

Abstract
Background
Point mutations of the PRKAR1A gene are a genetic cause of Carney complex (CNC) and primary pigmented nodular adrenocortical disease (PPNAD), but in 30% of the patients no mutation is detected.
Objective
Set up a routine-based technique for systematic detection of large deletions or duplications of this gene and functionally characterize these mutations.
Methods
Multiplex ligation-dependent probe amplification (MLPA) of the 12 exons of the PRKAR1A gene was validated and used to detect large rearrangements in 13 typical CNC and 39 confirmed or putative PPNAD without any mutations of the gene. An in-frame deletion was characterized by western blot and bioluminescence resonant energy transfer technique for its interaction with the catalytic subunit.
Results
MLPA allowed identification of exons 3–6 deletion in three patients of a family with typical CNC. The truncated protein is expressed, but rapidly degraded, and does not interact with the protein kinase A catalytic subunit.
Conclusions
MLPA is a powerful technique that may be used following the lack of mutations detected by direct sequencing in patients with bona fide CNC or PPNAD. We report here one such new deletion, as an example. However, these gene defects are not a frequent cause of CNC or PPNAD.
Introduction
Inactivating mutations of the type 1a regulatory subunit of cAMP-dependent protein kinase A gene (PRKAR1A) cause an autosomal dominant disorder, Carney complex (CNC) (1, 2). This syndrome is a multiple endocrine neoplasia syndrome associated not only with primary pigmented nodular adrenocortical disease (PPNAD), pituitary, thyroid, and ovarian tumors but also with cardiac and cutaneous myxomas and lentiginosis (3). PRKAR1A mutations are found in ~60% of the cases of CNC. Occasional mutations led to apparently isolated PPNAD (4, 5). The frequency of PRKAR1A mutations in sporadic PPNADs is not known but appears to be lower than that in classical CNC.
More than 120 different mutations of the PRKAR1A gene have been identified so far (recently reviewed in (6, 7)) and most are frameshift, splice site, or nonsense mutations (84%) resulting in a rapid degradation of the mutated mRNA by nonsense-mediated mRNA decay.
It is intriguing that less than three-quarters of patients with classic CNC, and even fewer of those with sporadic PPNAD, have a mutation in the PRKAR1A gene. This may suggest that other genes are involved, such as the recently identified PDE8 and PDE11 genes (8, 9). But, there is also the possibility that some other sequence defects of the gene are not identified by direct sequencing, such as large rearrangements of the gene. Only three large deletions of the gene have been identified so far (10, 11), but in the absence of a systematic approach to search for them, their true frequency in CNC or PPNAD is unknown.
In this study, a routine-based technique was developed for the detection of large deletions or duplications of the PRKAR1A gene using multiplex ligation-dependent probe amplification (MLPA). This technique was applied to a series of patients with CNC and PPNAD, who were negative for any PRKAR1A mutations by Sanger sequencing. A large in-frame deletion of exons 3–6 was identified in a family with CNC and the expressed deletion mutant was structurally and functionally characterized.
Subjects and methods
Patients
The PRKAR1A gene sequence was analyzed in 46 cases with CNC and 53 with isolated PPNAD in our oncogenetic laboratory from January 2003 to June 2011. The diagnosis of CNC was made according to standard criteria (12). Patients with isolated myxoma or lentiginosis were not included in the study.
The diagnosis of PPNAD was made on the basis of either histological observations after adrenalectomy or association of an ACTH-independent Cushing’s syndrome with normal or micronodular aspect of the adrenal glands upon imaging with bilateral adrenal uptake of iodocholesterol scintigraphy when this examination was performed. Patients with suspected bilateral adrenal macronodular hyperplasia or unilateral tumors were excluded from the study.
