

Abstract
Receptor activator of nuclear factor κB ligand (RANKL) is expressed by a number of cell types to participate in diverse physiological functions. We have previously identified 10 distal RANKL enhancers. Earlier studies have shown that RL-D5 is a multifunctional RANKL enhancer. Deletion of RL-D5 from the mouse genome leads to lower skeletal and lymphoid tissue RANKL, causing a high bone mass phenotype. Herein, we determine the physiological role and lineage specificity of 2 additional RANKL enhancers, RL-D6 and RL-T1, which are located 83 and 123 kb upstream of the gene's transcriptional start site, respectively. Lack of RL-D6 or RL-T1 did not alter skeletal RANKL or bone mineral density up to 48 weeks of age. Although both RL-D5 and RL-T1 contributed to activation induction of T-cell RANKL, RL-T1 knockout mice had drastically low lymphocyte and lymphoid tissue RANKL levels, indicating that RL-T1 is the major regulator of lymphocyte RANKL. Moreover, RL-T1 knockout mice had lower circulating soluble RANKL, suggesting that lymphocytes are important sources of circulating soluble RANKL. Under physiological conditions, lack of RL-D6 did not alter RANKL expression. However, lack of RL-D5 or RL-D6, but not of RL-T1, blunted the oncostatin M and lipopolysaccharide induction of RANKL ex vivo and in vivo, suggesting that RL-D5 and RL-D6 coregulate the inflammation-mediated induction of RANKL in osteocytes and osteoblasts while lack of RL-D6 did not alter secondary hyperparathyroidism or lactation induction of RANKL or bone loss. These results suggest that although RL-D5 mediates RANKL expression in multiple lineages, other cell type– or factor-specific enhancers are required for its appropriate control, demonstrating the cell type–specific and complex regulation of RANKL expression.
Receptor activator of nuclear factor κB ligand (RANKL) is a multifunctional TNF-like cytokine that is expressed in a wide array of cell types (1–6). It is indispensable for osteoclast differentiation and survival (3, 5). Thus, mice lacking this cytokine cannot produce osteoclasts, which leads to a lack of bone resorption and a severely osteopetrotic phenotype (3, 6, 7). In addition to impaired bone resorption, lack of the RANKL has been shown to cause defects in B-cell lymphopoiesis (3) and mammary gland (4) and lymph node development (3, 7). Defects in RANKL-null mice point out RANKL's diverse roles in organ development (1, 3, 4), immune function (1, 3), and skeletal remodeling (3, 6). Consistent with its multifunctionality, expression of this factor from the Tnfsf11 gene locus is regulated by an array of local and systemic factors (8–11) during physiological as well as pathophysiological conditions, including but not limited to lactation (12–14), secondary hyperparathyroidism (13, 15), and inflammatory bone loss (16).
Via in vitro genome-wide analysis, we have previously identified 10 distal RANKL enhancers (Figure 1A), which are located up to 155 kb upstream of the Tnfsf11 transcriptional start site (TSS) (17–22). One of these enhancers, RL-D5, is located 76 kb upstream of the RANKL TSS (18, 23). RL-D5 is a multifunctional enhancer, to which a variety of transcription factors, including vitamin D receptor (VDR) (17, 23), phosphorylated cAMP response element-binding protein (pCREB) (18, 23), signal transducer and activator of transcription 3 (STAT3) (20), and c-FOS (22), bind to regulate RANKL expression in response to numerous hormones and cytokines. On the other hand, other RANKL enhancers located 23, 88, and 123 kb upstream of the gene's TSS, designated RL-D2 (18), RL-D6 (20), and RL-T1 (22), respectively, provide transcriptional regulation in response to a unique hormone or cytokine (Figure 1A). Regulation of RANKL by multiple enhancers that have overlapping roles may suggest coregulation of this gene through multiple enhancers or an evolutionarily conserved redundancy in regulatory regions to ensure control of this important gene. For instance, previous genome-wide studies have shown PTH-induced pCREB binding to both RL-D2 and RL-D5 (18). Recently, we deleted RL-D2 from the mouse genome and showed that RL-D2 coregulates RANKL expression in response to PTH stimulation and contributes significantly to basal RANKL levels in osteoblast lineage cells (24). Importantly, lack of either RL-D2 (24) or RL-D5 (25) leads to a high bone mass phenotype, indicating a functional requirement for both enhancers in vivo. As indicated by this example, although in vitro models are great tools for identifying enhancers, in vivo models are necessary to address the functional interactions and the physiological role of these enhancers.
Figure 1.

Deletion of RL-D6 or RL-T1 RANKL enhancers does not affect bone mass accrual. A, Schematic structure of the Tnfsf11 gene indicating the previously identified enhancer regions (D1–D7 and T1–T3), transcription factors (pCREB, STAT3/STAT5, and cFOS) and nuclear receptors (VDR/retinoid X receptor [RXR]) capable of binding to these enhancers, as well as signaling pathways stimulating these interactions (shown in parentheses). B–E, Body weights (B) and serial dual x-ray absorptiometry BMDs (C–E) of female D6KO (purple line) and T1KO (dark blue line) mice and their littermate controls (D6WT [pink line] and T1WT [light blue line]) were measured up to 24 weeks (W) of age. At each time point, the values represent the mean ± SD of 8 to 11 mice/group. *, P < .05 vs littermate WT group by t test. 1,25(OH)2D3), 1,25-dihydroxyvitamin D3.
RANKL is expressed in multiple cell types ranging from osteoblast lineage cells to lymphocytes (3, 26, 27). Cell type–specific deletions of this cytokine have shown that RANKL produced by different cell types has different physiological functions. For example, osteocyte RANKL is the major functional source for bone remodeling in adult mice (26, 28), whereas B-cell RANKL has been shown to play a role in cancellous bone loss induced by estrogen deficiency but not in bone mass accrual or maintenance (27). The distinct roles of RANKL produced by different cell types highlight the importance of its cell type–specific expression and regulation. A comparison of histone modifications and transcriptional factor binding in genome-wide studies using different cell lines provides clues as to cell type specificity. The multifunctional enhancer of RANKL, namely RL-D5, has regulatory activity in both osteoblast cell lines as well as T-cell lines (19, 21, 22), suggesting that it is a multilineage enhancer. Consistent with this, deletion of RL-D5 from the mouse genome leads to decreased RANKL expression in skeletal tissues as well as lymphoid tissues (25). In contrast, RL-D2 and RL-D6 show regulatory activity only in osteoblast cell lines (17, 18, 20, 21), and RL-T1 shows regulatory activity only in T-cell lines (21, 22), suggesting that these enhancers may be providing lineage-specific regulation of RANKL (Figure 1A). Although these in vitro studies strongly suggest cell type specificity of certain RANKL enhancers, RANKL expression is variable and differentiation stage and passage dependent in different osteoblast cell lines (29–31), pointing to the need for in vivo models to address cell type specificity.
In the last 30 years, advances in the osteoimmunology field and our understanding of the interactions between bone cells, lymphocytes, and inflammatory cytokines have expanded beyond inflammatory bone diseases. We now know that lymphocytes and cytokines including IL-6–type cytokines play roles in many pathophysiological conditions including atherosclerosis and loss of sex steroid– and hyperparathyroidism-induced bone loss (27, 32–36). Whether it is the induction of RANKL by IL-6 (11) or lymphocytes as a source of RANKL (27, 37), RANKL has been one of the cornerstones of immune system–bone cell interactions. Thus, understanding its cell type– and factor-specific expression has great importance. As previously mentioned, genome-wide analysis has suggested that RL-D6 (20) and RL-T1 (22) may be regulating RANKL in a cell type–specific manner in response to IL-6–type cytokines (20) and T-cell activation (22), respectively. Herein, we delete RL-T1 or RL-D6 from the mouse genome and determine the tissue specificity, regulatory, and physiological roles of these enhancers. These murine models demonstrate that individual Tnfsf11 enhancers exhibit unique capabilities relative to cell type–specific RANKL expression and regulation and will be valuable in assessing the contribution of unique cell types to pathophysiological conditions such as atherosclerosis and inflammation- and ovariectomy-induced bone loss.
