

Abstract
Objective
We sought to evaluate the cardiovascular impact of coding variants in the apolipoprotein L1 gene APOL1 that protect against trypanosome infection but have been associated with kidney disease among African-Americans.
Approach and Results
As part of the Cardiovascular Health Study, a population-based cohort of Americans aged ≥65 years, we genotyped APOL1 polymorphisms rs73885319 and rs71785153 and examined kidney function, subclinical atherosclerosis, and incident cardiovascular disease and death over 13 years of follow-up among 91 African-Americans with two risk alleles, 707 other African-Americans, and 4964 white participants. The high-risk genotype with two risk alleles was associated with two-fold higher levels of albuminuria and lower ankle-brachial indices but similar carotid intima-media thickness among African-Americans. Median survival among high-risk African-Americans was 9.9 years (95% confidence interval [CI] 8.7-11.9), compared with 13.6 years (95% CI, 12.5-14.3) among other African-Americans and 13.3 (95% CI, 13.0-13.6) among whites (p 0.03). The high-risk genotype was also associated with increased risk for incident myocardial infarction (adjusted hazard ratio 1.8; 95% CI, 1.1-3.0) and mortality (adjusted hazard ratio 1.3; 95% CI 1.0-1.7). Albuminuria and risk for myocardial infarction and mortality were nearly identical between African-Americans with 0-1 risk alleles and whites.
Conclusion
APOL1 genotype is associated with albuminuria, subclinical atherosclerosis, incident myocardial infarction, and mortality in older African-Americans. African-Americans without two risk alleles do not differ significantly in risk of myocardial infarction or mortality from whites. APOL1 trypanolytic variants may account for a substantial proportion of the excess risk of chronic disease in African-Americans.
Keywords: genetics, kidney, apolipoproteins, epidemiology
Introduction
African-American (AA) adults are at risk for several morbidities including hypertension, peripheral artery disease, and diabetes.1-3 They are at particularly high risk for several types of chronic kidney disease (CKD), even accounting for associated risk factors.4 Although these findings suggest that genetic variants associated with CKD may exist in AA populations, this hypothesis remains elusive.5
Recent mapping studies among AA implicate a chromosome 22 locus in a surprising range of CKD among AA.6, 7 Subsequent studies have linked specific variants in the gene encoding apolipoprotein L1 (APOL1) to focal segmental glomerulosclerosis, human immunodeficiency virus-associated nephropathy, and hypertension-attributed end-stage renal disease.8-10 Furthermore, these variants appear to have originated within the last few thousand years and been subjected to strong selection pressure.11 These variants are absent in non-AA populations, and their gene products lyse Trypanosoma brucei rhodesiense in vitro while wild-type apolipoprotein L1 does not.8 Because these variants occur on over 30% of AA chromosomes, they potentially explain a large proportion of the excess CKD among AA. Indeed, AA participants without two risk alleles appear to have nearly the same rates of albuminuria and CKD as whites.12, 13
Given the strong associations of diminished estimated glomerular filtration rate (eGFR) and albuminuria with risks of subclinical atherosclerosis, cardiovascular disease, and death in older adults,14-17 the potential burden of morbidity and mortality related to these APOL1 variants may be substantial and extend well beyond CKD. Indeed, in the Jackson Heart Study and Women’s Health Initiative, individuals with two risk variants appeared to have roughly double the risk of cardiovascular disease, although the two studies had no information on mortality and included only 12 cases of myocardial infarction (MI) among high-risk subjects.18 Paradoxically, those studies also found that high-risk genotype was associated with less coronary artery calcification, but had no information on ankle-brachial index (ABI), which is strongly related to AA race.18
To better understand the associations of these variants with a full range of cardiovascular and renal outcomes and death, we examined APOL1 variants in a community-living population of older adults with detailed measures of clinical and subclinical vascular disease.
Materials and Methods are available in the online-only Data Supplement.
