inhibitor (TCI+1)-treated mice show effective suppression of phospho-p38 MAPK (p-p38MAPK) levels after SB203580 treatment. The gels are representative of two animals at each time point from one of three separate experiments. Equal loading of proteins in each lane was confirmed by reprobing the same blot with total p38 MAPK antibody. (C) Immunohistochemical analysis of in vivo changes in phospho-p38 MAPK expression in gastrocnemius muscles from control (C), testosterone (T), T+ inhibitor (T+I), and I only treated mice. Note the reduction in the number of phospho-p38 MAPK-positive nuclei in the T+I group compared with the T alone group. Scale bar=50 μm. (D) Morphometric analysis shows a significant (P<0.01) increase in the number of phospho-p38 MAPK-positive nuclei 2 week after T treatment when compared with controls and that can be fully prevented by SB203580 treatment.
Figure 8.
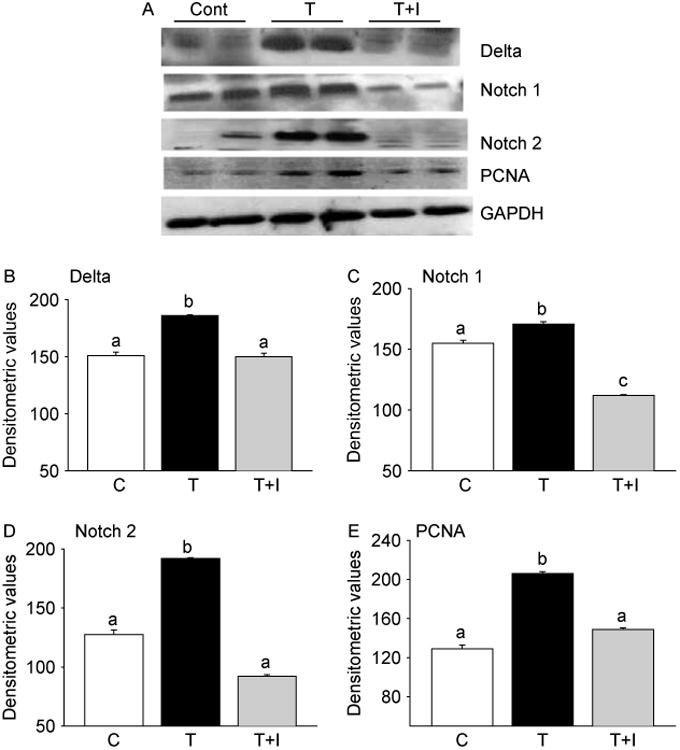
SB203580 effectively prevents T-induced activation of Notch signaling and muscle cell proliferation. (A) Representative western blots of muscle lysates from control (C), testosterone (T), and testosterone + inhibitor (T+I) treated mice show effective suppression of T-induced Delta 1, Notch 1, Notch 2, and PCNA levels by SB203580. The gels are representative of two animals at each time point from one of three separate experiments. GAPDH in the immunoblot is shown as a loading control. Densitometric analysis shows a significant increase in the expression of these proteins 2 week after T treatment and which can be significantly suppressed by inhibition of p38 MAPK (B–E). Values are mean±s.e.m. of six animals. Means with unlike superscripts are significantly (P<0.001) different.
Discussion
Given that many aspects of the signal transduction pathways are not feasible to study directly in humans, in this study, using a mouse model, we elucidated the molecular mechanisms by which T induces muscle growth. In concert with previous results in humans (Sinha-Hikim et al. 2002, 2003, 2006), here we show that T induces muscle fiber hypertrophy in mice. This is consistent with earlier works demonstrating that both T and dihydrotestosterone equally stimulated muscle mass in both orchidectomized wild type as well as in growth hormone receptor knockout mice even in comparison with respective sham-operated or untreated mice (Venken et al. 2007). In this context, it is important to note that, we found in our pilot study a 17.7% decrease, though not statistically significant, in muscle fiber CSA after GnRH-A treatment compared with normal controls. The present findings of deferential susceptibility of gastrocnemius muscles during T withdrawal and T supplementation possibly indicate involvement of different mechanisms during muscle atrophy and hypertrophy. This possibility clearly merits further investigations.
