

Abstract
Cardinal events in atherogenesis are the retention of apolipoprotein B-containing lipoproteins in the arterial wall and the reaction of macrophages to these particles. My laboratory has been interested in both the cell biological events producing apolipoprotein B-containing lipoproteins, as well as in the reversal of the damage they cause in the plaques formed in the arterial wall. In the 2013 George Lyman Duff Memorial Lecture, as summarized in this review, I covered 3 areas of my past, present, and future interests, namely, the regulation of hepatic VLDL production by the degradation of apolipoprotein B100, the dynamic changes in macrophages in the regression of atherosclerosis, and the application of nanoparticles to both image and treat atherosclerotic plaques.
Keywords: lipids and lipoprotein metabolism, autophagy, atherosclerosis, nanoparticle, macrophage
Introduction
It was a great honor to have been selected by my peers in the ATVB Council to deliver the 2013 George Lyman Duff Memorial Lecture at the Scientific Sessions of the American Heart Association in Dallas. Because this award is meant not only to commemorate the many important contributions of Dr. Duff to the field of atherosclerosis, but also to recognize the career achievements of the recipient, I arranged the lecture to cover 3 main themes that have been important long-term foci of my research program, namely the secretory regulation of apolipoprotein B (apoB; particularly of the apoB100 form) and its associated hepatic lipoprotein very low density lipoprotein (VLDL), the regression of atherosclerosis, and “theranostics”- the imaging and treatment of atherosclerosis by nanoparticles. Because these 3 themes include studies on the production of atherogenic lipoproteins and on the regression of atherosclerosis through increases in reverse cholesterol transport, the Lecture (and article) title has tried to capture this spectrum.
The published work in each area served as an introduction to the sections, with the emphasis in the presentation being on our most recent studies. This was to avoid giving the impression that a qualification for the Duff Lecture Award was to already have become a duffer! In some cases, then, extensive presentation of the primary data will not be possible in this article in order not to interfere with subsequent publication of research reports.
Part I: The secretory regulation of apoB and VLDL
VLDL is assembled in the liver and serves a number of important roles in lipid and lipoprotein metabolism, one of which is the transport from the liver to the circulation of triglycerides (TG) and cholesterol. The TG is used in peripheral tissues either for energy or energy storage. After VLDL particles are depleted of TG, they are remodeled to cholesterol-enriched “remnants”, some of which are removed from the circulation (mainly by the liver, but also by peripheral cells, including macrophages in atherosclerotic plaques), with the others becoming low density lipoprotein (LDL) particles. Although lipid transport to peripheral tissues is an essential metabolic function of VLDL, that it is the precursor of LDL, the strongest risk factor for coronary artery disease (CAD), has also provided an impetus to better understand VLDL assembly and secretion.
Although VLDL has a number of apolipoprotein species on its surface (e.g., apoE, apoCI, CII, CIII), there is a specific requirement for apoB for the assembly and secretion of VLDL. Besides being an important structural component, the apoB100 form has regulatory functions by its being a ligand for the LDL receptor. In humans, the form of apoB made in the liver is apoB100, whereas in the intestine, it is apoB48. ApoB48 arises from the translation of an apoB100 mRNA that has been edited to alter a codon for glutamine to be a translational stop codon, resulting in a protein 48% of the size of apoB100.
Rodents have relatively high levels of the editing complex in their livers, so they produce a VLDL particle with either one apoB100 or one apoB48 associated with it. Given the goal to understand human VLDL metabolism through the window of animal and in vitro models, we have typically focused on the apoB100 species produced by rodent hepatic cells. An obvious question is- why not use as a model system HepG2 or Huh7 cells, given that they produce exclusively apoB100 and they are of human origin?
Unfortunately, these cells are not competent to secrete appreciable quantities of VLDL, instead assembling the apoB100 into lipoproteins of considerably greater density1. Thus, rodent hepatic cells (including primary rat and mouse hepatocytes and the rat hepatoma line, McA-RH7777 (“McA”)), which secrete most of their apoB100 on VLDL particles with characteristics similar to those of humans, have long served investigators interested in VLDL assembly and secretion. The use of these models will continue, but with iPS technologies, the production of human and humanized hepatocytes for study and manipulation is rapidly developing to provide additional model systems in vitro.
In the mid 1980’s, Roger Davis, Sven Olofsson, and their colleagues showed that the number of apoB-associated lipoprotein particles secreted by hepatic cells was regulated by the degradation of apoB (reviewed in2). This major finding was in stark contrast to what was known about the bulk of hepatic proteins, namely that their secretion was driven primarily by the level of synthesis. Their work turned the attention of many investigators to the nature of this degradation process. I started my own laboratory around the time of these discoveries and joined the hunt shortly after I was introduced to VLDL metabolism by Julian Marsh.
