

Abstract
Background
The etiology of liver disease remains elusive in some adults presenting with severe hepatic dysfunction.
Methods and results
Here we describe a woman of Pakistani descent who had elevated aminotransferases at age 23. She developed muscle weakness in her mid-20s, and was diagnosed with hepatocellular carcinoma at age 29. She died without a diagnosis at age 32 after having a liver transplant. Exome sequencing revealed that she was homozygous for a missense mutation (R49H) in AHCY, the gene encoding S-adenosylhomocysteine (SAH) hydrolase. SAH hydrolase catalyzes the final step in conversion of methionine to homocysteine and inactivating mutations in this enzyme cause a rare autosomal recessive disorder, SAH hydrolase deficiency, that typically presents in infancy. An asymptomatic 7-year old son of the proband is also homozygous for the AHCY-R49H mutation and has elevated serum aminotransferase levels, as well as markedly elevated serum levels of SAH, S-adenosylmethionine (SAM), and methionine, which are hallmarks of SAH hydrolase deficiency.
Conclusion
This report reveals several new aspects of SAH hydrolase deficiency. Affected women with SAH hydrolase deficiency can give birth to healthy children. SAH hydrolase deficiency can remain asymptomatic in childhood, and the disorder can be associated with early onset hepatocellular carcinoma. The measurement of serum amino acids should be considered in patients with liver disease or hepatocellular carcinoma of unknown etiology.
Keywords: Hepatocellular carcinoma, Methionine, AHCY, Exome sequencing, Genetics
1. Introduction
The liver plays a key role in the metabolism of methionine, an essential amino acid in metazoa [1,2]. In mammals, the two pathways for methionine metabolism, the methionine cycle and the transsulfuration sequence, share the first three reactions in common. In both pathways, methionine is converted to S-adenosylmethionine, which functions as a methyl donor in diverse transmethylation reactions. These reactions yield methylated DNA, RNA and proteins plus S-adenosylhomocysteine, which is then cleaved to homocysteine and adenosine by S-adenosylhomocysteine (SAH) hydrolase. Approximately 50% of the methionine in humans is metabolized in the liver [1,2]. The medical importance of these pathways is revealed by the effects of genetic mutations that disrupt them in humans [3–6]. Such disruptive mutations lead to the accumulation of upstream metabolites, deficiencies in downstream metabolites, and a wide spectrum of clinical phenotypes, including neurological abnormalities, liver disease, and muscle weakness. A total of eight cases of SAH hydrolase deficiency has been described [6–12] and in all cases, the disease has manifested in infancy or early childhood.
Here we describe a family with a highly atypical presentation of SAH hydrolase deficiency. The proband is the offspring of a consanguineous mating who presented with liver disease of unknown etiology at age 23. She required liver transplantation at age 30 years and subsequent to her surgery, she developed severe muscle weakness and died at age 32 years. Exome sequencing performed post-mortem revealed homozygosity for a missense mutation in the gene encoding SAH hydrolase (AHCY). One of her two children, an asymptomatic 7-year old boy, is homozygous for the same mutation. Here, we describe the clinical course of the mother and the initial management of her affected son.
2. Patients and methods
2.1. Human subjects
The study protocol was approved by the Institutional Review Board of the University of Texas Southwestern Medical Center. Venous blood was collected from the proband after obtaining written informed consent. Genomic DNA was extracted from the leukocytes using an Autopure LS DNA extractor (Qiagen, Germantown, MD). Plasma and serum were isolated, aliquoted and stored at −80 °C. The proband’s medical history and family medical history were obtained from medical records and from interviews with relatives. Blood samples were also collected from her husband and two sons, who were referred to the pediatric metabolism clinic for clinical testing and treatment. No tissue samples were available from any of the other relatives of the proband.
2.2. Genotyping and DNA sequencing
The proband was genotyped using the Human-Omni5-4 BeadChip microarray (Illumina, San Diego, CA). Allele calls from the array data were generated using GenomeStudio. Of the 4,292,096 SNPs assayed, 4,283,655 (99.8%) were successfully called (GenCall Score > 0.15).
For exome sequencing, 3 μg of genomic DNA was sonicated using a Covaris S2 ultrasonicator (Covaris, Woburn, MA), purified, and assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). DNA was end-repaired, and 3′ ends were adenylated and barcoded with truncated adapters. PCR amplified libraries were purified with AmpureXP beads (New England Biolabs, Ipswich, MA) and assayed using an Agilent 2100 Bioanalyzer. A 750 ng aliquot of the fragment library was concentrated by vacuum to 3.5 μL and hybridized and captured with a SureSelect Human All Exon V4 kit (Agilent Technologies, Santa Clara, CA). Following hybridization the captured library was amplified and index tags were added to the adapters. DNA was again purified with AmpureXP beads, and fragment sizes were assayed using the Agilent 2100 Bioanalyzer. Paired-end sequencing (150 basepairs) was performed using an Illumina Hiseq 2000. Sequences were aligned to the human reference genome b37, and variants were called using the Genome Analysis Toolkit (GATK) HaplotypeCaller [13].
Homozygosity mapping was performed using PLINK. Runs of homozygosity (ROHs) were detected by a sliding window algorithm with a window size of 100 SNPs [14,15]. ROHs longer than 1 Mb were used to assist in filtering the variants identified by exome sequencing, since runs of this length are uncommon in the general population [16].
