

Abstract
Numerous studies have suggested that ketamine administration can induce neuroapoptosis in primary cultured cortical neurons. Neurosteroids modulate neuronal function and serve important roles in the central nervous system, however the role of neurosteroids in neuroapoptosis induced by ketamine remains to be elucidated. The present study aimed to explore whether neurosteroidogenesis was a pivotal mechanism for neuroprotection against ketamine-induced neuroapoptosis, and whether it may be selectively regulated under ketamine-induced neuroapoptosis conditions in primary cultured cortical neurons. To study this hypothesis, the effect of ketamine exposure on neurosteroidogenesis in primary cultured cortical neurons was investigated. Cholesterol, a substrate involved in the synthesis of neurosteroids, was added to the culture medium, and neurosteroids were quantified using high-performance liquid chromatography-tandem mass spectrometry analysis. The data demonstrated that cholesterol blocked ketamine-induced neuroapoptosis by promoting the synthesis of various neurosteroids, and the pathway of neurosteroid testosterone conversion into estradiol was inhibited by ketamine exposure. These data suggest that endogenous neurosteroids biosynthesis is critical for neuroprotection against ketamine-induced neuroapoptosis and inhibiting the biosynthesis of neuroprotective-neurosteroid estradiol is of notable importance for ketamine-induced neuroapoptosis.
Keywords: ketamine, neurosteroids, neuroapoptosis, 17β-estradiol, testosterone
Introduction
Ketamine, a noncompetitive N-methyl-D-aspartate receptor (NMDAR) ion channel blocker, is a commonly used anesthetic in pediatric patients (1). Previous studies have suggested that ketamine induces neuroapoptosis in developing animal brains (2,3) and primary cultured neurons (4,5), resulting in persistent cognitive deficits as the animal matures (2,3). Thus far, the mechanisms underlying ketamine-induced neuroapoptosis in the developing brain remain elusive.
Steroid hormones and their metabolites present within the central nervous system (CNS) are commonly defined as neuro-active steroids or neurosteroids (6). They can be synthesized de novo from cholesterol in the CNS by glial cells and neurons, or in the periphery by the adrenal and gonadal glands. The concentration of neurosteroids is higher in the CNS than in the periphery (6). They are regulators of CNS function, and serve important roles in mood, behavior, reproduction and cognition, and act as protective agents in models of injury and disease, such as experimental models of traumatic brain injury, including Alzheimer's disease (AD), autism, stroke and mood disorders (6–8). In addition, certain neurosteroids exert important regulatory and protective roles in the fetal brain (9). Li et al (5) suggested that 17β-estradiol is able to protect primary cultured cortical neurons against ketamine-induced neuroapoptosis. Reduced concentrations or the absence of neurosteroids during development and in adults may be associated with neurodevelopmental, psychiatric or behavioral disorders or neurodegeneration (10). A previous study demonstrated that neurotoxicity induced by the NMDAR blocker MK801 is associated with reduced levels of 17β-estradiol (11). Ethanol, another NMDAR blocker, has been reported to alter certain neurosteroid levels with prenatal exposure, and this alteration may be associated with the pathogenesis of ethanol-induced neurodevelopmental disorders and fetal alcohol syndrome in the developing rat brain (12). Trickler et al (13), suggested that ketamine may attenuate 17β-estradiol levels in the early life stages of zebrafish, and ketamine has been indicated to be neurotoxic to zebrafish embryos (14).
Therefore, it is reasonable to assume that neurosteroid biosynthesis is an important mechanism underlying the neuro-protection against ketamine-induced neuroapoptosis, and that it may be regulated or markedly affected in primary cultured cortical neurons. In the present study, the main aim was to investigate the effects of ketamine exposure on the biosynthesis of neurosteroids under ketamine-induced neuroapoptosis conditions in primary cultured cortical neurons.
Materials and methods
Reagents
Gibco Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), neurobasal medium and B27 supplement were purchased from Thermo Fisher Scientific, Inc. (Waltham, MA, USA). Ketamine hydrochloride was purchased from Fujian Gutian Yuanhang Medical Company Ltd., Co. (Ningde, China). 17β-Estradiol, pregnenolone (PREG), methyltestosterone (MT), dansyl chloride, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Testosterone was provided by the National Institute for Control of Pharmaceutical and Biological Products (Beijing, China). Ethyl acetate-n-hexane was provided by Concord Technology Co., Ltd. (Tianjin, China). Trypsin and Hoechst 33258 were purchased from Beijing Solarbio Science & Technology Co., Ltd. (Beijing, China). The in situ cell death detection kit was from Roche Applied Science (Mannheim, Germany).
Primary culture of cortical neurons
Cortical neurons from rats were cultured as previously described (15). Briefly, cerebral cortices from newborn Sprague-Dawley rat pups [obtained from Hebei Medical University (Shijiazhuang, China)], <24 h old, were dissected and placed in ice-cold DMEM, then mechanically dissociated and digested at 37°C for 15 min with trypsin (1.25 g/l). The cells were distributed and seeded on polylysine-coated plates (Beyotime Institute of Biotechnology, Shanghai, China) in DMEM supplemented with 10% FBS at 37°C in a humidified 5% CO2 air atmosphere incubator. DMEM was replaced by neurobasal medium containing B27 supplement 24 h later. From then, half the volume of culture medium was replaced with fresh medium every other day. Neurons cultured for 7 days were used for experimental purposes. The neurons were divided into four groups as follows: Vehicle control group, treated with an equal volume of neurobasal medium containing B27 supplement; cholesterol-treated group, treated with 5 µM cholesterol; ketamine-treated group, treated with 100 µM ketamine; and cholesterol + ketamine-treated group, treated with 5 µM cholesterol and 100 µM ketamine. All experiments were performed in accordance with the Ethics Review Committee for Animal Experimentation of Bethune International Peace Hospital of Chinese PLA (Shijiazhuang, China).