Not surprisingly, the regulation of apoB degradation has turned out to be complex with different proteolytic processes depending on the metabolic state of the hepatic cell. The current summary is depicted in Figure 1. There are 3 main pathways of apoB100 degradation, one at each end of VLDL assembly and secretion, and the third somewhere in between. My lab and our collaborators have made primary discoveries in all 3 pathways, which have been integrated into the fabric of the field woven by a host of investigators. Limited space precludes an in depth survey of apoB/VLDL research, even restricting the focus to Figure 1, so only the contributions of my laboratory to the early and middle pathways of apoB100 degradation will be discussed, as in the Lecture.
Figure 1. ApoB Degradation and the Pathway to VLDL.

The nascent apoB100 polypeptide becomes associated with lipids transferred by MTP during its translocation across the ER membrane. When there is an insufficient level of either lipid synthesis or MTP activity, much of apoB100 gets shunted to the ERAD/proteasome pathway. The immature VLDL particles (pre-VLDL) are transported in vesicles to the Golgi, where they either 1) complete their maturation to fully lipidated VLDL particles, or 2) VLDL particles that fail to normally mature (because of metabolic circumstances, such as treatment with fish oils or insulin) get shunted from the Golgi to autophagosomes, which eventually fuse with lysosomes as part of autophagy. There is yet another opportunity to “intercept” VLDL particles before they enter the systemic circulation- namely at the cell surface through interactions with either LDL receptors or HSPGs, in a re-uptake process.
MTP, microsomal triglyceride transfer protein; PDI, protein disulfide isomerase; PERPP, post-ER, pre-secretory proteolysis; TG, triglyceride; VLDL, very low density lipoproteins.
The early pathway is proteasomal-ER-associated degradation (ERAD), particularly relevant whenever lipidation of apoB100 polypeptides being translocated into the ER lumen is insufficient because of low levels of either TG synthesis or of microsomal triglyceride transfer protein (MTP) activity3. This results in the failure to stabilize the conformation of apoB100 polypeptides by co-translocational lipidation, and the aberrantly folded molecules become engaged by the quality control machinery of the ER and targeted by ERAD to the proteasome (shown in collaboration with Henry Ginsberg and Jeffrey Brodsky2, 3). While this pathway is particularly active in HepG2 and Huh7 cells, likely because they synthesize sub-normal amounts of TG, in rat hepatoma and primary rat and mouse hepatocytes, ERAD is less active, yet substantial degradation of apoB100 can occur. A particular example of this is the reduction of VLDL-TG and apoB100 secretion by diets rich in fish oils, which are used clinically in patients with hypertriglyceridemia. We showed that the active fatty acids in fish oil (DHA and EPA) stimulated apoB100 degradation in primary rat and mouse hepatocytes as well as in McA cells (e.g.,4), but this did not involve ERAD. Rather, engaged was a post-ER pre-secretory proteolysis pathway that Kevin J. Williams and I dubbed PERPP5. As shown in Figure 1, PERPP turned out to be autophagy6.
There is an accumulating list of examples of apoB100 post-ER autophagic degradation induced by diverse metabolic stimuli, which in addition to fish oils, include post-prandial levels of insulin7, 8. What appears to be common among these settings is a failure to complete successfully post-ER maturation of VLDL, thought to occur in the Golgi apparatus (reviewed in9. Thus, though most protein quality control has been studied in the ER, it makes sense that there is a parallel mechanism for complexes that were competent to exit the ER, but judged incompetent to exit the Golgi. Just how fish oils make VLDL Golgi “incompetent” is the subject of 2 of our papers6, 10, and will not be reviewed here.
In the Duff Lecture, I proposed that the effects of niacin are another example of post-ER autophagic apoB100 degradation regulating VLDL secretion. Like fish oils, niacin has been used to treat patients with hypertriglyceridemia, and its mechanism of action has been largely attributed to events outside of the liver (e.g., in the adipocyte) or after VLDL is secreted (e.g., hydrolysis of the TG). The studies that have gone into the most detail about possible effects of niacin at the level of the hepatocyte have focused on effects on TG metabolism, particularly their synthesis or transfer to apoB100 by MTP11. There are at least 2 problems, however, in generalizing those results. First, many data are from HepG2 cells, which as noted above, make little true VLDL. Second, if this was the mechanism- lipid insufficiency- as reviewed above and in Figure 1, niacin should have stimulated proteasomal ERAD of apoB100, which in our studies in primary mouse hepatocytes or McA cells does not occur (L. Guo and E. Fisher, unpublished). I presented in Dallas data showing that niacin treatment of rodent hepatic cells and of wild type mice decreased VLDL-TG and apoB100 production, and that these effects were attenuated by genetic or pharmacological deficiency of autophagy. A metabolite of niacin, nicotinic acid adenine dinucleotide phosphate (NAADP) can increase autophagy12 and in our current studies we are focusing on this as the mechanistic basis in the liver for increased apoB100 degradation and reduced VLDL secretion upon niacin treatment.