Materials and Methods
Reagents
αMEM and Hanks' balanced salt solution were purchased from Mediatech/Cellgro, fetal bovine serum, and penicillin-streptomycin-l-glutamine were purchased from HyClone, and ascorbic acid was purchased from Sigma-Aldrich. Primary cell cultures were treated with forskolin (Fsk) purchased from Sigma-Aldrich, oncostatin M (OSM) purchased from R&D Systems, or lipopolysaccharides (LPSs purchased from Sigma-Aldrich. PTH(1–84) used for the in vivo injections was purchased from Bachem California Inc. Dietary calcium-deficient (0.01% calcium) and control (0.516% calcium) diets used for dietary calcium deficiency experiments were purchased from MP Biomedicals.
Generation of mutant mice
RL-D6 knockout (D6KO) and RL-T1 knockout (T1KO) mice were produced by genOway with a strategy similar to that for production of RL-D2 knockout mice (24). In brief, D6KO mice were created by replacing the 1-kb RL-D6 enhancer or 7.4-kb RL-T1 enhancer with a targeting vector consistent of a floxed Neo selection cassette flanked by 5.6-kb-long and 1.6-kb-short homology arms or 6-kb-long and 2.2-kb-short homology arms, respectively. The homology arms, amplified from genomic C57BL/6 embryonic stem (ES) cell DNA, were cloned into a vector to produce the targeting vector containing the Neo selection cassette flanked by the long and short homology arms. The targeting vector was linearized and electroporated into ES cells. Positive selection using G418 treatment, PCR confirmation, and Southern blot analysis of the homology arms was used to identify the ES cells successfully targeted by homologous recombination. These ES cell clones were then injected into C57BL/6 blastocysts to create chimeric mice, and the injected blastocysts were reimplanted into OF1 pseudopregnant females and allowed to develop. The neocassette was removed by crossing the germ line–transmitting chimeric mice with mice ubiquitously expressing Cre recombinase. The D6KO mice were genotyped using the primers 5′-GGTGGAGGTACAGCAAGGGACCA-3′, 5′-TGGCCCACATTCAAATTCCCGGGGT-3′, and 5′- CTGGGGGTATGGGCAGAAGGCA-3′, and the T1KO mice were genotyped using the primers 5′-GGGGCTCATGGTTCAGGCAAGG-3′, 5′-GCGGCCGCTCTAGTATAACTTCG-3′, and 5′-AGGCCGAAGGCTAAGATGGGTT-3′. RL-D6 and RL-T1 heterozygous mice (D6+/− and T1+/−) were used as breeders to obtain the wild-type (WT) and knockout (KO) experimental mice. In all experiments, WT littermates were used as controls for enhancer KO mice.
Animal studies
Mice were housed in high-density ventilated caging in the Animal Research Facility of the University of Wisconsin–Madison. The husbandry rooms were monitored for 12-hour light/dark cycles, 72°F temperature, and 45% humidity. To induce RANKL expression in vivo, animals were injected intraperitoneally with 230 ng/g body weight (bw) PTH(1–84) (in PBS), 10 mg/kg bw LPS (in PBS), or vehicle (PBS). For these injections, the animals were stratified into groups according to their bw. Animals were killed, and tissues were collected 1 hour after PTH injection and 6 hours after LPS injection. For secondary hyperparathyroidism and lactation experiments, animals were stratified into groups based on bone mineral density (BMD). To induce secondary hyperparathyroidism, 1- or 6-month-old mice were fed a dietary calcium-deficient or control diet for 7 days (1-month-old mice) or 30 days (6-month-old mice). For the lactation study, 5-month-old virgin female mice were mated with C57BL6 males and allowed to lactate for 12 days. Animals were killed, and tissues were collected on day 7 or day 30 of dietary calcium deficiency and on day 12 of lactation. All experiments and tissue collections were performed in the procedure rooms in the Research Animal Facility of the University of Wisconsin–Madison. All animal studies were reviewed and approved by the Research Animal Care and Use Committee of the University of Wisconsin–Madison.
Primary stromal cell cultures
Bone marrow was isolated from femurs of female mice, and primary cultures were established as described previously (24). In brief, isolated cells were seeded in 12-well plates at a density of 5 × 106 cells/well and differentiated into osteoblastic stromal cells using osteogenic culture medium. At ∼90% confluence, the differentiated cells were washed with PBS, and the medium was replaced with fresh culture medium containing vehicle or treatments. To induce RANKL expression, differentiated osteoblastic stromal cell cultures were treated with OSM (25 ng/mL) in PBS, LPS (1 μg/mL) in PBS, or Fsk (10−6 M) in dimethylsulfoxide for 24 hours. After 24 hours of treatment, RNA was isolated from cultured cells using TriZOL reagent (Life Technologies) according to the manufacturer's instructions.
BMD analysis
BMDs were measured and analyzed by dual x-ray absorptiometry with a PIXImus densitometer (GE-Lunar Corp) as described previously (38). BMDs of D6KO mice, T1KO mice, and their littermates were measured up to 48 weeks of age. For dietary calcium deficiency, BMD measurements were taken the day before the start of the diet (day 0) and at the end of diet duration, namely day 7 for 1-month-old and day 30 for the 6-month-old animals. For the lactation study, BMDs were measured on day 4 and day 12 of lactation.
Gene expression
Dissected tissues were frozen immediately in liquid nitrogen and stored at −80°C. Frozen tissues were homogenized in TriZOL reagent (Life Technologies), and RNA was isolated according to the manufacturer's instructions. For the in vivo lymphocyte gene expression studies, T and B cells were isolated via positive selection, and their RNA was recovered as described previously (38). For the ex vivo T-cell activation experiments, spleen CD4+ T cells were isolated by negative selection using an EasySep mouse CD4+ T-cell isolation kit (Stemcell Technologies) according to the manufacturer's directions. Isolated CD4+ T cells were plated in 24-well plates with a density of 1 × 106 cells/well and treated 3 hours with vehicle (PBS) or 25 μL/well of CD3/CD28 mouse T-activator Dynabeads (Gibco by Life Technologies). RNA (1 μg) was used as a template to synthesize cDNA using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). RNA isolation and cDNA production was performed in a blinded fashion. Relative mRNA levels were determined via multiplex TaqMan quantitative RT-PCR using VIC-labeled mouse ACTB and FAM-labeled TaqMan gene expression assays (Applied Biosystems). The following TaqMan gene expression probes (Applied Biosystems) were used for RT-PCR: RANKL (Tnfsf11, Mm00441906), IL-6 (Il6, Mm00446190), IL-2 (Il2, Mm00434256_m1), and mouse ACTB (Actb, 4352341E). Relative mRNA levels were calculated using the ΔCt method (39).
Blood chemistry
Cardiac blood was collected at the time of killing. Collected blood was maintained at room temperature for 30 minutes followed by centrifugation at 6000 rpm for 12 minutes to obtain serum. The soluble RANKL (sRANKL) content in the serum was measured using an R&D Systems Quantikine Mouse RANKL kit (catalog no. MTR00) according to the manual provided by the manufacturer.
Statistical evaluation
Data were analyzed using GraphPad Prism 4 software (GraphPad Software, Inc). All values are reported as the means ± SD, and differences between group means were evaluated using two-way ANOVA or Student t test as indicated in the figure legends.