Variant alleles with a minor allele frequency (MAF) of less than 1% in the 1000 Genomes Project (http://www.1000genomes.org) and the Exome Aggregation Consortium (ExAC) [17] databases that were homozygous in the proband were collated and filtered for potentially pathogenic variants (missense, nonsense, splice-site, or frameshift) located within ROH intervals >1 Mb. Selected mutations were confirmed by Sanger DNA sequencing PCR-amplified fragments containing the nucleotide substitution.
The oligonucleotides used to confirm the mutation identified in AHCY were the following: 5′-TGCGGTGACAGAGTGCTAAG-3′ and 5′-ACCGAGTGAGAGGGAGGAAC-3′.
2.3. Clinical chemistry
Serum amino acids, homocysteine, and aminotransferases were measured using standard biochemical methods at Children’s Medical Center, University of Texas Southwestern Medical Center. SAH and S-adenosylmethionine (SAM) levels were measured by tandem mass spectrometry (Shimadzu Nexera LC System interfaced with a 5500QTRAP® Sciex) at the Institute for Metabolic Disease at Baylor University Medical Center, Dallas, TX as previously described [18].
3. Results
3.1. Hepatocellular carcinoma of unknown etiology
The proband (III.2, Fig. 1) was a 32 year old woman of Pakistani descent, whose parents were first-cousins. She was the product of a term pregnancy and developed normally. She had a self-limited episode of jaundice at age 7 that was attributed to hepatitis A, though no clinical testing was performed at the time. She was not a good student, starting in elementary school. She never underwent psychometric or intelligence testing. At age 23 she married and migrated to the United States where she worked in the home. Soon thereafter she developed pneumonia and was found to have elevated aminotransferase (AST 56 U/L [ref. range 10–40 U/L], ALT 107 U/L [ref. range 7–56 U/L]), and serum creatine phosphokinase levels (615 to 1256 U/L [ref. range: <145 U/L]) with a normal level of bilirubin (0.7 mg/dL [ref. range 0.2–1.5 mg/dL]) and reduced plasma albumin level (2.1 g/dL [ref. range 3.5–5.5 g/dL]). Her coagulation studies were abnormal. Her PT was 37 s (ref. range 9.5–13.5 s) with an INR of 3.7 (ref. range 0.8–1.3). Her PTT was 25 s (ref. range 25–35 s). She tested negative for hepatitis B surface Ag and core IgM, hepatitis C Ab, and hepatitis A IgM. She tested positive for hepatitis A IgG, thus indicating a prior exposure to this virus. Computed tomography (CT) and ultrasonography of her liver revealed no abnormalities. Six months later, her INR was 1.7, PT was 19 s, PTT was 25 s, and GGT was 13 U/L (ref. range <30 U/L).
Fig. 1.
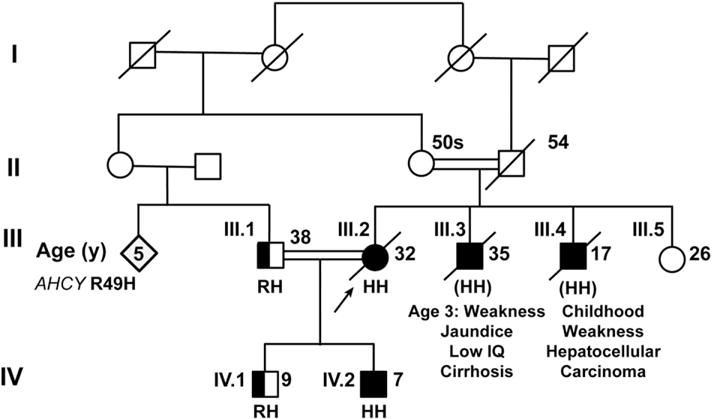
Pedigree of Pakistani family with SAH hydrolase deficiency. The arrow indicates the proband (III.2). The ages provided are current ages or the age at time of death. Filled-in boxes and circles represent affected or presumably affected males and females, respectively. Half filled-in boxes represent unaffected heterozygous carriers for R49H. Unfilled boxes and circles represent unaffected family members. The amino acid present at amino acid 49 is provided (R, arginine or H, histidine) for all those in whom genomic DNA was available. (HH) indicates that the mutation was inferred in III.3 and III.4.
At age 24, when she was 20 weeks pregnant, she presented with thrombocytopenia accompanied by a hypochromic, microcytic anemia. Her reticulocyte count was normal but her serum aminotransferase levels were elevated (AST 56 U/L, ALT 99 U/L). Two months later, her AST was 159 U/L, ALT was 98 U/L and her plasma albumin was 3.0 g/dL (ref. range in the third trimester 2.3–4.2 g/dL), which fell further to 2.5 g/dL during the pregnancy. Abdominal ultrasound showed a normal liver with moderate splenomegaly. After delivery, her aminotransferase levels returned to baseline, but her plasma albumin level remained slightly decreased (3.4 g/dL). Therefore, she underwent a liver biopsy, which revealed normal histology without any evidence of fibrosis or inflammation. She gave birth to a second son at age 26. After the birth, she noted increasing muscle weakness and fatigue, though she did not have difficulty walking or taking care of her children.