Part II. Atherosclerosis regression
Work from my lab has focused on regression, and since 2001 we have introduced a number of models and molecular approaches to study this clinically important goal. Briefly, these include 1) a transplant model, in which plaque-bearing aortic segments are transferred into normolipidemic mice13, 2) a “genetic switch” Reversa model, developed with Stephen Young, in which LDL production can be conditionally reduced in Ldlr−/− mice14, 3) acute treatment models, in which Apoe−/− mice are injected either with apoA1 (in collaboration with Stan Hazen and Jonathan Smith15), a MTP inhibitor16, or anti-miR-33 (in collaboration with Kathryn Moore17, 18).
Figure 2 is taken from our recent review19 and shows some of the events in the arterial wall during atherosclerosis progression and regression based on results from these and other models. Our early results with Gwen Randolph from the aortic transplant model showed, surprisingly, that part of the regression process could involve changes in macrophage emigration from the plaques20, 21. We and others (e.g.,22,18, 23, 24) have also shown regulation of monocyte recruitment and macrophage retention can contribute to the decreased content of macrophages in regressing plaques. It is becoming clear that the quantitative impact of each kinetic arm of monocyte/macrophage trafficking is dependent on the regression model used, as well as on the specific metabolic conditions, and the reasons for the variations are a focus in a number of laboratories, including our own.
Figure 2. Macrophage-related events in the arterial wall during atherosclerosis progression and regression.
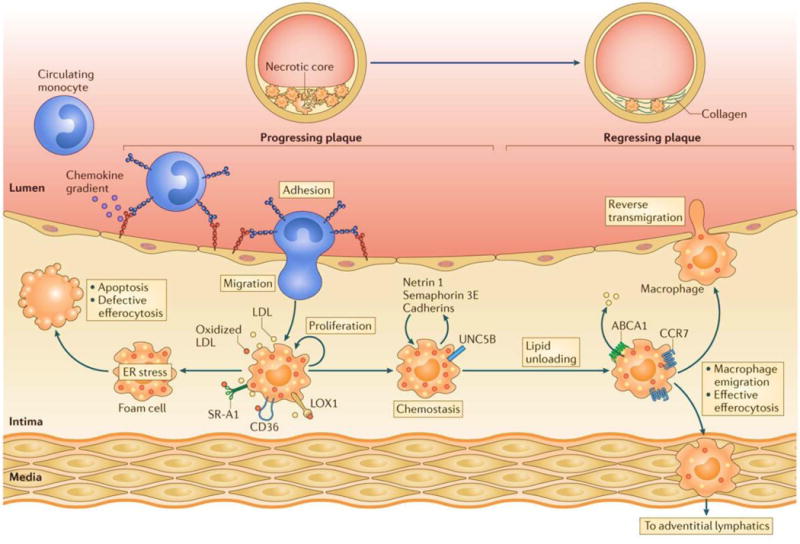
Hyperlipidemia increases the number of circulating GR1+LY6Chi monocytes, which constitute 80% of the monocytes recruited to mouse atherosclerotic plaques in a multi-step proccess involving chemokine–chemokine receptor pairs, endothelial adhesion molecules, including selectins, and adhesion molecules 1. The recruited monocytes differentiate into macrophages or dendritic cells in the intima, where they take up atherogenic lipoproteins via macropinocytosis or scavenger receptor-mediated pathways. The resulting foam cells secrete pro-inflammatory cytokines and chemokines, as well as retention factors (such as netrin 1, semaphorin 3E, and cadherins) that amplify the inflammatory response and promote macrophage chemostasis. These accumulating macrophages experience endoplasmic reticulum (ER) stress, which, if prolonged, results in apoptosis. This cell death, coupled with defective efferocytosis, results in the formation of the necrotic core that is characteristic of advanced plaques. The mechanisms that promote lipid unloading of the foam cell, including the factors that upregulate ATP-binding cassette subfamily A member 1 (ABCA1) expression on plaque macrophages and cholesterol efflux, reverse the accumulation of these foam cells. In some mouse models, this plaque regression is characterized by an upregulation of CC-chemokine receptor 7 (CCR7) on myeloid-derived cells, a decrease in the expression of retention factors, and reduced monocyte recruitment. The accumulating evidence summarized in our recent review (ref. 19) supports the idea that the regulation of macrophage the retention/migration factors contributes to macrophage emigration from the plaque through reverse transmigration to the lumen or through trafficking to the adventitial lymphatics. LDL, low-density lipoprotein; LOX1, lectin-like oxidized LDL receptor 1; SR-A1, scavenger receptor A1; UNC5B, a receptor for netrin 1. Figure and much of this legend were originally published in ref. 19 and is used by permission.