Results
Among AA participants, the numbers of participants homozygous wild-type, heterozygous, and homozygous variants at G1 were 513, 249, and 36 (minor allele frequency 0.20; Hardy-Weinberg p=0.41). The corresponding figures for G2 were 604, 173, and 19 (minor allele frequency 0.13; Hardy-Weinberg p=0.12). A total of 91 (11%) AA participants had two risk alleles. As expected, no individual had more than two risk alleles.
Table 1 shows participant characteristics according to APOL1 status. There were no significant differences between AA participants with and without two risk alleles in the characteristics shown, including fasting lipids and C-reactive protein. Consistent with expectation, AA participants had a greater prevalence of hypertension and diabetes and higher body-mass index than whites.
Table 1.
Baseline characteristics of CHS participants according to APOL1 genotype.
| AA 2 Risk Alleles (N=91) |
AA 0-1 Risk Alleles (N=707) |
White (N=4964) |
|
|---|---|---|---|
| Age, years | 73.5 (5.9) | 72.7 (5.6) | 72.8 (5.6) |
| Female | 53 (58) | 450 (64) | 2812 (57) |
| Current smoking | 14 (15) | 111 (16) | 555 (11) |
| Body-mass index, kg/m2 | 28.5 (4.9) | 28.6 (5.6) | 26.3 (4.5) |
| Prevalent coronary heart disease |
16 (18) | 141 (20) | 975 (20) |
| Prevalent congestive heart failure |
9 (10) | 45 (6) | 215 (4) |
| Hypertension | 69 (76) | 516 (73) | 2780 (56) |
| Diabetes | 26 (29) | 170 (25) | 725 (15) |
| Total cholesterol, mg/dl | 207 (35) | 210 (40) | 212 (39) |
| High-density lipoprotein cholesterol, mg/dl |
57 (17) | 58 (15) | 54 (16) |
| Triglycerides, mg/dl | 102 (75-127) | 102 (79-136) | 124 (95-170) |
| C-reactive protein, mg/L | 3.7 (1.8-9.2) | 3.3 (1.6-6.9) | 2.4 (1.2-4.3) |
| Creatinine, mg/dl | 1.1 (0.3) | 1.1 (0.6) | 1.1 (0.3) |
| eGFR, ml/min/1.73 m2 | 76 (21) | 80 (22) | 77 (19) |
Entries in table are N (%) for categorical variables and mean (standard deviation) for continuous variables, excepting triglycerides and C-reactive protein (median, interquartile range). eGFR indicates estimated glomerular filtration rate.
Kidney Function and Subclinical Atherosclerosis
Table 2 shows results for kidney function and subclinical atherosclerosis. We observed no difference in mean eGFR between AA participants with zero or one versus two risk alleles, nor a difference in the rate of change of eGFR between the groups. Analyses using baseline eGFR or creatinine (rather than cystatin) yielded similar results. There was also no significant association between APOL1 genotype and eGFR below 60 among AA individuals (adjusted odds ratio 1.4; 95% confidence interval [CI], 0.8-2.4; p=0.29). In contrast, APOL1 genotype was strongly associated with mean albuminuria, with two-fold higher levels among AA participants with two risk alleles but similar levels among other AA and white participants. We also observed a strong positive association of having two risk alleles with ascending categories of micro- and macroalbuminuria (adjusted odds ratio 2.9; 95% CI, 1.6-5.1; p<0.001).
Table 2.
Measures of kidney function and subclinical atherosclerosis according to APOL1 genotype.
