

Abstract
Self-assembly enables exquisite control at the smallest scale and generates order amongst macromolecular building blocks that remain too small to be manipulated individually. Environmental cues, such as heating, can trigger the organization of these materials from individual molecules to multiparticle assemblies with a variety of compositions and functions. Synthetic as well as biological polymers have been engineered for these purposes; however, biological strategies can offer unparalleled control over the composition of these macromolecular building blocks. Biologic polymers are macromolecules, themselves composed of monomeric units that can be precisely tailored at the genetic level; furthermore, they can often utilize endogenous biodegradation pathways, which may enhance their potential clinical applications. DNA (nucleotides), polysaccharides (carbohydrates), and proteins (amino acids) have all been engineered to self-assemble into nanostructures in response to a change in temperature. This focus article reviews the growing body of literature exploring temperature-dependent nano-assembly of these biological macromolecules, summarizes some of their physical properties, and discusses future directions.
Keywords: protein polymer, nanoparticle, hyperthermia, elastin-like polypeptide, self assembly
Introduction
Multifunctional biomaterials offer the possibility to engineer systems with mechanical, chemical, and physical properties that respond to environmental changes in nuanced ways1-3. In the pursuit of these biomaterials, enhanced control is needed over their assembly at the nanoscale. The rational design of self-assembling nanoscale systems is one strategy of fabricating these materials from the bottom-up. In the medical arena, self-assembled nanostructures are under evaluation as both diagnostics4 and therapeutics5. Beyond medical applications, nanoscale self assembly is being used in electronics to create clusters of molecules that act as insulators, semiconductors, and even superconductors6. Non-covalent forces drive the formation of ordered structures and patterns at the nanoscale, imbuing novel functions to systems that would not occur in the disordered state7. The shapes of higher order nanostructures or patterns are largely determined by the individual parts comprising the system. Therefore, the ability to engineer precision macromolecular parts is expected to enable the rational design of multifunctional biomaterials.
The most complex examples of self-assembly are biological. Through the evolution of life, interconnected networks of proteins, sugars, lipids, and oligonucleotides have been genetically ‘programmed’ to assemble the multifunctional systems essential for biology. Inspired by these natural systems, we have entered a new era of engineering, where intrinsic properties of these natural building blocks can be reengineered for diverse applications. In addition to harnessing the physical properties inspired by natural biomaterials, biologically derived building blocks can result in more uniform synthesis, better biocompatibility8, and utilization of endogenous biodegradation pathways9. Biologically derived systems can often be processed by endogenous nucleases, proteases, and glycosylases, after which the degraded products are naturally excreted or reutilized by the body9, 10.
Many polymers and biological materials undergo thermally-mediated assembly processes, which have been proposed as strategies to enhance the assembly and functionality of biomaterials1, 2, 11. Assembly of nanostructures from synthetic polymers such as poly N-isopropylacrylamide (PNIPAM) and poly lactic acid (PLA) have been extensively studied and reviewed12, 13. These ‘smart’ materials have been engineered into temperature responsive hydrogels, micelles, and vesicles14-16. However, an emerging body of work now characterizes the use of biopolymers to self-assemble structures using temperature as an environmental cue. The purpose of this focus article is to review the current status of thermally responsive biopolymers. For this manuscript, biopolymers are divided into three broad categories: nucleic acids, polysaccharides, and proteins (Figure 1). Due to limitations on space, it is impossible to delve deeply into each category. Instead we introduce unique properties arising from each category of biopolymer. Examples have been identified that produce structural features that might be engineered to assemble or respond to temperature (Table 1). By juxtaposition of these three biopolymers, it may be possible to stimulate cross-fertilization of their applications.
Figure 1.
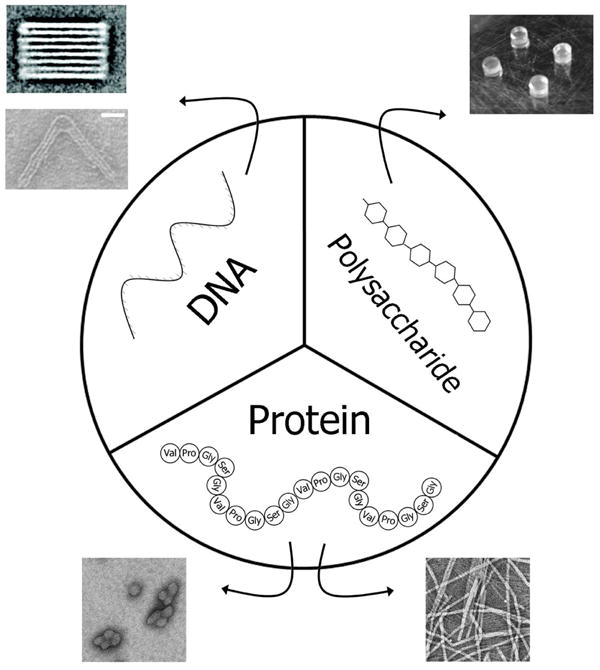
Biological polymers represent a rich source for the engineering of nanostructures. Three classes of biopolymers are evaluated within this review: DNA oligonucleotides, polysaccharides, and protein-based sequences. Each has the ability to specifically self-associate, which can modulate the formation of a wide variety of nanostructures. As their association depends on kinetics and/or thermodynamics, these structures are responsive to temperature. All produced from biological sources, they can be engineered by manipulation at the genetic level to varying degrees. The exquisite control exerted by biological synthetic pathways suggests that they are excellent candidates to engineer useful biomaterials. Adapted with permission from the American Chemical Society28, 79, 81. Adapted with permission from John Wiley and Sons41. Adapted with permission from the Centre National de la Recherche Scientifique and The Royal Society of Chemistry61.
Table 1. Engineered biopolymers and the structures they assemble.
| Biopolymer | Response to heating | Nature of assembly | Reversible | Structures formed | Applications | References |
|---|---|---|---|---|---|---|
| Oligonucleotides | ||||||
| DNA | Disassemble | ssDNA to dsDNA | Yes | Origami | Scaffolding, Encapsulation, Bioreactors | 23-26, 28 |
|
| ||||||
| Polysaccharides | ||||||
| Chitosan | Disassemble | Conferred by PNIPAM | Yes | Nanogel | Drug delivery, Food science | 32-35 |
| κ-carrrageenan | Disassemble | Double helix to random coil | Yes | Nanosphere, Nanogel | Drug delivery, Thickening agent | 37-39 |
| Gellan Gum | Disassemble | Double helix to random coil | Yes | Hydrogel | Tissue engineering | 40, 41 |
| Xanthan Gum | Disassemble | Ordered helix to disordered coil | Yes | Hydrogel | Drug delivery | 31, 42, 43 |
|
| ||||||
| Polypeptides | ||||||
| Coiled-coil | Disassemble | Alpha helix to random coil | Yes | Nanorope, Origami | Nanoreactor, Drug targeting, Drug delivery | 49, 52, 77, 78 |
| Collagen-like | Disassemble | Triple helix to random coil | No | Nanofiber, Nanosheet | Tissue engineering | 54-56 |
| Elastin-like | Assemble | Random coil to β-turn spiral | Yes | Micelle, Vesicle, Nanofiber, Hydrogel | Protein purification, Drug delivery, Intracellular switches | 1, 58-63, 66, 67, 79 |
| Silk-like | Assemble | β-sheet | Yes/No* | Hydrogel, Micelle, | Drug delivery | 68-71, 80 |
| Resilin | Disassemble/Assemble | Spherical network to particles | Yes | Spherical nanoparticle | Biosensors, Drug delivery | 46, 72 |
|
| ||||||
| Polypeptides (de novo) | ||||||
| 17-H-6 | Disassemble/Assemble | Folded α-helix to unfolded β-sheet | Yes/No | Multimer, Nanofiber | Drug delivery | 73, 74 |
| YEHK | Disassemble | Antiparallel β-sheet to random coil | Yes | Nanofiber | Nanoelectronics | 75, 76 |
Silk-like polymer reversibility is contingent on ratio of silk to elastin polymer ratio
DNA Nanostructures
The high fidelity base-pairing specificity of nucleotides has been exploited by molecular biologists for almost 30 years through applications of the polymerase chain reaction (PCR)17. As part of PCR, small oligonucleotides are used to recognize and pair with a very specific sequence of melted single stranded DNA. A nucleobase on one DNA strand binds its complementary nucleobase on the opposite strand18. Purine bases hydrogen bond with pyrimidines and vice versa. Adenine bonds with thymine and cytosine bonds with guanine. While having only 2 types of bonds appears a limitation, the linear combinations of 4 nucleotides of length, n, yield 4n possible permutations. A family of oligonucleotides with 10 nucleobases, could thus recognize and bind to over one million unique sequences of DNA. This means that short oligonucleotides can encode for the many unique intersections required to engineer specific nanostructures. Since nucleotide bases pair non-covalently, oligonucleotide-stabilized structures assemble easily with a decrease in temperature. The melt temperature for each of these oligonucleotides can be tuned by adjusting the guanine-cytosine content, the oligonucleotide length, or by introducing mismatches into the recognition sequence. Thus, oligonucleotides possess thermally responsive properties, tunability, and the ability to multiplex a large number of specific interactions within the same structure.
