

Abstract
Photoreceptor rod cells contain a unique tetraspanin fusion protein known as peripherin/rds. This protein is important in membrane fusion events hypothesized to be essential to disk membrane morphogenesis and disk shedding. In vivo and in vitro fusogenic activity has been mapped to the C-terminal domain of peripherin/rds. Moreover, a fusion peptide domain localized to a 15 amino acid long region (residues 311–325) is essential for mediating lipid bilayer fusion of model membranes. To address the functional and structural properties required for peripherin/rds dependent membrane fusion, constructs of the entire C-terminal domain (residues 284–346) were generated and polypeptides expressed. A wild type-peripherin/rds C-terminal GST fusion construct that included the entire C-terminus (PERCTER) or a C-terminal truncation mutant (PERCTN) were engineered with a thrombin cleavage site. Protein expression was induced in E. coli with IPTG, expressed proteins cleaved from the GST with thrombin and purified to homogeneity on a Superdex 75 column. Purity was confirmed by SDS–PAGE and Western blot analysis. The purified wt C-terminal protein resolved as a monomer under reducing conditions on SDS–PAGE (15%) and was immunoreactive with anti peripherin/rds antibody 2B6 (gift from Dr R. Molday). The purified polypeptide promoted the requisite steps of fusion, membrane destabilization, lipid mixing and aqueous contents mixing. Conversely, the truncation mutant lacking a portion of the fusion domain was unable to promote these steps. A common feature of most membrane fusion proteins is a change in conformation upon membrane association. Structural changes in the C-terminal polypeptide were investigated using far UV CD. The far UV CD spectra of the purified C-terminal polypeptide indicated substantial α-helical content in the wt peptide in isotonic aqueous buffer. An increase in intensity of 208 and 222 nm CD bands upon addition of DPC vesicles indicated an increase in α-helical content of the polypeptide. These results demonstrate that a purified soluble form of the C-terminus of peripherin/rds can interact with biological phospholipids; moreover, this interaction promotes a conformational change that is most consistent with an increase in α-helical content.
Keywords: peripherin/rds, membrane fusion, photoreceptors, GST-fusion protein
1. Introduction
Membrane fusion is an essential component of cellular events as diverse as endocytosis, exocytosis, fertilization, viral entry into host cells and trancosytosis in polarized epithelial cells (Pecheur et al., 1999; Wasserman, 1999; White 1992; Hernandez et al., 1996). Membrane fusion events also play a critical role in the function of photoreceptor rod cells, the cells that are responsible for vision under low levels of light. These cells have an inner segment and a highly specialized outer segment region. The specialized outer segment contains a stack of closed flattened membranous sacs known as disks that contain the phototransduction protein rhodopsin. In these post-mitotic cells, an active membrane renewal process is required to maintain structural integrity and thereby normal physiological function. The outer segment region maintains a constant number of disk membranes through the coordinated processes of disk morphogenesis at the base and a compensatory shedding of older disks at the apical tip (Young, 1976). Both of these processes require membrane fusion (for review see Boesze-Battaglia and Goldberg, 2001); at the disk base, evaginations of the ROS plasma membrane fuse to form a closed disk membrane (Steinberg et al., 1976); at the apical tip a series of older disks are delineated from the remainder of the outer segment in disk packets by a fusion of the disks with the plasma membrane (Matsumoto and Besharse, 1985).
Previous work from this laboratory has identified a unique photoreceptor specific fusion protein, peripherin/rds (a.k.a. rds/peripherin and peripherin-2), proposed to mediate fusion between membranes within the ROS (Boesze-Battaglia et al., 1997; 1998). Peripherin/rds is a member of the tetraspanin superfamily. It is localized exclusively to the rim region of the disks (Arikawa et al., 1992) and it is oriented in the disk membranes such that its N- and C-termini are exposed to the cytoplasm (Connell and Molday, 1990). Studies investigating the effect of peripherin/rds mutations on fusion of phospholipid bilayers identified amino acid residues 311–325, located within the cytoplasmic C-terminus of peripherin/rds, as the minimal region required for membrane binding and subsequent fusion (Boesze-Battaglia et al., 1998). A peptide corresponding to this fusion region was found to be an amphiphilic α-helix by FTIR (Boucheix and Rubinstein, 2001; Boesze-Battaglia et al., 1997; 1998; 2000; Muller-Weeks et al., 2002). Further studies of fusion in phospholipid bilayers utilized a peptide analogue of the fusion domain, called PP-5. These studies showed that fusion competency requires a tetrameric arrangement of the fusion peptide and by inference of the entire C-terminus (Boesze-Battaglia et al., 2000). Recently, a highly conserved region upstream of the fusion peptide domain (Stefano et al., 2002) that includes a proline at position 295 was shown to participate in the transition from a pre-fusion to a fusion competent state of this protein (Stefano et al., 2002). Similar regions have been identified as essential for conformational changes of fusion proteins in intracellular fusion, neurotransmitter release and viral infectivity (Boucheix and Rubinstein, 2001; Pecheuer et al., 2000; Weimbs et al., 1997).
Biological membrane fusion may be mediated by a single fusion protein or by fusion protein complexes specific to the cell (Pecheur et al., 1999; Wasserman, 1999; White, 1992; Hernandez et al., 1996; Pecheur et al., 2000; Weimbs et al., 1997; Jones et al., 1998). Common properties of these fusion proteins include multiple regions including a fusion peptide domain (Young, 1976; Pecheur et al., 2000), a ‘fusion trigger’ region and a hairpin-like domain necessary for the formation of a fusion pore (Weimbs et al., 1997). A comprehensive analysis of the fusion proteins involved in these processes and the processes themselves has provided a paradigm having four clearly defined steps: cell adhesion, close approach of the two opposing membrane, localized membrane destabilization and mixing of the aqueous contents (Bentz and Ellens, 1998). These steps require a series of conformational changes within the fusion protein complexes, as well as within the fusion peptide domain (Bentz, 2000).
In this study we expressed the C terminal region of peripherin/rds (residues 284–346) as well as a truncated version (residues 284–318), which does contain an intact fusion peptide. The soluble full-length C terminal polypeptide mediated the steps of membrane fusion while the truncated version did not. These fusion studies indicate that the properties inherent in the longer polypeptide are sufficient to support its biological role as a membrane fusion protein in the absence of anchorage to the membrane. Finally, we show that the secondary structure of the peptide is sensitive to the presence of a lipid environment in that the α-helical content increases in the presence of the lipid. This type of structural change is consistent with the formation of a coiled–coiled intermediate as part of the molecular mechanism of the fusion process. This work is of particular significance since peripherin/rds is the first tetraspanin proposed to directly mediate biological membrane fusion (Pecheur et al., 1999).