To elucidate the mechanisms by which T induces muscle fiber hypertrophy, we examined the potential contributions of ERK, JNK, and p38 MAPK to T-induced muscle growth. We found that T supplementation not only triggers p38 MAPK activation but also concurrently suppresses JNK activation within 2 weeks of treatment. T-induced muscle growth is, however, independent of ERK. Thus, activation of p38 MAPK together with the inactivation of JNK may be critical for muscle fiber hypertrophy in response to T treatment. The observed activation of p38 MAPK after T treatment is consistent with its pivotal role, through regulation of myogenic regulatory factors (MRFs), in skeletal muscle remodeling and growth (Lluis et al. 2006). The MRFs have the unique property of converting non-muscle cells to muscle lineage. We knew from our earlier studies that satellite cells play a critical role in skeletal muscle adaptation to hypertrophic stimuli such as T, resulting in fiber hypertrophy (Sinha-Hikim et al. 2003, 2006). When activated, satellite cells undergo proliferation and differentiation and commit to a myoblast cell fate, which either join preexisting fibers causing hypertrophy, or migrate and make new fibers (Wagers & Conboy 2005). The mechanisms by which T promotes activation of p38 MAPK are not known. One intriguing possibility is that T could activate p38 MAPK through inactivation of myostatin. Indeed, there is evidence that T negatively regulates myostatin levels in skeletal muscles of rats and mice (Kawada et al. 2006, Mendler et al. 2007). There have also been studies indicating that myostatin abrogates the phenylephrine-induced cardiac myocyte growth via inactivation of p38 MAPK signaling (Morissette et al. 2006). Thus, the signal for p38 MAPK activation possibly emanates from T-induced suppression of myostatin levels. We are also intrigued by the observation that T-induced muscle growth is associated with suppression of JNK signaling. Given that the JNK signaling can be activated by myostatin, it is likely that the observed suppression of JNK could result from T-induced suppression of myostatin levels (Kawada et al. 2006, Mendler et al. 2007). We have previously demonstrated that the JNK signaling pathway constitutes a critical component of apoptotic signaling in skeletal muscles after injury (Sinha-Hikim et al. 2007) or in aging (Braga et al. 2008). Therefore, it is possible that T-mediated inhibition of JNK could lead to suppression of muscle cell apoptosis, resulting in fiber growth. It is also possible that suppression of JNK could activate cellular proliferation and, in turn, muscle growth by attenuating myostatin-induced upregulation of p21 (Thomas et al. 2000, Huang et al. 2007), a potent inhibitor of various cyclin-dependent kinase activities (Child & Mann 2006). In this context, it is pertinent to note here that blocking JNK signaling pathway by pretreatment with SP600126 effectively attenuated myostatin-induced upregulation of p21 and abolished the growth inhibitor role of myostatin in C2C12 cells (Huang et al. 2007). Clearly, one implication of these observations is that T can promote muscle growth, not only through regulation of MRFs by activating p38 MAPK signaling, but also by augmenting cellular proliferation and/or inhibiting apoptosis by suppressing JNK activation.
The Notch signaling pathway is essential for the activation, proliferation, and myogenic progression of satellite cells necessary for muscle regeneration and repair (Conboy & Rando 2002, Conboy et al. 2003, 2005). Our recent studies on elderly men (Sinha-Hikim et al. 2006) indicate the involvement of Notch signaling in T-mediated muscle cell proliferation. Consistent with a role for Notch signaling in muscle growth, in the present study, we found increased expression of Notch 1 and 2 and their ligand Delta 1 in muscle lysates by immunoblotting, as early as within 2 weeks of T treatment. Interestingly, we found upregulation of Delta 1 at 2 and 8 weeks during activation. By contrast, there was almost no upregulation of Delta 1 at 4 weeks. A possible explanation of this finding is uncertain and can not be explained by changes in circulating T levels, since no significant changes in T levels were detected among various time intervals after treatment.