Assessment of cell viability
The MTT assay was used to quantify the cell viability, performed as previously described (5). In the current experiment, cells were treated with ketamine (Fujian Gutian Yuanhang Medical Company Ltd., Co.) and/or cholesterol (Sigma-Aldrich) according to the experimental design. Following treatment, 10 µl MTT (5 mg/ml) was added to each well and incubated for 4 h at 37°C. The MTT culture medium was discarded and replaced with 200 µl DMSO to dissolve the formazan crystals. The absorbance of each sample was measured with a microplate reader (Molecular Devices LLC, Sunnyvale, CA, USA) at 570 nm. Results were expressed as the percentage of MTT reduction, assuming the absorbance of vehicle control is 100%.
Apoptosis assays
Apoptosis was determined by using two different assays. Firstly, apoptotic neurons were determined by the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) method. The TUNEL assay was performed using an in situ apoptosis detection kit according to the manufacturer's instructions. Briefly, neurons cultured on coverslips were rinsed with phosphate-buffered saline [PBS (pH 7.2–7.4); Beijing noble Ryder Technology Co., Ltd., Beijing, China) and fixed with 4% paraformaldehyde (PFA) in PBS for 30 min at room temperature. Terminal deoxynucleotidyl transferase (Thermo Fisher Scientific, Inc., Waltham, MA, USA), a template-independent polymerase, was used to incorporate nucleotides at the sites of DNA breaks. Nuclei were stained with Invitrogen TO-PRO-3 [1:1,000 (1 µM) in PBS; Thermo Fisher Scientific, Inc.). Then, the cells were incubated with diaminobenzidine substrate (Shanghai Yesbio Biological Technology Co., Ltd., Shangai, China) to produce a dark brown precipitate. Apoptotic cells were identified according to the following criteria: Condensed chromatin, reduced size and dark brown nucleus. The TUNEL-positive and -negative cells were counted in five randomly selected microscopic fields with a BX41 light microscope (Olympus Corporation, Tokyo, Japan).
The second method used was Hoechst 33258 staining. Neurons were stained with Hoechst 33258 nuclear dye according to the manufacturer's instructions. Briefly, the cells were washed in PBS, fixed with 4% PFA for 30 min at 4°C, then incubated with 10 µg/ml Hoechst 33258 for 8 min at room temperature and images were captured under a BX41 fluorescence microscope (Olympus Corporation). Morphological alterations in chromatin that are characteristic of apoptosis, including condensation and fragmentation, were observed. The percentage of positive cells was calculated in five randomly selected microscopic fields using the light microscope.
Quantification of neurosteroids
The samples (~1 ml) were subjected to MT (30 ng/ml, internal standards) and were extracted three times with 2 ml ethyl acetate-n-hexane (9:1, v/v). The organic phases were combined and dried with a gentle stream of nitrogen in a water bath (50°C). The samples were precipitated with dansyl chloride (Shanghai Huicheng technology, Ltd., Shanghai, China) in a water bath (60°C, for 40 min), concentrated via centrifugation (16,000 × g, for 10 min at 4°C) and then transferred to autosampler vials prior to high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) analysis.
The HPLC-MS/MS system (Thermo Fisher Scientific, Inc.) consisted of a Surveyor MS Pump Plus, Surveyor AS Plus, TSQ Quantum Access Triple-Quadrupole Mass Spectrometer and Xcalibur software (version 2.1). Separation was achieved on an Eclipse XDB C18 analytical column (4.6×50 mm; Agilent Technologies Inc, Palo Alto, CA, USA) fitted with a XDB C18 guard column (4.6×12 mm). The HPLC mobile phases were H2O/0.1% formic acid (solvent-A) and MeOH/0.1% formic acid (solvent-B), and the gradient (flow rate 0.5 ml/min) was as follows: T0, 36, T6.5, 36, T6.6, 30, T16, 10, T17, 10, and T18, 36% solvent-A. The column temperature was 40°C, and the injection volume was 30 µl. An MSD quadrupole mass spectrometer equipped with an atmospheric pressure chemical ionization source (Thermo Fisher Scientific, Inc.) was used for the detection of analytes in the positive ion mode. The quantification was performed using a multiple-reaction monitoring method with transitions of 299.03-280.9 m/z for PREG, 254.97-158.9 m/z for 17β-estradiol, 289-97.2 m/z for testosterone and 303.1-109.06 m/z for MT.
Statistical analysis
All data are expressed as the mean ± standard deviation. Data analyses were performed with SPSS software, version 13.0 (SPSS, Inc., Chicago, IL, USA). Data were subjected to statistical analysis using one-way analysis of variance followed by the post-hoc Duncan's test. P<0.05 was considered to indicate a statistically significant difference.
Results
Cholesterol attenuates ketamine-induced neuronal toxicity in cultured cortical neurons
The MTT assay was performed to determine the protective role of cholesterol against ketamine-induced neuronal toxicity. Compared with the control group, neurons exposed to 100 µM ketamine exhibited a significant reduction in viability (P<0.01; Fig. 1). The combination of 100 µM ketamine and 5 µM cholesterol treatments resulted in a significantly increased cell survival, compared with the cholesterol-treated group (P<0.05; Fig. 1).
Figure 1.

Effects of cholesterol treatments on cell viability of ketamine-treated cells. Neurons were treated with 100 µM ketamine for 24 h in the absence or presence of 5 µM cholesterol. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assays were used to determine cell viability. Data are presented as the percentage of values in control cultures, and as the mean ± standard deviation (n=3). **P<0.01 vs. the control group, ##P<0.01 vs. the ketamine-treated group and +P<0.05 vs. the cholesterol-treated group.