| AA 2 Risk Alleles (N=91) |
AA 0-1 Risk Alleles (N=707) |
White (N=4964) |
P-value (0-1 vs. 2 Risk Alleles) |
|
|---|---|---|---|---|
| Kidney Function | ||||
| Mean eGFR (ml/min/1.73m2) | 76.3 (72.2-80.4) | 78.6 (77.1-80.1) | 71.7 (71.2-72.2) | 0.31 |
| Decline in eGFR per year (ml/min/1.73m2) |
1.5 (0.6-2.4) | 1.6 (1.3-1.9) | 1.5 (1.4-1.6) | 0.76 |
| Mean albuminuria (mg/g creatinine) |
27.9 (19.8-39.5) | 14.6 (12.8-16.6) | 13.5 (12.8-14.2) | <0.001 |
| Subclinical Atherosclerosis | ||||
| Ankle brachial index (mmHg/mmHg) |
0.98 (0.94-1.01) | 1.02 (1.01-1.03) | 1.06 (1.06-1.07) | 0.02 |
| Composite carotid intima-media thickness (SD units) |
0.16 (0.03-0.35) | 0.15 (0.08-0.22) | −0.02 (−0.05 - 0.002) | 0.90 |
Similarly, we observed an association of ABI, but not carotid IMT, with APOL1 genotype. AA participants had lower mean ABI values than did whites, regardless of genotype, but those with two risk variants had significantly lower values than did those with fewer. The prevalence of an ABI below 0.9 was higher among those AA participants with two risk alleles than those with fewer (adjusted odds ratio 1.6; 95% CI, 1.0-2.7; p=0.001).
Incident Cardiovascular Disease and Mortality
Figure 1 illustrates the association of APOL1 genotype with mortality. Individuals with two APOL1 variants had the greatest mortality, while mortality was virtually identical among white and AA participants with one or fewer variants. Median survival among AA individuals with two risk alleles was 9.9 years (95% CI, 8.7-11.9), compared with 13.6 (95% CI, 12.5-14.3) among other AA and 13.3 (95% CI, 13.0-13.6) among whites (log-rank p 0.03).
Figure 1.

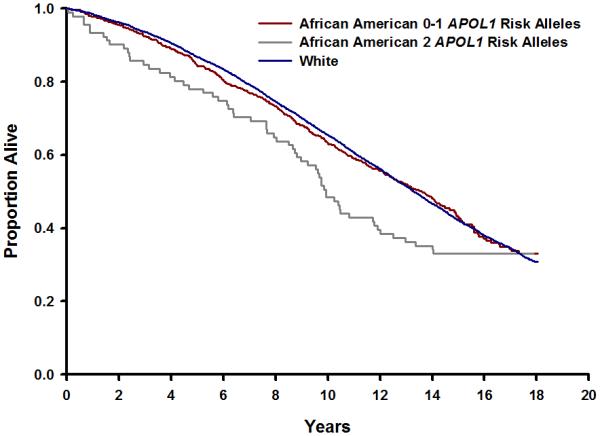
Kaplan-Meier estimates of survival are shown according to APOL1 genotype. The blue line indicates white participants, the red line African-American participants with 0-1 risk alleles, and the gray line African-American participants with two risk alleles. The curves differed significantly from each other (log-rank p 0.03). The number of participants at risk in each stratum in five-year increments is shown below.
Table 2 illustrates the adjusted associations of genotype with incident cardiovascular events and death over a median of 13 years of follow-up. MI risk was approximately 80% higher among participants with two risk alleles than other AA participants; whites and AA participants with fewer than two risk alleles had comparable risk. Similarly, risk of death was approximately 30% higher among those participants with two risk alleles compared with other AA participants. The magnitude of higher risk was modestly higher for non-cardiovascular than cardiovascular deaths, but the estimates did not differ significantly when modeled simultaneously in competing risk analyses (p=0.72). As with MI, cause-specific mortality was essentially identical between white and AA participants with fewer than two risk alleles. For both MI and mortality, the observed hazard ratios were not attenuated by adjustment for eGFR. Hazard ratios for specific causes among non-cardiovascular deaths are shown in Supplemental Table III. We did not observe differences in rates of total stroke or congestive heart failure according to APOL1 genotype. There were also no significant difference between AA participants with two or fewer risk alleles in risk of ischemic stroke (n=799 cases; adjusted hazard ratio 1.1; 95% CI, 0.6-2.0), but only 102 cases of hemorrhagic stroke occurred overall and none among AA participants with two risk alleles (p=0.41 for comparison across three race-APOL1 groups). Similarly, we documented 309 incident cases of clinically significant peripheral artery disease, of which 9 occurred among high-risk APOL1 individuals. Nonetheless, results largely concord with those seen for ABI. Compared with lower-risk AA, hazard ratios were 1.5 (95% CI, 0.7-3.1) for AA with two risk alleles and 0.7 (95% CI, 0.5-0.9) for white participants.