Our first transcriptome profiling (in collaboration with Michael Garabedian and Oscar Puig) of macrophages in progressing and regressing plaques disclosed arginase I, a commonly used murine marker of M2 macrophages25, 26, as the most upregulated gene in regression in the aortic transplant model. We have since established that independent of the models we have used (see19 for a review), a general feature of the regressing plaques is enrichment of macrophages with features of the M2 state. In progressing plaques, the predominant phenotype of macrophages is the activated, inflammatory M1 state. In a number of contexts, M2 macrophages are considered to be anti-inflammatory (e.g., they secrete IL-10) and tissue remodeling (e.g., they are integral to heal wounds), but at the time of our transcriptome report, little was known about their presence or roles in regressing plaques. Logically, the presence of M2 or M2-like macrophages would make sense in that the resolution of inflammation and remodeling of an atherosclerotic plaque to a more normal state is akin to wound healing. Also consistent with M2 macrophages having anti-inflammatory properties are the data that IL-4 and IL-13-based treatments (which polarize macrophages to the M2 state in vitro) retard plaque progression, as assessed by the content of activated macrophages27, 28.
In the Duff Lecture, I extended this finding by presenting unpublished results on the role and source of M2 polarized macrophages in regressing plaques. Perspective on these results has greatly benefitted by my time in David Greaves’ laboratory at Oxford during my Eastman Professorship, and has led to the goal of determining whether the M2 enrichment represents: a) the conversion of M1 macrophages to a M2-like state when the plaque environment is changed, b) the expansion of a pre-existing population of M2-like resident cells in the plaque, or c) the recruitment of new monocytes that become M2-like. If it is the last possibility, then a related issue is whether the subset of circulating cells belongs to the LyC6hi or Ly6Clo class. As frequently reviewed, the Ly6Chi monocyte subset is thought to be the precursor of M1 macrophages, and they are therefore sometimes referred to as “inflammatory monocytes”, whereas the Ly6Clo cells are thought to be precursors of M2 macrophages (e.g.,29). Recent studies, however, have shown that in some settings of inflammation (e.g.,30), M2 macrophages can be derived from the LyC6hi subset. To approach this experimentally, we have begun to test the possibility that the M2 macrophages are derived from circulating monocytes, given that we know that even after lipid lowering, monocytes continue to enter the plaque21. Experimentally, this has involved doing aortic transplants from Apoe−/− mice with advanced atherosclerosis into normolipidemic mice that were wild type or with deficiency in CCR2 or CCR5, chemokine receptors used for the recruitment into tissues, including plaques, of Ly6Chi and Ly6Clo monocytes, respectively (e.g., see31). As presented in the Lecture, the picture that is forming supports the origin of the M2-like cells from circulating monocytes recruited after lipid lowering, and that the source of the cells is the Ly6Chi pool. Furthermore, in the absence of M2-enrichment, regression was severely impaired, pointing out the parallel between regressing a plaque and healing a wound. Besides rigorously establishing these findings, studies in the near term, in collaboration with Png Loke and Kathryn Moore, will include identifying the type(s) and source(s) of the signals that cause the newly recruited Ly6Chi monocytes to assume M2-like properties.
Part III. Atherosclerosis “theranostics”
Theranostics is a term that refers to agents that have the dual capability of diagnosing and treating a disease. A little over 10 years ago, my involvement in cardiovascular theranostics began when I envisioned that HDL particles could be adapted to deliver gadolinium (Gd) to improve the imaging of atherosclerotic plaques by magnetic resonance (MR). I had been working with Zahi Fayad, who spearheaded the first studies to show that MR imaging could detect plaques in atherosclerotic mice32, and we wished to increase the sensitivity. As shown in Figure 3A, the form of Gd typically used for imaging is incorporated in the form of a chelate with a phospholipid molecule, and we were able to reconstitute HDL particles containing this form. Their use significantly enhanced the quality of the images (Figure 3B and33).
Figure 3. Building theranostic HDL particles and their applications.