Cholesterol treatment reduces ketamine-induced cell apoptosis in cultured cortical neurons
The TUNEL assay was utilized to detect whether the ketamine-induced cell death is apoptotic. Apoptotic cells were characterized by the appearance of intensely stained nuclei. The results demonstrated that few apoptotic cells were present in the control and cholesterol-treated groups (Fig. 2Aa and b, respectively), while the number of apoptotic cells significantly increased in the ketamine-treated group (Fig. 2Ac), compared with the control group (P<0.01; Fig. 2B). Cholesterol treatment reduced the number of apoptotic cells compared with the ketamine exposure group (P<0.05; Fig. 2Ad and B).
Figure 2.
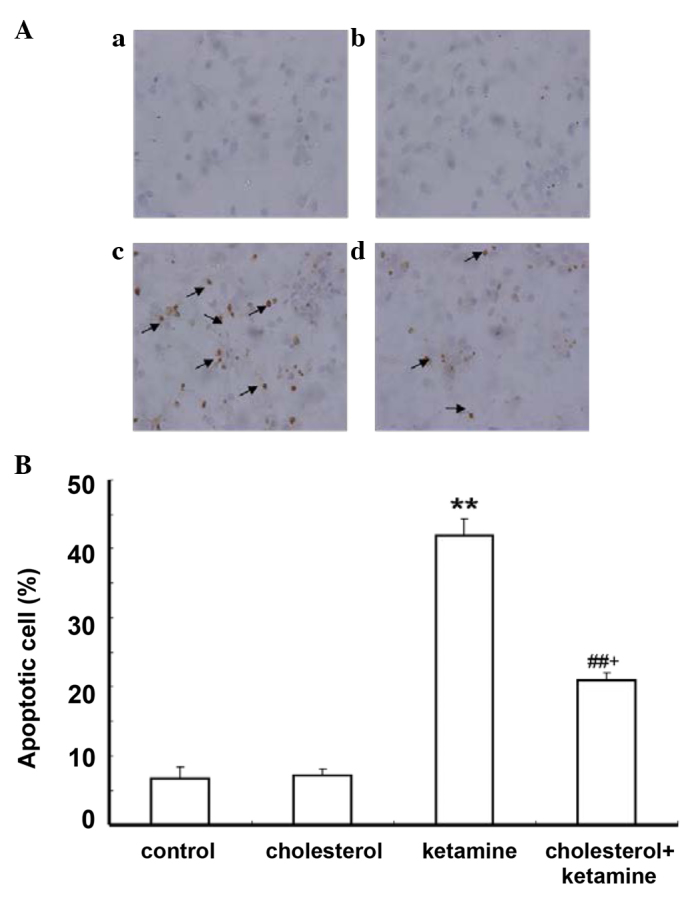
Cholesterol treatment attenuates ketamine-induced neuroapoptosis detected. (A) TUNEL labeling was performed to visualize the extent of apoptosis. TUNEL-positive cells indicate cellular apoptosis. Arrows in all panels indicate neurons with apoptotic morphology (magnification, ×400). (Aa) Control; (Ab) 5 µM cholesterol treatment; (Ac) 100 µM ketamine treatment; (Ad) 100 µM ketamine + 5 µM cholesterol treatment. (B) Quantification of the percentage of apoptotic cells present following different treatments. Data are presented as the mean ± standard deviation (n=3). **P<0.01 vs. the control group, ##P<0.01 vs. the ketamine-treated group and +P<0.05 vs. the cholesterol-treated group. TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
Hoechst 33258 staining was conducted to further investigate the anti-apoptotic effect of cholesterol. As demonstrated, in the control and cholesterol-treated groups, a small number of apoptotic cells was present (Fig. 3Aa and b). The ketamine-treated group (Fig. 3Ac) exhibited increased numbers of apoptotic cells compared with the control group (P<0.01; Fig. 3B). A similar effect to the previous assay was observed for the cholesterol and ketamine-treated group (Fig. 3A), compared with the cholesterol-treated group (P<0.05; Fig. 3B).
Figure 3.
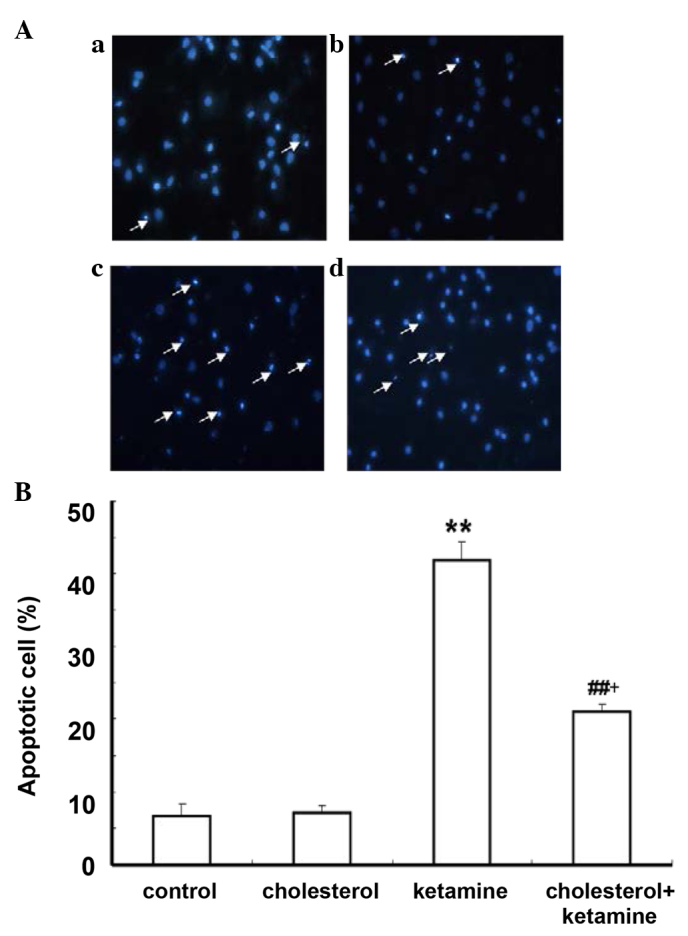
Cholesterol treatment attenuates ketamine-induced neuroapoptosis. (A) Hoechst 33258 staining was performed to visualize the extent of apoptosis. Fragmented nuclei labeled by Hoechst 33258 indicate cellular apoptosis. Arrows in all panels indicate neurons with apoptotic morphology. (Aa) The control; (Ab) 5 µM cholesterol-treated sample; (Ac) 100 µM ketamine-treated sample; (Ad) 100 µM ketamine + 5 µM cholesterol-treated sample (magnification, ×400). (B) Quantification of the percentage of apoptotic cells present following each treatment. Data are presented as the mean ± standard deviation (n=3). **P<0.01 vs. the control group, ##P<0.01 vs. the ketamine-treated group and +P<0.05 vs. the cholesterol-treated group.