Despite these opportunities to engineer oligonucleotide nanostructures, their full power to assemble diverse structures has only recently exploded as a source of tunable and biodegradable nanostructures. In part, this has been facilitated by the recent advancement of DNA synthesis technology, which now allows for the production of kilobase strands of nucleotides19. When the design of these base pairings is aided by a computer, a vast array of nanostructures can be constructed, including cubes, spheres, and even a map of the western hemisphere20. Typically these structures are formed using bonding schemes between single stranded DNA (ssDNA) oligonucleotides21, long ssDNA scaffolds ‘stapled’ with shorter oligonucleotides20, and also through the multimerization of branched DNA tiles22-24.
Nucleic acid-base pairing and any resulting structural assembly is a thermodynamic process, which makes them amenable to thermal stimulation. Each annealed oligonucleotide sequence has a specific melting temperature, which can easily be monitored using intercalating fluorescent probes23. The oligonucleotide melting temperature occurs where half of all the nucleotide strands are paired with their complementary strand, which can adopt a double helix that is relatively rigid in comparison with ssDNA. Sacca and coworkers characterized the first and second temperature transitions of DNA strands that self-assembled into DNA tiles23. In one example, oligonucleotides were mixed at equimolar concentrations and melted at 90 °C. While the samples were slowly cooled, a first transition was observed around 60 °C. At this point, single stranded nucleic acid sequences hybridize and form double helices. After a controlled cooling step, these DNA double helices further organize based into tiles with ordered three-dimensional structures determined by their sequence. Both annealing processes are fully reversible.
Large scale DNA origami structures have recently been described that are comprised of megadalton sized DNA-origami tiles24. Previous work describes the formation of polyhedra using a three arm ‘tripod’ tile22. The edges of the tile are comprised of 3 double helix structures, which are connected by single stranded hinges. These tiles form shapes such as nanoprisms and buckeyballs. However, scaling up this scheme to megadalton sized tiles initially failed to produce well-formed shapes. Iinuma and coworkers circumvented this size limitation by creating tripod tiles out of 3 distinct DNA strands24. DNA strands were mixed with p8064, a circular ssDNA scaffold, and heated to 80 °C. The mixture anneals when the system was rapidly cooled from 80 to 65 °C over 1 hour. This was followed by a slow annealing step, where the sample was cooled from 62 to 24 °C over 42 hours. The shape formed by the DNA origami ‘tripod’ tiles was determined by the angle between the tripod legs. For example, tetrahedron formation requires monomeric tripod tiles with 60° - 60° - 60° angles between the tripod legs while cubes require 90° - 90° - 90° angles between tripod legs. This scheme produced 20 MD tetrahedrons, 40 MD cubes, and 60 MD hexagonal prisms. The edges on these polyhedra were approximately 100 nm.
As mentioned in the introduction, a thermally responsive system can be created by the addition of a thermally responsive element. For example, functionalizing a molecule or particle with a thermally responsive DNA strand will impart thermal sensitivity to the parent system. Qi and coworkers describe a system where DNA-functionalized hydrogels self assemble into structures as large as 1 mm in length25. The system was engineered by conjugating a short DNA primer to a polyethylene glycol(PEG-)NHS molecule. This DNA-PEG-NHS solution was mixed with polyethylene glycol diacrylate (PEGDA), and exposed to UV light. PEGDA undergoes photo-initiated crosslinking; therefore, the mixture conforms to a mold when activated by UV light. This process yields hydrogel cubes with short DNA primers extending from one face or many, depending on the pattern of DNA functionalization achieved with photolithography. Using a circular DNA as a template, the rolling circle amplification technique was able to extend DNA primers mounted in the hydrogel26. This resulted in a cube with sequences of ‘giant DNA’ extending off faces of the hydrogel. Mixing giant-DNA functionalized hydrogels with hydrogels functionalized using complementary DNA further yielded even larger nanostructures. The shape of these nanostructures can be engineered by protecting faces of the hydrogel during functionalization with giant DNA as well as by varying complementary pairing of DNA. This method has generated hydrogel cuboids that self-assemble into 1 mm long chains as well as 2 × 2 hydrogel squares. As with all DNA-mediated assembled structures, a sharp increase in heat is expected to disassemble the structure; however, it remains to be seen if the assembly temperature for these nanostructures can be tuned across a range of relevant temperature. Some of these three-dimensional DNA origami systems are even being evaluated as drug delivery systems27.
Much of the literature on DNA nanostructure has described complex 2-D and 3-D designs, however all of these are rigid, fixed structures. In contrast, Zhou and coworkers introduce a method for creating DNA nanostructures with tunable mechanical properties28. Using the same annealing process described above, they designed a DNA structure with two arms made up of 3 bundles of double stranded DNA flanking an inner structure composed of only 6 DNA helices and more flexible ssDNA oligonucleotides. At the center of the structure, a fixed DNA strand serves as the hinge while a flexible ssDNA spring pulls tension on the two arms. By adjusting the length of the ssDNA the resulting structures adopt different angles (Figure 2). A short ssDNA spring pulls the joint into a decreased angle. A longer ssDNA spring relaxes the tension put on the bundle of double stranded DNA and results in a wider, more linear angle. Such a mechanically flexible system can ultimately be used to control motion of nanomechanical devices; furthermore, as these materials are designed from oligonucleotide base paring, it is plausible that the angle adopted by these hinges might one day be engineered to be temperature dependent.
Figure 2. Oligonucleotide origami uses single stranded DNA (ssDNA) springs to engineer precision joints.
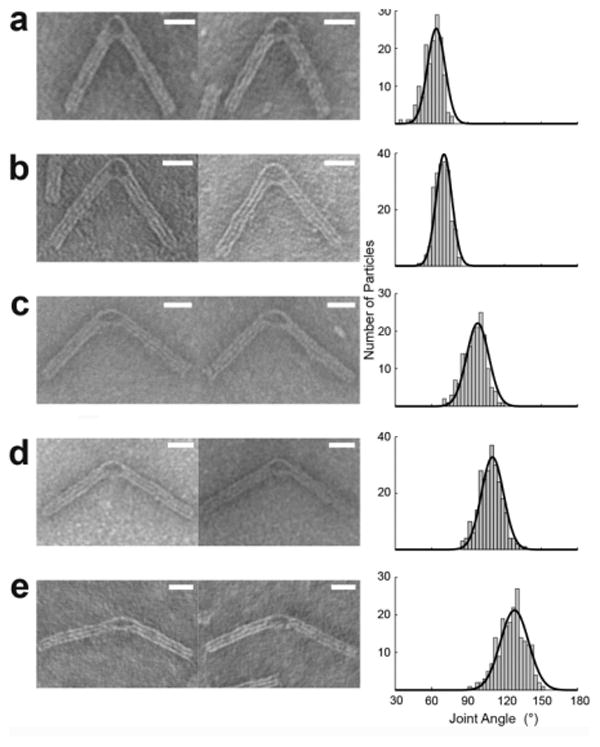
Two arms composed of 3 bundles of double stranded DNA (18 helices total) were linked by a single rigid bundle (6 helices) as well as a ssDNA spring. The length of the spring was modified to produce different angles. Conformational analysis was performed using TEM imaging. Typical particles and histogram distribution are presented with A) 0, B) 11, C) 32, D) 53, and E) 74 bases in the ssDNA springs. The black lines show Gaussian fits to the data. The angles corresponding to the peak values of Gaussian fits were 56.5° (n = 154), 70.2° (n = 213), 97.9° (n = 169), 110.0° (n = 252), 128.2° (n = 204). Bars = 20 nm. Adapted with permission from the American Chemical Society28.
Polysaccharides
Polysaccharides are polymeric carbohydrates obtained or inspired from a broad array of sources including animals, plants, and microorganisms29. The majority of polysaccharides are anionic with the exception of cationic chitosan30. While their chemical structure is not directly determined by the genetic code, they are biologically synthesized via well-coordinated biochemical pathways resulting from conserved polysaccharide synthases. Thermal responsiveness has most frequently been conferred to polysaccharides using synthetic polymers such as PNIPAM; however, gellan and xanthan gums, produced by microorganisms, are intrinsically thermally responsive30. This thermal responsiveness is derived from a conformational change in structure; as the temperature increases, the polysaccharide goes from ordered helix to disordered coil31.