2. Materials and methods
Materials
N-methyl dioleoyl phosphatidylethanolamine (N-methyl-DOPE), dodecylphosphatidylcholine (DPC), phosphatidylcholine (egg), phosphatidylserine (egg) and cholesterol were purchased from Avanti Polar Lipids (Birmingham, AL). N-(7-nitro-2,1,3-benzoxadiazol-4-yl) phosphatidylethanolamine (NBD-PE) and N-(lissamine Rhodamine B sulfonyl) phosphatidylethanolamine (Rh-PE), 8-aminonaphtalene-1,3,6-trisulfonic acid sodium salt (ANTS) and p-xylenebis(pyridinium) bromide (DPX) were purchased from Molecular Probes (Eugene, USA).
Preparation of GST-C-terminal peripherin/rds construct and purified protein
A glutathione S-transferase (GST) fusion protein encoding the predicted 63 C-terminal amino acids of bovine peripherin/rds, FPCTER, was produced using conventional methology (Maniatus et al., 1989). A 130 bp PCR amplification product was obtained using oligonucleotides primers (5′-TGTTGGATCCCGC-TACCTGCACACG-3′ and 5′-GGAGTACTAGTAAC-CCTGGCCCCAGTCACGACGTTGTAA-3′), the coding sequence of bovine peripherin/rds C-terminus (Connell and Molday, 1990), and Taq polymerase. The PCR product was digested with BamHI, purified, and ligated into BamHI cut and purified pGEX-2T (Smith and Johnson, 1988) to generate pFPCTER. Proper insert orientation was selected by restriction mapping; insert and junction sequences were verified by double-stranded DNA sequencing, using a Big Dye terminator cycle sequencing kit (Applied Biosystems, Inc.). Expression of the predicted 32 kDa fusion protein was confirmed by induction of a TOP10F′ bacterial culture with 1 mM IPTG for 2 hr at 37°C.
A truncated version of the C-terminal fusion protein, FPCTCN, was created by digesting pFPCTER with Stu I and Sma I, then purifying and self-ligating the 5.1 kb fragment. The completed construct was verified by double-stranded DNA sequencing, using a Big Dye terminator cycle sequencing kit (Applied Biosystems, Inc.) The pFPCTCN plasmid encodes a GST fusion protein joined to the N-terminal 34 amino acids of the peripherin/rds C-terminus. This truncation interrupts the fusogenic helix (Boesze-Battaglia et al., 1997; Boesze-Battaglia et al., 1998) with a stop codon at its approximate midpoint. Expression of the predicted 29 kDa fusion protein was confirmed by induction of TOP10F′ bacterial culture with 1 mM IPTG for 2 hr at 37°C.
Cleavage of purified GST-C-terminal fusion proteins
The full-length GST-C-terminal peripherin/rds fusion protein and the GST-C-terminal truncation mutant were engineered with a thrombin cleavage site. The isolated GST-fusion proteins were treated with thrombin as described (Smith and Johnson, 1988). Briefly, the GST-C-terminal proteins were diluted into thrombin cleavage buffer; 50 mM Tris–HCl, 150 NaCl, 2.5 mM CaCl2, pH 7.5 and incubated overnight at either 37°C or room temperature with thrombin in a 1:1 (wt/wt) ratio. The reaction was stopped with the addition of 2 × SDS-sample buffer for electrophoresis and Western blot analysis or quick frozen in liquid nitrogen for depolarization studies, fusion assays and column chromatography.
Purification of GST-C-terminal fusion protein products
The full-length peripherin/rds C-terminal fusion protein was cleaved with thrombin as described above. The cleaved C-terminal polypeptide was purified using size exclusion FPLC. Routinely, 2 ml of sample was loaded onto a Hi-load 16/60 Superdex preparatory grade column (diameter 16 mM; length 62 cm; Amersham Pharmacia, Uppsala, Sweden). The sample was eluted with 0.25 M NaCl; in 0.02 M Na2 PO4, pH 7.4 at a flow rate of 1 ml min−1 and absorbance monitored at 214 nm. The identity of the peaks was confirmed using dot blot analysis with monoclonal antibody 2B6 and purity confirmed with Coomasie staining of 15% SDS–PAGE based on the system of Laemmli (Laemmli, 1970), and Western blot analysis (Boesze-Battaglia et al., 1998). The relative sizes of the complexes were determined by comparison to known standards; BSA, (87 kDa), thrombin, (33.5 kDa), GST (27.5 kDa), trypsin, (24 kDa); cytochrome c (12.4 kDa), angiotensin (1.25 kDa), met-and leu enkephalin (0.56 kDa) and the tripeptide, Val-Tyr-Val (0.38 kDa).
Vesicle preparation
Phospholipids were purchased from Avanti Polar Lipids (Birmingham, AL, USA). Phosphatidylcholine (PC), phosphatidylserine (PS) and cholesterol (Chol) were co-solubilized in cholorform in a 4:4:1molar ratio, dried under N2 and lyopholized. Large unilamellar vesicles (LUVs) were prepared by extrusion essentially as described (Hope et al., 1985). Briefly, dried lipid films were lyophilized for 4 hr and the resulting film resuspended in 5 mM Hepes, 100 mM NaCl (pH 7.4). The lipid dispersions were subjected to 10 freeze-thaw cycles in liquid nitrogen prior to 10 extrusion cycles. Extrusions were performed with a Nucleopore filtration device through either a membrane of 0.1 or 0.4 μm, depending on the types of assays. When the LUVs were to be used for aqueous contents mixing assays the fluorophores were encapsulated into the vesicles as described below. Phosphate was determined as described by Bartlett (1959) and modified by Litman (1973).
Resonance energy transfer (RET) assay depicting membrane mixing
The fusion between PC/PS/CHOL vesicles containing fluorescent probes NBD-PE and Rh-PE was monitored as a mixing of membrane components using a fluorescence resonance energy transfer method as described originally by Struck et al. (1981). Using a mixture of fluorescently labeled and unlabeled vesicles the assay is based on the dilution of a resonance energy transfer pair, Rh-PE and NBD-PE. Dilution due to membrane lipid mixing results in an increase in NBD-PE fluorescence. Vesicles containing 0.6% (mole%) Rh-PE and 0.6% (mole%) NBD-PE were mixed with unlabeled vesicles in a 1:1 molar ratio. At this concentration of fluorophores, the NBD-PE fluorescence increases linearly with the dilution of the probe. Emission of NBD- fluorescence was monitored at 530 nm, with the excitation wavelength set at 465 nm (rhodamine excitation). A 515 nm cutoff filter was used between the sample and the emission monochrometer to decrease interference from scattering.