Effect of ketamine on neurosteroid biosynthesis under ketamine-induced neuroapoptosis condition in primary cultured cortical neurons
Neurosteroids are synthesized from cholesterol in the CNS by means of a series of enzymatic processes, using the pathway 'cholesterol to pregnenolone to dehydroepiandrosterone to androstenedione to testosterone to estradiol' (16). To investigate the impact of ketamine on neurosteroid biosynthesis, 5 µM cholesterol was added into the culture medium. The concentration of neurosteroids was detected by the HPLC-MS/MS assay. Neurosteroids were not detected in the control and ketamine-treated groups, due to the lack of a synthetic substrate for cholesterol (data not shown). The cholesterol and cholesterol + ketamine-treated groups presented no significant difference in PREG content levels (P>0.05; Fig. 4A). Compared with the cholesterol-treated group, the 17β-estradiol content levels were reduced in the cholesterol + ketamine group (P<0.01; Fig. 4B). In addition, the testosterone content levels were increased in the dual treatment group, compared with the cholesterol-only group (P<0.01; Fig. 4C). These results indicate that ketamine is a potent suppressor of the conversion of testosterone into 17β-estradiol.
Figure 4.
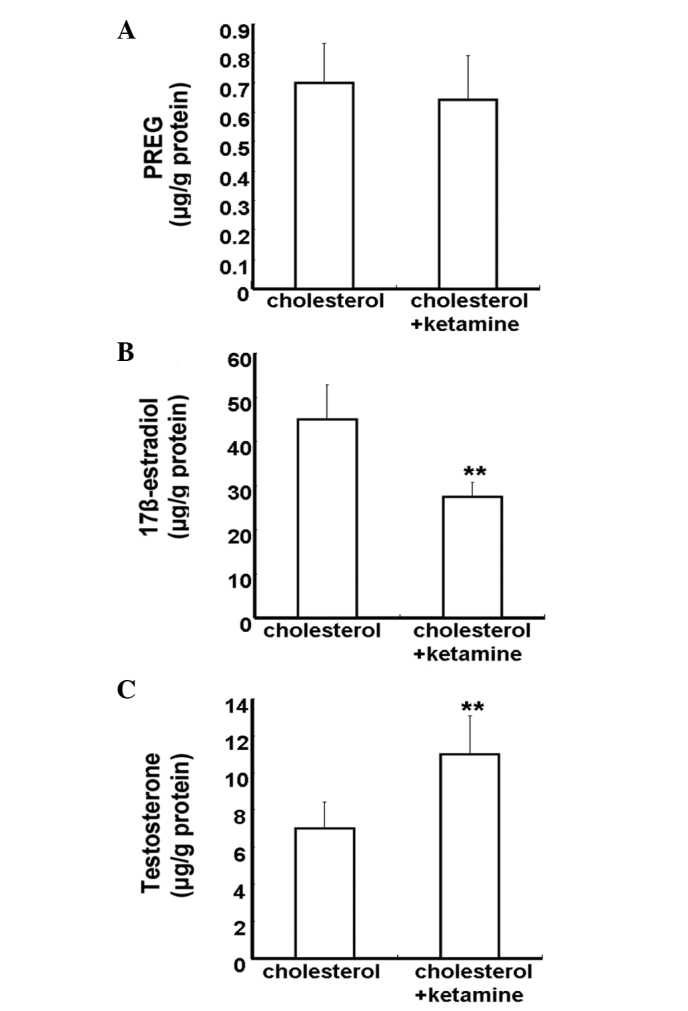
Quantitative analysis of the effect of ketamine on neurosteroid biosynthesis. (A) PREG, (B) 17β-estradiol and (C) testosterone. Neurons were treated with 5 µM cholesterol for 24 h in the absence or presence of 100 µM ketamine. Data are presented as the mean ± standard deviation (n=3).**P<0.01 vs. the cholesterol-treated group. PREG; pregnenolone.
Discussion
The results of the present study suggest that cholesterol protects against ketamine-induced neuroapoptosis by synthesizing certain neurosteroids. The results of the present study indicate that cholesterol protects against ketamine-induced neuro-apoptosis by synthesizing certain neurosteroids; in addition, ketamine selectively regulates the pathway of neurosteroid biosynthesis, which manifests in terms of a reduced level of 17β-estradiol and an increased level of testosterone. Data obtained in the current study may provide the underlying mechanism of ketamine-induced neuroapoptosis in primary cultured cortical neurons and suggest an adjunctive neuroprotective measure for ameliorating the toxicity of ketamine in the developing brain.