We also found that activation of Notch signaling is associated with muscle cell proliferation and differentiation as demonstrated by increased expression of PCNA and myogenin levels. Most importantly, we further show colocalization of PCNA and PAX3/7, suggesting an increase in satellite cell number as well as differentiation (Wagers & Conboy 2005, Hyatt et al. 2008). Together, these data indicate that Notch signaling, like human (Sinha-Hikim et al. 2006), also plays an important role in T-induced muscle fiber hypertrophy in mice.
The mechanisms by which T stimulates Notch signaling and, in turn, muscle hypertrophy are not known. One intriguing possibility is that T could stimulate Notch signaling through activation of p38 MAPK signaling. Indeed, earlier studies have shown the involvement of p38 MAPK in the activation of Notch signaling in some cell lines (Weijzen et al. 2002). Most importantly, we show that inhibition of p38 MAPK with SB203580 effectively blocked Notch signaling, as demonstrated by decreased levels of Delta 1, Notch 1, and Notch 2. We further show that SB203580 significantly suppressed PCNA levels. Thus, p38 MAPK signaling is necessary for activation of Notch signaling to promote T-induced muscle growth in mice.
In summary, we have provided new insights into the molecular mechanisms by which T-induces muscle fiber hypertrophy. Our data indicate that p38 MAPK mediated Notch signaling constitutes a critical component of signal transduction pathways by which T promotes muscle fiber hypertrophy in mice. These findings further emphasize the usefulness of mice as a suitable model to study the molecular mechanisms by which T promotes muscle growth. This model with additional manipulation (such as using transgenic or gene-ablated mice) can be used to further study the genetic regulation of muscle growth and devlopment. An understanding of the mechanistic pathways that mediate T-induced muscle fiber hypertrophy may unveil novel targets for the development of anabolic therapies in aging and in various neuromuscular disorders.
Acknowledgments
Funding: This work was supported by MBRS grant 5 SO6-GM068510 (to I S-H) and RCMI Clinical Research Infrastructure Initiative grant RR-011145 (to K N) from the National Institutes of Health.
Footnotes
Declaration of interest: The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- Bhasin S, Calof OM, Storer TW, Lee ML, Mazer NA, Jasuja R, Montori VM, Gao W, Dalton JT. Drug insight: testosterone and selective androgen receptor modulators as anabolic therapies for chronic illness and aging. Nature Clinical Practice Endocrinology & Metabolism. 2006;2:146–159. doi: 10.1038/ncpendmet0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga M, Sinha Hikim AP, Datta S, Ferrini M, Brown D, Kovacheva EL, Gonzalez-Cadavid NF, Sinha-Hikim I. Involvement of oxidative stress and caspase 2-mediated intrinsic pathway signaling in age-related increase in muscle cell apoptosis in mice. Apoptosis. 2008;13:822–832. doi: 10.1007/s10495-008-0216-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughlan A, Newhouse K, Namgung U, Xia Z. Chlorpyrifos induces apoptosis in rat cortical neurons that is regulated by a balance between p38 and ERK/JNK MAP kinases. Toxicological Science. 2004;78:125–134. doi: 10.1093/toxsci/kfh038. [DOI] [PubMed] [Google Scholar]
- Child ES, Mann DJ. The intricacies of p21 phosphorylation: protein/protein interactions, subcellular localization and stability. Cell Cycle. 2006;5:1313–1319. doi: 10.4161/cc.5.12.2863. [DOI] [PubMed] [Google Scholar]
- Conboy IM, Rando TA. The regulation of Notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Developmental Cell. 2002;3:397–409. doi: 10.1016/s1534-5807(02)00254-x. [DOI] [PubMed] [Google Scholar]