ANTS–DPX contents mixing assays
ANTS–DPX fusion assays were carried out to confirm protein induced aqueous contents mixing as described by Ellens et al. (1985). LUVs encapsulting either ANTS or DPX were prepared as described by Szoka et al. (1980) and in further detail by Ellens et al. (1985). Briefly, N-methyl-DOPE was hydrated on ice, under nitrogen in 25 mM ANTS, 45 mM NaCl, 10 mM glycine, pH 9·5 or 90 mM DPX, 10 mM glycine, pH 9·5 for 3 hr. Subsequent to the freeze-thaw and extrusion steps described above, the encapsulated fluorophores were separated from the unencapsulated fluorophores on a Sephadex G-50 column, with 100 mM NaCl, 10 mM Glycine, 0.1 mM EDTA, pH 9.5 as the elution buffer. The vesicles were used immediately for fusion assays. These assays were carried out in a final volume of 3 ml, with a 9:1 molar ratio of DPX-containing LUVs to ANTS containing LUVs. Fusion was initiated with a decrease in pH from 9.5 to 4.5 with the addition of 100 μl of 2 M Na acetate/acetic acid buffer. Fluorescence intensity was monitored with λex = 380 nm and λem = 510 nm: The mixing of the aqueous contents results in the formation of an ANTS–DPX complex with a reduced quantum yield and a quenching of fluorescence. This decrease in fluorescence was monitored continuously for 5–10 min. The initial rate of fusion (IRF) between the vesicles was calculated (Szoka et al., 1980; Ellens et al., 1985) from the slope of the fluorescence decay curve during the period immediately following the initiation of fusion (first 1–2 min). Hundred percent fluorescence intensity was taken to be the initial fluorescence intensity before the decrease in pH.
Membrane permeability studies
Membrane bilayer destabilization was inferred from membrane depolarization studies and measured fluorimetrically with the cation sensitive dye, diS-C2-5, as a collapse in a valinomycin induced diffusion potential (Kliger et al., 1997). SUVs composed of phosphatidylcholine/phosphatidylserine/cholesterol (PC/PS/CHOL) in a 4:4:1 molar ratio were prepared by probe sonication in the presence of K+ containing buffer (50 mM K2SO4, 10 mM Hepes-SO4, pH 6.8). An aliquot of the SUVs (phospholipid concentration = 36 nmol) was added to 10 ml of isotonic buffer (50 mM Na2SO4, 10 mM Hepes-SO4, pH 6.8) containing 10 μl of the fluorescent probe diS-C2-5 (stock 1 mM) and incubated at 37°C until stable baseline fluorescence was established. The addition of valinomycin (final concentration = 10−7 M), selectively permeabilized the vesicles to K+, creating a negative diffusion potential inside the vesicles resulting in the quenching of the dye’s fluorescence. GST-C-terminal fusion proteins or purified C-terminal polypeptide from a 1 mg ml−1 stock in either dH2O or K+ -free buffer were added in 25, 75, or 100 μl aliquots. Fluorescence was subsequently recorded at λex = 620 nm and λem = 670 nm for 90 min. An increase in fluorescence intensity upon the addition of proteins was indicative of a dissipation of the diffusion potential due to polypeptide induced destabilization. The percent fluorescence recovery was calculated as described (Kliger et al., 1997), using the equation below
It is the fluorescence after the addition of the peptide, at time t, Io is fluorescence after the addition of valinomycin, If = fluorescence intensity prior to the addition of valinomycin. Mellitin (final concentration = 9.0 μg ml−1) was added to confirm that the increase in fluorescence observed was due to a collapse in the diffusion potential. In all of the permeability studies mellitin was able to dissipate the valinomycin induced diffusion potential after the addition of those proteins which had no effect.
Fluorimetric assays
Fluorescence measurements were conducted in a Perkin–Elmer LS50B spectrofluorimeter, at 37°C with continuous stirring. In all assays the kinetics of the change in fluorescence was recorded continuously. Data symbols are included in the figures to allow for identification of the curves.
2.1. Circular dichroism studies
Purified full-length peripherin/rds C-terminal polypeptide samples were prepared in 50 mM phosphate buffer (pH 7) for CD studies at 0.1 mg ml−1. To determine the effect of dodecylphosphatidylcholine (DPC), additional samples were prepared as above, in which the lipid was present in a 100:1 ratio to the polypeptide. Spectra were recorded on a Jasco J-715 spectropolarimeter using a 1 nm bandwidth and a 0.2 cm path length. The instrument was programmed to average three spectra taken from 190 to 260 nm. The predicted structure was based on algorithms using wavelengths between 205 and 260 nm. No detectable change in intensity was observed with DPC vesicles alone. All samples were run at least three times at room temperature. Total protein was determined using a Bio-Rad microassay procedure (Sigma, St Louis, MO, USA).
3. Results
3.1. Purification of the C-terminal peripherin/rds polypeptide
Peripherin/rds has been shown to promote membrane fusion in cell free assay systems (Boesze-Battaglia et al., 1997; Boesze-Battaglia et al., 1998). Residues 311–325 of the C-terminus of peripherin/rds have been identified as the minimal region required to promote lipid bilayer fusion (Boesze-Battaglia et al., 1997; Boesze-Battaglia et al., 1998; Boesze-Battaglia et al., 2000). To further investigate the functional and structural properties of the C-terminus, polypeptides corresponding to the entire C-terminal region were expressed. In these studies, a bovine peripherin/rds GST-C-terminal fusion protein, depicted schematically in Fig. 1, was expressed in E. coli. The entire C-terminus of peripherin/rds from residues 284 to 346 was assembled into a construct engineered with a thrombin cleavage site. This peptide will be referred to as the full-length peripherin/rds C-terminal polypeptide (abbreviated PERCTER). A second similarly engineered construct corresponding to residues 284–318 was also generated. This truncation mutant lacks half of the fusion domain (residues 311–325) of peripherin and will be referred to as the C-terminal truncation polypeptide (abbreviated PERCTN). The GST-C-terminal fusions were expressed in E. coli by IPTG induction at 37°C for 3 hr and purified as described in Section 2.
Fig. 1.