The effect of anesthetics on the developing brain is of an important clinical and research interest. Although ketamine is a widely used anesthetic, in certain instances, studies have implicated that ketamine exposure can induce neuroapoptosis in the developing brain (1,17,18). In cases of severe damage, the developmental neuroapoptosis has adverse effects on brain function and development as the animal matures (2,3). Previously, numerous studies investigated the mechanisms of, and protective strategies from ketamine-induced neuroapoptosis (4,19,20). It was postulated that the ketamine-induced neuroapoptosis may be associated with a compensatory upregulation of the NMDA receptor subunits and subsequent overstimulation of the glutamatergic system by endogenous glutamate (3). Although previous results have suggest a direct association between blockade of NMDA receptors and neuroapoptosis (4), the underlying mechanisms remain largely unclear.
Neurosteroids are synthesized de novo from cholesterol in the brain (21). Neurosteroids are produced locally in neuronal and glial cells, which appear to modulate neurodevelopment by autocrine and/or paracrine actions, and evidence indicates that neurosteroids are neuroprotective and important during neurodevelopment (9). The imbalance of neurosteroid androgens and estrogens is associated with the toxic effect of MK801, another NMDA receptor blocker (11). A reduction of estradiol release from astrocytes previously contributed to the neurodegeneration in a model of Niemann-Pick disease type C (22). Previously, it was demonstrated that ketamine attenuated 17β-estradiol levels in the early stages of zebrafish life (13), and exerted neurotoxic effects on the development of zebrafish embryos (14). The main aim of the present study was to investigate whether or not endogenous neurosteroid synthesis was selectively regulated in primary cultured cortical neurons during ketamine-induced neuroapoptosis. The results suggested that cholesterol may protect against ketamine-induced neuroapoptosis in vitro, which may be associated with the conversion of cholesterol into certain neurosteroids, as cholesterol is a steroid precursor and can increase the levels of brain neurosteroids, including estradiol (16). Enzymes required for neurosteroidogenesis are abundant in numerous brain regions (23), which leads to the possibility that cholesterol was locally converted into certain neurosteroids. For example, 17β-estradiol can be synthesized in the cortex region (24). Previous studies suggested that continuous neurosteroid synthesis is important for normal transmission in the hippocampus (25). In addition, cholesterol has been suggested to be essential for synaptic activity (26,27) and dendritic differentiation (28). Cholesterol may inhibit stress-induced dendritic retraction through conversion into 17β-estradiol (29). One previous study provided a similar conclusion; that chronic treatment with 17β-estradiol or cholesterol inhibited stress-induced hippocampal CA3 dendritic retraction in ovariectomized female rats (30). In addition, another study demonstrated that a high-cholesterol diet may remarkably increase 17β-estradiol serum levels and improve cognition deficits in ovariectomized mice (31). Grewal et al (32) reported that carbamazepine exerted a neuroprotective effect against ischemia-reperfusion injury, which was due to the increase in synthesis of neurosteroids. Estrogens can exert neuroprotective effects and enhance the survival of neurons (33). Previous studies demonstrated that 17β-estradiol protects against ketamine-induced neuroapoptosis in primary cultured cortical neurons and in the developing rat brain (3,5). These studies suggest that endogenous neurosteroid biosyn-thesis, including synthesis of estradiol, may be important to generate a protective and trophic environment for cortical neurons.
Furthermore, the data suggested that the synthesis of neurosteroid 17β-estradiol may be disrupted and 17β-estradiol depletion contributes to the ketamine-induced neuroapoptosis in primary cultured cortical neurons. A previous study demonstrated that numerous pathogenic factors may induce neurodegenerative injuries by downregulating the neuroprotective neurosteroid biosynthesis in nerve cells (34). A number of studies identified the levels of certain neurosteroids in the brain to be markedly reduced in patients with AD and Niemann-Pick disease type C (21,35,36). Schaeffer et al (37) reported that estradiol synthesis was selectively inhibited by H2O2-treatment in SH-SY5Y cells and that pretreatment with estradiol may protect against H2O2-induced cell death. The present study identified that ketamine-induced neuroapoptosis may be associated with reduced endogenous 17β-estradiol secretion, and that treatment with 17β-estradiol may protect against ketamine-induced neuroapoptosis in the developing rat brain (3). Another study suggested that the inhibition of neurosteroid estradiol biosynthesis resulted in the reduction of long-term potentiation, dephosphorylation of cofilin and resulting synapse loss (38).
In theory, testosterone can be converted into estradiol through cytochrome p450 aromatase (16). Previous studies have demonstrated that ketamine reduced cytochrome p450 aromatase activity (13,39). However, the exact molecular mechanism underlying ketamine-induced inhibition of the cytochrome p450 aromatase gene remains largely unknown. In the brain, aromatase has been demonstrated to regulate neural plasticity by stimulating cell growth and migration, protecting against neurodegeneration and brain injury and influencing learning and memory processes (40). Furthermore, a previous study suggested that the inhibition of the cytochrome p450 aromatase expression in the brain exacerbates neuronal death induced by different forms of mild neurodegenerative stimuli (41). Morale et al (42) suggested that the inhibition of cytochrome p450 aromatase function contributes to Parkinson's disease. Others have demonstrated that female mice are infertile when the aromatase gene is silenced, as their reproductive organs cannot develop properly (43), that the function of the amygdala and hypothalamus in these mice is impaired (44) and that apoptosis in the frontal cortex is increased (45). Numerous studies on aromatase knockout mice have suggested that estrogen is important for neurodevelopment (46). Another study reported that aromatase suppression in hippocampal dispersed cells inhibited the proliferation and induced apoptosis in granule cells (47). One previous study demonstrated that H2O2 induced SH-SY5Y cell apoptosis by inhibiting aromatase activity and reducing endogenous estradiol formation (37). Another study suggested that an aromatase inhibitor had an effect on cognitive performance in peripubertal boys who had been treated over a period of two years with letrozole, a drug that inhibits estrogen synthesis (48). Furthermore, a previous study suggested that aromatase and estradiol exerts a potential neuroprotective effect against kainic acid-induced cytotoxicity (49–51). The present study identified that among the neurosteroidogenic pathways starting from the precursor, PREG and leading to the distal metabolite, estradiol, only the estradiol biosynthesis was markedly inhibited in ketamine-induced neuroapoptosis. These data suggest that cytochrome p450 aromatase activity may be selectively affected by ketamine, leading to the inhibition of estradiol biosynthesis and inducing neuroapoptosis in primary cultured cortical neurons. This should be further investigated to confirm the inhibition of ketamine on cytochrome p450 aromatase activity.