Chitosan, which is generated by the partial deacetylation of chitin, is the second most abundant natural polysaccharide after cellulose32. It is found in bacterial and fungal cell walls as well as in exoskeletons of crustaceans and insects. Chitosan undergoes a thermally induced gelation based on the degree of deacetylation. The more deacetyled the chitosan chain, the lower the temperature needed for gelation. This parameter allows bioengineers to tune the formation of chitosan hydrogels to occur in the body. Temperature sensitivity has also been conferred to chitosan using thermally responsive molecules. For example, complexes of chitosan with ovalbumin create stable, self-assembled nanogels33. To form these gels, a mixture of chitosan and ovalbumin is complexed at pH 5.4, and the mixture is heated to 70 °C, past the denaturation temperature of ovalbumin. Once the solution reaches 70 °C, ovalbumin undergoes a conformational change from an α-helix to a mixed β-sheet and coil structure. Additionally, it is believed that temperatures above 70 °C induce both intermolecular hydrophobic association and the formation of disulfide bonds, which stabilize the chitosan-ovalbumin association. When optimized, this strategy results in monodisperse chitosan-ovalbumin nanogels with a 50 nm radius, as confirmed using TEM imaging. Alternatively, thermal sensitivity has been conferred to chitosan using PNIPAM and Pluronic34, 35.
Thermo-reversible assembly is commonly explored in hydrogel systems. Many of these are mediated by synthetic polymers such as PNIPAM, which confer lower critical solution temperature properties to a protein or peptide36. Extensively used in the food industry as a thickening and gelating agent, κ-carrageenan has recently gained prominence as a thermosensitive material for nanogel formulations37. Extracted from red seaweed, κ-carrageenan is a high molecular weight (200 to 800 kDa) polysaccharide made up of repeating galactose and 3,6 anhydrogalactose units. Above the gelation temperature of carrageenan, the carrageenan adopts a random coil conformation. Cooling below the gelation point causes the polysaccharide to undergo a conformational change into a double helix, which ultimately self assembles. Gelation temperature can be tuned by the concentration of polymer in the system. This gelation has been utilized to form 100 nm nanoparticles that can be induced to release drug above their gel to solution transition temperature, engineered to occur at physiological temperatures (37 -45 °C)38. κ-carrageenan has also been mixed with methylcellulose to create a three-phase system. While κ-carrageenan solubilizes when heated, methylcellulose undergoes gelation when heated. By varying the concentrations of κ-carrageenan and methylcellulose from 1-2 wt%, Tomsic and coworkers engineered the gel-sol-gel transition to occur in the physiologic range, from 30-60 °C39, paving the way for entrapment and release of therapeutic colloids.
In addition to carrageenan, polysaccharide-based hydrogels have recently been explored in the tissue engineering community using gellan gum. Gellan gum is derived from the fermentation of bacterium Sphingomonas elodea40. It is comprised of tetrasaccharide repeats of glucose, D-glucuronic acid, glucose, and rhamnose. At high temperatures, gellan gum polysaccharide forms random coils in solution with a low viscosity. Decreasing the temperature allows the gellan to cross link through formation of double helices, which are stabilized by the carboxylic acids on the glucuronic acid moieties. The carboxylic acid participates in internal hydrogen bonds that stabilize antiparallel double helices. When in an antiparallel double helix conformation, gellan gum's viscosity increases dramatically, forming a gel. The mechanical properties of the gel can be tuned by controlling the degree of acetylation. Highly acetylated gellan gum forms soft, elastic, pliable hydrogels. However, gellan gum that is non-acetylated forms more brittle hydrogels. Pereira and coworkers explored mixtures of gellan gum with two degrees of acetylation, which together can form microparticles40. Mixing highly acetylated gellan gum with non-acetylated gellan gum at a ratio of 1:1 resulted in microparticles 741 μm in diameter, larger than when either acetylated or non-acetylated gellan gum are mixed in a 3:1 ratio. Oliveira and coworkers utilized gellan gum's thermal responsiveness to form hydrogels in a broad range of geometrical forms (Figure 3)41. The gels were formed by heating to 90 °C, which disperses the polysaccharide. CaCl2 was added before slowly cooling the solution to 50 °C, at which point the solution was cast into cylindrical molds and cooled to room temperature. This formed a hydrogel, which was thermally reversible upon heating. Depending on the mold, the gel can maintain a variety of geometries ranging from 10 mm discs, membranes, and porous scaffolds.
Figure 3. Structural versatility of biomaterials generated from gellan gum polysaccharides.
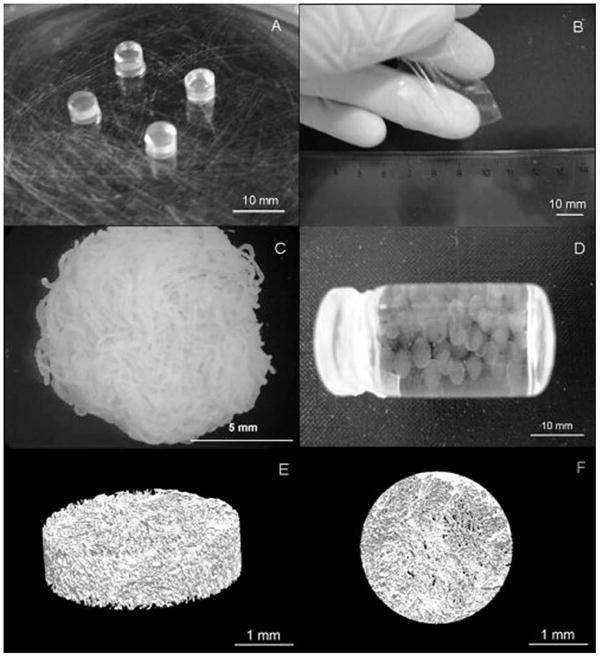
A) discs; B) membranes; C) fibers; D) microparticles; E) and F) 3D lyophilized scaffolds. Adapted with permission from John Wiley and Sons41.
Another example of a thermally responsive polysaccharide hydrogel comes from xanthan gum. Xanthan gum is a branched, high molecular weight exopolysaccaride produced by Xanthomonas campestris42. It is composed of D-glycosyl, D-mannosyl, and D-glucuronyl acid in 2:2:1 molar ratio along with variable amounts of O-acetyl and pyruvyl residues43. A thermal stimulus causes xanthan gum to undergo a conformational change in aqueous solution from an ordered helix, stabilized by non-covalent bonds, to disordered coil. This order-disorder transition is reversible and can be modulated to occur at a range of temperatures by altering the salt concentration and side chain substitution. Sereno and coworkers used xanthan gum to form particles through extrusion31. Based on microcalorimetry, when extruded below their order-disorder transition temperature these particles lose their helical structure and adopt more amorphous non-helical regions. This results in intermolecular cross-linking, which creates large interconnected networks. The extrusion process also aligns xanthan gum during particle formation. Similar to other polysaccharides, this cross-linking property of xanthan can be reversed by reheating the system past its order-disorder transition. Dropping the temperature back down, xanthan re-adopts its original helical conformation and stabilizes non-covalent bonds.
Proteins
Like polysaccharides and oligonucleotides, proteins display a range of thermally- responsive properties that are often strongly associated with their assembly of specific secondary structures. Most notably, the biological machinery responsible for protein translation can be used to generate high molecular weight (8 to 80 kDa) repetitive amino acid sequences called protein polymers. Protein polymers can be genetically engineered into a multitude of compositions by varying their encoding DNA sequence. By selection of the repetitive motif, the protein polymer can be designed to adopt specific secondary structures, which can be used to mediate assembly. Multiple classes of thermally responsive protein polymers are under exploration for material sciences applications, which include leucine zippers, collagen motifs, elastin-like polypeptides, resilin biomimetics, and protein polymers rationally designed de novo. While elastin-like polypeptides stabilize into ordered structures with the application of heat, leucine zippers and collagen motifs destabilize into smaller particles under increased temperatures44. Resilin mimetic polymers exhibit both of these properties: they assemble at low and high temperatures with a window from 6 -70 °C where they are soluble45, 46. Another physical property of protein polymers is the reversibility of this physical change. Elastin-like polypeptides, leucine-zippers, and resilin polymers undergo this conformational change reversibly, while collagen motifs undergo conformational changes irreversibly44, 46.