Results
Lack of RL-D6 or RL-T1 does not alter BMD
To address the physiological role of the RANKL enhancers RL-T1 and RL-D6, we deleted these enhancers from the mouse genome. D6KO and T1KO mice were similar in size and body weight to their littermate controls (Figure 1B and Supplemental Figure 1, A–D). It has been shown that lack of RANKL causes defects in mammary gland development (3, 4); however, D6KO or T1KO mice reproduced and lactated normally, suggesting appropriate mammary gland development. Tooth eruption was disrupted in RANKL-null mice (3), whereas all enhancer KO mice have normal tooth eruption (data not shown).
We have previously shown that lack of the multifunctional RL-D5 or PTH regulator RL-D2 leads to high bone mass phenotypes that progress with age (24, 25). Although lack of IL-6 does not affect bone mass (34, 40), deletion of different IL-6–type cytokines or their receptors has been shown to exhibit various skeletal phenotypes (40–42). As previously indicated, RL-D6 mediates not only IL-6 regulation of RANKL expression but also that of other IL-6–type cytokines (20). However, D6KO mice retained BMDs that were similar to those of their WT littermates up to 48 weeks of age (Figure 1, C–E, and Supplemental Figure 2, A and B). On the other hand, consistent with the absence of a skeletal phenotype in mice lacking T- or B-cell RANKL (27), lack of RL-T1 did not alter BMD up to 48 weeks of age (Figure 1, C–E, and Supplemental Figure 2, C and D).
Lack of RL-T1, but not of RL-D6, decreases lymphoid tissue and lymphocyte RANKL
We have previously shown that lack of RL-D5 or RL-D2 leads to a decrease in bone RANKL expression (24, 25). To determine whether RL-D6 or RL-T1 contribute to skeletal RANKL, we measured mRNA levels in tibia, lumbar vertebra 5, and femur shafts of 8-, 24- and 48-week-old enhancer KO mice and their littermate controls. Lack of RL-D6 or RL-T1 did not alter RANKL mRNA levels in any of the skeletal tissues measured (Figure 2, A–D, and Supplemental Figure 3, A–F).
Figure 2.

Lack of RL-T1, but not of RL-D6, results in lower RANKL levels in lymphoid tissues. A–D, RANKL mRNA levels of tibia (Tib), lumbar vertebra 5 (L5), femur shaft (FS), thymus (Thy), spleen (Spl), lung (Lng), and bone marrow (BM) were measured by RT-PCR in 8-week-old (A and C) and 24-week-old (B and D) female D6KO mice (A and B), T1KO mice (C and D), and their WT littermates. The values represent the means ± SD of 8 to 13 mice/group. *, P < .05 vs littermate WT group by t test.
It has been shown that RL-D5 knockout (D5KO) mice have 50% and 25% less spleen RANKL than WT mice at 4 and 20 weeks of age, respectively (25). Lack of RL-D6 did not alter lymphoid tissue RANKL expression at 8, 24, or 48 weeks of age (Figure 2, A and B, and Supplemental Figure 3, A–C). However, lack of RL-T1 resulted in a significant reduction in RANKL expression in the thymus (70%), spleen (50%), and lung (90%) (Figure 2, C and D, and Supplemental Figure 3, D–F). Although lack of RL-T1 did not alter RANKL mRNA levels in the bone marrow of 8-week-old mice (Figure 2C and Supplemental Figure 3D), bone marrow RANKL of male T1KO mice was significantly lower than that of WT littermates at 24 and 48 weeks of age (Supplemental Figure 3, E and F). At 24 weeks of age, there was a trend for a decrease in bone marrow RANKL in female T1KO mice; however, owing to variance, this difference was not significant (Figure 2D). We (27) and others (43) have previously shown that estrogen levels mediate bone marrow B-cell numbers, which contribute to overall bone marrow RANKL. Thus, the estrous cycling–dependent differences in estrogen levels and B-cell numbers of female mice may be one possible explanation for the higher variation in bone marrow RANKL of female mice.
Lymphocytes are proposed to be the major source of RANKL in spleen and thymus. Thus, we next determined whether the decrease in RANKL levels in T1KO lymphoid tissues was due to lower lymphocyte RANKL expression. T and B lymphocytes were isolated from bone marrow and spleens of 4- to 6-month-old enhancer KO mice and their littermate controls. Lack of RL-D6 did not alter lymphocyte RANKL expression (Figure 3, A–D). Lack of RL-T1 led to drastically lower RANKL mRNA levels in B cells (Figure 3, A and B) and T cells (Figure 3, C and D) residing in bone marrow or spleen, whereas lack of RL-D5 only slightly decreased spleen lymphocyte RANKL expression (Figure 3, A–D). These results suggest that although both RL-D5 and RL-T1 contribute to lymphocyte RANKL expression, RL-T1 is the major regulator of RANKL expression in lymphocytes.
Figure 3.

RL-T1 and RL-D5 enhancer regions regulate RANKL expression in lymphocytes. A–D, B cells (A and B) and T cells (C and D) were isolated by positive selection from bone marrow (A and C) and spleens (B and D) of 4- to 6-month-old male D5KO, D6KO, and T1KO mice and their littermate controls. RANKL mRNA levels of the isolated lymphocytes were measured by RT-PCR analysis. The values represent the means ± SD of 3 to 8 mice/group. *, P < .05 vs littermate WT group by t test.
RL-T1 and RL-D5 contribute to activation induction of T-cell RANKL
Having established that RL-T1 is the major regulator of lymphocyte RANKL expression under physiologically normal conditions, we next sought to identify the enhancers responsible for activation induction of T-cell RANKL. During T-cell activation, antigen is recognized by the T-cell antigen–specific receptor/CD3 complex and the costimulatory molecules; this engagement leads to activation of MAPKs, protein kinase C, and calcineurin signaling pathways that activate transcription factors NFAT, NF-κB, and AP1 (FOS/Jun). These transcription factors then mediate gene expression to induce cellular proliferation, differentiation, and cytokine production including expression of RANKL. We have previously shown that upon T-cell activation cFOS binds to RL-D5 and RL-T1, and mutations in AP1 (FOS/Jun) binding sites of RL-D5 or RL-T1 in reporter constructs decreases the activation induction of reporter constructs (22). To test whether RL-D5 and RL-T1 mediate activation induction of T-cell RANKL in a genomic context, we isolated CD4+ T cells from spleen and activated them with CD3/CD28 mouse T-cell activation beads for 3 hours. IL-2 mRNA levels increased to a similar extent in enhancer KO and WT mice, suggesting that the cells were isolated and activated equivalently (Figure 4, A–C). Similar to that observed in pan T cells (Figure 3D), lack of RL-T1 or RL-D5, but not of RL-D6, led to decreased RANKL expression in vehicle-treated CD4+ T cells (Figure 4, D–F). As expected, activation of T cells increased RANKL mRNA levels in all WT cells (Figure 4, D–F). Although lack of RL-D6 did not have an effect (Figure 4D), the absence of RL-T1 or RL-D5 blunted the activation-induced increase in RANKL expression (Figure 4, E and F).
Figure 4.

Lack of RL-T1 or RL-D5 blunts the activation-induced increase in RANKL expression of CD4+T cells. A–F, CD4+ T cells were isolated by negative selection from spleens of 4-month-old female mice. Isolated CD4+ T cells were treated with vehicle (Veh) or activated (Act) with CD3/CD28 Dynabeads for 3 hours. A–F, IL-2 (A–C) and RANKL (D–F) mRNA levels were measured by RT-PCR. All values represent mean fold changes (±SD) relative to that of the WT vehicle-treated group (n = 4–6 wells/group). Statistical comparisons were performed using two-way ANOVA *, P < .05 effect of treatment within genotype; #, P < .05 effect of genotype within treatment.