In conclusion, data from the present study provided evidence that cholesterol may partially block ketamine-induced neuroapoptosis in vitro by synthesizing numerous neurosteroids. Additionally, ketamine reduced neurosteroid 17β-estradiol levels while elevating testosterone levels in primary cultured cortical neurons. These data suggest that neurosteroid 17β-estradiol depletion may be associated with ketamine-induced neuroapoptosis in primary cultured cortical neurons. Further research is required to examine the potential neuroprotective effects of neurosteroids, including 17β-estradiol, against ketamine-induced neuroapoptosis in the developing brain, using a well-designed animal model.
References
- 1.Mellon RD, Simone AF, Rappaport BA. Use of anesthetic agents in neonates and young children. Anesth Analg. 2007;104:509–520. doi: 10.1213/01.ane.0000255729.96438.b0. [DOI] [PubMed] [Google Scholar]
- 2.Brambrink AM, Evers AS, Avidan MS, Farber NB, Smith DJ, Martin LD, Dissen GA, Creeley CE, Olney JW. Ketamine-induced neuroapoptosis in the fetal and neonatal rhesus macaque brain. Anesthesiology. 2012;116:372–384. doi: 10.1097/ALN.0b013e318242b2cd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li J, Wang B, Wu H, Yu Y, Xue G, Hou Y. 17β-estradiol attenuates ketamine-induced neuroapoptosis and persistent cognitive deficits in the developing brain. Brain Res. 2014;1593:30–39. doi: 10.1016/j.brainres.2014.09.013. [DOI] [PubMed] [Google Scholar]
- 4.Liu F, Patterson TA, Sadovova N, Zhang X, Liu S, Zou X, Hanig JP, Paule MG, Slikker W, Jr, Wang C. Ketamine-induced neuronal damage and altered N-methyl-D-aspartate receptor function in rat primary forebrain culture. Toxicol Sci. 2013;131:548–557. doi: 10.1093/toxsci/kfs296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li J, Wu H, Xue G, Wang P, Hou Y. 17β-Oestradiol protects primary-cultured rat cortical neurons from ketamine-induced apoptosis by activating PI3K/Akt/Bcl-2 signalling. Basic Clin Pharmacol Toxicol. 2013;113:411–418. doi: 10.1111/bcpt.12124. [DOI] [PubMed] [Google Scholar]
- 6.Melcangi RC, Garcia-Segura LM, Mensah-Nyagan AG. Neuroactive steroids: State of the art and new perspectives. Cell Mol Life Sci. 2008;65:777–797. doi: 10.1007/s00018-007-7403-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Panzica GC, Balthazart J, Frye CA, Garcia-Segura LM, Herbison E, Mensah-Nyagan AG, McCarthy MM, Melcangi RC. Milestones on steroids and the nervous system: 10 years of basic and translational research. J Neuroendocrinol. 2012;24:1–15. doi: 10.1111/j.1365-2826.2011.02265.x. [DOI] [PubMed] [Google Scholar]
- 8.Schumacher M, Hussain R, Gago N, Oudinet JP, Mattern C, Ghoumari AM. Progesterone synthesis in the nervous system: Implications for myelination and myelin repair. Front Neurosci. 2012;6:10. doi: 10.3389/fnins.2012.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hirst JJ, Kelleher MA, Walker DW, Palliser HK. Neuroactive steroids in pregnancy: Key regulatory and protective roles in the foetal brain. J Steroid Biochem Mol Biol. 2014;139:144–153. doi: 10.1016/j.jsbmb.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 10.Mellon SH. Neurosteroid regulation of central nervous system development. Pharmacol Ther. 2007;116:107–124. doi: 10.1016/j.pharmthera.2007.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Olmos S, Bueno A, Bender C, Lorenzo A, de Olmos J. Sex differences and influence of gonadal hormones on MK801-induced neuronal degeneration in the granular retro-splenial cortex of the rat. Brain Struct Funct. 2008;213:229–238. doi: 10.1007/s00429-008-0186-0. [DOI] [PubMed] [Google Scholar]
- 12.Caldeira JC, Wu Y, Mameli M, Purdy RH, Li PK, Akwa Y, Savage DD, Engen JR, Valenzuela CF. Fetal alcohol exposure alters neurosteroid levels in the developing rat brain. J Neurochem. 2004;90:1530–1539. doi: 10.1111/j.1471-4159.2004.02686.x. [DOI] [PubMed] [Google Scholar]
- 13.Trickler WJ, Guo X, Cuevas E, Ali SF, Paule MG, Kanungo J. Ketamine attenuates cytochrome p450 aromatase gene expression and estradiol-17β levels in zebrafish early life stages. J Appl Toxicol. 2014;34:480–488. doi: 10.1002/jat.2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kanungo J, Cuevas E, Ali SF, Paule MG. Ketamine induces motor neuron toxicity and alters neurogenic and proneural gene expression in zebrafish. J Appl Toxicol. 2013;33:410–417. doi: 10.1002/jat.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang C, Sadovova N, Fu X, Schmued L, Scallet A, Hanig J, Slikker W. The role of the N-methyl-D-aspartate receptor in ketamine-induced apoptosis in rat forebrain culture. Neuroscience. 2005;132:967–977. doi: 10.1016/j.neuroscience.2005.01.053. [DOI] [PubMed] [Google Scholar]