Leucine-Zippers and Collagen Motifs
Leucine zippers, a class of coiled coils, are structural motifs that function as transcription factor binding domains47. The name leucine zipper is derived from regularity of hydrophobic amino acids, such as leucine. Typically, leucine is found every seven residues on the peptide sequence, which allows for the formation of an amphipathic alpha helix with the leucine residues forming a hydrophobic region on one side47. This hydrophobic leucine region drives dimer formation between the two strands, creating a coiled coil of parallel alpha helices48. Leucine zippers reversibly unfold at elevated temperatures. Dimers dissociate when the polymer secondary structure changes from an alpha helix to a random coil. One example of a leucine-zipper derived polymer motif used in the formation of structures is a parallel coiled coil derived from the yeast transcription factor GCN449. This peptide takes two GCN4 sequences and separates them with 2 alanine inserts. The insertion produces a phase shift in the GCN4 repeats that creates two hydrophobic ridges 200° apart. The addition of cosolutes, or an increase in temperature causes the peptides to disassociate from large nanofibrils into smaller, more discreet nanoropes. At 4 °C, the molecular weight of the nanostructures is 1,800 kDa, but a shift in temperature up to 25 °C causes a decrease in polymer weight to 420 kDa. Atomic force microscopy imaging illustrates the formation of nanoropes from these peptides (Figure 4). The leucine zipper motif's ability to heterodimerize has recently been exploited to drive the localization of cytosolic proteins in E. coli50. Additionally, leucine zipper domains have been used to drive formation of hydrogels with mechanical properties strong enough to serve as scaffolds for tissue engineering applications51. Indicating the potential for crossover applications between different biomaterials, the DNA origami concept described above has recently been co-opted to fold an expressed polypeptide into a tetrahedron52. Gradišar and coworkers linked 6 distinct complementary coiled-coils domains together, one of which is based on the GCN4 sequence. The denatured polypeptides formed perfect nanostructures upon either dialysis and/or controlled cooling.
Figure 4. Peptide-based leucine zippers from GCN4 form nanoropes.
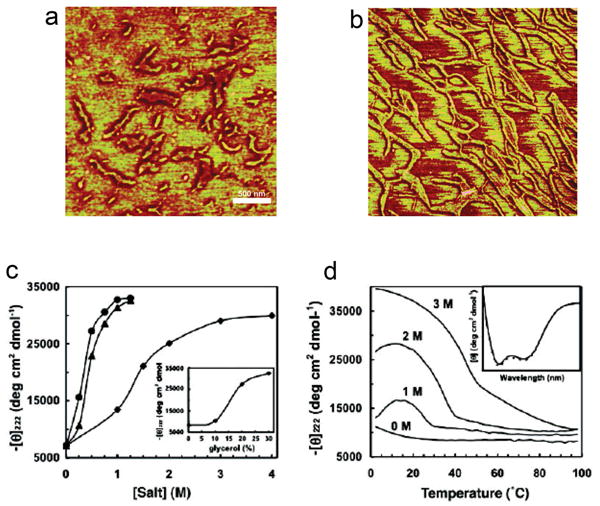
Nanorope formation is temperature and cosolute dependent. A) Phase image using tapping mode AFM of nanoropes formed in 1.5 M NaCl. The scale bar represents 500 nm and the z-scale represents 25 nm. B) Phase image of nanoropes formed in 0.75 M (NH4)2SO4 shows even longer structures than those in A). C,D) Circular dichroism was used to study the formation of alpha helices (-Θ222 nm for 144 μM peptide in 10 mM Tris, pH 8.0) as a function of C) cosolute concentration at 25 °C including Na2SO4 (●), (NH4) 2SO4 (▲), and NaCl (◆) and glycerol (inset graph) and D) temperature as a function of NaCl concentration, which shows that salt stabilizes the alpha helices until a higher temperature. Adapted with permission from PNAS49.
One of the most abundant proteins in the body, collagen has a unique tertiary structure: a right-handed triple helix made up of three helical peptide strands. Collagen is comprised of the amino acid sequence Xaa-Yaa-Gly with proline being the most abundant Xaa and L-hydroxyproline being the most abundant Yaa53. Multiple collagen-mimetic peptides have been described, however one of the best characterizations of a self-assembling collagen mimetic is done on (Pro-Lys-Gly)4(Pro-Hyp-Gly)4(Asp-Hyp-Gly)4, a collagen-mimetic polypeptide that replaces arginine and glutamate residues with lysine and aspartate54. This peptide adopts a triple helix secondary structure, which mediates assembly of nanofibers and a hydrogel. The assembly of this gel is temperature sensitive; furthermore, above 40 °C the collagen triple helix unfolds into a random coil and the hydrogel disassembles55.
Most of the collagen mimetic peptides are able to crosslink into larger hydrogels; however, recently collagen-mimetic peptides were tuned to assemble two-dimensional assemblies referred to as nanosheets56. Using 3 blocks with differing electrostatic properties, each block comprised of amino acid triads repeated 4 times, allowed Conticello and coworkers to uniaxially orient collagen- mimetic fibrils. By replacing the original positively charged arginine residue with the non-cannonical amino acid aminoproline, the resulting polypeptides form a collagen triple helical conformation with thermal stability up to 32 °C56. Altering the original collagen-mimetic formulation further, the central block of triads was lengthened from 4 to 7 repeats of (Pro-Hyp-Gly), which increased the thermal stability up to 60 °C. The resulting collagen-mimetic assembles into multi-layered nanosheets with well-defined morphology. This is the first collagen formulation to form two-dimensional structures without the need for a non-native structural interaction such as metal promoted crosslinking. Since self-assembling collagen motifs are derived from endogenous collagens, a number of applications relating to tissue regeneration using collagen motifs have emerged57.
Elastin- like polypeptides
Elastin-like polypeptides, derived from the human gene for tropoelastin, are explored in a variety of applications ranging from protein purification, drug delivery, to hydrogel formation, due in part to their temperature responsive properties58-60. At a critical temperature, termed an inverse transition temperature (Tt), ELPs undergo a conformational change from random coils to type II β-turn spirals; furthermore, this change in secondary structure precedes phase separation. The ELP transition temperature can be rationally tuned by varying the amino acid composition and polymer molecular weight. ELPs are comprised of the amino acid repeat Val-Pro-Gly-Xaa-Gly, where the hydrophobicity of the guest residue, Xaa, largely determines the transition temperature. For example, a hydrophilic amino acid such as alanine endows an ELP with a higher transition temperature than an equivalent length ELP with a more hydrophobic guest residue such as valine. Additionally, molecular weight has similar effects. An ELP with a molecular weight of 76 kDa will undergo a conformational change and phase separation at a temperature below that of an otherwise equivalent ELP with a molecular weight of 40 kDa61.
The ability of ELPs to abruptly form insoluble assemblies in response to heating has been exploited by our group to form intracellular microdomains. A plasmid encoding for an ELP that phase separates near a physiologically relevant temperature is first transfected into cultured cells62, 63. These cells produce ELPs or ELP fusion proteins in their cytosol. In one example, cells transfected with a 28 kDa ELP with valine guest residues, undergo intracellular phase separation at 33 °C. This conformational change results in the formation of genetically engineered protein microdomains (GEPMs), which can be visualized in living cells by attachment to fluorescent proteins (Figure 5a). Microdomain formation is reversible with a decrease in temperature. Alternatively, by attaching an ELP to a functional protein, such as the clathrin light chain, microdomains associate with a target protein complex sequestered inside. In one example64, these microdomains sequester clathrin heavy chain, which functionally knocks-down clathrin-mediated endocytosis (Figure 5b,c). This strategy results in a rapid, reversible blockage of target intracellular pathways, and may have broader applications to processes ranging from cellular trafficking to signaling pathways.
Figure 5. Intracellular assembly and function of elastin-like polypeptide microdomains.
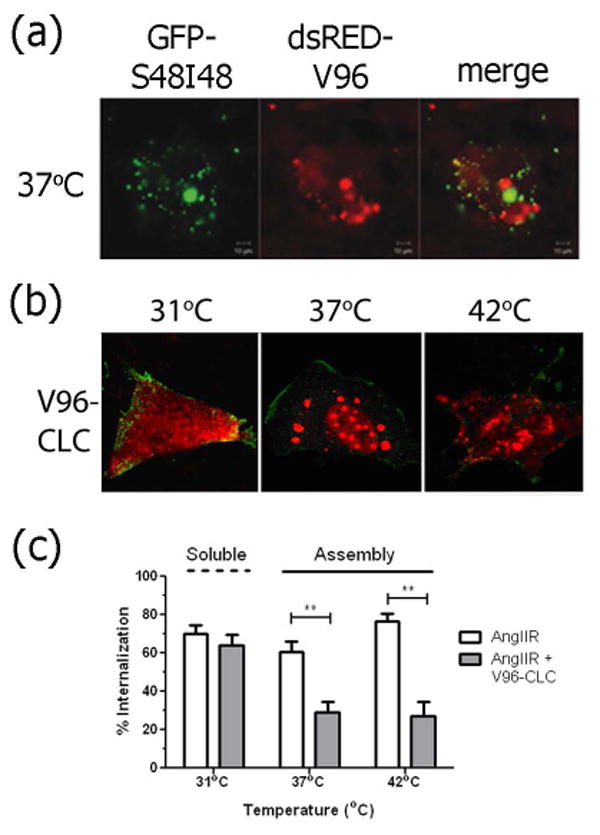
A) An amphiphilic ELP (GFP-S48I48) and an ELP monoblock (dsRED-V96) with similar transition temperatures and molecular weight sort into separate microdomains in live cells. B) An ELP fused with the clathrin-light chain (V96-CLC) is soluble at 31 °C but assembles into V96-CLC microdomains above 37 °C. Red = ELP, Green = the angiotensin II receptor (AngIIR) at the cellular membrane. C) V96-CLC microdomains sequester the machinery of clathrin-mediated endocytosis and inhibit the internalization of AngIIR at 37 and 42 °C. The V96-CLC fusion remains soluble at 31 °C and does not affect receptor internalization. (**p<0.0001) Mean ± 95% confidence interval (n=3). Adapted with permission from Wiley–VCH Verlag GmbH & Co 63, 64.