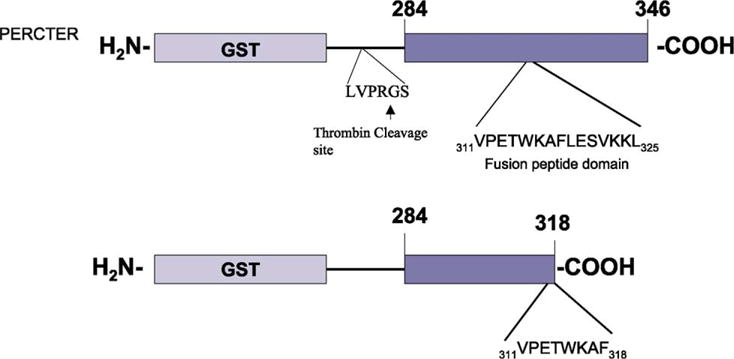
Schematic representation of GST-fusion Proteins. The full-length C-terminus of peripherin/rds (PERCTER) corresponding to residues 284–346 is shown with the fusion domain indicated. The GST C-terminal truncation mutant protein (PERCTN) is also shown from residues 284 to 318, with the truncated form of the fusion peptide domain indicated. GST refers to glutathione-S-transferase. The thrombin cleavage site is indicated by an arrow.
Expressed protein eluted with glutathione from GST-Sepharose was characterized by immunobloting. An anti-GST and anti-peripherin/rds mAb 2B6 immunoreactive band with a molecular mass of 36 kDa, corresponding to the predicted size of the GST-peripherin/rds C terminal polypeptide was isolated in the eluant. The GST-C-terminal polypeptide was cleaved with thrombin and the proteins in the cleavage reaction separated by size exclusion chromatography (Fig. 2). The six isolated fractions (A–F) were run on a 15% SDS–PAGE gel, transferred to nitrocellulose and immunoblotted with anti-peripherin/rds mAb 2B6 and anti-GST. The results of the anti-peripherin/rds blots are shown in the inset to Fig. 2. Peak E was found to contain an 8 kDa band that was immunoreactive with anti-peripherin/rds mAb 2B6, no additional proteins were detected in this fraction as shown by Coomasie staining (data not shown). In addition a 35.5 kDa band was detected in peak B corresponding to uncleaved protein (Fig. 2 inset). No detectable immunoreactivity with anti peripherin/rds mAb 2B6 was observed in Peak A or F.
Fig. 2.
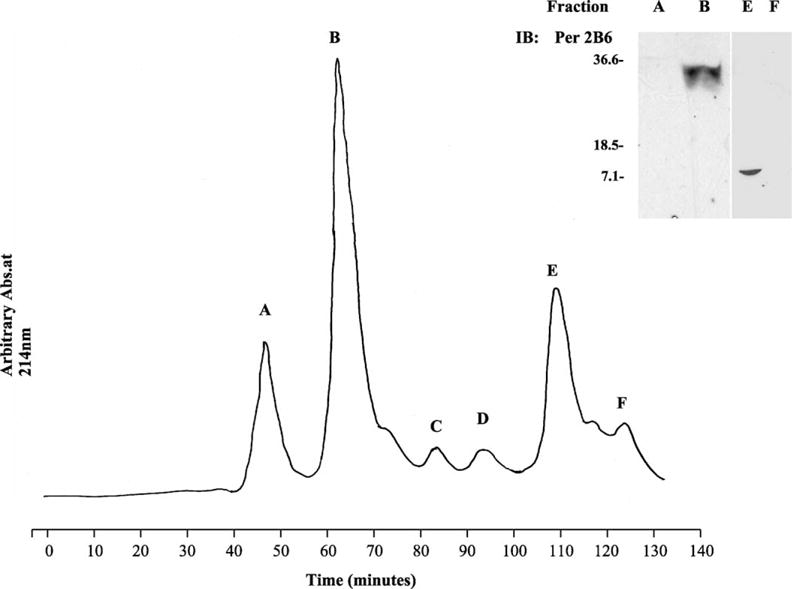
Purification of full-length peripherin/rds C-terminal polypeptide from GST-fusion protein. The GST-C-terminal polypeptide was cleaved with thrombin. The resultant proteins were purified on a Hi Load Superdex column eluted with 0.025 M NaCl, 0.02 M Na2PO4 pH 7.4 at a flow rate of 1 ml min−1. The eluant was monitored at 214 nm and the peaks designated as A–F. The elution volume, which is equivalent to the elution time is indicated below the peak designation. Inset. Immunoblot analysis of proteins in the fractions corresponding to Peaks A, B, E and F. Proteins were separated on a 15% SDS–PAGE, transferred to nitrocellulose and immunoblotted with anti-peripherin/rds mAb 2B6.
3.2. The full-length C-terminal polypeptide promotes the steps of fusion
To investigate the ability of the purified C-terminus to mediate lipid bilayer fusion, the final steps of membrane fusion were assayed. First, the destabilization of the membrane was inferred from membrane depolarization studies, and second, membrane fusion was assessed both by mixing of the bilayer lipids and mixing of the aqueous contents (Bentz and Ellens, 1988). Biochemical and biophysical techniques demonstrating each of these steps have been used in previous studies to show that the synthetic peptide (PP-5) that corresponds to the peripherin/rds fusion peptide (residues 311–325), can promote membrane depolarization and membrane fusion in phospholipid vesicles (Boesze-Battaglia et al., 1997; Boesze-Battaglia et al., 1998; Boesze-Battaglia et al., 2000). Since we have identified the minimal region necessary to promote fusion we now wished to investigate how the entire C-terminal domain participates in this process.
3.3. Membrane depolarization
Membrane destabilization can be indirectly measured as an increase in membrane permeability that is monitored experimentally as a dissipation of a diffusion potential in a model membrane system. Phospholipid vesicles consisting of PC/PS/CHOL were prepared in a K+ containing buffer as described in Section 2. Upon addition of valinomycin, K+ selectively exits the vesicles and establishes a membrane potential across the bilayer. The K+ gradient can be detected as a change in the fluorescence of the cation sensitive dye, diS-C2-5. Membrane destabilization, inferred from the collapse of this gradient can be detected as an increase in fluorescence.
The ability of the C-terminal polypeptide and the truncated polypeptide to destabilize the bilayer was tested by addition of each of these to a suspension of vesicles in which a K+ gradient was established in the presence of diSC2-5. A polypeptide that promotes membrane depolarization likely does so by destabilizing the bilayer thus increasing its permeability. Fig. 3(A) shows representative depolarization experiments. The purified, peripherin/rds C-terminal (PERCTER) polypeptide was able to promote membrane depolarization as indicated by the increase in fluorescence. During the same time period the truncated C-terminal polypeptide (PERCTN) did not promote membrane depolarization. Therefore, membrane depolarization (indicative of destabilization) required the fusion peptide domain, residues 311–325. The ability to depolarize the model membranes was found to be correlated to the amount of purified polypeptide added as shown in Fig. 3(B). The highest percent depolarization was observed at a 50:50 mix of phospholipid to polypeptide (mole/mole). The basal levels of depolarization, change in fluorescence in the absence of added peptide corresponds to less than 2% depolarization (data not shown).
Fig. 3.