As stated above, AP1 proteins and cFOS regulate gene expression necessary to induce cell proliferation and differentiation. Consistent with this, cFOS levels decrease significantly as osteoblasts differentiate into postmitotic osteocytes in vitro (44) and in vivo (45), making it less likely for cFOS to play an import role in osteocytes. Moreover, lack of cFOS does not affect the ability of cFOS−/− osteoblasts to support osteoclastogenesis (46), suggesting that lack of cFOS does not alter osteoblast RANKL levels. Thus, because of lack of an effect on osteoblast RANKL in cFOS-null mice, low cFOS levels in osteocytes, cellular processes cFOS plays a role in, and lack of active enhancer histone signatures, we do not expect cFOS binding to RL-T1 to mediate RANKL expression in osteoblast lineage cells.
Lymphocytes contribute significantly to sRANKL in circulation
RANKL is produced as a membrane-bound protein that can be cleaved to soluble form (47, 48). Although measurement of sRANKL levels is common practice, the physiological role or the source of circulating sRANKL is not known. Two possible sources of sRANKL are osteoblast lineage cells and lymphocytes. It has previously been shown that sRANKL levels were normal in mice lacking osteocyte RANKL (24, 26). Because RL-T1 is the major regulator of lymphocyte RANKL expression, we used this mouse model to determine whether lymphocytes contribute to circulating sRANKL levels. To this end, we measured sRANKL levels from sera of enhancer KO mice and showed that sRANKL was 45% lower in the circulation of T1KO and 30% lower in circulation of D5KO mice (Figure 5). Taken together, these results suggest that lymphocytes, but not osteocytes, are a significant source of circulating sRANKL levels. Thus, in mice lacking lymphocyte or osteocyte RANKL circulating sRANKL levels do not correlate with the physiological state of bone remodeling, indicating the physiological importance of local RANKL rather than circulating levels. However, further studies are required to examine the physiological role of soluble vs membrane-bound RANKL.
Figure 5.
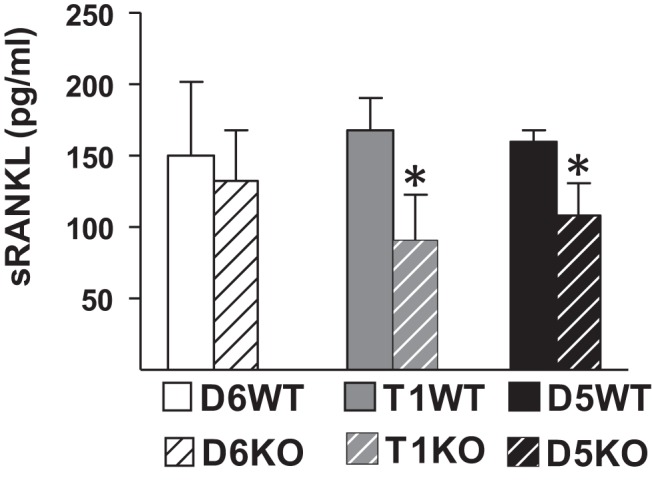
Lack of RL-T1 or RL-D5 decreases circulating RANKL levels. Circulating sRANKL levels of 8-week-old female D6KO, T1KO, and D5KO mice and their littermate controls were measured. The values represent the means ± SD of 3 to 9 mice/group. *, P < .05 vs littermate WT group by t test.
RL-D6 and RL-D5, but not RL-T1, contribute to IL-6–type cytokine induction of RANKL
IL-6–type cytokines increase RANKL expression in osteoblast lineage cells (11). We have previously shown that both IL-6 and OSM induce STAT3 binding to RL-D5 and RL-D6 in osteoblast lineage cells (20), whereas RL-D6 did not show any enhancer activity in T cells (22). In Figure 2, we show that unlike other RANKL enhancers (24, 25), RL-D6 does not contribute to RANKL expression under normal physiological conditions. However, RL-D6 may still be responsible for transcriptional regulation of RANKL in response to IL-6–type cytokine stimulation under pathophysiological conditions such as chronic inflammation. To address this possibility, we treated osteoblastic primary stromal cells with 25 ng/mL OSM, 1 μg/mL LPS, 10−6 M Fsk, or vehicle for 24 hours (Figure 6). OSM, LPS, and Fsk increased IL-6 mRNA levels similarly in WT and KO mice, suggesting that the differentiation and treatments were equivalent (Figure 6, A–C). Consistent with our observations at the whole bone level, only RL-D5–lacking cells had lower RANKL levels than WT cells in the vehicle-treated group (Figure 6, D–F). Lack of RL-D5 and RL-D6 blunted the OSM- and LPS-induced increase in RANKL expression (Figure 6, D and E). Lack of RL-T1 did not alter RANKL expression in response to any of the treatments, suggesting that RL-T1 does not play a role in RANKL regulation in osteoblast lineage cells (Figure 6F).
Figure 6.

OSM and LPS increase RANKL expression via RL-D5 and RL-D6 enhancers. A–F, Ex vivo primary stromal cultures of D5KO (A and D), D6KO (B and E), and T1KO (C–F) cells and their WT controls were treated with vehicle (Veh), 25 ng/mL OSM, 1 μg/mL LPS, or 10−6 M Fsk for 24 hours. A–F, IL-6 (A–C) and RANKL (D–F) mRNA expression in the treated cultures was measured by quantitative RT-PCR and normalized to β-actin mRNA levels. All values represent mean fold changes (±SD) relative to those for the WT vehicle-treated group (n = 3–4 wells/group). Statistical comparisons were accomplished using Student t test. *, P < .05 effect of treatment within the same genotype.
To determine the contribution of RANKL enhancers under in vivo inflammatory conditions, we injected mice with 10 mg/kg LPS or vehicle and measured gene expression 6 hours after a single injection. As expected, LPS injection increased IL-6 mRNA levels similarly in WT and KO mice of all murine lines (Figure 7, A–C), pointing out equivalent injection in all mice. Lack of RL-D5 or RL-D6, but not of RL-T1, blunted the LPS-induced increase in RANKL expression in lumbar vertebrae 5 (Figure 7, D–F) and femur shafts enriched in osteoblast and osteocytes (Figure 7, G–J). These results suggest that both RL-D5 and RL-D6 contribute to IL-6–type cytokine regulation of RANKL expression in osteoblasts and osteocytes and that RL-T1–mediated lymphocyte RANKL does not contribute to the LPS-induced increase in RANKL expression in bone.
Figure 7.

Lack of RL-D5 or RL-D6 blunts the LPS-induced increase in RANKL expression in vivo. A–J, 2- to 4-month-old female D5KO (A, D, and G), D6KO (B, E, and H), and T1KO (C, F, J) mice or their control littermates were injected with vehicle (V) or 10 mg/kg LPS. Tissues were collected 6 hours after a single injection. A–J, IL-6 (A–C) and RANKL (D–F) mRNA levels of lumbar vertebra 5 and RANKL (G–J) mRNA levels of femur shafts were measured by RT-PCR analysis. All values represent mean fold changes (±SD) relative to those for the WT vehicle-treated group (n = 4–7 animals/group). Statistical comparisons were performed using two-way ANOVA. *, P < .05 effect of treatment within genotype; #, P < .05 effect of genotype within treatment.
RL-D6 does not play a role in secondary hyperparathyroidism– or lactation-induced bone loss
PTH increases IL-6 production, and IL-6 has been proposed as an indirect mechanism by which PTH induces bone resorption (49). However, hyperparathyroidism induction of RANKL, bone resorption, or bone loss was not affected in growing IL-6 KO mice (50). Nonetheless, PTH also increases other IL-6–type cytokines and their receptors, which are also inducers of RANKL expression (11, 40, 51). To determine whether the PTH-stimulated increase in IL-6–type cytokines contributes to PTH induction of RANKL as a secondary mechanism, we injected D6KO mice and their littermates with 230 ng/g bw PTH. In 1 hour, PTH significantly increased IL-6 and RANKL mRNA levels in whole bones (Figure 8, A and B) and bone marrow (Figure 8, C and D). However, lack of RL-D6 did not alter the PTH-induced increase in RANKL mRNA levels (Figure 8, A and C).