While the thermally-mediated assembly of ELP microdomains appears to have potential applications, genetic engineering with these polymers also allows for the construction of amphiphilic block copolymers capable of assembling nanoparticles65. Ligating in tandem two ELPs of varying hydrophobicity results in the formation of stable protein nanoparticles. The ELP with the more hydrophobic amino acids undergoes a conformational change at a temperature below that of the more hydrophilic ELP block. Phase separation of the more hydrophobic block mediates the assembly of a nanoparticle core, with the soluble hydrophilic ELP block forming the corona. Interestingly, the critical micelle temperature for ELP diblock copolymers depends mostly upon the properties of the more hydrophobic ELP; furthermore, this can be predicted by a quantitative model61. This scheme has been used to load ELP micelles with therapeutics for drug delivery applications66, 67. Of potential utility in the manipulation of intracellular ELP microdomains, these nanoparticles (GFP-S48I48) appear to sort from ELP monoblocks (dsRED-V96) co-expressed in the same cell (Figure 5a).
In other reports, ELPs have been incorporated into hybrid systems along with silk-like polypeptide (SLP) domains to confer thermal responsiveness and decrease crystallinity of SLP domains10. Like ELPs, SLPs are composed of repetitive sequences of amino acids, namely glycine and alanine. The most commonly used SLP motif is Gly-Ala-Gly-Ala-Gly-Ser, derived from the silkworm Bombix Mori68. Alone, GAGAGS domains assemble into insoluble, β-sheets, making them difficult to use in aqueous environments. Incorporating blocks of ELP allows for increased solubility of the silk-elastin-like polypeptide (SELP).
While SELPs have been explored for gelation upon tumor injection69, the Kaplan group has recently engineered SELP micelles for systemic administration70. The SELP nanoparticles described by Xia and coworkers undergo a two-step thermal transition71. Above 20 °C, but below 40 °C, SELP polymers with a 1:8 silk to elastin ratio and 55.7 kDa spontaneously form spherical structures with a 60 nm radius. Soluble ELP domains are expected to localize at the corona of the micelle with silk domains stabilizing the core with intermolecular hydrogen bonding. The second transition occurs above 60 °C when interactions between SELP particles result in larger coacervate formations with a 241 nm radius. While this assembly is reversible, it is interesting to note that upon cooling to 20 °C, SELP morphology varies based on the silk to elastin ratio. For example, after cooling a 1:8 ratio silk to elastin polymer returns to solution as a monomer. However, a 1:4 silk to elastin ratio, upon cooling adopts a worm-like nanostructure composed of small spherical particles.
Resilin
While elastin-like polypeptides are derived from the repetitive region of the mammalian gene tropoelastin, the resilin-mimetic polymer rec1-resilin is derived from the repeat sequence found in the Drosophila melanogaster CG15920 gene46. Resilin is highly elastic. For example, Drosophila melanogaster only expresses resilin during its pupa stage, yet it remains functional throughout the insect's lifetime46. Rec1-resilin is composed of 18 copies of the 15-residue repeat sequence Gly-Gly-Arg-Pro-Ser-Asp-Ser-Tyr-Gly-Ala-Pro-Gly-Gly-Gly-Asn72. Like an ELP, this sequence is rich in Gly and Pro residues; however, in contrast to an ELP it lacks aliphatic residues with bulky side chains. As a stimulus responsive biopolymer, rec1-resilin is of interest because it possesses not only lower critical solution temperature (LCST) behavior observed for ELPs, but also an upper critical solution temperature (UCST). Additionally, rec1-resilin maintains a self-assembled form both above and below both critical solution temperatures. Below its UCST at 6 °C, recl-resilin is turbid in solution. Cryo-TEM images show rec1-resilin assembles a high-density network of spherical particles approximately 5.4 nm in diameter (Figure 6). As the temperature is increased above the UCST, the rec1-resilin solution becomes transparent. Cryo-TEM images show discreet nanoparticles 9.5 nm in diameter, and DLS confirms this by characterizing rec1-resilin as having a Dh of 11 nm with low polydispersity in the 15 to 70 °C temperature span. Above rec1-resilin's 70 °C LCST, rec1-resilin forms larger discreet spherical nanoparticles between 100 and 130 nm in diameter. Since rec1-resilin is a 28.5 kDa protein, the diameters observed between the UCST and LCST using DLS and cryo-TEM are consistent with a population with a low aggregation number.
Figure 6. Resilin: A genetically engineered protein responsive to multiple stimuli.

Cryo-TEM micrographs of rec1-resilin for solutions of A) Vitrified rec1-resilin at 20 °C has only dispersed spherical particles approximately 9.5 nm in diameter with poor contrast, which is consistent with soluble proteins. B) Vitrified rec1-resilin just below UCST (4 °C), shows a high-density network of well-dispersed interconnected spherical particles with 5.4 nm diameters and excellent contrast. C) Above LCST, large discrete spherical aggregates with a size range of 100 nm to 130 nm diameters form. D) Increasing solution concentration to 10 mg ml-1 causes the formation of an interconnected gel particle network at the UCST. Adapted with permission from John Wiley and Sons72.
De Novo Protein Polymers
In addition to naturally occurring or biomimetic polymers such as ELPs and leucine zipper domains, a number of stimulus responsive polypeptides have been rationally designed de novo. One such polypeptide, 17-H-6, is an alanine-rich helical polypeptide. Amino acids like alanine, glutamine, and glutamic acid are commonly used in de novo polypeptide designs because of their high helical propensity73. The 17-H-6 alanine-rich polypeptide is comprised of Ala-Ala-Ala-Gln-Glu-Ala-Ala-Ala-Ala-Gln-Ala-Ala-Ala-Gln-Ala-Glu-Ala-Ala-Gln-Ala-Ala-Gln. At acidic pH and low temperature, 17-H-6 adopts an alpha-helical, folded conformation, which was confirmed using circular dichroism. This results in a globular nanostructure between 10 and 20 nm in diameter. The increase in pH to 7.4 dissociates the polypeptide. However, while maintaining an acidic pH yet increasing the temperature to 80 °C for 18 hr, the polypeptide dissociated and unfolds before undergoing a conformational change to β-sheet 2 stabilized fibrils 5-10 nm in diameter74. While the conformational change induced by a low temperature and increase in pH is reversible, the temperature mediated conformation change into nanofibrils is not.
Another example of thermally responsive fibrillar structures is the 687 amino acid polypeptide pioneered by the Welch group, termed YEHK. The YEHK polypeptide contains the repeating units (Gly-Ala)3Gly-Tyr(Gly-Ala)3Gly-Glu(Gly-Ala)3Gly-His(Gly-Ala)3Gly-Lys. The GA residues drive the antiparallel β sheet formation with YEHK residues designed to induce turns. At room temperature, the polypeptide forms face to face antiparallel β-sheets, stabilized by internal hydrogen bonds, that result in a fibrillar structure 15 nm in width and 10 to 1000 nm in length. At 90 °C, the polypeptide reversibly denatures and adopts a random coil conformation75, 76. YEHK's ability to reversibly unfold and refold due to temperature stimuli makes it an excellent model to study protein-folding mechanisms of globular β-sheet proteins.