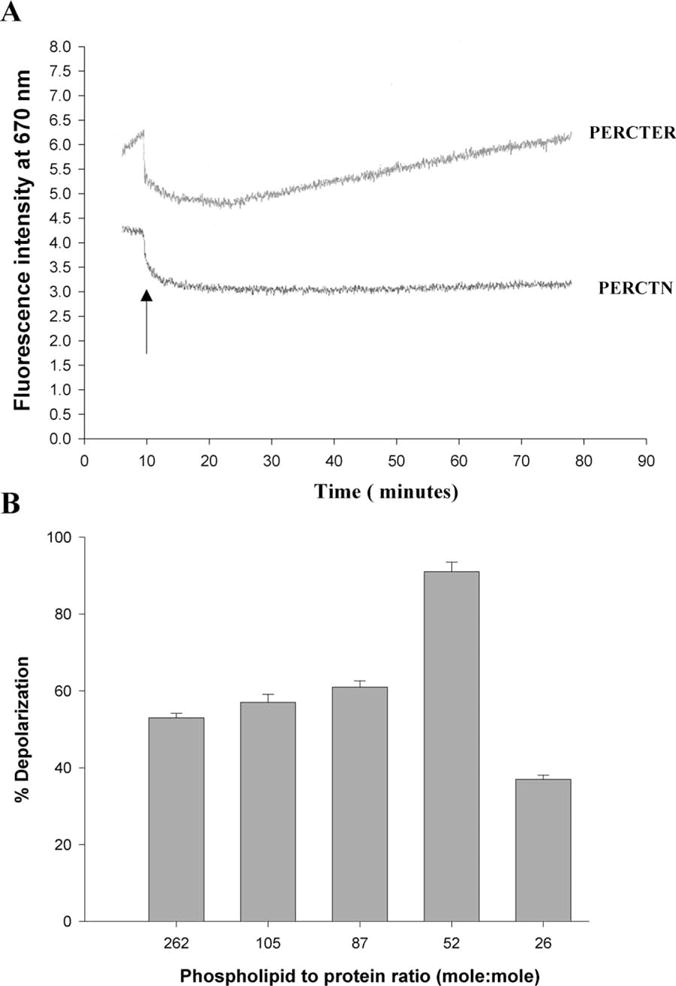
C-terminal polypeptide induced dissipation of valinomycin induced diffusion potential in phosphatidylcholine/phosphatidylethanolamine/cholesterol SUVs. (A) The fluorescence intensity of diS-C2-5, at λex = 620 nm and λem = 670 nm was followed over time (minutes). The addition of either the full-length purified C-terminal polypeptide PERCTER or the truncation mutant PERCTN is shown by an arrow. The results shown are of a representative experiment. The phospholipid to polypeptide ratio in this sample was 50:1 (mole/mole). (B) The extent of depolarization as a function of phospholipid to pure C-terminal polypeptide ratio as indicated. The results are an average ± SD of four independent experiments.
Complete membrane fusion requires that localized membrane destabilization is followed by mixing of both the bilayer lipids and the aqueous contents of the vesicles. The ability of the GST-C-terminus and the purified C-terminal polypeptide to promote fusion was determined using two different assays; one assay measured lipid mixing the other aqueous contents mixing.
3.4. Lipid bilayer mixing
Mixing of the bilayer lipids of large unilamellar PC/PS/CHOL vesicles was monitored using a fluorescence resonance energy transfer. Vesicles were prepared that contained the lipid soluble fluorescent probes, Rh-PE and NBD-PE at sufficiently high concentration in the bilayer to quench their fluorescence. These labelled vesicles were then mixed with unlabeled vesicles. Mixing of the lipids from the labelled and unlabeled vesicles was detected as an increase in NBD-PE fluorescence. In Fig. 4(A) the initial rate of fusion in response to the addition of purified C-terminal polypeptide is shown. The purified C-terminal peptide was found to promote fusion as detected by bilayer lipid mixing. In contrast, addition of the truncated C-terminal (PERCTN) polypeptide resulted in no change in fluorescence intensity (data not shown, suggesting that it is unable to promote lipid mixing.
Fig. 4.
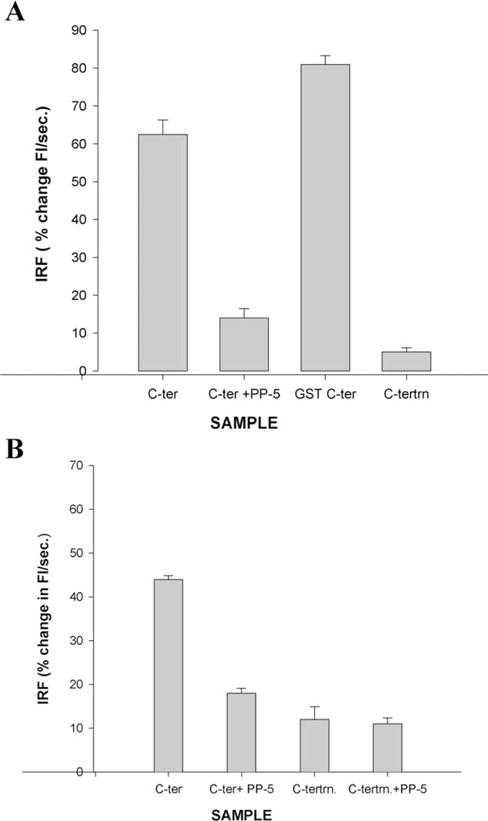
The purified C-terminal polypeptide (PERCTER) promotes on NBD-PE, Rh-PE lipid mixing and ANTS–DPX aqueous contents mixing. The assessment of fusogenic function of full-length C-terminal polypeptide is shown. (A) Lipid mixing detected using resonance energy transfer between NBD-PE and Rh-PE and is quantitated as the initial rate of fusion (IRF),% change in fluorescence intensity per second. Fusion was initiated with the addition of either the pure C-terminal polypeptide (C-ter) to which PP-5 was added (C-ter + PP-5) or GST-C-terminal polypeptide (GST C-ter). (B) Aqueous contents mixing detected as a decrease in fluorescence as a complex between vesicles containing two fluorescent probes ANTS and DPX is formed. The vesicles were incubated with, pure C-terminal polypeptide (c-ter), C-terminus + PP-5 (c-ter PP-5) or the truncation mutant polypeptide (c-tertrn) or truncation mutant + polypeptide (c-tertrn + PP-5) as indicated at 37°C. The phospholipid to peptide ratio is 50–60:1.