Figure 8.
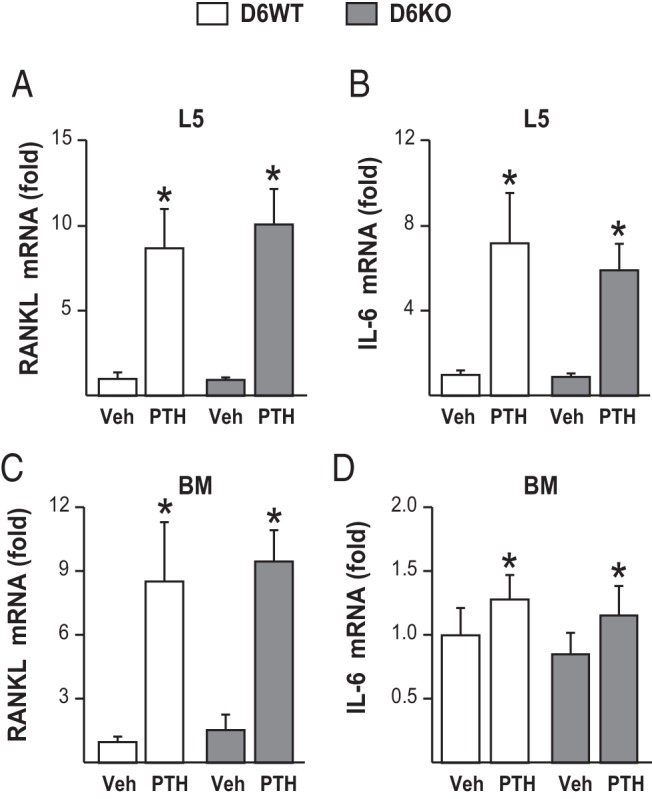
Lack of RL-D6 does not alter the PTH-induced increase in RANKL expression in vivo. Five-month-old female D6KO mice and their WT littermates (D6WT) were injected with 230 ng/g bw PTH or vehicle (Veh). The animals were killed, and tissues were collected 1 hour after injection. A–D, RANKL (A and C) and IL-6 (B and D) mRNA levels in lumbar vertebra 5 (L5) (A and B) and bone marrow (BM) (C and D) were measured by quantitative RT-PCR and normalized to β-actin mRNA levels (n = 4 animals/group). All RT-PCR values represent mean fold changes relative to those for the corresponding WT vehicle-injected control group. All statistical comparisons were performed using two-way ANOVA. *, P < .05 effect of injection within the same genotype; #, P < .05 effect of genotype within the same injection.
We have previously shown that Fsk and OSM act synergistically on RL-D6 to increase RANKL expression in vitro (20). To test whether this synergistic regulation of RANKL via RL-D6 is present in vivo, we examined the role of RL-D6 in 2 bone loss conditions in which both IL-6–type cytokines and PTH/PTHrP signaling are increased, namely secondary hyperparathyroidism and lactation. To induce secondary hyperparathyroidism, 1-month-old and 6-month old mice were fed a calcium-deficient diet for 7 or 30 days, respectively. Dietary calcium deficiency increased RANKL expression similarly in RL-D6 wild-type (D6WT) and D6KO mice at both ages (Figure 9, A and C). Consistent with this, lack of RL-D6 did not alter dietary calcium deficiency–induced bone loss in growing or adult mice (Figure 9, B and D). We then determined whether the RL-D6-mediated increase in RANKL contributes to lactation-induced bone loss. D6KO and D6WT mice had similar numbers of pups and were able to lactate (data not shown). Lactation increased RANKL expression (Figure 10A) and led to bone loss (Figure 10B) equivalently in both D6KO and D6WT mice. These results suggest that unlike our observations in vitro, RL-D6 does not mediate a synergistic control of RANKL by IL-6–type cytokines and PTH/PTHrP signaling in vivo.
Figure 9.

Lack of RL-D6 does not alter the dietary calcium deficiency–induced increase in RANKL expression or bone loss. A–D, 1-month-old female (A and B) and 6-month-old male (C and D) D6KO and control mice were fed a calcium-deficient diet (ca-def diet) or control diet for 7 or 30 days, respectively. A and C, RANKL mRNA levels of lumbar vertebra 5 (L5) were measured with RT-PCR at day 7 in 1-month-old mice (A) and at day 30 in 6-month-old mice (C). B and D, BMDs were measured at the beginning and at the end of diet change and are represented here as percent change. All RT-PCR values represent mean fold changes relative to those for the corresponding WT control diet group. All statistical comparisons were performed using two-way ANOVA. *, P < .05 effect of diet within the same genotype; #, P < .05 effect of genotype within the same diet.
Figure 10.

Lack of RL-D6 does not alter the lactation-induced bone loss. Four-month-old D6WT and D6KO lactating mice were compared with their nulliparous littermates. A, Mice were killed at day 12 of lactation, and RANKL mRNA levels of lumbar vertebra 5 were measured with RT-PCR. B, BMDs were measured at day 4 and day 12 of lactation and are represented here as percent change. All RT-PCR values represent mean fold changes relative to those for the corresponding WT nulliparous expression. All statistical comparisons were accomplished using two-way ANOVA. *, P < .05 effect of lactation within the same genotype; #, P < .05 effect of genotype within lactation state.
Discussion
Transcriptional control of Tnfsf11 is mediated by a complex set of distal enhancers (17–23, 38, 52, 53). RL-D5, which is 76 kb upstream of RANKL TSS, has been deleted from the mouse genome, and the phenotype of the mice lacking RL-D5 has been extensively examined (25). To determine the cell type specificity and physiological role of RL-D6 and RL-T1 enhancers, which are located 88 and 123 kb upstream of the RANKL TSS (20, 22), respectively, we deleted these enhancers from the mouse genome as well. Unlike the D5KO mice, which had lower skeletal RANKL expression and high bone mass phenotype (25), mice that lack RL-D6 or RL-T1 exhibited skeletal RANKL and bone mass that were similar to those of their WT littermates. However, we show here that lack of RL-T1 leads to a drastic decrease in the expression of lymphocyte and lymphoid tissue RANKL in T1KO mice. In addition, RL-T1 did not exhibit any activity in osteoblast lineage cells ex vivo or in vivo, suggesting that it is a hematopoietic lineage–specific RANKL enhancer. While RL-D6 did not regulate RANKL under physiologically normal conditions, lack of RL-D6 and RL-D5 blunted the OSM- and LPS-induced RANKL expression in osteoblast lineage cells ex vivo and in vivo, suggesting that RL-D6 is the IL-6–type cytokine mediator of RANKL expression in osteoblast and osteocytes. PTH increases IL-6–type cytokine expression (11); thus, IL-6–type cytokines may constitute an indirect path by which RANKL is increased in response to PTH (49). However, lack of RL-D6 did not alter hyperparathyroidism- or lactation-induced increases in RANKL expression or bone loss, suggesting that IL-6–type cytokine regulation of RANKL via RL-D6 does not contribute to increased RANKL or bone loss in these conditions. Taken together, these data reveal the complex and cell type–specific regulation of RANKL expression and provide new murine models to study the physiological and pathophysiological role of lymphocyte RANKL and IL-6–type cytokine regulation of RANKL.