Future directions
Biopolymers composed of carbohydrates, nucleic acids, and amino acids have excellent potential to impact a wide variety of biomedical applications, which have been mostly dominated by synthetic materials. As biopolymers are derived from biological sources, they can utilize endogenous biodegradation pathways, often be encoded at the genetic level, and provide a level of nanoscale control found only in biology. The biopolymers described above are being explored to construct hydrogels for tissue engineering and nanoreactors for biomanufacturing. Alternatively, biopolymers are being explored in drug delivery to confer spatial control over targeting moieties and to trigger drug encapsulation and release. In these applications, there remain fundamental limitations to current technology. For example, in drug delivery the goal of creating a safe/responsive/targeted carrier remains elusive. Akin to a viral particle, it is likely that multiple or even many distinct functionalities will need to be engineered onto a nanostructure on the order of 10-100 nm to realize this goal. Biopolymers offer unique opportunities to utilize the peptide, carbohydrate, and nucleotide polymerization machinery to pattern functional macromolecules in the most optimal shape and orientation at the nanoscale. Since these biopolymers can all be generated in a cell, they can conceivably be fused into hybrid systems during biosynthesis. When our community understands synthetic biology well enough, it may become feasible to create true nanomachines that run assays on their environment, move with purpose toward targets, carry out programmed functions, and report their findings. Traditional top-down approaches such as photolithography have been extraordinarily powerful; however, it is unlikely that they will ever be able to pattern multiple mechanical and electronic functions into particles smaller than 100 nm. Current bottom-up approaches based on lipid and polymeric amphiphiles are also powerful; however, neither approach has scratched the surface of what has been achieved through eons of biological evolution. We contend that the biopolymers presented herein are the tools with which the next generation of multifunctional nanomachines can be engineered.
To realize this futuristic goal, substantial work will be required to explore the properties of these biopolymers. In particular, better control over their expression, assembly, tunability, and safety will be needed to engineer these biopolymers to overcome current limitations. For instance, the number of independent functions that can be engineered into a single expressed biopolymer have yet to be determined. More amenable to multifunctionality, DNA and proteins can be directly engineered with high fidelity using chemically synthesized genes. In contrast, modification of carbohydrate may be more challenging to control within an organism. The abundance of glycoproteins in nature suggests there may be advantages to developing hybrids between these biopolymers. Despite this, methods to coassemble distinct peptides, oligonucleotides, and carbohydrates together into a hybrid system remain largely unexplored. To design nanomachines that process environmental information, such as temperature or the presence of biological analytes, a much tighter control over the factors that determine their tunability/responsiveness will be required. Lastly, as with any biomaterial, they must be engineered to be biocompatible from the immunologic, mutagenic, teratogenic, and cytotoxic perspective. A deeper understanding of biopolymer properties and their interactions with biological systems is needed to facilitate future achievements.
Conclusions
Creating structures at the nanoscale requires self-assembling materials whose monomeric composition dictates the ultimate multimolecular structure. Current technologies for manufacturing biologic materials offer unprecedented control over the composition of these monomeric units; furthermore, there now appear to be opportunities to directly assemble these nanostructures within the living cell. Future advances in biopolymer engineering are expected to further propel the field of molecular self-assembly into multifunctional and biologically relevant nanomaterials.
Acknowledgments
This work was made possible by the University of Southern California, the National Institute of Health R21EB012281 to J.A.M., and the American Foundation for Pharmaceutical Education Predoctoral Fellowship and the Grayson and Judith Manning Predoctoral Fellowship to M.K.P.
References
- 1.Shi P, Gustafson JA, MacKay JA. Genetically engineered nanocarriers for drug delivery. Int J Nanomedicine. 2014;9:1617–1626. doi: 10.2147/IJN.S53886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aluri S, Janib SM, Mackay JA. Environmentally responsive peptides as anticancer drug carriers. Adv Drug Deliv Rev. 2009;61:940–952. doi: 10.1016/j.addr.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Janib SM, Moses AS, MacKay JA. Imaging and drug delivery using theranostic nanoparticles. Adv Drug Deliv Rev. 2010;62:1052–1063. doi: 10.1016/j.addr.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mi P, Kokuryo D, Cabral H, Kumagai M, Nomoto T, Aoki I, Terada Y, Kishimura A, Nishiyama N, Kataoka K. Hydrothermally synthesized PEGylated calcium phosphate nanoparticles incorporating Gd-DTPA for contrast enhanced MRI diagnosis of solid tumors. J Control Release. 2014;174:63–71. doi: 10.1016/j.jconrel.2013.10.038. [DOI] [PubMed] [Google Scholar]
- 5.Wu C, Han D, Chen T, Peng L, Zhu G, You M, Qiu L, Sefah K, Zhang X, Tan W. Building a multifunctional aptamer-based DNA nanoassembly for targeted cancer therapy. J Am Chem Soc. 2013;135:18644–18650. doi: 10.1021/ja4094617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fendler JH. Chemical self-assembly for electronic applications. Chemistry of Materials. 2001;13:3196–3210. [Google Scholar]
- 7.Zhao H, Guo X, He S, Zeng X, Zhou X, Zhang C, Hu J, Wu X, Xing Z, Chu L, et al. Complex self-assembly of pyrimido[4,5-d]pyrimidine nucleoside supramolecular structures. Nat Commun. 2014;5:3108. doi: 10.1038/ncomms4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Urry DW, Parker TM, Reid MC, Gowda DC. Biocompatibility of the Bioelastic Materials, Poly(Gvgvp) and Its Gamma-Irradiation Cross-Linked Matrix - Summary of Generic Biological Test-Results. Journal of Bioactive and Compatible Polymers. 1991;6:263–282. [Google Scholar]
- 9.Shah M, Hsueh PY, Sun G, Chang HY, Janib SM, MacKay JA. Biodegradation of elastin-like polypeptide nanoparticles. Protein Sci. 2012;21:743–750. doi: 10.1002/pro.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cappello J, Crissman J, Dorman M, Mikolajczak M, Textor G, Marquet M, Ferrari F. Genetic engineering of structural protein polymers. Biotechnol Prog. 1990;6:198–202. doi: 10.1021/bp00003a006. [DOI] [PubMed] [Google Scholar]
- 11.Mackay JA, Chilkoti A. Temperature sensitive peptides: engineering hyperthermia-directed therapeutics. International journal of hyperthermia : the official journal of European Society for Hyperthermic Oncology, North American Hyperthermia Group. 2008;24:483–495. doi: 10.1080/02656730802149570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sierra-Martin B, Retama JR, Laurenti M, Fernandez Barbero A, Lopez Cabarcos E. Structure and polymer dynamics within PNIPAM-based microgel particles. Adv Colloid Interface Sci. 2014;205C:113–123. doi: 10.1016/j.cis.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 13.Roy D, Brooks WL, Sumerlin BS. New directions in thermoresponsive polymers. Chem Soc Rev. 2013;42:7214–7243. doi: 10.1039/c3cs35499g. [DOI] [PubMed] [Google Scholar]
- 14.Xu F, Yan TT, Luo YL. Studies on micellization behavior of thermosensitive PNIPAAm-b-PLA amphiphilic block copolymers. J Nanosci Nanotechnol. 2012;12:2287–2291. doi: 10.1166/jnn.2012.5705. [DOI] [PubMed] [Google Scholar]
- 15.Doherty NM. In vitro evaluation of resin-retained extracoronal precision attachments. Int J Prosthodont. 1991;4:63–69. [PubMed] [Google Scholar]