3.5. Aqueous contents mixing
It is not sufficient to demonstrate fusion by lipid mixing only. A number of fusion proteins, such as the hemagglutinin, have been shown to induce hemifusion, that is lipid mixing in the absence of complete aqueous contents mixing (Kliger et al., 1997). To assess whether the purified C-terminal polypeptide could promote aqueous contents mixing, two sets of large unilamellar vesicles, one containing the soluble fluorophore, ANTS and the other containing the soluble fluorophore, DPX were mixed. If the aqueous contents of the two vesicles mix an ANTS–DPX complex is formed and the fluorescence is quenched. As shown in Fig. 4(B), the purified C-terminal polypeptide PERCTER was found to promote aqueous contents mixing by this criterion. Similar results were obtained when the C-terminus of native purified peripherin/rds was cleaved by trypsin proteolysis and isolated in a supernatant fraction. This C-terminal polypeptide has previously been shown to promote aqueous contents mixing (Boesze-Battaglia et al., 1997; Boesze-Battaglia et al., 1998). The fusion peptide analogue, PP-5, was shown to inhibit both lipid mixing and aqueous contents mixing at concentrations similar to its inhibitory effect on native peripherin/rds in cell free assay systems (Boesze Battaglia et al., 1997; Boesze-Battaglia et al., 1998).
Collectively, these results demonstrate that the purified C-terminal polypeptide is properly folded as defined by the ability to promote depolarization and both lipid mixing and aqueous contents mixing, i.e. complete fusion.
3.6. Analysis of secondary structure by circular dichroism
Peptides corresponding to regions of intact proteins have been shown to exhibit the secondary structural elements present in the intact protein. Furthermore, the fusion peptide domain has been shown to exhibit α-helical structure (Boesze-Battaglia et al., 1998). Far ultraviolet circular dichroism (CD) was used to investigate the secondary structure present in the peripherin/rds C-terminal polypeptide. As shown in Fig. 5 the C-terminal region exhibits considerable secondary structure. The elements of the secondary structure were predicted based on the CD spectrum using algorithms as described (Bohm et al., 1992). The results of these studies are shown in Table 1. An α-helical region in the polypeptide is consistent with the structure observed for the fusion peptide, PP-5, in solution detected using FTIR (Arikawa et al., 1992).
Fig. 5.
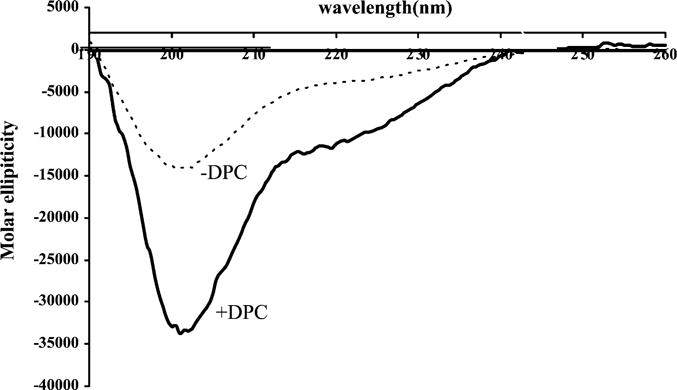
The effect of DPC on the CD spectrum of full-length peripherin/rdsC-terminal polypeptide (PERCTER). The CD spectrum of PERCTER (0.1 mg ml−1) was determined in 50 mM phosphate buffer, pH 7.0 (⋯). The CD spectrum of PERCTER at the same concentration was then determined in the presence of DPC (—). The mole ratio of DPC to PERCTER was 100:1. The spectrum of DPC at the same concentration used in the later determination is also shown (- - -). The spectra are presented as molar ellopticity versus wavelength. The secondary structure was calculated as described in Section 2 usingwavelengths between 205 and 260 nm and is summarized in tabular form in Table 1.
Table 1.
Secondary structural predictions based on CD spectra. The secondary structure of the C-terminal polypeptide was predicted as described in Section 2 and is indicated as the percent of the total in the absence (−DPC) and presence (+DPC) of a lipid surface
| Predicted secondary structural characteristics | −DPC (%) | +DPC (%) |
|---|---|---|
| Anti-parallel β-sheet | 26 | 14 |
| Parallel β-sheet | 4 | 4 |
| α-Helix | 9 | 19 |
| β-Turn | 26 | 26 |
| Random structure | 37 | 36 |
The values listed are the average of two independent experimental determinations.
To investigate the effect of a membrane surface on the secondary structure of the polypeptide the CD spectrum of the C-terminal polypeptide was determined in the presence of the lipid surface provided by DPC vesicles. As shown in Fig. 5, in the presence of a lipid surface the minima detected at 222 and 208 nm are more pronounced than in the absence of the lipid. The CD spectrum was analyzed for secondary structure using algorithms again as described (Bohm et al., 1992) and the results are shown in Table 1. Based on this analysis the α-helical content of the C-terminus is markedly increased in the presence of the lipid DPC while the anti-parallel β-sheet decreases. Other structural elements remain unchanged. This increased α-helical content of the C-terminal polypeptide has been observed in other fusion peptides and further analysis of the tertiary structure is necessary to predict the orientation and structural motif of the C-terminus.
4. Discussion
Rod and cone cells contain a unique tetraspanin membrane fusion protein, peripherin/rds, the C-terminal domain of which is essential in mediating lipid bilayer fusion. Previous studies demonstrated that a peptide corresponding to a conserved sequence in the C-terminal could promote bilayer fusion. However, by analogy to other membrane fusion proteins it is likely that additional regions within the C-terminus are required for regulation of fusion and recognition of the appropriate membrane surfaces (White, 1992; Hernandez et al., 1996; Pecheur et al., 1999; Wasserman, 1999). To investigate the structure and function of this larger protein domain we have generated a C-terminal peripherin/rds polypeptide (PERCTER) that includes the fusion domain. This polypeptide can promote each of the requisite steps in membrane fusion. In contrast, a polypeptide that includes only a portion of the fusion domain (PERCTN) cannot mediate fusion events. The work herein demonstrates that the C-terminal polypeptide interacts with a lipid surface, changes conformation in the presence of a membranous medium and induces contents and lipid mixing of model membranes.
Moreover, this work provides compelling evidence that the C-terminus of peripherin/rds is able to promote membrane fusion; both lipid mixing and aqueous contents mixing, without anchorage to the bilayer. These results suggest that peripherin/rds dependent fusion may proceed through a mechanism different from that of HIV, and HA induced fusion, processes that require anchorage of the fusion protein to the host membrane (Kliger et al., 1997). Recently, a highly conserved region upstream of the fusion peptide domain (Smith and Johnson, 1988) that includes a proline at position 295 was shown to enhance fusion the mechanism of this enhancement is presently unknown (Stefano et al., 2002). Similar regions have been identified as essential for conformational changes of fusion proteins in intracellular fusion, neurotransmitter release and viral infectivity (Laemmli, 1970; Hope et al., 1985). Collectively the data presented herein in combination with previous studies further supports the hypothesis that regions in addition to the 15 amino acid long fusion peptide region participate in peripherin/rds dependent fusion.