Recent advances in our understating of the diverse functions of RANKL produced by different cell types has drawn attention to the importance of cell type–specific regulation of RANKL expression (26–28, 54). Recently, we have shown that a transgene spanning 46 kb downstream to 178 kb upstream of the Tnfsf11 TSS is capable of producing RANKL mRNA in a cell type–specific fashion in vivo (38), suggesting that the DNA elements providing cell type specificity to RANKL are present within the region contained in the transgene. RL-D5 has been shown to regulate RANKL expression in osteoblast lineage cells (23, 25). Lack of RL-D5 has decreased lymphoid tissue RANKL, suggesting that RL-D5 may regulate RANKL expression in lymphocytes (25). Herein, we show that RL-D5 contributed to basal lymphocyte RANKL expression. However, lack of RL-T1 led to a drastic decrease in lymphocyte and lymphoid tissue RANKL expression, much greater than that observed in D5KO mice, suggesting that RL-T1 is the major regulator of basal lymphocyte RANKL expression. In our recent studies, we have also shown that lack of RL-D2 also results in lower RANKL expression in osteoblast lineage cells (24). Taken together, these results suggest that although RL-D5 regulates RANKL expression in response to a wide array of hormones and cytokines regardless of the cell type, other RANKL enhancers such as RL-D2 and RL-T1 are required for maintaining cell type–specific RANKL expression under physiological conditions.
Control of RANKL expression by a wide range of hormones and cytokines has been proposed to play a role in many physiological and pathophysiological conditions (15, 16, 27, 37, 55–57). It was previously shown that in the absence of RL-D5, induction of RANKL by PTH and 1,25-dihydroxyvitamin D3 were blunted in vivo (25). Herein, we show that RL-D5 also contributes to in vivo LPS induction of RANKL and activation induction of T-cell RANKL. However, the contribution of RL-D5 to control of RANKL in response to these factors is incomplete. In a recent study, we have shown that RL-D2 also regulates RANKL expression in response to PTH (24). And herein, we show that RL-D6 and RL-T1 contribute to IL-6–type cytokine and activation induction of RANKL, respectively. Taken together, these results show that RL-D5 collaborates with other cell type– and factor-specific enhancers, such as RL-D2, RL-D6, and RL-T1, to coregulate RANKL expression in a cell type–specific and fine-tuned manner.
Mediation of cell type expression of genes by cell type–specific enhancers depends on the accessibility of the enhancer to transcription factors and the presence of signal-dependent transcription factors in that cell type (58). For example, in osteoblast cell lines RL-D6 is marked as an active enhancer by the histone modification histone H4 acetylation and the presence of RNA polymerase II and is thus accessible to STAT3 binding upon IL-6–type cytokine treatment (20). However, RL-D6 is an inactive enhancer that is not accessible to transcription factors in T cells, as suggested by the lack of histone modifications, RNA polymerase II or transcription factor binding (22). Thus, despite the presence of the IL-6-gp300-STAT3 pathway in T cells (59, 60), we do not expect T-cell RANKL to be mediated by IL-6–type cytokines via RL-D6.
We have previously shown that RL-T1 is marked as an active enhancer by histone modifications and is accessible for transcriptional factor binding in T cells (22). We have previously shown that cFOS binds to RL-T1 and RL-D5 upon T-cell activation and mutating cFOS binding sites blunts the activity of these enhancers in reporter constructs (22). Herein, we show that RL-T1 not only mediates the activation induction of T-cell RANKL but also is the master regulator of T- and B-cell RANKL under physiologically normal conditions. Thus, it would be reasonable to assume that blunting of the activation induction of T-cell RANKL in D5KO or T1KO mice is due to lack of cFOS binding to RL-T1 or RL-D5. However, because cFOS or NFAT is not bound to the RANKL locus in untreated/vehicle-treated T cells, we do not know which transcription factors and signaling pathways mediate the RANKL expression via RL-T1 in T or B cells under physiologically normal conditions. In our future studies, we will use the CRISPR/Cas9 gene editing methodology to determine the mechanism of basal regulation of lymphocyte RANKL expression via RL-T1 in T and B cells.
Cell type–specific deletion of RANKL from different stages of osteoblast lineage (15, 26), synovial fibroblasts (54), and lymphocytes (27, 54) has shown that production of RANKL from different cell types has diverse physiological and pathophysiological roles. We and others have previously deleted RANKL specifically from T cells or B cells and showed that lack of RANKL from either cell type does not alter basal bone remodeling (27, 54). Consistent with these findings, the drastic decrease in RANKL expression in both B and T cells of T1KO mice did not alter bone mass up to 12 months of age, once again indicating that lymphocyte RANKL is not required for bone remodeling under physiologically normal conditions. We also show that lack of RL-D6 does not alter skeletal RANKL or bone mass, which suggests that IL-6–type cytokine mediation of RANKL expression, at least via RL-D6, is not required for bone remodeling under physiologically normal conditions. However, because both RL-D5 and RL-D6 seem to contribute equivalently to IL-6–type cytokine control of osteoblast and osteocyte RANKL expression, IL-6–type cytokine control of RANKL via RL-D5 may be sufficient or compensatory for the lack of RL-D6.
Increases in RANKL expression have been proposed to play a role in a variety of diseases such as dietary calcium deficiency, inflammatory bone loss, and athrosclerosis. Deletions of cell type– or factor-specific enhancers of RANKL have been useful tools in determining the physiological (26–28) and pathophysiological (13, 15, 27, 54) roles of RANKL from different sources, as well as in addressing the necessity of the increase in RANKL expression under these conditions. For instance, we have previously shown that lack of RL-D5 blunts lactation- and dietary calcium deficiency–induced RANKL expression, but not bone loss (13), suggesting that given that the basal levels of RANKL are maintained, the increase in RANKL expression is dispensable for bone loss under these conditions. Herein, we show that lack of RL-D6 does not alter lactation- and dietary calcium deficiency–induced RANKL expression or bone loss, indicating that in these conditions RL-D6-mediated control of RANKL expression by IL-6–type cytokines is also not essential for increased RANKL expression or the bone loss.
In summary, transcriptional control of RANKL is mediated by a complex set of enhancers including RL-D2, RL-D6, and RL-T1, which provide cell type– and factor-specific regulation, and RL-D5, which provides regulation in response to an array of hormones and cytokines regardless of cell type. Because lymphocyte RANKL and IL-6–type cytokine regulation of RANKL have been proposed to contribute to the pathology of inflammatory diseases such as rheumatoid arthritis (16, 37, 54, 61, 62) and atherosclerosis (63–65), T1KO, D5KO, and D6KO mice could be used to establish the role of lymphocytes or IL-6–type cytokines in these disease models, and we are currently investigating the physiological role of RL-T1– and RL-D5–regulated RANKL in atherosclerosis. Thus, these enhancer deletion models not only expand our understanding of the complexity of RANKL's transcriptional control but also provide us with new in vivo murine models to study the role of RANKL regulation in different cell types in health and disease.
Acknowledgments
We thank Charles A. O'Brien for proving the D5KO mice and for helpful discussions. We thank members of the University of Wisconsin Biochemistry Animal Support Staff for their contributions to this effort.
Author contributions: study design, M.O. and J.W.P.; study conduct, M.O.; data collection, M.O., H.C.S., A.L.D., E.M.R., and J.W.M.; data analysis, M.O., data interpretation, M.O. and J.W.P.; drafting manuscript, M.O. and J.W.P.; revising manuscript content, M.O. and J.W.P.; approving final version of manuscript, J.W.P. J.W.P. takes responsibility for the integrity of the data analysis.
Current address for H.C.S.: Proteovista LLC, 510 Charmany Drive, Madison, WI 53719.
Current address for A.L.D.: Northwestern Health Sciences University, Bloomington, MN 55431.
Current address for E.M.R.: Northwestern University, 2145 Sheridan Road, Evanston, IL 60208.