Sensitivity Analyses
We examined the dose-response relationships of APOL1 variants with albuminuria, ABI, and risks of MI and mortality. In all four instances, higher risk was limited to individuals with two risk alleles (data not shown). Although power was limited, we also observed generally similar associations with albuminuria and ABI for homozygosity at either G1 or G2.
We repeated our primary analyses within subgroups defined by age or diabetes and observed generally consistent positive associations (Supplemental Table I). Similarly, genotype did not interact with age or diabetes on albuminuria (p>0.10) or with age on ABI (p=0.99), but did so nominally with diabetes on ABI (p=0.02). The difference in ABI between AA participants with two risk alleles and those with fewer was 0.00±0.02 units among non-diabetic individuals (p=0.99) and 0.11±0.03 among diabetic participants (p<0.001).
In analyses of categorical outcomes (albuminuria, low ABI, MI, and death) adjusted for commonly collected clinical characteristics (Supplemental Table II), we found no consistent attenuation of the associations across outcomes with this additional adjustment. We also repeated our analyses among the subset of 781 AA participants with available markers of ancestry. We found no attenuation with adjustment for albuminuria. Estimates for ABI, MI, and death were attenuated by ~20% with adjustment.
APOL1 genotype was not consistently associated with retinal vascular caliber; it was marginally associated with smaller arteriolar diameter but not venular diameter, although both differed significantly between AA and white participants (Supplemental Table IV).
Lastly, we compared the associations with mortality of two APOL1 variant alleles with two APOE4 alleles. Among 5396 participants with available data on both loci, the mutually adjusted hazards ratios for mortality were 1.3 (1.0-1.7) for APOL1 and 1.4 (1.1-1.8) for APOE4 (p=0.78 for comparison of APOL1 and APOE).
Discussion
In this prospective study of older adults over two decades, APOL1 genotype was related to albuminuria, peripheral atherosclerosis, and risk of MI and death. AA participants with two risk alleles had a median survival approximately three years lower than other AA individuals or whites, and AA participants with fewer risk alleles had levels of albuminuria and risk of MI essentially identical to whites.
Humans are innately immune to Trypanosoma brucei infection for all but two subspecies - gambiense and rhodesiense – which cause African sleeping sickness. This immunity results from apolipoprotein L1, a Bcl-2-like protein carried on dense high-density lipoprotein particles that enters the parasitic lysosome after endocytosis,19 where it forms disruptive, trypanolytic pores.20 T. Brucei rhodesiense produces serum resistance associated protein, which interacts with the C-terminal helix of apolipoprotein L1 in the lysosome to limit its pore-forming activity and induce resistance.21 As a consequence, T. brucei infection poses a serious burden to individuals at or below child-bearing age in Africa.22
APOL1 variants appear to have emerged relatively recently and become prevalent among African populations because they may protect against sleeping sickness. Much like those hemoglobinopathies that confer protection from malarial infection,23 the protein products of these APOL1 variants retain trypanolytic activity against T. brucei rhodesiense (but not gambiense) species, albeit at an apparent cost of long-term kidney and related chronic disease. The G2 variant, which encodes a two amino-acid deletion in the C-terminal domain, prevents serum resistance associated protein from binding to apolipoprotein L1 in vitro. The mechanism by which the G1 variant retains pore-forming activity is less clear, but both amino-acid substitutions appear to be necessary for maximal effect.8
Our results expand the risks associated with APOL1 genotype. Previously, the G1 and G2 variants at this locus had been associated in AA with hypertension-associated end-stage renal disease, focal segmental glomerulosclerosis, and human immunodeficiency virus-nephropathy.8, 9 Among non-diabetic individuals in the Dallas Heart Study, APOL1 genotype was also associated with microalbuminuria and eGFR <60 ml/min per 1.73 m2, with a prevalence of non-diabetic CKD among AA without two risk alleles (1.7%) that matched that of whites (1.5%).12 Among AA with end-stage renal disease, APOL1 risk variants appear to confer a lower age at initiation of dialysis24 and more rapid progression of existing CKD.13 The mechanism by which these variants contribute to kidney disease remains uncertain, although indirect genetic evidence suggests that the APOL1 locus is likely to be causal.11
In CHS, APOL1 genotype was strongly associated with albuminuria, a strong risk factor for mortality.15 It was not associated with mean eGFR, either cross-sectionally nor longitudinally, much as AA race is associated with albuminuria but not with eGFR in CHS,5 although we observed a trend toward higher risk of eGFR <60 ml/min per 1.73 m2. We previously observed a parallel finding in the Dallas Heart Study, with a strong association with low eGFR but not with mean eGFR.12 Other studies have also found a lack of association with eGFR,25 suggesting that albuminuria is a more sensitive marker of the nephrotoxic effects of APOLI genotype.