Membrane fusion is a thermodynamically unfavourable event that requires a change in normal membrane bilayer structure. Fusion proteins act by perturbing local membrane bilayer structure. The mechanism by which a fusion protein carries out this function has been the subject of intense interest and is currently unresolved.
Numerous biochemical and biophysical studies have led investigators to propose that membrane fusion proteins exist in two major conformations; a fusogenic or stable conformation and a pre-fusion or metastable conformation (Carr et al., 1997). Structural changes within fusion proteins are required to allow a fusion protein to go from the pre-fusion to the fusion competent state and to mediate the formation of a fusion pore (Mellman, 1995; Pecheur et al., 1999; Bentz, 2000). Peptide-modeling studies suggest that sperm fertilin α (Muga et al., 1994), measles virus F1 (Epand et al., 1992) and the S protein from hepatitis B virus (Rodriguez-Crespo et al., 1995; Rodriguez-Crespo et al., 1996) adopt a β-sheet structure in a lipid environment (Pecheur et al., 1999). Alternatively at higher lipid-to-protein ratios fertilin has been shown to prefer an α-helical conformation with a helix that inserts obliquely into the membrane bilayer, a topology that requires the presence of negatively charged lipids (Martin et al., 1998). An α-helical conformation and an oblique membrane orientation is also observed for fusion peptides of SIV (Blacklow et al., 1995), and HIV (Carr and Kim, 1993). The increase in α-helical content has lead to suggestions that a coiled-coil intermediate (Bentz, 2000) is the favoured conformation for a fusion pore. Such a structural requirement is clearly supported by the crystal structure of the membrane fusion protein HA (Carr and Kim, 1993; Bullough et al., 1994), the HIV ectodomain (Chan et al., 1996; Tan et al., 1997; Weissenhorn et al., 1998) and the synaptobrevin–syntaxin complex (Sutton et al., 1998). Most striking is the observation that both highly hydrophobic viral fusion peptide domains and charged fusion peptides within fertilin both appear to adopt a coiled-coiled intermediate.
The above studies indicate that fusion proteins undergo changes in secondary structure during the process of membrane fusion. Numerous other studies have shown that polypeptides that correspond to protein domains exhibit secondary structure that corresponds to that of the intact protein. Therefore, having established that the C-terminus is able to promote fusion we investigated the secondary structure of this polypeptide. In aqueous solution the C-terminus exhibits a far UV circular dichroism (CD) spectrum characteristic of a peptide with substantial secondary structure, as shown in Fig. 5 and in Table 1.
A fusion mediating protein must interact with lipid bilayers. Therefore it is reasonable to propose a conformational change to the fusion competent state in the presence of a lipid surface. The CD spectrum of purified wt peripherin/rds C-terminal polypeptide PERCTER (Fig. 5) exhibits such an increased intensity. Analysis of the CD spectrum indicates a striking increase in α-helical secondary structure content when a lipid surface is provided. It is particularly interesting to note that the structural elements corresponding to β-turn, parallel β-sheet and random structure remain unchanged upon interaction with the lipid surface. The anti-parallel β-sheet is approximately halved while the α-helix is approximately doubled. This leads to the speculation of a localized, dramatic conformational change. We propose that this change in secondary structure of the C-terminal PERCTER polypeptide is associated with the transition from a pre-fusion to a fusion competent state. Earlier results based upon synthetic peptides to the fusion domain suggested that fusion competency requires a tetrameric arrangement of amphiphilic α-helical peptides (Boesze-Battaglia et al., 2002). It is tempting to propose that the C-terminus of peripherin/rds may likewise form a fusion competent coiled-coil tetramer by disruption of an anti-parallel β-sheet region.
Since a number of mutations leading to various forms of retinal degenerative disease are located within the C-terminal domain of peripherin/rds a thorough analysis of the structural and functional properties of the C-terminus will provide valuable insight into the etiology of these diseases and allow genotype-phenotype correlations. An understanding of the factors that determine the specificity and stability of structural changes within peripherin/rds is essential to understand of the mechanisms of photoreceptor renewal processes, specifically disk morphogenesis and disk shedding. While it is clear that membrane fusion is important in each of these processes, the mechanism of the fusion has not been elucidated. Finally, the analysis of structural changes within the C-terminus has more far reaching implications, given that it has been recently proposed that the localization signal for the targeting of this protein to the ROS also resides within this domain (Tam et al., 2001, 2002).
Acknowledgments
This work was supported by grants from the National Eye Institute, EY 10420 (K.B.-B.), EY 13246 (A.F.X.G.) and EY 03328 (A.D.A.). K.B.-B. is a recipient of an E. Matilda Ziegler Vision Award. G.E. is a recipient of a GSBS Summer undergraduate research fellowship. The authors would like to thank Dr R. Molday for anti peripherin/rds mAb 2B6 and Dr R. Schimmel for critical reading of the manuscript.