- Conboy IM, Conboy MJ, Smythe GM, Rando TA. Notch-mediated restoration of regenerative potential to aged muscle. Science. 2003;302:1575–1577. doi: 10.1126/science.1087573. [DOI] [PubMed] [Google Scholar]
- Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005;433:760–764. doi: 10.1038/nature03260. [DOI] [PubMed] [Google Scholar]
- Cuenda A, Rouse J, Doza YN, Meier R, Cohen P, Gallagher TF, Young PR, Lee JC. SB203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Letters. 1995;364:229–233. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- Huang Z, Chen D, Zhang K, Yu B, Chen X, Meng J. Regulation of myostatin signaling by c-Jun N-terminal kinase in C2C12 cells. Cellular Signalling. 2007;19:2286–2295. doi: 10.1016/j.cellsig.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Hyatt JPK, McCall GE, Kander EM, Zhong H, Roy RR, Huey KA. Pax3/7 expression coincides with myoD during chronic skeletal muscle overload. Muscle & Nerve. 2008;38:861–866. doi: 10.1002/mus.21006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- Johnson C, Jia Y, Wang C, Lue Y, Swerdloff RS, Zhang XS, Hu ZY, Li YC, Liu YX, Sinha Hikim AP. Role of caspase 2 in apoptotic signaling of primate and murine germ cells. Biology of Reproduction. 2008;79:806–814. doi: 10.1095/biolreprod.108.068833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawada S, Okuno M, Ishii N. Testosterone causes decrease in the content of skeletal muscle myostatin. International Journal of Sport and Health Sciences. 2006;4:44–48. [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sulivan R, Jobling WA, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- Lahti A, Kankaanranta H, Moilanen E. p38 mitogen-activated protein kinase inhibitor SB203580 has a bi-directional effect on iNOS expression and NO production. European Journal of Pharmacology. 2002;454:115–123. doi: 10.1016/s0014-2999(02)02490-1. [DOI] [PubMed] [Google Scholar]
- Liang Q, Mollkentin JD. Redefining the roles of p38 and JNK signaling in cardiac hypertrophy: dichotomy between cultured myocytes and animal models. Journal of Molecular and Cellular Cardiology. 2003;35:1385–1394. doi: 10.1016/j.yjmcc.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Lin A, Dibling B. The true face of JNK activation in apoptosis. Aging Cell. 2002;1:112–116. doi: 10.1046/j.1474-9728.2002.00014.x. [DOI] [PubMed] [Google Scholar]
- Lluis F, Perdiguero E, Nebreda AR, Munoz-Canoves P. Regulation of skeletal muscle gene expression by p38 MAP kinases. Trends in Cell Biology. 2006;16:36–44. doi: 10.1016/j.tcb.2005.11.002. [DOI] [PubMed] [Google Scholar]
- McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass by a new TGF-beta superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- Mendler L, Baka Z, Kovacs-Simon A, Dux L. Androgens negatively regulate myostatin expression in an androgen-dependent skeletal muscle. Biochemical and Biophysical Research Communications. 2007;361:237–242. doi: 10.1016/j.bbrc.2007.07.023. [DOI] [PubMed] [Google Scholar]
- Morgan JE, Partridge TA. Cells in focus: muscle satellite cells. International Journal of Biochemistry and Cell Biology. 2003;35:1151–1156. doi: 10.1016/s1357-2725(03)00042-6. [DOI] [PubMed] [Google Scholar]
- Morissette MR, Cook SA, Foo SY, McKoy G, Ashida N, Novikov M, Scherrer-Crosbie M, Li L, Matsui T, Brooks G, et al. Myostatin regulates cardiomyocyte growth through modulation of Akt signaling. Circulation. 2006;99:15–24. doi: 10.1161/01.RES.0000231290.45676.d4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisz-Porszasz S, Bhasin S, Artaza JN, Shen R, Sinha-Hikim I, Hogue A, Fielder TJ, Gonzalez-Cadavid NF. Lower skeletal muscle mass in male transgenic mice with muscle specific overexpression of myostatin. American Journal of Physiology-Endocrinology and Metabolism. 2003;285:E876–E888. doi: 10.1152/ajpendo.00107.2003. [DOI] [PubMed] [Google Scholar]