The prevalence and magnitude of effect of APOL1 genotype is noteworthy. The combined minor allele frequency was approximately one-third, and more than 10% of AA individuals had a high-risk genotype, suggesting that over 4 million Americans may have this genotype. Further, the high-risk two-allele genotype was associated with approximately 80% higher risk of MI, higher than any single nucleotide polymorphism identified in genome-wide association studies to our knowledge.26 The magnitude of the association with albuminuria also exceeds that observed for polymorphisms found in genome-wide association studies.27 Thus, APOL1 appears to parallel APOE as among the few loci that harbor common polymorphisms with strong effects on the widespread chronic diseases of older age.
Our results suggest that a potentially large proportion of the excess risk of morbidity and mortality among AA beyond kidney disease alone relates to APOL1 genotype. The 90% of AA participants with no or one risk allele had virtually identical cardiovascular and non-cardiovascular mortality and kidney function to white participants. However, our results suggest that APOL1 genotype does not fully explain the excess risk of peripheral atherosclerosis observed among AA adults,28 for ABI remained higher even among those without two risk alleles, and genotype was not associated with carotid IMT, hypertension, or congestive heart failure. Furthermore, genotype clearly does not explain racial disparities in access to health care29 or risk of mortality unrelated to chronic disease,30, 31 and hence race remains more than a simple genetic construct32 even when race-specific disease loci are identified.
Although we genotyped nearly 800 AA individuals, the number of high-risk AA participants limited some analyses. For example, because we measured albuminuria four years after most AA participants enrolled, we could not reliably estimate whether excess mortality associated with APOL1 genotype was mediated by albuminuria; the high-risk stratum included 54 individuals and 27 subsequent deaths at that point. Clarification of that point will be important in future studies, as proteinuric kidney disease remains a likely mechanism by which APOL1 genotype might influence vascular disease and mortality. Similarly, we had limited ability to examine subgroups in which genotype might be most important, although we found no clear differences by age or diabetes.
In summary, APOL1 genotype was strongly associated with albuminuria, ABI, and risks of MI and mortality in older AA adults. Mean albuminuria and risks for MI and mortality did not differ between AA with no or one risk alleles and whites, suggesting that this locus may account for much of the excess risk of these conditions among AA adults.
Supplementary Material
Table 3.