References
- Arikawa K, Molday L, Molday RS, Williams D. J Cell Biol. 1992;116:659–667. doi: 10.1083/jcb.116.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett GR. J Biol Chem. 1959;234:466–473. [PubMed] [Google Scholar]
- Bentz J. Biophys J. 2000;78:886–900. doi: 10.1016/S0006-3495(00)76646-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentz J, Ellens H. Colloids Surf. 1988;30:65–112. [Google Scholar]
- Blacklow SC, Lu M, Kim PS. Biochemistry. 1995;34:14955–14962. doi: 10.1021/bi00046a001. [DOI] [PubMed] [Google Scholar]
- Boesze-Battaglia K, Goldberg AFX. Int Rev Cytol. 2002;217:183–226. doi: 10.1016/s0074-7696(02)17015-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boesze-Battaglia K, Kong F, Lamba OP, Stefano FP, Williams DS. Biochemistry. 1997;36:6835–6846. doi: 10.1021/bi9627370. [DOI] [PubMed] [Google Scholar]
- Boesze-Battaglia K, Lamba OP, Napoli A, Sinha S, Guo Y. Biochemistry. 1998;37:9477–9487. doi: 10.1021/bi980173p. [DOI] [PubMed] [Google Scholar]
- Boesze-Battaglia K, Stefano FP, Fenner M, Napoli AA. Biochim Biophys Acta. 2000;1463:343–354. doi: 10.1016/s0005-2736(99)00226-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohm G, Muhr R, Jaenicke R. Protein Eng. 1992;5:191–195. doi: 10.1093/protein/5.3.191. [DOI] [PubMed] [Google Scholar]
- Boucheix C, Rubinstein E. Cell Mol Life Sci. 2001;58:1189–1205. doi: 10.1007/PL00000933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullough PA, Highson FM, Skehel JJ, Wiley DC. Nature. 1994;371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- Carr CM, Chaudry C, Kim PS. Proc Nat Acad Sci USA. 1997;94:14306–14313. doi: 10.1073/pnas.94.26.14306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr CM, Kim PS. Cell. 1993;73:823–832. doi: 10.1016/0092-8674(93)90260-w. [DOI] [PubMed] [Google Scholar]
- Chan DC, Fass D, Berger M, Kim PS. Nat Struct Biol. 1996;3:465–469. doi: 10.1038/nsb0596-465. [DOI] [PubMed] [Google Scholar]
- Connell G, Molday RS. Biochemistry. 1990;29:4691–4698. doi: 10.1021/bi00471a025. [DOI] [PubMed] [Google Scholar]
- Ellens H, Bentz J, Szoka FC. Biochemistry. 1985;24:3099–3106. doi: 10.1021/bi00334a005. [DOI] [PubMed] [Google Scholar]
- Epand RM, Cheetham JJ, Epand RF, Yeagle PL, Richardson CD, Rockwell A, De Grado WF. Biopolymers. 1992;32:309–314. doi: 10.1002/bip.360320403. [DOI] [PubMed] [Google Scholar]
- Hernandez LD, Hoffman LR, Wolfsberg TG, White JR. Ann Rev Cell Dev Biol. 1996;12:627–661. doi: 10.1146/annurev.cellbio.12.1.627. [DOI] [PubMed] [Google Scholar]
- Hope MJ, Bally MB, Webb G, Cullis PR. Biochim Biophys Acta. 1985;1147:223–236. doi: 10.1016/0005-2736(85)90521-8. [DOI] [PubMed] [Google Scholar]
- Jones TA, Korte T, Blumenthal R. J Biol Chem. 1998;273:404–409. doi: 10.1074/jbc.273.1.404. [DOI] [PubMed] [Google Scholar]
- Kliger Y, Aharoni A, Rapaport D, Jones P, Blumenthal R, Shai Y. J Biol Chem. 1997;272:13496–13505. doi: 10.1074/jbc.272.21.13496. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Nature. 1970;277:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Litman BJ. Biochemistry. 1973;13:2545–2554. doi: 10.1021/bi00737a028. [DOI] [PubMed] [Google Scholar]
- Maecker HT, Todd SC, Levy S. FASEB J. 1997;11:428–442. [PubMed] [Google Scholar]
- Maniatus T, Frisch EF, Sambrock MD. Molecular Cloning (A Laboratory Manual) Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. [Google Scholar]
- Martin I, Epand R, Ruysschaert JM. Biochemistry. 1998;37:17030–17039. doi: 10.1021/bi980909i. [DOI] [PubMed] [Google Scholar]
- Matsumoto B, Besharse JC. Invest Ophthalmol Vis Sci. 1985;26:628–635. [PubMed] [Google Scholar]
- Mellman I. Cell. 1995;82:869–872. doi: 10.1016/0092-8674(95)90018-7. [DOI] [PubMed] [Google Scholar]
- Muga A, Neugebauer W, Hirama T, Surewicz WK. Biochemistry. 1994;33:4444–4448. doi: 10.1021/bi00181a002. [DOI] [PubMed] [Google Scholar]
- Muller-Weeks S, Boesze-Battaglia K, Fitzgerald C. Exp Eye Res. 2002;75:143–157. doi: 10.1006/exer.2002.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecheur EI, Martin I, Bienvenue A, Ruysschaert JM, Hoekstra D. J Biol Chem. 2000;275:3936–3942. doi: 10.1074/jbc.275.6.3936. [DOI] [PubMed] [Google Scholar]; Bartlett GR. J Biol Chem. 1959;234:466–473. [PubMed] [Google Scholar]
- Pecheur EI, Sainte-Marie J, Bienvenue A, Hoekstra D. J Memb Biol. 1999;167:1–17. doi: 10.1007/s002329900466. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Crespo I, Gomez-Gutierrez J, Encinar JA, Gonzalez-Ros JM, Albar JP, Peterson DL, Gavilanes F. Eur J Biochem. 1996;242:243–248. doi: 10.1111/j.1432-1033.1996.0243r.x. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Crespo I, Nunez E, Gomez-Gutierrez J, Yelamos B, Albar JP, Peterson DL, Gavilanes F. J Gen Virol. 1995;76:301–308. doi: 10.1099/0022-1317-76-2-301. [DOI] [PubMed] [Google Scholar]
- Smith DB, Johnson KS. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- Stefano FP, Krouse J, Marta P, Boesze-Battaglia K. Exp Eye Res. 2002;74:267–283. doi: 10.1006/exer.2001.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg RH, Fisher SK, Anderson DH. Invest Ophthalmol Vis Sci. 1976;15:700–721. [PubMed] [Google Scholar]
- Struck DK, Hoekstra D, Pagano RE. Biochemistry. 1981;20:4093–4099. doi: 10.1021/bi00517a023. [DOI] [PubMed] [Google Scholar]
- Sutton RB, Fasshauer D, Jahn R, Brunger AT. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- Szoka F, Olson F, Heath T, Vail W, Mayhew E, Papahadjopoulos d. Biochim Biophysic Acta. 1980;601:559–571. doi: 10.1016/0005-2736(80)90558-1. [DOI] [PubMed] [Google Scholar]
- Tam BM, Moritz OL, Hurd LB, Papermaster D. Invest Ophthalmol Vis Sci. 2001;42(4) Abstract no. [PubMed] [Google Scholar]
- Tam BM, Moritz OL, Hurd LB, Papermaster D. Invest Ophthalmol Vis Sci. 2002;43(4) Abstract no. [PubMed] [Google Scholar]
- Tan K, Liu J, Wang JH, Shen S, Lu M. Proc Nat Acad Sci USA. 1997;94:12303–12308. doi: 10.1073/pnas.94.23.12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman P. Cell. 1999;96:175–183. doi: 10.1016/s0092-8674(00)80558-9. [DOI] [PubMed] [Google Scholar]
- Weimbs T, Low SH, Chapin SJ, Mostov KE. Trends Cell Biol. 1997;7:393–399. doi: 10.1016/S0962-8924(97)01130-6. [DOI] [PubMed] [Google Scholar]
- Weissenhorn W, Carfi A, Lee KH, Skehel JJ, Wiley DC. Nature. 1998;387:426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- White J. Science. 1992;258:917–924. doi: 10.1126/science.1439803. [DOI] [PubMed] [Google Scholar]
- Young R. Invest Ophthalmol Vis Sci. 1976;15:700–710. [PubMed] [Google Scholar]