- Sciote JJ, Horton MJ, Zyman Y, Pascoe G. Differential effects of diminished oestrogen and androgen levels on development of skeletal muscle fibers in hypogonadal mice. Acta Physiologica Scandinavica. 2001;172:179–187. doi: 10.1046/j.1365-201x.2001.00854.x. [DOI] [PubMed] [Google Scholar]
- Sinha-Hikim I, Artaza J, Woodhouse L, Cadavid NG, Singh AB, Lee MI, Storer TW, Casaburi R, Shen R, Bhasin S. Testosterone-induced increase in muscle size in healthy young men is associated with muscle fiber hypertrophy. American Journal of Physiology-Endocrinology and Metabolism. 2002;283:E154–E164. doi: 10.1152/ajpendo.00502.2001. [DOI] [PubMed] [Google Scholar]
- Sinha-Hikim I, Roth SM, Lee MI, Bhasin S. Testosterone induced muscle hypertrophy is associated with an increase in satellite cell number in healthy, young men. American Journal of Physiology-Endocrinology and Metabolism. 2003;285:E197–E205. doi: 10.1152/ajpendo.00370.2002. [DOI] [PubMed] [Google Scholar]
- Sinha Hikim AP, Vera Y, Elhag RI, Lue Y, Cui YG, Pope V, Leung A, Atienza V, Wang C, Swerdloff RS. Mouse model of male germ cell apoptosis in response to a lack of hormonal stimulation. Indian Journal of Experimental Biology. 2005;43:1048–1057. [PubMed] [Google Scholar]
- Sinha-Hikim I, Cornford M, Gaytan H, Lee ML, Bhasin S. Effects of testosterone supplementation on skeletal muscle fiber hypertrophy and satellite cells in community dwelling, older men. Journal of Clinical Endocrinology and Metabolism. 2006;91:3024–3033. doi: 10.1210/jc.2006-0357. [DOI] [PubMed] [Google Scholar]
- Sinha-Hikim I, Braga M, Shen R, Sinha Hikim AP. Involvement of c-Jun NH2-terminal kinase and nitric oxide-mediated mitochondria-dependent intrinsic pathway signaling in cardiotoxin-induced muscle cell death: role of testosterone. Apoptosis. 2007;12:1965–1978. doi: 10.1007/s10495-007-0120-6. [DOI] [PubMed] [Google Scholar]
- Thomas M, Langley B, Berry C, Sharma M, Kirk S, Bass J, Kambadur R. Myostatin, a negative regulator of muscle growth, functions by inhibiting myoblast proliferation. Journal of Biological Chemistry. 2000;275:40235–40243. doi: 10.1074/jbc.M004356200. [DOI] [PubMed] [Google Scholar]
- Venken K, Moverare-Skrtic S, Kopchick J, Coschigano KT, Ohlsson C, Boonen S, Bouillion R, Vanderschueren D. Impact of androgens, growth hormone, and IGF-1 on bone and muscle in male mice during puberty. Journal of Bone and Mineral Research. 2007;22:72–82. doi: 10.1359/jbmr.060911. [DOI] [PubMed] [Google Scholar]
- Vianzof M, Ayub-Guerrieri D, Onofre PCG, Martinus PCM, Lopes VF, Zilberztajn D, Maria LS, Sell K, Yamamoto LU. Animal models for genetic neuromascular diseases. Journal of Molecular Neuroscience. 2008;34:241–248. doi: 10.1007/s12031-007-9023-9. [DOI] [PubMed] [Google Scholar]
- Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838–2849. doi: 10.1038/sj.onc.1207556. [DOI] [PubMed] [Google Scholar]
- Wagers A, Conboy IM. Cellular and molecular signatures of muscle regeneration: current concepts and controversies in adult myogenesis. Cell. 2005;122:659–667. doi: 10.1016/j.cell.2005.08.021. [DOI] [PubMed] [Google Scholar]
- Weijzen S, Rizzo P, Braid M, Vishnav R, Jonkheer SM, Zlobin A, Osborne BA, Gottipati S, Aster JC, Hahn WC, et al. Activation of Notch-1 signaling maintains neoplastic phenotype in human Ras-transformed cells. Nature Medicine. 2002;8:979–986. doi: 10.1038/nm754. [DOI] [PubMed] [Google Scholar]