Hazard ratios for incident cardiovascular disease and mortality according to APOL1 genotype.
| AA 2 Risk Alleles |
AA 0-1 Risk Alleles |
White (N=4964) |
P-value (0-1 vs. 2 Risk Alleles) |
|
|---|---|---|---|---|
| Individuals / Person-Years | 91 / 882 | 707 / 7831 | 4964 / 60526 | |
| Cardiovascular Disease | ||||
| Myocardial Infarction, N | 19 | 84 | 779 | |
| Adjusted Hazard Ratio (CI) | 1.8 (1.1-3.0) | Referent | 1.1 (0.9-1.4) | 0.02 |
| Stroke, N | 12 | 114 | 846 | |
| Adjusted Hazard Ratio (CI) | 0.9 (0.5-1.7) | Referent | 0.9 (0.7-1.1) | 0.80 |
| Congestive Heart Failure, N | 23 | 182 | 1407 | |
| Adjusted Hazard Ratio (CI) | 1.0 (0.6-1.5) | Referent | 0.9 (0.7-1.0) | 0.98 |
| Mortality | ||||
| Total Mortality, N | 61 | 401 | 3366 | |
| Adjusted Hazard Ratio (CI) | 1.3 (1.0-1.7) | Referent | 1.0 (0.9-1.1) | 0.05 |
| Cardiovascular Mortality, N | 23 | 159 | 1334 | |
| Adjusted Hazard Ratio (CI) | 1.3 (0.8-2.0) | Referent | 1.0 (0.8-1.1) | 0.31 |
| Non-Cardiovascular Mortality, N | 38 | 240 | 2026 | |
| Adjusted Hazard Ratio (CI) | 1.4 (1.0-1.9) | Referent | 1.0 (0.8-1.1) | 0.05 |
Significance.
Recently described APOL1 risk variants appear to protect against trypanosomiasis but have been associated with specific forms of kidney disease among individuals of African descent. In a population-based study of older adults, we find that rates of kidney disease, myocardial infarction, and total mortality were identical among whites and African-Americans who did not have the high-risk genotype, suggesting that potentially all of the excess risk of mortality, albuminuria, and MI among African-Americans may be attributable to their carriage of these alleles. Also, the high-risk genotype was associated with both total mortality and peripheral atherosclerosis, highlighting the importance of this locus for cardiovascular health among African-Americans.
Acknowledgements
A full list of principal CHS investigators and institutions can be found at http://www.chs-nhlbi.org. Dr. Mukamal had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Sources of Funding
This research was supported by contracts HHSN268201200036C, HHSN268200800007C, N01 HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083, N01HC85086, and grants HL080295 and HL094555 from the National Heart, Lung, and Blood Institute, with additional contribution from the National Institute of Neurological Disorders and Stroke. Additional support was provided by AG023629 from the National Institute on Aging and MD007092 from the National Institute on Minority Health and Health Disparities.
Abbreviations
- AA
African-American
- ABI
Ankle-brachial index
- CHS
Cardiovascular Health Study
- IMT
Carotid intima-media thickness
- CKD
Chronic kidney disease
- eGFR
Estimated glomerular filtration rate
- MI
Myocardial infarction
- SNP
Single nucleotide polymorphism
Footnotes
Disclosures
D.J.F., M.R.P., and the Beth Israel Deaconess Medical Center have filed for patents related to APOL1. There are no other conflicts of interest to disclose.