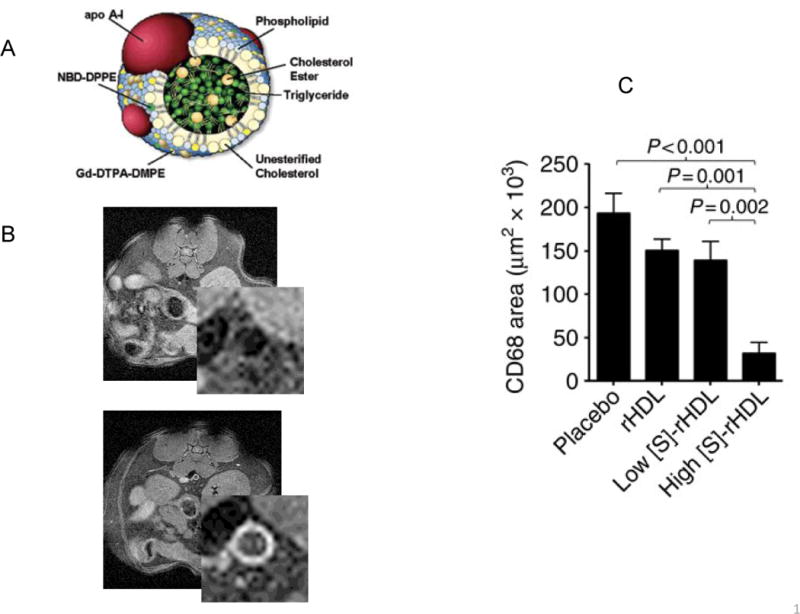
A) Shown in cross section is a typical spherical HDL particle. The MR imaging agent Gd has been incorporated as a chelate with DTPA conjugated to PE. There is also a fluorescent dye (NBD), also incorporated as a conjugate with PE. Other imaging agents (such as quantum dots, gold, and iron oxide particles), as well as drugs (statin, LXR agonists) can be placed in the core, where the lipid esters usually are. B) ApoE-deficient mice were fed the western diet for 48 weeks, then imaged by MR without (pre) or 24 h after injection with HDL reconstituted with Gd. The lower power MR images are abdominal cross sections, the higher power inserts show more clearly the aortae. The bright signals in the Gd-HDL injected mouse correlated with plaque macrophage content on subsequent immunohistochemistry. C) ApoE-deficient mice were fed the western diet for 27 weeks, after which they received every other day injections over a week of the indicated materials (rHDL, reconstituted control HDL; low or high [S]-rHDL; HDL reconstituted with simvastatin, but administered at 2 different dose levels).
ApoA-I, apolipoprotein A-I; DMPE, di-myristoyl PE; DTPA, diethylene triamine penta-acetic acid; Gd, gadolinium; NBD, nitro-2,1,3-benzoxadiazol; PE, phosphatidylethanolamine.
Panels A and B are reprinted with permission from ref. 19 (copyright 2004, American Chemical Society), and panel C is from ref. 21, also used with permission.
This was the “diagnostic” aspect; HDL infusions in animal models and in humans were shown to regress plaques (reviewed in34), suggesting that the administration of the Gd-HDL particles would also be therapeutic. Furthermore, by further adapting HDL to carry both imaging agents and drugs, we envisioned that the therapeutic efficacy could be improved. Indeed, in studies directed by Willem Mulder, whose laboratory incorporated simvastatin into HDL particles, we recently showed that plaque regression resulting from infusion of HDL was dramatically enhanced by the incorporation of statin (see35 and Figure 3C). Along with parallel advances in incorporating imaging agents for modalities besides MR (reviewed in36), the potential for HDL to become a theranostic agent has been amply supported.
In the Duff Lecture, I presented studies in which we showed how a nanoparticle approach could overcome undesirable effects of liver-x receptor (LXR) agonists. While LXR agonists are thought to have major benefits at the level of the plaque by promoting cholesterol efflux from macrophages and by having anti-inflammatory activities, in mammals, including humans, they also stimulate hepatic lipid synthesis and can cause steatosis and hypertriglyceridemia. As shown in Figure 4, when LXR agonist GW3965 was incorporated into nanoparticles (details about their composition are given in37), steatosis was avoided, but beneficial effects on plaques were observed. Currently we are testing whether reconstituted HDL containing GW3965 has similar effects. If so, then there will be multiple nano-formulations, based on HDL and other platforms, from which to identify the best candidates to use in human imaging and therapeutic trials.
Figure 4. Nanoparticles (NPs) containing LXR agonist GW3965 favorably affect the plaque while sparing the liver.