At age 29, she presented with elevated levels of aminotransferases (AST 63 U/L, ALT 105 U/L), an increased INR (3.4) and reduced plasma levels of albumin (1.9 g/dL). She had a markedly elevated level of serum alpha-fetoprotein (1426 ng/mL [ref. range <50 ng/mL]). Her rheumatoid factor was 36 U/mL (ref. range <15 U/mL), and her plasma levels of copper, ceruloplasmin and α1-antitrypsin were normal. She did not have any detectable antibodies to mitochondria, Jo-1, centromeres, dsDNA, SCL-70, SSA, SSB, RNP, Smith antigen and smooth muscle antigen. Ultrasonography, CT scan, and magnetic resonance imaging (MRI) of her abdomen showed several focal hepatic lesions. Biopsies of one of the lesions revealed a well-differentiated hepatocellular carcinoma. Histology was negative for PAS and iron staining. She underwent liver transplantation at age 30.
Over the ensuing 14 months, she developed muscle weakness that was initially attributed to her immunosuppressive therapy, which included glucocorticoids. She could walk only short distances. Cessation of glucocorticoid treatment failed to improve her muscle strength. Her creatine kinase level was 3069 U/L 6 months after liver transplantation, and two weeks later the level remained elevated at 639 U/L. Electromyography and nerve-conduction studies showed a myopathic pattern in the proximal musculature with no irritative features, and a mild sensorimotor neuropathy with primarily axonal features. MRI of the legs showed muscular atrophy, but no inflammation. A muscle biopsy from the soleus was performed and showed myofiber type 2 atrophy and a possible very low-grade neuropathic process. There was no myocyte necrosis, myocyte regeneration, inflammatory infiltrates, or evidence of vasculitis. No ragged red fibers were seen. Electron microscopy was unremarkable; no dystrophic processes or myelin bodies were seen.
At age 31, the proband developed malaise and shortness of breath and was admitted to an intensive care unit for 3 weeks. Four months later, she fell and suffered a hip fracture and remained confined to her bed until she died 8 months later at age 32.
The proband (III.2) and her husband (III.1) were both from Punjab, Pakistan and their mothers were sisters (Fig. 1). The proband had two brothers; the older brother (III.3) developed muscle weakness starting at age 3 and had multiple episodes of jaundice during childhood. A muscle biopsy was suggestive of a mitochondrial myopathy. He never learned to read, write, or drive a car and died in Pakistan of cirrhosis at age 35. The proband’s younger brother (III.4) had muscle weakness during childhood and died at age 17 years in Pakistan of hepatocellular carcinoma. Her younger sister (III.5) was healthy at age 26, living in Pakistan.
3.2. Homozygosity mapping and exome sequencing identified a mutation in AHCY
SNP analysis using the HumanOmni5-4 BeadChip kit revealed 42 ROHs greater than 1 Mb in the proband, with the longest segment being ~23.0 Mb. Exome sequencing was performed as described in the methods. The median depth of coverage was 107-fold, and 97.5% of the exome was read at ≥20×. Based on the pedigree and family history, an autosomal recessive mutation was considered the most likely cause of her disease. A total of 33 rare (frequency < 1% in the ExAC database), potentially pathogenic (missense, nonsense, splice-site, or frameshift) homozygous mutations were identified and 23 of these variants were located in ROHs. One of these was a missense mutation in AHCY (c.146G > A, p.R49H), which was located in an ROH of 7.4 Mb. The missense mutation was confirmed by Sanger sequencing.
AHCY encodes SAH hydrolase, a 432 amino acid protein that catalyzes the hydrolysis of SAH to yield adenosine and homocysteine (Fig. 2A). S-Adenosylhomocysteine binds the protein between residues 28 and 132. The enzyme has a split catalytic domain (AA 1–181 and AA 352–402) that flanks a nicotinamide adenine dinucleotide (NAD)-binding domain (AA 182–351) (Fig. 2B) [19]. The mutation results in the substitution of histidine for arginine in residue 49, which is located in the SAH binding domain. The arginine at this position is highly conserved among different species (Fig. 2C). The mutation was present in a single individual in the ExAC browser, a database of exonic variants identified by exome sequencing of 60,706 individuals [17].
Fig. 2.
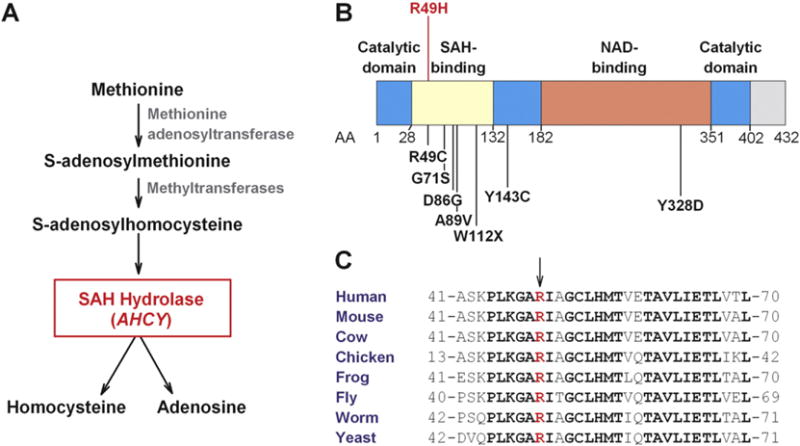
The metabolic pathway (A), structural domains (B), and amino acid sequence conservation (C) of SAH hydrolase. A) Methionine is converted to S-adenosylmethionine (SAM) and then to S-adenosylhomocysteine (SAH). SAH hydrolase, which is encoded by AHCY, hydrolyzes S-adenosylhomocysteine to form homocysteine and adenosine. Inactivation of SAH hydrolase results in accumulation of SAH, SAM, and methionine in tissues. B) Schematic of SAH hydrolase with the location of the mutations that have been identified in patients with SAH hydrolase deficiency (see Table 2). The functional domains have been mapped as described [19]. AA, amino acid; NAD, nicotinamide adenine dinucleotide. C) Alignment of amino acid sequences from different species in the region of SAH hydrolase that flanks R49H. Bold letters denote residues that are conserved among all species shown. The R49 residue is red.