References
- 1.Mensah GA, Mokdad AH, Ford ES, Greenlund KJ, Croft JB. State of disparities in cardiovascular health in the united states. Circulation. 2005;111:1233–1241. doi: 10.1161/01.CIR.0000158136.76824.04. [DOI] [PubMed] [Google Scholar]
- 2.Ix JH, Allison MA, Denenberg JO, Cushman M, Criqui MH. Novel cardiovascular risk factors do not completely explain the higher prevalence of peripheral arterial disease among african americans. The san diego population study. J Am Coll Cardiol. 2008;51:2347–2354. doi: 10.1016/j.jacc.2008.03.022. [DOI] [PubMed] [Google Scholar]
- 3.Cowie CC, Rust KF, Byrd-Holt DD, Eberhardt MS, Flegal KM, Engelgau MM, Saydah SH, Williams DE, Geiss LS, Gregg EW. Prevalence of diabetes and impaired fasting glucose in adults in the u.S. Population: National health and nutrition examination survey 1999-2002. Diabetes Care. 2006;29:1263–1268. doi: 10.2337/dc06-0062. [DOI] [PubMed] [Google Scholar]
- 4.Halevy D, Radhakrishnan J, Appel GB. Racial and socioeconomic factors in glomerular disease. Semin Nephrol. 2001;21:403–410. doi: 10.1053/snep.2001.23775. [DOI] [PubMed] [Google Scholar]
- 5.Peralta CA, Ziv E, Katz R, Reiner A, Burchard EG, Fried L, Kwok PY, Psaty B, Shlipak M. African ancestry, socioeconomic status, and kidney function in elderly african americans: A genetic admixture analysis. J Am Soc Nephrol. 2006;17:3491–3496. doi: 10.1681/ASN.2006050493. [DOI] [PubMed] [Google Scholar]
- 6.Kao WH, Klag MJ, Meoni LA, et al. Myh9 is associated with nondiabetic end-stage renal disease in african americans. Nature genetics. 2008;40:1185–1192. doi: 10.1038/ng.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kopp JB, Smith MW, Nelson GW, et al. Myh9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nature genetics. 2008;40:1175–1184. doi: 10.1038/ng.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic apol1 variants with kidney disease in african americans. Science. 2010;329:841–845. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kopp JB, Nelson GW, Sampath K, et al. Apol1 genetic variants in focal segmental glomerulosclerosis and hiv-associated nephropathy. Journal of the American Society of Nephrology : JASN. 2011;22:2129–2137. doi: 10.1681/ASN.2011040388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tzur S, Rosset S, Shemer R, Yudkovsky G, Selig S, Tarekegn A, Bekele E, Bradman N, Wasser WG, Behar DM, Skorecki K. Missense mutations in the apol1 gene are highly associated with end stage kidney disease risk previously attributed to the myh9 gene. Hum Genet. 2010;128:345–350. doi: 10.1007/s00439-010-0861-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Genovese G, Friedman DJ, Pollak MR. Apol1 variants and kidney disease in people of recent african ancestry. Nature reviews. Nephrology. 2013;9:240–244. doi: 10.1038/nrneph.2013.34. [DOI] [PubMed] [Google Scholar]
- 12.Friedman DJ, Kozlitina J, Genovese G, Jog P, Pollak MR. Population-based risk assessment of apol1 on renal disease. Journal of the American Society of Nephrology : JASN. 2011;22:2098–2105. doi: 10.1681/ASN.2011050519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parsa A, Kao WH, Xie D, et al. Apol1 risk variants, race, and progression of chronic kidney disease. N Engl J Med. 2013;369:2183–2196. doi: 10.1056/NEJMoa1310345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shlipak MG, Sarnak MJ, Katz R, Fried LF, Seliger SL, Newman AB, Siscovick DS, Stehman-Breen C. Cystatin c and the risk of death and cardiovascular events among elderly persons. N Engl J Med. 2005;352:2049–2060. doi: 10.1056/NEJMoa043161. [DOI] [PubMed] [Google Scholar]
- 15.Cao JJ, Biggs ML, Barzilay J, Konen J, Psaty BM, Kuller L, Bleyer AJ, Olson J, Wexler J, Summerson J, Cushman M. Cardiovascular and mortality risk prediction and stratification using urinary albumin excretion in older adults ages 68-102: The cardiovascular health study. Atherosclerosis. 2008;197:806–813. doi: 10.1016/j.atherosclerosis.2007.07.029. [DOI] [PubMed] [Google Scholar]