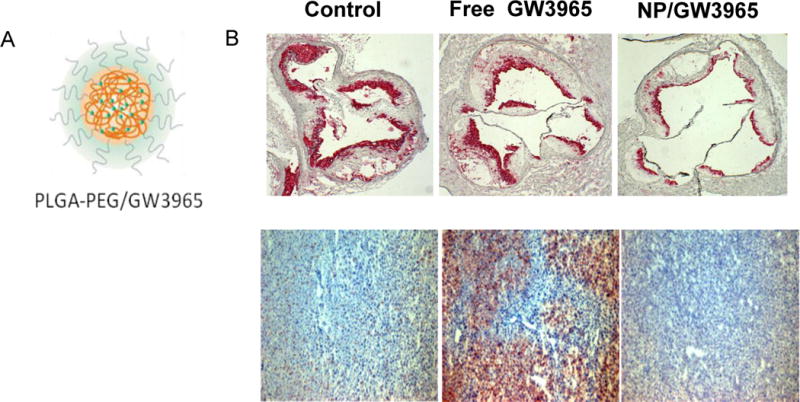
A) Chemical structure of GW3965-containing NP’s. The particle consists of an outer PEG surface, and a biodegradable polymer matrix (PLGA, golden wavy lines) loaded with GW3965 (green circles). B) Top: ApoE-deficient mice were fed western diet for 16 weeks, then treated 3X/week for 2 weeks with buffer (control), free GW3965, or NPs containing GW3965 at the same dose. The aortic roots were sectioned and stained for macrophages (CD68+ cells, in red). Note in the representative images that both free drug and NP-GW3965 decreased plaque macrophage content. Bottom: In contrast, when liver sections were stained from the corresponding mice, the oil Red O positive area, which represents triglyceride content, was obviously higher in the free GW3965 treatment group.
PEG, polyethylene glycol; PLGA, poly lactic-co-glycolic acid
Concluding Remarks
To review the past, describe the present, and speculate on the future in one 30 minute lecture was an interesting experience, especially in Dallas during the week of the 50th anniversary of the assassination of President Kennedy, as well as of my own Bar Mitzvah! As noted earlier, I started my independent career focused on the cell and molecular biological aspects of hepatic lipoprotein metabolism, later adding the area of atherosclerosis, specifically its regression and imaging. While there are many other exciting topics tempting me to investigate, my inner voice says that it will be ambitious enough to definitively answer some of the important questions raised by the results shared with you in the Duff Lecture and in this review. I look forward to reporting on our progress and hearing about yours as well.
Acknowledgments
Needless to say, all of the research in my laboratory has benefitted by the contributions of many mentors, trainees, research technicians, and collaborators. Space limitations do not allow me to list them individually, but their names (and the complete Lecture slide set) can be accessed at https://www.med.nyu.edu/medicine/cardiology/center-prevention-cv-disease/edward-fisher-md-phd-profile. I am particularly indebted to Jan Breslow, who as a young assistant professor gambled by taking me on as his first graduate student. Deep thanks are also due to my family and friends.
Sources of funding: The work presented has been supported over the years by grants primarily from the NIH and AHA, with some funding also coming from investigator-initiated programs of Merck, Takeda, Pfizer, and Astra Zeneca.
Disclosures: I have research grants from the pharmaceutical companies listed in the Sources of Funding.
Footnotes
Subject codes: Animal models of human disease, Atherosclerosis, Imaging, Inflammation, Lipids and cholesterol.
References
- 1.Meex SJ, Andreo U, Sparks JD, Fisher EA. Huh-7 or hepg2 cells: Which is the better model for studying human apolipoprotein-b100 assembly and secretion? J Lipid Res. 2011;52:152–158. doi: 10.1194/jlr.D008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ginsberg HN, Fisher EA. The ever-expanding role of degradation in the regulation of apolipoprotein b metabolism. J Lipid Res. 2009;50(Suppl):S162–166. doi: 10.1194/jlr.R800090-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brodsky JL, Fisher EA. The many intersecting pathways underlying apolipoprotein b secretion and degradation. Trends Endocrinol Metab. 2008;19:254–259. doi: 10.1016/j.tem.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang H, Chen X, Fisher EA. N-3 fatty acids stimulate intracellular degradation of apoprotein b in rat hepatocytes. J Clin Invest. 1993;91:1380–1389. doi: 10.1172/JCI116340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fisher EA, Pan M, Chen X, Wu X, Wang H, Jamil H, Sparks JD, Williams KJ. The triple threat to nascent apolipoprotein b. Evidence for multiple, distinct degradative pathways. J Biol Chem. 2001;276:27855–27863. doi: 10.1074/jbc.M008885200. [DOI] [PubMed] [Google Scholar]