Diallelic inactivating mutations in AHCY cause SAH hydrolase deficiency (OMIM: 180960). Mutations are clustered in the SAH-binding domain (Fig. 2B). The same residue (R49) that was mutated in the proband was previously found to be mutated to a cysteine residue in three patients with SAH hydrolase deficiency, [8,9] including two sisters from Texas of unreported ethnicity who were born with severe hypotonia, fetal hydrops and brain anomalies [9]. The two sisters died at 1 and 4 months of age. Another patient from the Czech Republic was a compound heterozygote for this mutation plus a substitution of serine for glycine at residue 71; she presented with hypotonia and developmental delay during infancy and with hepatic decompensation at 4.5 years [8].
3.3. Biochemical confirmation of SAH hydrolase deficiency in a son of the proband
The family of the proband was screened for the presence of the missense mutation in AHCY. Her husband (III.1), who was her first cousin, and her 9 year-old son (IV.1) were both heterozygous for the AHCY-49H variant. Her 7-year-old son (IV.2) was homozygous for the substitution (Fig. 1).
Both sons were referred to a pediatrician for evaluation. The 9 year-old son, who is heterozygous for the mutation, had normal serum levels of aminotransferases and methionine (22 μmol/L, ref. range 6–60 μmol/L) but mildly elevated levels of SAH (33 nmol/L, ref. range 13–28 nmol/L) and SAM (105 nmol/L, ref. range 33–95 nmol/L). The younger son, who was homozygous for the R49H mutation, had moderately elevated serum levels of ALT and AST (167 U/L and 155 U/L, respectively), and reduced albumin (3.0 g/dL, ref. range in 7–9 year olds: 3.7–5.6 g/dL). Serum levels of SAH were markedly elevated (3260 nmol/L), as were levels of SAM (1930 nmol/L) and methionine (528 μmol/L) (Table 1). This son was the product of an uncomplicated delivery and had no medical problems except for mild jaundice at birth that resolved without medical intervention. He has been a good student and is currently in grade school and participates in sports without difficulty.
Table 1.
Serum chemistries of affected son (IV.2).
| Initial | 4 weeks on low methionine diet | 2 weeks after dietary compliance deteriorated | Reference range | |
|---|---|---|---|---|
| Methionine (μmol/L) | 528 | 10 | 10 | 6–60 |
| Homocysteine (μmol/L) | 21.2 | 8.6 | – | 4–15 |
| SAM (nmol/L) | 1930 | 162 | 406 | 33–95 |
| SAH (nmol/L) | 3260 | 133 | 850 | 13–28 |
| ALT (U/L) | 167 | 31 | 66 | 7–56 |
| AST (U/L) | 155 | 34 | 71 | 10–40 |
Abbreviations: SAM: S-adenosylmethionine. SAH: S-adenosylhomocysteine. ALT: alanine aminotransferase.AST: aspartate aminotransferase.
On physical exam the affected child was an alert, well developed, well-nourished boy in no acute distress. He weighed 27.3 kg and was 127 cm tall (body mass index = 17 kg/m2). He was anicteric and had no hepatosplenomegaly. He had normal muscle mass, muscle tone and strength. No neurologic deficits or localized weakness was found.
3.4. Treatment of SAH hydrolase deficiency in the affected son
The son was started on a low methionine diet. The target protein content of the diet was 5–10 g/day with 40 g/day of protein supplementation that contained no methionine. He initially took half of the protein supplement that was prescribed, significantly reducing the protein in his diet. Blood samples were obtained 4 weeks after initiation of the dietary treatment. His aminotransferases and methionine levels normalized and his SAH and SAM levels dropped remarkably (Table 1).
Thereafter, the father reported that he was no longer able to get his son to take even a quarter of the prescribed amount of medical protein. The aminotransferases, and SAH and SAM increased but the methionine level remained within normal limits (Table 1). A gastrostomy tube is being placed with plans to implement direct gastric feeding.
4. Discussion
4.1. SAH hydrolase deficiency cases in the literature
The family described in this study extends the phenotype of SAH hydrolase deficiency and raises important questions regarding clinical management of this rare disorder. The 32-year old proband is, to our knowledge, the oldest person identified with SAH hydrolase deficiency and the first who had hepatocellular carcinoma. The development of hepatocellular carcinoma in her 17 year old brother suggests that hepatic malignancy may be a major complication in SAH hydrolase-deficient patients who survive infancy. The proband’s 7 year-old son is the first patient to be diagnosed pre-symptomatically with SAH hydrolase deficiency. His biochemical response to, and subsequent inability to comply with, dietary intervention highlights potential therapeutic approaches and problems of treating this disorder.