- 16.Cao JJ, Barzilay JI, Peterson D, Manolio TA, Psaty BM, Kuller L, Wexler J, Bleyer AJ, Cushman M. The association of microalbuminuria with clinical cardiovascular disease and subclinical atherosclerosis in the elderly: The cardiovascular health study. Atherosclerosis. 2006;187:372–377. doi: 10.1016/j.atherosclerosis.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 17.Fung ET, Wilson AM, Zhang F, Harris N, Edwards KA, Olin JW, Cooke JP. A biomarker panel for peripheral arterial disease. Vasc Med. 2008;13:217–224. doi: 10.1177/1358863X08089276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ito K, Bick AG, Flannick J, et al. Increased burden of cardiovascular disease in carriers of apol1 genetic variants. Circ Res. 2014;114:845–850. doi: 10.1161/CIRCRESAHA.114.302347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vanhollebeke B, Pays E. The trypanolytic factor of human serum: Many ways to enter the parasite, a single way to kill. Mol Microbiol. 2010;76:806–814. doi: 10.1111/j.1365-2958.2010.07156.x. [DOI] [PubMed] [Google Scholar]
- 20.Pays E, Vanhollebeke B, Vanhamme L, Paturiaux-Hanocq F, Nolan DP, Perez-Morga D. The trypanolytic factor of human serum. Nat Rev Microbiol. 2006;4:477–486. doi: 10.1038/nrmicro1428. [DOI] [PubMed] [Google Scholar]
- 21.Lecordier L, Vanhollebeke B, Poelvoorde P, Tebabi P, Paturiaux-Hanocq F, Andris F, Lins L, Pays E. C-terminal mutants of apolipoprotein l-i efficiently kill both trypanosoma brucei brucei and trypanosoma brucei rhodesiense. PLoS Pathog. 2009;5:e1000685. doi: 10.1371/journal.ppat.1000685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matemba LE, Fevre EM, Kibona SN, Picozzi K, Cleaveland S, Shaw AP, Welburn SC. Quantifying the burden of rhodesiense sleeping sickness in urambo district, tanzania. PLoS Negl Trop Dis. 2010;4:e868. doi: 10.1371/journal.pntd.0000868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richer J, Chudley AE. The hemoglobinopathies and malaria. Clin Genet. 2005;68:332–336. doi: 10.1111/j.1399-0004.2005.00503.x. [DOI] [PubMed] [Google Scholar]
- 24.Kanji Z, Powe CE, Wenger JB, et al. Genetic variation in apol1 associates with younger age at hemodialysis initiation. Journal of the American Society of Nephrology : JASN. 2011;22:2091–2097. doi: 10.1681/ASN.2010121234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bentley AR, Doumatey AP, Chen G, et al. Variation in apol1 contributes to ancestry-level differences in hdlc-kidney function association. Int J Nephrol. 2012;2012:748984. doi: 10.1155/2012/748984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kessler T, Erdmann J, Schunkert H. Genetics of coronary artery disease and myocardial infarction - 2013. Curr Cardiol Rep. 2013;15:368. doi: 10.1007/s11886-013-0368-0. [DOI] [PubMed] [Google Scholar]
- 27.Boger CA, Chen MH, Tin A, et al. Cubn is a gene locus for albuminuria. Journal of the American Society of Nephrology : JASN. 2011;22:555–570. doi: 10.1681/ASN.2010060598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manolio TA, Burke GL, Psaty BM, Newman AB, Haan M, Powe N, Tracy RP, O'Leary DH. Black-white differences in subclinical cardiovascular disease among older adults: The cardiovascular health study. Chs collaborative research group. J Clin Epidemiol. 1995;48:1141–1152. doi: 10.1016/0895-4356(94)00240-q. [DOI] [PubMed] [Google Scholar]
- 29.Kelley E, Moy E, Stryer D, Burstin H, Clancy C. The national healthcare quality and disparities reports: An overview. Medical care. 2005;43:I3–8. doi: 10.1097/00005650-200503001-00002. [DOI] [PubMed] [Google Scholar]
- 30.Hauck FR, Tanabe KO, Moon RY. Racial and ethnic disparities in infant mortality. Semin Perinatol. 2011;35:209–220. doi: 10.1053/j.semperi.2011.02.018. [DOI] [PubMed] [Google Scholar]
- 31.Hennekens CH, Drowos J, Levine RS. Mortality from homicide among young black men: A new american tragedy. The American journal of medicine. 2013;126:282–283. doi: 10.1016/j.amjmed.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 32.Schwartz RS. Racial profiling in medical research. N Engl J Med. 2001;344:1392–1393. doi: 10.1056/NEJM200105033441810. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.