- 6.Pan M, Maitin V, Parathath S, Andreo U, Lin SX, St Germain C, Yao Z, Maxfield FR, Williams KJ, Fisher EA. Presecretory oxidation, aggregation, and autophagic destruction of apoprotein-b: A pathway for late-stage quality control. Proc Natl Acad Sci U S A. 2008;105:5862–5867. doi: 10.1073/pnas.0707460104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andreo U, Guo L, Chirieac DV, Tuyama AC, Montenont E, Brodsky JL, Fisher EA. Insulin-stimulated degradation of apolipoprotein b100: Roles of class ii phosphatidylinositol-3-kinase and autophagy. PLoS One. 2013;8:e57590. doi: 10.1371/journal.pone.0057590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sparks JD, O’Dell C, Chamberlain JM, Sparks CE. Insulin-dependent apolipoprotein b degradation is mediated by autophagy and involves class i and class iii phosphatidylinositide 3-kinases. Biochem Biophys Res Commun. 2013;435:616–620. doi: 10.1016/j.bbrc.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olofsson SO, Boren J. Apolipoprotein b secretory regulation by degradation. Arterioscler Thromb Vasc Biol. 2012;32:1334–1338. doi: 10.1161/ATVBAHA.112.251116. [DOI] [PubMed] [Google Scholar]
- 10.Maitin V, Andreo U, Guo L, Fisher EA. Docosahexaenoic acid impairs the maturation of very low density lipoproteins in rat hepatic cells. J Lipid Res. 2014;55:75–84. doi: 10.1194/jlr.M043026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamanna VS, Kashyap ML. Mechanism of action of niacin. Am J Cardiol. 2008;101:20B–26B. doi: 10.1016/j.amjcard.2008.02.029. [DOI] [PubMed] [Google Scholar]
- 12.Pereira GJ, Hirata H, Fimia GM, do Carmo LG, Bincoletto C, Han SW, Stilhano RS, Ureshino RP, Bloor-Young D, Churchill G, Piacentini M, Patel S, Smaili SS. Nicotinic acid adenine dinucleotide phosphate (naadp) regulates autophagy in cultured astrocytes. J Biol Chem. 2011;286:27875–27881. doi: 10.1074/jbc.C110.216580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reis ED, Li J, Fayad ZA, Rong JX, Hansoty D, Aguinaldo JG, Fallon JT, Fisher EA. Dramatic remodeling of advanced atherosclerotic plaques of the apolipoprotein e-deficient mouse in a novel transplantation model. J Vasc Surg. 2001;34:541–547. doi: 10.1067/mva.2001.115963. [DOI] [PubMed] [Google Scholar]
- 14.Feig JE, Parathath S, Rong JX, Mick SL, Vengrenyuk Y, Grauer L, Young SG, Fisher EA. Reversal of hyperlipidemia with a genetic switch favorably affects the content and inflammatory state of macrophages in atherosclerotic plaques. Circulation. 2011;123:989–998. doi: 10.1161/CIRCULATIONAHA.110.984146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hewing B, Parathath S, Barrett T, et al. Effects of native and myeloperoxidase-modified apolipoprotein a-i on reverse cholesterol transport and atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2014;34:779–789. doi: 10.1161/ATVBAHA.113.303044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hewing B, Parathath S, Mai CK, Fiel MI, Guo L, Fisher EA. Rapid regression of atherosclerosis with mtp inhibitor treatment. Atherosclerosis. 2013;227:125–129. doi: 10.1016/j.atherosclerosis.2012.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rayner KJ, Sheedy FJ, Esau CC, Hussain FN, Temel RE, Parathath S, van Gils JM, Rayner AJ, Chang AN, Suarez Y, Fernandez-Hernando C, Fisher EA, Moore KJ. Antagonism of mir-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. The Journal of clinical investigation. 2011;121:2921–2931. doi: 10.1172/JCI57275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Distel E, Barrett TJ, Chung K, Girgis NM, Parathath S, Essau CC, Murphy AJ, Moore KJ, Fisher EA. Mir33 inhibition overcomes deleterious effects of diabetes mellitus on atherosclerosis plaque regression in mice. Circ Res. 2014;115:759–769. doi: 10.1161/CIRCRESAHA.115.304164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: A dynamic balance. Nature reviews Immunology. 2013;13:709–721. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Llodra J, Angeli V, Liu J, Trogan E, Fisher EA, Randolph GJ. Emigration of monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc Natl Acad Sci U S A. 2004;101:11779–11784. doi: 10.1073/pnas.0403259101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trogan E, Feig JE, Dogan S, Rothblat GH, Angeli V, Tacke F, Randolph GJ, Fisher EA. Gene expression changes in foam cells and the role of chemokine receptor ccr7 during atherosclerosis regression in apoe-deficient mice. Proc Natl Acad Sci U S A. 2006;103:3781–3786. doi: 10.1073/pnas.0511043103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Potteaux S, Gautier EL, Hutchison SB, van Rooijen N, Rader DJ, Thomas MJ, Sorci-Thomas MG, Randolph GJ. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of apoe−/− mice during disease regression. J Clin Invest. 2011;121:2025–2036. doi: 10.1172/JCI43802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Gils JM, Derby MC, Fernandes LR, et al. The neuroimmune guidance cue netrin-1 promotes atherosclerosis by inhibiting the emigration of macrophages from plaques. Nat Immunol. 2012;13:136–143. doi: 10.1038/ni.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagareddy PR, Murphy AJ, Stirzaker RA, et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 2013;17:695–708. doi: 10.1016/j.cmet.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feig JE, Vengrenyuk Y, Reiser V, Wu C, Statnikov A, Aliferis CF, Garabedian MJ, Fisher EA, Puig O. Regression of atherosclerosis is characterized by broad changes in the plaque macrophage transcriptome. PloS one. 2012;7:e39790. doi: 10.1371/journal.pone.0039790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolfs IM, Stoger JL, Goossens P, et al. Reprogramming macrophages to an anti-inflammatory phenotype by helminth antigens reduces murine atherosclerosis. FASEB J. 2014;28:288–299. doi: 10.1096/fj.13-235911. [DOI] [PubMed] [Google Scholar]
- 28.Cardilo-Reis L, Gruber S, Schreier SM, Drechsler M, Papac-Milicevic N, Weber C, Wagner O, Stangl H, Soehnlein O, Binder CJ. Interleukin-13 protects from atherosclerosis and modulates plaque composition by skewing the macrophage phenotype. EMBO molecular medicine. 2012;4:1072–1086. doi: 10.1002/emmm.201201374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Woollard KJ, Geissmann F. Monocytes in atherosclerosis: Subsets and functions. Nat Rev Cardiol. 2010;7:77–86. doi: 10.1038/nrcardio.2009.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Egawa M, Mukai K, Yoshikawa S, Iki M, Mukaida N, Kawano Y, Minegishi Y, Karasuyama H. Inflammatory monocytes recruited to allergic skin acquire an anti-inflammatory m2 phenotype via basophil-derived interleukin-4. Immunity. 2013;38:570–580. doi: 10.1016/j.immuni.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 31.Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, van Rooijen N, Lira SA, Habenicht AJ, Randolph GJ. Monocyte subsets differentially employ ccr2, ccr5, and cx3cr1 to accumulate within atherosclerotic plaques. The Journal of clinical investigation. 2007;117:185–194. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fayad ZA, Fallon JT, Shinnar M, Wehrli S, Dansky HM, Poon M, Badimon JJ, Charlton SA, Fisher EA, Breslow JL, Fuster V. Noninvasive in vivo high-resolution magnetic resonance imaging of atherosclerotic lesions in genetically engineered mice. Circulation. 1998;98:1541–1547. doi: 10.1161/01.cir.98.15.1541. [DOI] [PubMed] [Google Scholar]
- 33.Frias JC, Williams KJ, Fisher EA, Fayad ZA. Recombinant hdl-like nanoparticles: A specific contrast agent for mri of atherosclerotic plaques. J Am Chem Soc. 2004;126:16316–16317. doi: 10.1021/ja044911a. [DOI] [PubMed] [Google Scholar]
- 34.Feig JE, Hewing B, Smith JD, Hazen SL, Fisher EA. High-density lipoprotein and atherosclerosis regression: Evidence from preclinical and clinical studies. Circulation research. 2014;114:205–213. doi: 10.1161/CIRCRESAHA.114.300760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duivenvoorden R, Tang J, Cormode DP, et al. A statin-loaded reconstituted high-density lipoprotein nanoparticle inhibits atherosclerotic plaque inflammation. Nat Commun. 2014;5:3065. doi: 10.1038/ncomms4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cormode DP, Frias JC, Ma Y, Chen W, Skajaa T, Briley-Saebo K, Barazza A, Williams KJ, Mulder WJ, Fayad ZA, Fisher EA. Hdl as a contrast agent for medical imaging. Clin Lipidol. 2009;4:493–500. doi: 10.2217/clp.09.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang XQ, Even-Or O, Xu X, van Rosmalen M, Lim L, Gadde S, Farokhzad OC, Fisher EA. Nanoparticles containing a liver x receptor agonist inhibit inflammation and atherosclerosis. Adv Healthc Mater. 2015;4:228–236. doi: 10.1002/adhm.201400337. [DOI] [PMC free article] [PubMed] [Google Scholar]