- 16.Motornov M, Roiter Y, Tokarev I, Minko S. Stimuli-responsive nanoparticles, nanogels and capsules for integrated multifunctional intelligent systems. Progress in Polymer Science. 2010;35:174–211. [Google Scholar]
- 17.Saiki RK, Bugawan TL, Horn GT, Mullis KB, Erlich HA. Analysis of enzymatically amplified beta-globin and HLA-DQ alpha DNA with allele-specific oligonucleotide probes. Nature. 1986;324:163–166. doi: 10.1038/324163a0. [DOI] [PubMed] [Google Scholar]
- 18.Dohno C, Nakatani K. Control of DNA hybridization by photoswitchable molecular glue. Chem Soc Rev. 2011;40:5718–5729. doi: 10.1039/c1cs15062f. [DOI] [PubMed] [Google Scholar]
- 19.Hughes RA, Miklos AE, Ellington AD. Gene synthesis: methods and applications. Methods Enzymol. 2011;498:277–309. doi: 10.1016/B978-0-12-385120-8.00012-7. [DOI] [PubMed] [Google Scholar]
- 20.Rothemund PW. Folding DNA to create nanoscale shapes and patterns. Nature. 2006;440:297–302. doi: 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- 21.Winfree E, Liu F, Wenzler LA, Seeman NC. Design and self-assembly of two-dimensional DNA crystals. Nature. 1998;394:539–544. doi: 10.1038/28998. [DOI] [PubMed] [Google Scholar]
- 22.He Y, Ye T, Su M, Zhang C, Ribbe AE, Jiang W, Mao C. Hierarchical self-assembly of DNA into symmetric supramolecular polyhedra. Nature. 2008;452:198–201. doi: 10.1038/nature06597. [DOI] [PubMed] [Google Scholar]
- 23.Sacca B, Meyer R, Niemeyer CM. Temperature-dependent FRET spectroscopy for the high-throughput analysis of self-assembled DNA nanostructures in real time. Nat Protoc. 2009;4:271–285. doi: 10.1038/nprot.2008.220. [DOI] [PubMed] [Google Scholar]
- 24.Iinuma R, Ke Y, Jungmann R, Schlichthaerle T, Woehrstein JB, Yin P. Polyhedra Self-Assembled from DNA Tripods and Characterized with 3D DNA-PAINT. Science. 2014 doi: 10.1126/science.1250944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qi H, Ghodousi M, Du Y, Grun C, Bae H, Yin P, Khademhosseini A. DNA-directed self-assembly of shape-controlled hydrogels. Nat Commun. 2013;4:2275. doi: 10.1038/ncomms3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schopf E, Chen Y. Attomole DNA detection assay via rolling circle amplification and single molecule detection. Anal Biochem. 2010;397:115–117. doi: 10.1016/j.ab.2009.09.023. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Q, Jiang Q, Li N, Dai LR, Liu Q, Song LL, Wang JY, Li YQ, Tian J, Ding BQ, et al. DNA Origami as an In Vivo Drug Delivery Vehicle for Cancer Therapy. Acs Nano. 2014;8:6633–6643. doi: 10.1021/nn502058j. [DOI] [PubMed] [Google Scholar]
- 28.Zhou L, Marras AE, Su HJ, Castro CE. DNA origami compliant nanostructures with tunable mechanical properties. ACS Nano. 2014;8:27–34. doi: 10.1021/nn405408g. [DOI] [PubMed] [Google Scholar]
- 29.Sutherland IW. Polysaccharides from microorganisms, plants and animals. Biopolymers Online. 2005 [Google Scholar]
- 30.Alvarez-Lorenzo C, Blanco-Fernandez B, Puga AM, Concheiro A. Crosslinked ionic polysaccharides for stimuli-sensitive drug delivery. Adv Drug Deliv Rev. 2013;65:1148–1171. doi: 10.1016/j.addr.2013.04.016. [DOI] [PubMed] [Google Scholar]
- 31.Sereno NM, Hill SE, Mitchell JR. Impact of the extrusion process on xanthan gum behaviour. Carbohydr Res. 2007;342:1333–1342. doi: 10.1016/j.carres.2007.03.023. [DOI] [PubMed] [Google Scholar]
- 32.Zohuriaan MJ, Shokrolahi F. Thermal studies on natural and modified gums. Polymer Testing. 2004;23:575–579. [Google Scholar]
- 33.Yu S, Hu J, Pan X, Yao P, Jiang M. Stable and pH-sensitive nanogels prepared by self-assembly of chitosan and ovalbumin. Langmuir. 2006;22:2754–2759. doi: 10.1021/la053158b. [DOI] [PubMed] [Google Scholar]
- 34.Chen JP, Cheng TH. Thermo-responsive chitosan-graft-poly(N-isopropylacrylamide) injectable hydrogel for cultivation of chondrocytes and meniscus cells. Macromol Biosci. 2006;6:1026–1039. doi: 10.1002/mabi.200600142. [DOI] [PubMed] [Google Scholar]
- 35.Park KM, Lee SY, Joung YK, Na JS, Lee MC, Park KD. Thermosensitive chitosan-Pluronic hydrogel as an injectable cell delivery carrier for cartilage regeneration. Acta Biomater. 2009;5:1956–1965. doi: 10.1016/j.actbio.2009.01.040. [DOI] [PubMed] [Google Scholar]
- 36.Zhang X, Wu D, Chu CC. Synthesis and characterization of partially biodegradable, temperature and pH sensitive Dex-MA/PNIPAAm hydrogels. Biomaterials. 2004;25:4719–4730. doi: 10.1016/j.biomaterials.2003.11.040. [DOI] [PubMed] [Google Scholar]
- 37.Michel AS, Mestdagh MM, Axelos MAV. Physico-chemical properties of carrageenan gels in presence of various cations. International Journal of Biological Macromolecules. 1997;21:195–200. doi: 10.1016/s0141-8130(97)00061-5. [DOI] [PubMed] [Google Scholar]
- 38.Daniel-da-Silva AL, Ferreira L, Gil AM, Trindade T. Synthesis and swelling behavior of temperature responsive kappa-carrageenan nanogels. J Colloid Interface Sci. 2011;355:512–517. doi: 10.1016/j.jcis.2010.12.071. [DOI] [PubMed] [Google Scholar]
- 39.Tomsic M, Prossnigg F, Glatter O. A thermoreversible double gel: characterization of a methylcellulose and kappa-carrageenan mixed system in water by SAXS, DSC and rheology. J Colloid Interface Sci. 2008;322:41–50. doi: 10.1016/j.jcis.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 40.Pereira DR, Silva-Correia J, Caridade SG, Oliveira JT, Sousa RA, Salgado AJ, Oliveira JM, Mano JF, Sousa N, Reis RL. Development of gellan gum-based microparticles/hydrogel matrices for application in the intervertebral disc regeneration. Tissue Eng Part C Methods. 2011;17:961–972. doi: 10.1089/ten.TEC.2011.0115. [DOI] [PubMed] [Google Scholar]
- 41.Oliveira JT, Martins L, Picciochi R, Malafaya PB, Sousa RA, Neves NM, Mano JF, Reis RL. Gellan gum: a new biomaterial for cartilage tissue engineering applications. J Biomed Mater Res A. 2010;93:852–863. doi: 10.1002/jbm.a.32574. [DOI] [PubMed] [Google Scholar]
- 42.Norton IT, Goodall DM, Frangou SA, Morris ER, Rees DA. Mechanism and dynamics of conformational ordering in xanthan polysaccharide. J Mol Biol. 1984;175:371–394. doi: 10.1016/0022-2836(84)90354-1. [DOI] [PubMed] [Google Scholar]
- 43.Bueno VB, Petri DF. Xanthan hydrogel films: molecular conformation, charge density and protein carriers. Carbohydr Polym. 2014;101:897–904. doi: 10.1016/j.carbpol.2013.10.039. [DOI] [PubMed] [Google Scholar]
- 44.Mackay JA, Chilkoti A. Temperature sensitive peptides: engineering hyperthermia-directed therapeutics. Int J Hyperthermia. 2008;24:483–495. doi: 10.1080/02656730802149570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lyons RE, Nairn KM, Huson MG, Kim M, Dumsday G, Elvin CM. Comparisons of recombinant resilin-like proteins: repetitive domains are sufficient to confer resilin-like properties. Biomacromolecules. 2009;10:3009–3014. doi: 10.1021/bm900601h. [DOI] [PubMed] [Google Scholar]
- 46.Elvin CM, Carr AG, Huson MG, Maxwell JM, Pearson RD, Vuocolo T, Liyou NE, Wong DC, Merritt DJ, Dixon NE. Synthesis and properties of crosslinked recombinant pro-resilin. Nature. 2005;437:999–1002. doi: 10.1038/nature04085. [DOI] [PubMed] [Google Scholar]
- 47.Landschulz WH, Johnson PF, McKnight SL. The leucine zipper: a hypothetical structure common to a new class of DNA binding proteins. Science. 1988;240:1759–1764. doi: 10.1126/science.3289117. [DOI] [PubMed] [Google Scholar]
- 48.Weiss MA. Thermal unfolding studies of a leucine zipper domain and its specific DNA complex: implications for scissor's grip recognition. Biochemistry. 1990;29:8020–8024. doi: 10.1021/bi00487a004. [DOI] [PubMed] [Google Scholar]