Eight cases of SAH hydrolase deficiency have been reported in the literature (Table 2). The disorder was first identified by Barić et al. [6] in 2004. The first patient (#1, Table 2) was a 5 month old Croatian infant who had delayed psychomotor development and hypotonia since birth accompanied by elevated serum aminotransferase levels. Liver biopsy revealed mild chronic inflammation and fibrosis. Electron microscopy of hepatocytes showed hyperplasia of the smooth endoplasmic reticulum, a scarcity of rough endoplasmic reticulum, and numerous small mitochondria with sparse cristae. Amino acid analysis revealed markedly elevated serum levels of methionine, SAH, and SAM. Inactivating mutations (Y143C and W112X) were identified in both his maternal and paternal SAH alleles. Biochemical assays in his red blood cells, cultured skin fibroblasts, and liver revealed a residual SAH hydrolase activity of 3% in liver and 5–10% in red blood cells and fibroblasts [6]. Two of the proband’s brothers (#2 and #3, Table 2) also had symptomatic SAH hydrolase deficiency diagnosed during infancy [10,12].
Table 2.
Patients with SAH hydrolase deficiency.
| # | Mutations | Age of onset | Sex | Country of origin | Liver | Muscle | Cognitive | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | W112X/Y143C | <1 month | M | Croatia | Hepatitis, hepatic fibrosis | Hypotonia/weakness | Delayed development | [6] |
| 2 | W112X/Y143C | <1 month | M | Croatia | ? | Hypotonia/weakness | Delayed development | [12] |
| 3 | W112X/Y143C | <1 month | M | Croatia | ? | Hypotonia/weakness | ? | [10] |
| 4 | R49C/D86G | <1 month (1 monthb) | F | USA (unreported ethnicity) | Likely liver disease | Hypotonia/weakness | ? | [9] |
| 5 | R49C/D86G | <1 month (4 monthsb) | F | USA (unreported ethnicity) | Liver disease | Hypotonia/weakness | ? | [9] |
| 6 | Y143C/Y328D | 8 months | F | USA (European descent) | Elevated aminotransferases | Hypotonia/weakness | Delayed development | [7] |
| 7 | R49C/G71S | <1 month | F | Czech Republic | Hepatic decompensation | Hypotonia/weakness | Delayed development | [8] |
| 8 | Y143C/A89V | <1 month | M | USA (unreported ethnicity) | At 14 months: liver biopsy ➔ large lipid droplets | Hypotonia/weakness | IQ 64 at age 20 years | [11] |
| 9 | R49H/R49H | 23 years (32b) | F | Pakistan | Hepatocellular carcinoma | Weakness | Low IQ | |
| 10 | (R49H/R49H)a | 3 years (35b) | M | Pakistan | Cirrhosis | Weakness | Low IQ | |
| 11 | (R49H/R49H)a | ? (17b) | M | Pakistan | Hepatocellular carcinoma | Weakness | ? | |
| 12 | R49H/R49H | Asymptomatic at 7 years | M | Pakistan | Elevated aminotransferases | Normal | Normal |
Cases 9–12 are described in the present report. Question marks indicate no information on phenotype available.
Mutations not determined, but likely to have been R49H homozygotes.
Died at indicated age.
An additional 5 patients from 4 families have been described with SAH hydrolase deficiency. Two sisters of unreported ethnicity from Texas (#4 and #5, Table 2) had severe hypotonia and fetal hydrops; they died at 1 and 4 months of age [9]. A Czech girl (#6, Table 2) had hypotonia and psychomotor retardation since birth and liver disease since infancy [8]. Recently, an 8 month-old girl (#7, Table 2) with developmental delay, hypotonia, and elevated aminotransferase levels due to SAH hydrolase deficiency was reported. Dietary methionine restriction reduced, but failed to normalize the levels of SAH and SAM in her serum so she underwent liver transplantation at 40 months of age [7]. Within six months, her serum SAH and SAM levels had normalized and her growth and development had appeared to accelerate.
The oldest patient with SAH hydrolase deficiency described prior to this report was a 26-year old man (#8, Table 2) who was heterozygous for two AHCY mutations: Y143C and A89V [11]. Unlike our proband, he presented in infancy with hypotonia and mild developmental delay. At 5 months of age he was noted to have elevated serum levels of methionine (800 μmol/L), CPK, LDH, and AST. Dietary methionine restriction resulted in a fall in his serum methionine to near-normal levels (between 10 and 60 μmol/L). The restricted diet was maintained for 5 years, but it did not improve the patient’s hypotonia or developmental delay, and was therefore discontinued. At age 20, his global IQ was 64. The SAH hydrolase activity of red blood cell lysates from this patient was ~20% of normal, and the in vitro enzymatic activity of recombinant SAH containing valine at position 89 was ~10% of the wildtype protein [11,20].