- 16.Kawato S. Endocrine disrupters as disrupters of brain function: A neurosteroid viewpoint. Environ Sci. 2004;11:1–14. [PubMed] [Google Scholar]
- 17.Hudson AE, Hemmings HC., Jr Are anaesthetics toxic to the brain? Br J Anaesth. 2011;107:30–37. doi: 10.1093/bja/aer122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vutskits L, Gascon E, Kiss JZ. Effects of ketamine on the developing central nervous system. Ideggyogy Sz. 2007;60:109–112. [PubMed] [Google Scholar]
- 19.Liu F, Paule MG, Ali S, Wang C. Ketamine-induced neurotoxicity and changes in gene expression in the developing rat brain. Curr Neuropharmacol. 2011;9:256–261. doi: 10.2174/157015911795017155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duan X, Li Y, Zhou C, Huang L, Dong Z. Dexmedetomidine provides neuroprotection: impact on ketamine-induced neuroapoptosis in the developing rat brain. Acta Anaesthesiol Scand. 2014;58:1121–1126. doi: 10.1111/aas.12356. [DOI] [PubMed] [Google Scholar]
- 21.Belelli D, Lambert JJ. Neurosteroids: Endogenous regulators of the GABA(A) receptor. Nat Rev Neurosci. 2005;6:565–575. doi: 10.1038/nrn1703. [DOI] [PubMed] [Google Scholar]
- 22.Chen G, Li HM, Chen YR, Gu XS, Duan S. Decreased estradiol release from astrocytes contributes to the neurodegeneration in a mouse model of Niemann-Pick disease type C. Glia. 2007;55:1509–1518. doi: 10.1002/glia.20563. [DOI] [PubMed] [Google Scholar]
- 23.Nagarajan G, Aruna A, Chang CF. Neurosteroidogenic enzymes and their regulation in the early brain of the protogynous grouper Epinephelus coioides during gonadal sex differentiation. Gen Comp Endocrinol. 2013;181:271–287. doi: 10.1016/j.ygcen.2012.10.013. [DOI] [PubMed] [Google Scholar]
- 24.McCarthy MM. Estradiol and the developing brain. Physiol Rev. 2008;88:91–124. doi: 10.1152/physrev.00010.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tanaka M, Sokabe M. Continuous de novo synthesis of neurosteroids is required for normal synaptic transmission and plasticity in the dentate gyrus of the rat hippocampus. Neuropharmacology. 2012;62:2373–2387. doi: 10.1016/j.neuropharm.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki S, Kiyosue K, Hazama S, Ogura A, Kashihara M, Hara T, Koshimizu H, Kojima M. Brain-derived neurotrophic factor regulates cholesterol metabolism for synapse development. J Neurosci. 2007;27:6417–6427. doi: 10.1523/JNEUROSCI.0690-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frank C, Rufini S, Tancredi V, Forcina R, Grossi D, D'Arcangelo G. Cholesterol depletion inhibits synaptic transmission and synaptic plasticity in rat hippocampus. Exp Neurol. 2008;12:407–414. doi: 10.1016/j.expneurol.2008.04.019. [DOI] [PubMed] [Google Scholar]
- 28.Goritz C, Mauch DH, Pfrieger FW. Multiple mechanisms mediate cholesterol-induced synaptogenesis in a CNS neuron. Mol Cell Neurosci. 2005;29:190–201. doi: 10.1016/j.mcn.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 29.Ortiz JB, McLaughlin KJ, Hamilton GF, Baran SE, Campbell AN, Conrad CD. Cholesterol and perhaps estradiol protect against corticosterone-induced hippocampal CA3 dendritic retraction in gonadectomized female and male rats. Neuroscience. 2013;246:409–421. doi: 10.1016/j.neuroscience.2013.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McLaughlin KJ, Wilson JO, Harman J, Wright RL, Wieczorek L, Gomez J, Korol DL, Conrad CD. Chronic 17beta-estradiol or cholesterol prevents stress-induced hippocampal CA3 dendritic retraction in ovariectomized female rats: Possible correspondence between CA1 spine properties and spatial acquisition. Hippocampus. 2010;20:768–786. doi: 10.1002/hipo.20678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li L, Xiao N, Yang X, Gao J, Ding J, Wang T, Hu G, Xiao M. A high cholesterol diet ameliorates hippocampus-related cognitive and pathological deficits in ovariectomized mice. Behav Brain Res. 2012;230:251–258. doi: 10.1016/j.bbr.2012.02.024. [DOI] [PubMed] [Google Scholar]
- 32.Grewal AK, Jaggi AS, Rana AC, Singh N. Effect of neurosteroid modulation on global ischaemia-reperfusion-induced cerebral injury in mice. Korean J Physiol Pharmacol. 2013;17:485–491. doi: 10.4196/kjpp.2013.17.6.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fiocchetti M, Ascenzi P, Marino M. Neuroprotective effects of 17β-estradiol rely on estrogen receptor membrane initiated signals. Front Physiol. 2012;3:73. doi: 10.3389/fphys.2012.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schaeffer V, Patte-Mensah C, Eckert A, Mensah-Nyagan AG. Modulation of neurosteroid production in human neuroblastoma cells by Alzheimer's disease key proteins. J Neurobiol. 2006;66:868–881. doi: 10.1002/neu.20267. [DOI] [PubMed] [Google Scholar]