This work was supported by the National Institutes of Health National Institute of Arthritis and Musculoskeletal and Skin Diseases (Grant AR-074993 to J.W.P.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- BMD
- bone mineral density
- bw
- body weight
- D5KO
- RL-D5 knockout
- D5WT
- RL-D5 wild-type
- D6KO
- RL-D6 knockout
- D6WT
- RL-D6 wild-type
- ES
- embryonic stem
- Fsk
- forskolin
- KO
- knockout
- LPS
- lipopolysaccharide
- OSM
- oncostatin M
- pCREB
- phosphorylated cAMP response element-binding protein
- RANKL
- receptor activator of nuclear factor κB ligand
- sRANKL
- soluble RANKL
- Stat3
- signal transducer and activator of transcription 3
- T1KO
- RL-T1 knockout
- T1WT
- RL-T1 wild-type
- TSS
- transcriptional start site
- VDR
- vitamin D receptor
- WT
- wild type.
References
- 1. Hess E, Duheron V, Decossas M, et al. RANKL induces organized lymph node growth by stromal cell proliferation. J Immunol. 2012;188:1245–1254. [DOI] [PubMed] [Google Scholar]
- 2. Hanada R, Leibbrandt A, Hanada T, et al. Central control of fever and female body temperature by RANKL/RANK. Nature. 2009;462:505–509. [DOI] [PubMed] [Google Scholar]
- 3. Kong YY, Yoshida H, Sarosi I, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–323. [DOI] [PubMed] [Google Scholar]
- 4. Fata JE, Kong YY, Li J, et al. The osteoclast differentiation factor osteoprotegerin-ligand is essential for mammary gland development. Cell. 2000;103:41–50. [DOI] [PubMed] [Google Scholar]
- 5. Lacey DL, Timms E, Tan HL, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. [DOI] [PubMed] [Google Scholar]
- 6. Kim N, Odgren PR, Kim DK, Marks SC, Jr, Choi Y. Diverse roles of the tumor necrosis factor family member TRANCE in skeletal physiology revealed by TRANCE deficiency and partial rescue by a lymphocyte-expressed TRANCE transgene. Proc Natl Acad Sci USA. 2000;97:10905–10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim D, Mebius RE, MacMicking JD, et al. Regulation of peripheral lymph node genesis by the tumor necrosis factor family member TRANCE. J Exp Med. 2000;192:1467–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yasuda H, Shima N, Nakagawa N, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci USA. 1998;95:3597–3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. O'Brien CA, Gubrij I, Lin SC, Saylors RL, Manolagas SC. STAT3 activation in stromal/osteoblastic cells is required for induction of the receptor activator of NF-κB ligand and stimulation of osteoclastogenesis by gp130-utilizing cytokines or interleukin-1 but not 1,25-dihydroxyvitamin D3 or parathyroid hormone. J Biol Chem. 1999;274:19301–19308. [DOI] [PubMed] [Google Scholar]
- 10. Lee SK, Lorenzo JA. Parathyroid hormone stimulates TRANCE and inhibits osteoprotegerin messenger ribonucleic acid expression in murine bone marrow cultures: correlation with osteoclast-like cell formation. Endocrinology. 1999;140:3552–3561. [DOI] [PubMed] [Google Scholar]
- 11. Palmqvist P, Persson E, Conaway HH, Lerner UH. IL-6, leukemia inhibitory factor, and oncostatin M stimulate bone resorption and regulate the expression of receptor activator of NF-κB ligand, osteoprotegerin, and receptor activator of NF-κ B in mouse calvariae. J Immunol. 2002;169:3353–3362. [DOI] [PubMed] [Google Scholar]
- 12. Ardeshirpour L, Brian S, Dann P, VanHouten J, Wysolmerski J. Increased PTHrP and decreased estrogens alter bone turnover but do not reproduce the full effects of lactation on the skeleton. Endocrinology. 2010;151:5591–5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Onal M, Galli C, Fu Q, et al. The RANKL distal control region is required for the increase in RANKL expression, but not the bone loss, associated with hyperparathyroidism or lactation in adult mice. Mol Endocrinol. 2012;26:341–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ardeshirpour L, Dann P, Adams DJ, et al. Weaning triggers a decrease in receptor activator of nuclear factor-κB ligand expression, widespread osteoclast apoptosis, and rapid recovery of bone mass after lactation in mice. Endocrinology. 2007;148:3875–3886. [DOI] [PubMed] [Google Scholar]
- 15. Xiong J, Piemontese M, Thostenson JD, Weinstein RS, Manolagas SC, O'Brien CA. Osteocyte-derived RANKL is a critical mediator of the increased bone resorption caused by dietary calcium deficiency. Bone. 2014;66:146–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nakashima T, Takayanagi H. RANKL signal and osteoimmunology [in Japanese]. Clin Calcium. 2011;21:1131–1140. [PubMed] [Google Scholar]
- 17. Kim S, Yamazaki M, Zella LA, Shevde NK, Pike JW. Activation of receptor activator of NF-κB ligand gene expression by 1,25-dihydroxyvitamin D3 is mediated through multiple long-range enhancers. Mol Cell Biol. 2006;26:6469–6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim S, Yamazaki M, Shevde NK, Pike JW. Transcriptional control of receptor activator of nuclear factor-κB ligand by the protein kinase A activator forskolin and the transmembrane glycoprotein 130-activating cytokine, oncostatin M, is exerted through multiple distal enhancers. Mol Endocrinol. 2007;21:197–214. [DOI] [PubMed] [Google Scholar]
- 19. Kim S, Yamazaki M, Zella LA, et al. Multiple enhancer regions located at significant distances upstream of the transcriptional start site mediate RANKL gene expression in response to 1,25-dihydroxyvitamin D3. J Steroid Biochem Mol Biol. 2007;103:430–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bishop KA, Meyer MB, Pike JW. A novel distal enhancer mediates cytokine induction of mouse RANKl gene expression. Mol Endocrinol. 2009;23:2095–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Martowicz ML, Meyer MB, Pike JW. The mouse RANKL gene locus is defined by a broad pattern of histone H4 acetylation and regulated through distinct distal enhancers. J Cell Biochem. 2011;112:2030–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bishop KA, Coy HM, Nerenz RD, Meyer MB, Pike JW. Mouse Rankl expression is regulated in T cells by c-Fos through a cluster of distal regulatory enhancers designated the T cell control region. J Biol Chem. 2011;286:20880–20891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fu Q, Manolagas SC, O'Brien CA. Parathyroid hormone controls receptor activator of NF-κB ligand gene expression via a distant transcriptional enhancer. Mol Cell Biol. 2006;26:6453–6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Onal M, St John HC, Danielson AL, Pike JW. Deletion of the distal Tnfsf11 RL-D2 enhancer that contributes to PTH-mediated RANKL expression in osteoblast lineage cells results in a high bone mass phenotype in mice. J Bone Miner Res. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Galli C, Zella LA, Fretz JA, et al. Targeted deletion of a distant transcriptional enhancer of the receptor activator of nuclear factor-κB ligand gene reduces bone remodeling and increases bone mass. Endocrinology. 2008;149:146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O'Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med. 2011;17:1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Onal M, Xiong J, Chen X, et al. Receptor activator of nuclear factor κB ligand (RANKL) protein expression by B lymphocytes contributes to ovariectomy-induced bone loss. J Biol Chem. 2012;287:29851–29860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nakashima T, Hayashi M, Fukunaga T, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. 2011;17:1231–1234. [DOI] [PubMed] [Google Scholar]
- 29. Udagawa N, Takahashi N, Akatsu T, et al. The bone marrow-derived stromal cell lines MC3T3–G2/PA6 and ST2 support osteoclast-like cell differentiation in cocultures with mouse spleen cells. Endocrinology. 1989;125:1805–1813. [DOI] [PubMed] [Google Scholar]