The proband described in this paper (III.2, Fig. 1) had a later onset of symptoms than any of the other patients reported with this disease (Table 2). Whereas all previous cases developed symptoms during infancy, the proband remained largely asymptomatic until her mid-20s, although we cannot rule out that the proband’s difficulties in school were not related to the SAH hydrolase deficiency. Her homozygous son was asymptomatic at age 7 despite having very high levels of SAH, SAM, and methionine. The proband’s two brothers with liver disease survived to 17 and 35 years of age without treatment, but both were symptomatic in childhood. Although we could not confirm molecularly that these two brothers both were homozygous for the same mutation in AHCY, their clinical course is compatible with them also having SAH hydrolase deficiency.
4.2. AHCY-mutations in SAH hydrolase deficiency
Most SAH hydrolase deficiency-causing mutations have resulted in nonconservative amino acid substitutions (R49C, Y143C, D86G, G71S, Y328D), or a premature stop codon (W112X). Unfortunately, no enzymatic assays of SAH hydrolase activity were performed in the proband. However, the later onset and milder phenotypes seen in the affected family members reported here may indicate that the R49H substitution has a less severe effect on enzyme function. The finding that substituting R49 with the small hydrophobic amino acid cysteine causes disease much earlier than the R49H substitution is consistent with this notion. Arginine and histidine are both positively charged amino acids, and are closer in size than are arginine and cysteine.(Fig. 2B) [8,9]. Measurement of SAH hydrolase activity in tissues from the proband’s affected son may be informative.
Several rare nonsynonymous variants in AHCY with minor allele frequencies of up to 0.5% are present in the ExAC database, including known SAH hydrolase deficiency-causing mutations (Y143C, Y328D, R49C, and R49H, MAF < 1 × 10−4) [17]. This raises the question as to how common SAH hydrolase deficiency is in the general population. We speculate that the disease is likely to be underdiagnosed. As evidenced by the present report, cases may remain asymptomatic until early adulthood. The proband’s symptoms and medical history were not especially suggestive of an inborn error of metabolism, and genetic testing was only undertaken on the basis of the family history. A similar case, without a family history to suggest a genetic cause, could easily remain undiagnosed.
4.3. Serum amino acids and SAH hydrolase deficiency
The diagnosis of SAH hydrolase deficiency would likely have been made in the proband if serum amino acids had been measured. Although serum methionine levels are elevated in hepatic insufficiency, the magnitude of the elevation is much greater in this disorder. Typically, patients with liver disease have ~1.5-fold elevations in serum methionine levels, whereas the levels in SAH hydrolase deficiency are typically more than 10-fold elevated, except in early infancy when the levels can be normal or only slightly elevated [21,22].
4.4. Disease mechanism and treatment options
What is the cause of the hepatic, musculoskeletal and neurological problems associated with SAH hydrolase deficiency? Is it due to the markedly elevated levels of methionine, SAM, or SAH? Dietary intake of methionine is associated with hepatotoxicity in rats, and MAT 1A-deficient mice with hypermethioninemia are at higher risk for liver injury, suggesting that hypermethioninemia may play a role in the liver disease seen in SAH hydrolase deficiency [23,24]. In humans, however, there is little evidence supporting hypermethioninemia causing liver disease; patients with MAT I/III deficiency do not have liver or muscle disease [25]. In contrast, high hypermethioninemia is known to cause neurological disease, and this might explain the neurological phenotypes seen in SAH hydrolase deficiency.
Elevated SAM levels are associated with liver cancer in several animal models and could have contributed to the development of hepatocellular carcinoma in our SAH hydrolase-deficient patient [26,27]. Increased SAH levels inhibit several methyltransferases, which may contribute to the pathogenesis of SAH hydrolase deficiency [3]. Interestingly, SAH hydrolase acts as a tumor suppressor in vitro, and loss of SAH hydrolase expression is a common feature of human tumors [28].
Dietary methionine restriction can substantially lower circulating levels of methionine in SAH hydrolase-deficient patients (Table 2) [6,12], including the son of the proband described here. Two other infant brothers with SAH hydrolase deficiency had some clinical improvement with dietary restriction despite their serum SAH and SAM concentrations only occasionally reaching the normal range [12]. In another affected child, normalization of serum methionine levels with dietary restriction initiated at 5 months of age did not improve the clinical course of the disease [11]. Thus, it remains unclear whether dietary methionine restriction is of therapeutic benefit in patients with SAH hydrolase deficiency.
Liver transplantation may be another treatment option for SAH hydrolase deficiency [7]. Only one patient with SAH hydrolase deficiency has undergone liver transplantation. In that patient, serum SAH and SAM levels were normalized within 6 months of transplantation, but further observation will be required to assess the effects of liver transplantation on the course of the disease [7]. The early development and fatal course of hepatocellular carcinoma in the proband and her brother raises the possibility that prophylactic screening of the liver may be indicated in these patients and that the possibility of developing a hepatic malignancy may be a further indication for liver transplantation.
4.5. Summary
In summary, we used exome sequencing to identify SAH hydrolase deficiency as the cause of adult-onset liver disease, hepatocellular carcinoma, and muscle weakness in a 32-year old woman. The patient was healthy enough to have children and her affected child is remarkably free of symptoms. With increasing clinical use of high-throughput, hypothesis-free methods to analyze the genome, it is likely that many similar cases of cryptic inborn errors of metabolism will be revealed. Timely identification of these atypical cases may allow for targeted intervention to forestall progression of the disease.
Acknowledgments
We thank the McDermott Center Sequencing and Bioinformatics Cores (especially Vanessa Schmid) for DNA sequencing and sequence analysis. We also thank Barbara Gilbert for excellent technical assistance.