- 35.Marx CE, Trost WT, Shampine LJ, Stevens RD, Hulette CM, Steffens DC, Ervin JF, Butterfield MI, Blazer DG, Massing MW, et al. The neurosteroid allopregnanolone is reduced in prefrontal cortex in Alzheimer's disease. Biol Psychiatry. 2006;60:1287–1294. doi: 10.1016/j.biopsych.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 36.Yue X, Lu M, Lancaster T, Cao P, Honda S, Staufenbiel M, Harada N, Zhong Z, Shen Y, Li R. Brain estrogen deficiency accelerates Abeta plaque formation in an Alzheimer's disease animal model. Proc Natl Acad Sci USA. 2005;102:19198–19203. doi: 10.1073/pnas.0505203102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schaeffer V, Patte-Mensah C, Eckert A, Mensah-Nyagan AG. Selective regulation of neurosteroid biosynthesis in human neuroblastoma cells under hydrogen peroxide-induced oxidative stress condition. Neuroscience. 2008;151:758–770. doi: 10.1016/j.neuroscience.2007.11.032. [DOI] [PubMed] [Google Scholar]
- 38.Vierk R, Brandt N, Rune GM. Hippocampal estradiol synthesis and its significance for hippocampal synaptic stability in male and female animals. Neuroscience. 2014;274:24–32. doi: 10.1016/j.neuroscience.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 39.Lupp A, Kerst S, Karge E. Evaluation of possible pro- or antioxidative properties and of the interaction capacity with the microsomal cytochrome P450 system of different NMDA-receptor ligands and of taurine in vitro. Exp Toxicol Pathol. 2003;54:441–448. doi: 10.1078/0940-2993-00280. [DOI] [PubMed] [Google Scholar]
- 40.Lephart ED, Adlercreutz H, Lund TD. Dietary soy phytoestrogen effects on brain structure and aromatase in Long-Evans rats. Neuroreport. 2001;12:3451–3455. doi: 10.1097/00001756-200111160-00015. [DOI] [PubMed] [Google Scholar]
- 41.Garcia-Ovejero D, Azcoitia I, Doncarlos LL, Melcangi RC, Garcia-Segura LM. Glia-neuron crosstalk in the neuroprotective mechanisms of sex steroid hormones. Brain Res Brain Res Rev. 2005;48:273–286. doi: 10.1016/j.brainresrev.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 42.Morale MC, L'Episcopo F, Tirolo C, Giaquinta G, Caniglia S, Testa N, Arcieri P, Serra PA, Lupo G, Alberghina M, et al. Loss of aromatase cytochrome P450 function as a risk factor for Parkinson's disease? Brain Res Brain Res Rev. 2008;57:431–443. doi: 10.1016/j.brainresrev.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 43.Simpson ER. Models of aromatase insufficiency. Semin Reprod Med. 2004;22:25–30. doi: 10.1055/s-2004-823024. [DOI] [PubMed] [Google Scholar]
- 44.Pierman S, Sica M, Allieri F, Viglietti-Panzica C, Panzica GC, Bakker J. Activational effects of estradiol and dihydrotestosterone on social recognition and the arginine-vasopressin immunoreactive system in male mice lacking a functional aromatase gene. Horm Behav. 2008;54:98–106. doi: 10.1016/j.yhbeh.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hill RA, Simpson ER, Boon WC. Evidence for the existence of an estrogen-responsive sexually dimorphic group of cells in the medial preoptic area of the 129SvEv mouse strain. Int J Impot Res. 2008;20:315–323. doi: 10.1038/ijir.2008.2. [DOI] [PubMed] [Google Scholar]
- 46.Sasahara K, Shikimi H, Haraguchi S, Sakamoto H, Honda S, Harada N, Tsutsui K. Mode of action and functional significance of estrogen-inducing dendritic growth, spinogenesis, and synaptogenesis in the developing Purkinje cell. J Neurosci. 2007;27:7408–7417. doi: 10.1523/JNEUROSCI.0710-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fester L, Ribeiro-Gouveia V, Prange-Kiel J, Von Schassen C, Böttner M, Jarry H, Rune GM. Proliferation and apoptosis of hippocampal granule cells require local oestrogen synthesis. J Neurochem. 2006;97:1136–1144. doi: 10.1111/j.1471-4159.2006.03809.x. [DOI] [PubMed] [Google Scholar]
- 48.Hero M, Maury S, Luotoniemi E, Service E, Dunkel L. Cognitive effects of aromatase inhibitor therapy in peripubertal boys. Eur J Endocrinol. 2010;163:149–155. doi: 10.1530/EJE-10-0040. [DOI] [PubMed] [Google Scholar]
- 49.Garcia-Segura LM, Veiga S, Sierra A, Melcangi RC, Azcoitia I. Aromatase: A neuroprotective enzyme. Prog Neurobiol. 2003;71:31–41. doi: 10.1016/j.pneurobio.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 50.Azcoitia I, Sierra A, Veiga S, Honda S, Harada N, Garcia-Segura LM. Brain aromatase is neuroprotective. J Neurobiol. 2001;47:318–329. doi: 10.1002/neu.1038. [DOI] [PubMed] [Google Scholar]
- 51.Veiga S, Azcoitia I, Garcia-Segura LM. Extragonadal synthesis of estradiol is protective against kainic acid excitotoxic damage to the hippocampus. Neuroreport. 2005;16:1599–1603. doi: 10.1097/01.wnr.0000179081.39659.7d. [DOI] [PubMed] [Google Scholar]