- 49.Wagner DE, Phillips CL, Ali WM, Nybakken GE, Crawford ED, Schwab AD, Smith WF, Fairman R. Toward the development of peptide nanofilaments and nanoropes as smart materials. Proc Natl Acad Sci U S A. 2005;102:12656–12661. doi: 10.1073/pnas.0505871102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Choi SL, Lee SJ, Yeom SJ, Kim HJ, Rhee YH, Jung HC, Lee SG. Controlled localization of functionally active proteins to inclusion bodies using leucine zippers. PLoS One. 2014;9:e97093. doi: 10.1371/journal.pone.0097093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang CC, Ravindran S, Yin Z, George A. 3-D self-assembling leucine zipper hydrogel with tunable properties for tissue engineering. Biomaterials. 2014;35:5316–5326. doi: 10.1016/j.biomaterials.2014.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gradisar H, Bozic S, Doles T, Vengust D, Hafner-Bratkovic I, Mertelj A, Webb B, Sali A, Klavzar S, Jerala R. Design of a single-chain polypeptide tetrahedron assembled from coiled-coil segments. Nat Chem Biol. 2013;9:362–366. doi: 10.1038/nchembio.1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gottlieb D, Morin SA, Jin S, Raines RT. Self-Assembled Collagen-like Peptide Fibers as Templates for Metallic Nanowires. J Mater Chem. 2008;18:3865. doi: 10.1039/b807150k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O'Leary LE, Fallas JA, Bakota EL, Kang MK, Hartgerink JD. Multi-hierarchical self-assembly of a collagen mimetic peptide from triple helix to nanofibre and hydrogel. Nat Chem. 2011;3:821–828. doi: 10.1038/nchem.1123. [DOI] [PubMed] [Google Scholar]
- 55.Morris NP, Watt SL, Davis JM, Bachinger HP. Unfolding intermediates in the triple helix to coil transition of bovine type XI collagen and human type V collagens alpha 1(2) alpha 2 and alpha 1 alpha 2 alpha 3. J Biol Chem. 1990;265:10081–10087. [PubMed] [Google Scholar]
- 56.Jiang T, Xu C, Liu Y, Liu Z, Wall JS, Zuo X, Lian T, Salaita K, Ni C, Pochan DJ, et al. Structurally Defined Nano-scale Sheets from Self-Assembly of Collagen-Mimetic Peptides. J Am Chem Soc. 2014 doi: 10.1021/ja412867z. [DOI] [PubMed] [Google Scholar]
- 57.Luo J, Tong YW. Self-assembly of collagen-mimetic peptide amphiphiles into biofunctional nanofiber. ACS Nano. 2011;5:7739–7747. doi: 10.1021/nn202822f. [DOI] [PubMed] [Google Scholar]
- 58.Bellucci JJ, Amiram M, Bhattacharyya J, McCafferty D, Chilkoti A. Three-in-one chromatography-free purification, tag removal, and site-specific modification of recombinant fusion proteins using sortase A and elastin-like polypeptides. Angew Chem Int Ed Engl. 2013;52:3703–3708. doi: 10.1002/anie.201208292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aluri SR, Shi P, Gustafson JA, Wang W, Lin YA, Cui H, Liu S, Conti PS, Li Z, Hu P, et al. A hybrid protein-polymer nanoworm potentiates apoptosis better than a monoclonal antibody. ACS Nano. 2014;8:2064–2076. doi: 10.1021/nn403973g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Asai D, Xu D, Liu W, Garcia Quiroz F, Callahan DJ, Zalutsky MR, Craig SL, Chilkoti A. Protein polymer hydrogels by in situ, rapid and reversible self-gelation. Biomaterials. 2012;33:5451–5458. doi: 10.1016/j.biomaterials.2012.03.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Janib SM, Pastuszka M, Aluri S, Folchman-Wagner Z, Hsueh PY, Shi P, Yi A, Cui H, Mackay JA. A quantitative recipe for engineering protein polymer nanoparticles. Polym Chem. 2014;5:1614–1625. doi: 10.1039/C3PY00537B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pastuszka MK, Janib SM, Weitzhandler I, Okamoto CT, Hamm-Alvarez S, Mackay JA. A tunable and reversible platform for the intracellular formation of genetically engineered protein microdomains. Biomacromolecules. 2012;13:3439–3444. doi: 10.1021/bm301090x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shi P, Lin YA, Pastuszka M, Cui H, Mackay JA. Triggered Sorting and Co-Assembly of Genetically Engineered Protein Microdomains in the Cytoplasm. Adv Mater. 2013 doi: 10.1002/adma.201303356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pastuszka MK, Okamoto CT, Hamm-Alvarez SF, MacKay JA. Flipping the Switch on Clathrin-Mediated Endocytosis using Thermally Responsive Protein Microdomains. Advanced Functional Materials. 2014;24:5340–5347. doi: 10.1002/adfm.201400715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dreher MR, Simnick AJ, Fischer K, Smith RJ, Patel A, Schmidt M, Chilkoti A. Temperature triggered self-assembly of polypeptides into multivalent spherical micelles. J Am Chem Soc. 2008;130:687–694. doi: 10.1021/ja0764862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shi P, Aluri S, Lin YA, Shah M, Edman M, Dhandhukia J, Cui H, MacKay JA. Elastin-based protein polymer nanoparticles carrying drug at both corona and core suppress tumor growth in vivo. J Control Release. 2013;171:330–338. doi: 10.1016/j.jconrel.2013.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shah M, Edman MC, Janga SR, Shi P, Dhandhukia J, Liu S, Louie SG, Rodgers K, Mackay JA, Hamm-Alvarez SF. A rapamycin-binding protein polymer nanoparticle shows potent therapeutic activity in suppressing autoimmune dacryoadenitis in a mouse model of Sjogren's syndrome. J Control Release. 2013;171:269–279. doi: 10.1016/j.jconrel.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chow D, Nunalee ML, Lim DW, Simnick AJ, Chilkoti A. Peptide-based Biopolymers in Biomedicine and Biotechnology. Mater Sci Eng R Rep. 2008;62:125–155. doi: 10.1016/j.mser.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Greish K, Araki K, Li D, O'Malley BW, Jr, Dandu R, Frandsen J, Cappello J, Ghandehari H. Silk-elastinlike protein polymer hydrogels for localized adenoviral gene therapy of head and neck tumors. Biomacromolecules. 2009;10:2183–2188. doi: 10.1021/bm900356j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xia XX, Wang M, Lin Y, Xu Q, Kaplan DL. Hydrophobic drug-triggered self-assembly of nanoparticles from silk-elastin-like protein polymers for drug delivery. Biomacromolecules. 2014;15:908–914. doi: 10.1021/bm4017594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xia XX, Xu Q, Hu X, Qin G, Kaplan DL. Tunable self-assembly of genetically engineered silk--elastin-like protein polymers. Biomacromolecules. 2011;12:3844–3850. doi: 10.1021/bm201165h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dutta NK, Truong MY, Mayavan S, Choudhury NR, Elvin CM, Kim M, Knott R, Nairn KM, Hill AJ. A genetically engineered protein responsive to multiple stimuli. Angew Chem Int Ed Engl. 2011;50:4428–4431. doi: 10.1002/anie.201007920. [DOI] [PubMed] [Google Scholar]
- 73.Pace CN, Scholtz JM. A helix propensity scale based on experimental studies of peptides and proteins. Biophys J. 1998;75:422–427. doi: 10.1016/s0006-3495(98)77529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Top A, Kiick KL, Roberts CJ. Modulation of self-association and subsequent fibril formation in an alanine-rich helical polypeptide. Biomacromolecules. 2008;9:1595–1603. doi: 10.1021/bm800056r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Topilina NI, Higashiya S, Rana N, Ermolenkov VV, Kossow C, Carlsen A, Ngo SC, Wells CC, Eisenbraun ET, Dunn KA, et al. Bilayer fibril formation by genetically engineered polypeptides: preparation and characterization. Biomacromolecules. 2006;7:1104–1111. doi: 10.1021/bm0509016. [DOI] [PubMed] [Google Scholar]
- 76.Lednev IK, Ermolenkov VV, Higashiya S, Popova LA, Topilina NI, Welch JT. Reversible thermal denaturation of a 60-kDa genetically engineered beta-sheet polypeptide. Biophys J. 2006;91:3805–3818. doi: 10.1529/biophysj.106.082792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ryadnov MG. A self-assembling peptide polynanoreactor. Angew Chem Int Ed Engl. 2007;46:969–972. doi: 10.1002/anie.200604014. [DOI] [PubMed] [Google Scholar]
- 78.Raman S, Machaidze G, Lustig A, Aebi U, Burkhard P. Structure-based design of peptides that self-assemble into regular polyhedral nanoparticles. Nanomedicine. 2006;2:95–102. doi: 10.1016/j.nano.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 79.Aluri S, Pastuszka MK, Moses AS, MacKay JA. Elastin-like peptide amphiphiles form nanofibers with tunable length. Biomacromolecules. 2012;13:2645–2654. doi: 10.1021/bm300472y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cappello J, Crissman J, Dorman M, Mikolajczak M, Textor G, Marquet M, Ferrari F. Genetic-Engineering of Structural Protein Polymers. Biotechnology Progress. 1990;6:198–202. doi: 10.1021/bp00003a006. [DOI] [PubMed] [Google Scholar]
- 81.Ke Y, Douglas SM, Liu M, Sharma J, Cheng A, Leung A, Liu Y, Shih WM, Yan H. Multilayer DNA origami packed on a square lattice. J Am Chem Soc. 2009;131:15903–15908. doi: 10.1021/ja906381y. [DOI] [PMC free article] [PubMed] [Google Scholar]