- 30. Takai H, Kanematsu M, Yano K, et al. Transforming growth factor-beta stimulates the production of osteoprotegerin/osteoclastogenesis inhibitory factor by bone marrow stromal cells. J Biol Chem. 1998;273:27091–27096. [DOI] [PubMed] [Google Scholar]
- 31. Kitazawa R, Kitazawa S, Maeda S. Promoter structure of mouse RANKL/TRANCE/OPGL/ODF gene. Biochim Biophys Acta. 1999;1445:134–141. [DOI] [PubMed] [Google Scholar]
- 32. Weitzmann MN. T-cells and B-cells in osteoporosis. Curr Opin Endocrinol Diabetes Obes. 2014;21:461–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bellido T, Jilka RL, Boyce BF, et al. Regulation of interleukin-6, osteoclastogenesis, and bone mass by androgens. The role of the androgen receptor. J Clin Invest. 1995;95:2886–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Poli V, Balena R, Fattori E, et al. Interleukin-6 deficient mice are protected from bone loss caused by estrogen depletion. EMBO J. 1994;13:1189–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Robinson JW, Li JY, Walker LD, et al. T cell-expressed CD40L potentiates the bone anabolic activity of intermittent PTH treatment. J Bone Miner Res. 2015;30:695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hedrick CC. Lymphocytes in atherosclerosis. Arterioscler Thromb Vasc Biol. 2015;35:253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Guerrini MM, Takayanagi H. The immune system, bone and RANKL. Arch Biochem Biophys. 2014;561:118–123. [DOI] [PubMed] [Google Scholar]
- 38. Onal M, Bishop KA, St John HC, et al. A DNA segment spanning the mouse Tnfsf11 transcription unit and its upstream regulatory domain rescues the pleiotropic biologic phenotype of the RANKL null mouse. J Bone Miner Res. 2015;30:855–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- 40. Sims NA, Jenkins BJ, Nakamura A, et al. Interleukin-11 receptor signaling is required for normal bone remodeling. J Bone Miner Res. 2005;20:1093–1102. [DOI] [PubMed] [Google Scholar]
- 41. Bozec A, Bakiri L, Hoebertz A, et al. Osteoclast size is controlled by Fra-2 through LIF/LIF-receptor signalling and hypoxia. Nature. 2008;454:221–225. [DOI] [PubMed] [Google Scholar]
- 42. Sims NA. gp130 signaling in bone cell biology: multiple roles revealed by analysis of genetically altered mice. Mol Cell Endocrinol. 2009;310:30–39. [DOI] [PubMed] [Google Scholar]
- 43. Kanematsu M, Sato T, Takai H, Watanabe K, Ikeda K, Yamada Y. Prostaglandin E2 induces expression of receptor activator of nuclear factor-κB ligand/osteoprotegrin ligand on pre-B cells: implications for accelerated osteoclastogenesis in estrogen deficiency. J Bone Miner Res. 2000;15:1321–1329. [DOI] [PubMed] [Google Scholar]
- 44. St John HC, Bishop KA, Meyer MB, et al. The osteoblast to osteocyte transition: epigenetic changes and response to the vitamin D3 hormone. Mol Endocrinol. 2014;28:1150–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Oyama M, Chiba J, Kato Y, et al. Distribution and expression of mRNAs for the proto-oncogenes c-fos and c-jun in bone cells in vivo. Histol Histopathol. 1998;13:671–678. [DOI] [PubMed] [Google Scholar]
- 46. Grigoriadis AE, Wang ZQ, Cecchini MG, et al. c-Fos: a key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science. 1994;266:443–448. [DOI] [PubMed] [Google Scholar]
- 47. Lum L, Wong BR, Josien R, et al. Evidence for a role of a tumor necrosis factor-alpha (TNF-α)-converting enzyme-like protease in shedding of TRANCE, a TNF family member involved in osteoclastogenesis and dendritic cell survival. J Biol Chem. 1999;274:13613–13618. [DOI] [PubMed] [Google Scholar]
- 48. Kanamaru F, Iwai H, Ikeda T, Nakajima A, Ishikawa I, Azuma M. Expression of membrane-bound and soluble receptor activator of NF-κB ligand (RANKL) in human T cells. Immunol Lett. 2004;94:239–246. [DOI] [PubMed] [Google Scholar]
- 49. Grey A, Mitnick MA, Masiukiewicz U, et al. A role for interleukin-6 in parathyroid hormone-induced bone resorption in vivo. Endocrinology. 1999;140:4683–4690. [DOI] [PubMed] [Google Scholar]
- 50. O'Brien CA, Jilka RL, Fu Q, Stewart S, Weinstein RS, Manolagas SC. IL-6 is not required for parathyroid hormone stimulation of RANKL expression, osteoclast formation, and bone loss in mice. Am J Physiol Endocrinol Metab. 2005;289:E784–793. [DOI] [PubMed] [Google Scholar]
- 51. Walker EC, Poulton IJ, McGregor NE, et al. Sustained RANKL response to parathyroid hormone in oncostatin M receptor-deficient osteoblasts converts anabolic treatment to a catabolic effect in vivo. J Bone Miner Res. 2012;27:902–912. [DOI] [PubMed] [Google Scholar]
- 52. Nerenz RD, Martowicz ML, Pike JW. An enhancer 20 kilobases upstream of the human receptor activator of nuclear factor-κB ligand gene mediates dominant activation by 1,25-dihydroxyvitamin D3. Mol Endocrinol. 2008;22:1044–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bishop KA, Wang X, Coy HM, Meyer MB, Gumperz JE, Pike JW. Transcriptional regulation of the human TNFSF11 gene in T cells via a cell type-selective set of distal enhancers. J Cell Biochem. 2015;116:320–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Danks L, Komatsu N, Guerrini MM, et al. RANKL expressed on synovial fibroblasts is primarily responsible for bone erosions during joint inflammation. Ann Rheum Dis. In press. [DOI] [PubMed] [Google Scholar]
- 55. Takayanagi H. Inflammatory bone destruction and osteoimmunology. J Periodontal Res. 2005;40:287–293. [DOI] [PubMed] [Google Scholar]
- 56. Komatsu N, Takayanagi H. Autoimmune arthritis: the interface between the immune system and joints. Adv Immunol. 2012;115:45–71. [DOI] [PubMed] [Google Scholar]
- 57. Xiong J, O'Brien CA. Osteocyte RANKL: new insights into the control of bone remodeling. J Bone Miner Res. 2012;27:499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Heinz S, Romanoski CE, Benner C, Glass CK. The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol. 2015;16:144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Takeda K, Kaisho T, Yoshida N, Takeda J, Kishimoto T, Akira S. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J Immunol. 1998;161:4652–4660. [PubMed] [Google Scholar]
- 60. Nishihara M, Ogura H, Ueda N, et al. IL-6-gp130-STAT3 in T cells directs the development of IL-17+ Th with a minimum effect on that of Treg in the steady state. Int Immunol. 2007;19:695–702. [DOI] [PubMed] [Google Scholar]
- 61. Robak T, Gladalska A, Stepie H, Robak E. Serum levels of interleukin-6 type cytokines and soluble interleukin-6 receptor in patients with rheumatoid arthritis. Mediators Inflamm. 1998;7:347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wong PK, Campbell IK, Egan PJ, Ernst M, Wicks IP. The role of the interleukin-6 family of cytokines in inflammatory arthritis and bone turnover. Arthritis Rheum. 2003;48:1177–1189. [DOI] [PubMed] [Google Scholar]
- 63. Sandberg WJ, Yndestad A, Øie E, et al. Enhanced T-cell expression of RANK ligand in acute coronary syndrome: possible role in plaque destabilization. Arterioscler Thromb Vasc Biol. 2006;26:857–863. [DOI] [PubMed] [Google Scholar]
- 64. Montecucco F, Steffens S, Mach F. The immune response is involved in atherosclerotic plaque calcification: could the RANKL/RANK/OPG system be a marker of plaque instability? Clin Dev Immunol. 2007;2007:75805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ait-Oufella H, Taleb S, Mallat Z, Tedgui A. Recent advances on the role of cytokines in atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31:969–979. [DOI] [PubMed] [Google Scholar]