Financial support
Supported by grants from the NIH: PO1 HL20948, RO1 DK090066 and UL1TR001105. Dr. Stender is supported by a Sapere Aude Research Talent grant from the Danish Medical Research Council (grant no. 4004-00398).
Abbreviations
- AA
amino acid
- CT
computed tomography
- MAF
minor allele frequency
- MRI
magnetic resonance imaging
- ROH
runs of homozygosity
- SAH
S-adenosylhomocysteine
- SAM
S-adenosylmethionine
Footnotes
Conflicts of interest
None of the authors have conflicts of interest.
Contributor Information
Stefan Stender, Email: Stefan.stender@utsouthwestern.edu.
Rima S. Chakrabarti, Email: Rima.shah@utsouthwestern.edu.
Chao Xing, Email: Chao.xing@utsouthwestern.edu.
Garrett Gotway, Email: Garrett.gotway@utsouthwestern.edu.
Jonathan C. Cohen, Email: Jonathan.cohen@utsouthwestern.edu.
References
- 1.Finkelstein JD. Methionine metabolism in mammals. J Nutr Biochem. 1990;1:228–237. doi: 10.1016/0955-2863(90)90070-2. [DOI] [PubMed] [Google Scholar]
- 2.Li T, Yu G, Guo T, Qi H, Bing Y, Xiao Y, Li C, Liu W, Yuan Y, He Y, Liu Z, Liu Q. The plasma S-adenosylmethionine level is associated with the severity of hepatitis B-related liver disease. Medicine (Baltimore) 2015;94:e489. doi: 10.1097/MD.0000000000000489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barić I. Inherited disorders in the conversion of methionine to homocysteine. J Inherit Metab Dis. 2009;32:459–471. doi: 10.1007/s10545-009-1146-4. [DOI] [PubMed] [Google Scholar]
- 4.Belužić R, Vugrek O. S-Adenosylhomocysteine hydrolase (AHCY) deficiency: a natural model system for methylation research. Rad Med Sci. 2010;35:77–92. [Google Scholar]
- 5.Mudd SH. Hypermethioninemias of genetic and non-genetic origin: a review. Am J Med Genet C: Semin Med Genet. 2011;157C:3–32. doi: 10.1002/ajmg.c.30293. [DOI] [PubMed] [Google Scholar]
- 6.Barić I, Fumić K, Glenn B, Ćuk M, Schulze A, Finkelstein JD, James SJ, Mejaški-Bošnjak V, Pažanin L, Pogribny IP, Radoš M, Sarnavka V, Sćukanec-Spoljar M, Allen RH, Stabler S, Uzelac L, Vugrek O, Wagner C, Zeisel S, Mudd SH. S-Adenosylhomocysteine hydrolase deficiency in a human: a genetic disorder of methionine metabolism. PNAS. 2004;101:4234–4239. doi: 10.1073/pnas.0400658101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strauss KA, Ferreira C, Bottiglieri T, Zhao X, Arning E, Zhang S, Zeisel SH, Escolar ML, Presnick N, Puffenberger EG, Vugrek O, Kovacevic L, Wagner C, Mazariegos GV, Mudd SH, Soltys K. Liver transplantation for treatment of severe S-adenosylhomocysteine hydrolase deficiency. Mol Genet Metab. 2015;116:44–52. doi: 10.1016/j.ymgme.2015.06.005. [DOI] [PubMed] [Google Scholar]
- 8.Honzík T, Magner M, Krijt J, Sokolová J, Vugrek O, Belužić R, Barić I, Hansíkova H, Elleder M, Veselá K, Bauerová L, Ondrušková N, Ješina P, Zeman J, Kožich V. Clinical picture of S-adenosylhomocysteine hydrolase deficiency resembles phosphomannomutase 2 deficiency. Mol Genet Metab. 2012;107:611–613. doi: 10.1016/j.ymgme.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 9.Grubbs R, Vugrek O, Deisch J, Wagner C, Stabler S, Allen R, Barić I, Rados M, Mudd SH. S-Adenosylhomocysteine hydrolase deficiency: two siblings with fetal hydrops and fatal outcomes. J Inherit Metab Dis. 2010;33:705–713. doi: 10.1007/s10545-010-9171-x. [DOI] [PubMed] [Google Scholar]
- 10.Ćuk M. The fourth S-adenosylhomocysteine hydrolase deficient patient: further evidence of congenital myopathy. Clin Chem Lab Med. 2007;45(A43) [Google Scholar]
- 11.Buist NR, Glenn B, Vugrek O, Wagner C, Stabler S, Allen RH, Pogribny I, Schulze A, Zeisel SH, Barić I, Mudd SH. S-Adenosylhomocysteine hydrolase deficiency in a 26-year-old man. J Inherit Metab Dis. 2006;29:538–545. doi: 10.1007/s10545-006-0240-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barić I, Ćuk M, Fumić K, Vugrek O, Allen RH, Glenn B, Maradin M, Pažanin L, Pogribny I, Radoš M, Sarnavka V, Schulze A, Stabler S, Wagner C, Zeisel SH, Mudd SH. S-Adenosylhomocysteine hydrolase deficiency: a second patient, the younger brother of the index patient, and outcomes during therapy. J Inherit Metab Dis. 2005;28:885–902. doi: 10.1007/s10545-005-0192-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.http://pngu.mgh.harvard.edu/purcell/plink/, accessed September 4th, 2015
- 16.Pemberton TJ, Absher D, Feldman MW, Myers RM, Rosenberg NA, Li JZ. Genomic patterns of homozygosity in worldwide human populations. Am J Hum Genet. 2012;91:275–292. doi: 10.1016/j.ajhg.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.http://exac.broadinstitute.org/, accessed September 4th, 2015
- 18.Arning E, Bottiglieri T. Quantification of S-adenosylmethionine and S-adenosylhomocysteine using liquid chromatography electrospray tandem mass spectrometry. Clinical Applications of Mass Spectrometry: Methods in Molecular Biology. 2015 doi: 10.1007/978-1-0716-2565-1_4. in press. [DOI] [PubMed] [Google Scholar]
- 19.Hu Y, Komoto J, Huang Y, Gomi T, Ogawa H, Takata Y, Fujioka M, Takusagawa F. Crystal structure of S-adenosylhomocysteine hydrolase from rat liver. Biochemistry. 1999;38:8323–8333. doi: 10.1021/bi990332k. [DOI] [PubMed] [Google Scholar]
- 20.Belužić R, Ćuk M, Pavkov T, Barić I, Vugrek O. S-Adenosylhomocysteine hydrolase (AdoHcyase) deficiency: enzymatic capabilities of human AdoHcyase are highly effected by changes to codon 89 and its surrounding residues. Biochem Biophys Res Commun. 2008;368:30–36. doi: 10.1016/j.bbrc.2008.01.042. [DOI] [PubMed] [Google Scholar]
- 21.Marchesini G, Bugianesi E, Bianchi G, Fabbri A, Marchi E, Zoli M, Pisi E. Defective methionine metabolism in cirrhosis: relation to severity of liver disease. Hepatology. 1992;16:149–155. doi: 10.1002/hep.1840160125. [DOI] [PubMed] [Google Scholar]
- 22.Bosy-Westphal A, Ruschmeyer M, Czech N, Oehler G, Hinrichsen H, Plauth M, Lotterer E, Fleig W, Müller MJ. Determinants of hyperhomocysteinemia in patients with chronic liver disease and after orthotopic liver transplantation. Am J Clin Nutr. 2003;77:1269–1277. doi: 10.1093/ajcn/77.5.1269. [DOI] [PubMed] [Google Scholar]
- 23.Gomez J, Caro P, Sanchez I, Naudi A, Jove M, Portero-Otin M, Lopez-Torres M, Pamplona R, Barja G. Effect of methionine dietary supplementation on mitochondrial oxygen radical generation and oxidative DNA damage in rat liver and heart. J Bioenerg Biomembr. 2009;41:309–321. doi: 10.1007/s10863-009-9229-3. [DOI] [PubMed] [Google Scholar]
- 24.Lu SC, Alvarez L, Huang ZZ, Chen L, An W, Corrales FJ, Avila MA, Kanel G, Mato JM. Methionine adenosyltransferase 1A knockout mice are predisposed to liver injury and exhibit increased expression of genes involved in proliferation. PNAS. 2001;98:5560–5565. doi: 10.1073/pnas.091016398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chien YH, Abdenur JE, Baronio F, Bannick AA, Corrales F, Couce M, Donner MG, Ficicioglu C, Freehauf C, Frithiof D, Gotway G, Hirabayashi K, Hofstede F, Hoganson G, Hwu WL, James P, Kim S, Korman SH, Lachmann R, Levy H, Lindner M, Lykopoulou L, Mayatepek E, Muntau A, Okano Y, Raymond K, Rubio-Gozalbo E, Scholl-Bürgi S, Schulze A, Singh R, Stabler S, Stuy M, Thomas J, Wagner C, Wilson WG, Wortmann S, Yamamoto S, Pao M, Blom HJ. Mudd’s disease (MAT I/III deficiency): a survey of data for MAT1A homozygotes and compound heterozygotes. Orphanet J Rare Dis. 2015;10:99. doi: 10.1186/s13023-015-0321-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martínez-Chantar ML, Corrales FJ, Martínez-Cruz LA, García-Trevijano ER, Huang ZZ, Chen L, Kanel G, Avila MA, Mato JM, Lu SC. Spontaneous oxidative stress and liver tumors in mice lacking methionine adenosyltransferase 1A. FASEB J. 2002;16:1292–1294. doi: 10.1096/fj.02-0078fje. [DOI] [PubMed] [Google Scholar]
- 27.Martínez-Chantar ML, Vázquez-Chantada M, Ariz U, Martínez N, Varela M, Luka Z, Capdevila A, Rodríguez J, Aransay AM, Matthiesen R, Yang H, Calvisi DF, Esteller M, Fraga M, Lu SC, Wagner C, Mato JM. Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatology. 2008;47:1191–1199. doi: 10.1002/hep.22159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leal JF, Ferrer I, Blanco-Aparicio C, Hernández-Losa J, Ramón Y, Cajal S, Carnero A, Lleonart ME. S-Adenosylhomocysteine hydrolase downregulation contributes to tumorigenesis. Carcinogenesis. 2008;29:2089–2095. doi: 10.1093/carcin/bgn198. [DOI] [PubMed] [Google Scholar]