MLPA of the PRKAR1A gene
In the absence of a commercially available kit for the PRKAR1A gene, a homemade kit was developed for the 12 exons of this gene and contained the classical reagents for the MLPA reaction provided by MRC Holland (Amsterdam, The Netherlands) and 12 MLPA probes, each corresponding to one exon and composed of two oligonucleotides (Table 1a). Owing to the presence of a pseudogene, which presents 89% homology with the coding sequence of hPRKAR1A (13), most of the designed probes overlapped the adjacent intronic sequences. These probes were strictly exonic for exons 3, 4, 7, 9, and 11. Two different MLPA reactions were set up due to the size limitation of the probes. MLPA mix A contains probes for exons 1a, 2, 4, 6, 8, and 10 and four control probes within the reference genes (Table 1c). MLPA mix B contains probes for exons 1b, 3, 5, 7, 9, and 11 and for the same four control probes. For each MLPA reaction, 0.1 nM of each oligonucleotide targeting PRKAR1A exons and references of either MLPA mix A or mix B were mixed with 250 to 500 ng of genomic DNA. Hybridization of the primers on genomic DNA was carried out for 16 h at 60 °C after the denaturation of genomic DNA (98 °C for 5 min). The ligation and PCR steps were performed according to MRC Holland standard protocols using the EK-kit containing buffers, ligase, polymerase, and primers necessary for ligation and PCRs. The ligation step (54 °C for 15 min) and 35 cycles of PCR (95 °C–30 s, 60 °C–30 s, and 72 °C–1 min) were processed in a GeneAmp PCR system (Applied Biosystems). The fragments were then separated on a Beckman CEQ 8000 DNA sequencer (Beckman Coulter Genomics, Danvers, MA, USA). Specific peaks, corresponding to each exon, were identified according to their migration relative to the molecular size standards and results were exported to a Microsoft Excel spreadsheet. First, peak intensity was normalized by slope correction according to a linear regression between the sizes and intensities of the peaks of the reference probes, in order to remove the contribution of probe size to its peak intensity. Second, assessment of copy number alterations was performed with the REX-MLPA (regression-enhanced MLPA) method. REX-MLPA is based on an iterative regression between case and control samples (three control samples were included in each experiment). Starting with control probes, the regression defines the threshold for which target probes are considered to have no change in copy number. Such probes are used to re-estimate the thresholds. Test probes that fall outside the confidence intervals are classified as having either a gain or a loss in copy number (14, 15). The spreadsheet templates used for the data analysis reported here are available upon request.
Table 1.
Pairs of oligonucleotides synthesized for the MLPA technique. In all cases, universal PCR primer tags are added at 5′-end of LHS (5′-GGGTTCCCTAAGGGTTGGA-3′) and at 3′-end of RHS (5′-TCTAGATTGGATCTTGCTGGCAC-3′).
|
Oligonucleotide sequences
|
Size
(bp) |
||
|---|---|---|---|
| Left hybridization sequence | Right hybridization sequence | ||
| a) PRKAR1A genea | |||
| EXON 1A | 5′-GGAAAGGAGGGAGAAAAGGCAGAGGCGTC-3′ | 5′-AAGGGAGGCCGGAGGGAGAGTGGGGTGGA-3′ | 100 |
| EXON 1B | 5′-GTTTCCGGTGGAGCTGTCGCCTAGCCGCT-3′ | 5′-ATCGCAGAGTGGAGCGGGGCTGGGAGCAA-3′ | 100 |
| EXON 2 | 5′-CAG GGAATACTTT GAGAGGTTGG AGAAGGTAAA-3′ | 5′-AATAAATGTGGGGAGATGATGAGGTGATTGTGA-3′ | 108 |
| EXON 3 | 5′-CCAGTGGTTAAAGGTAGGAGGCGACGAGGTGCT-3′ | 5′-ATCAGCGCTGAGGTCTACACGGAGGAAGATGCG-3′ | 108 |
| EXON 4 | 5′-CTAGGTTATACCAAAAGATTACAAGACAATGGCCGCT-3′ | 5′-TTAGCCAAAGCCATTGAAAAGAATGTGCTGTTTTCAC-3′ | 116 |
| EXON 5 | 5′-TGCCATGTTTTCGGTCTCCTTTATCGCAGGAGAGACTGT- GATTC-3′ |
5′-AGCAAGGTAAGGGCCTCTGGAGCATGCAAT-3′ | 116 |
| EXON 6 | 5′-CTCTTTTAGGTGATGAAGGGGATAACTTCTATGTGATT- GAT-3′ |
5′-CAAGGAGAGACGGATGTAAGATTTACCAATATCAAAAA- TAT-3 ′ |
124 |
| EXON 7 | 5′-GATTTATGGAACACCGAGAGCAGCCACTGTCAAAG- CAAAGA-3′ |
5′-CAAATGTGAAATTGTGGGGCATCGACCGAGACAGCTA- TAGA-3′ |
124 |
| EXON 8 | 5′-CGGAAGATGTATGAGGAATTCCTTAGTAAAGTCTCTATTT- TAGG T-3′ |
5′-GAGTTGTAAAGTGTGTTAACTTTGCTAGTATGTGAGA- TACCCCTG-3′ |
132 |
| EXON 9 | 5′-GTCTTACGGTAGCTGATGCATTGGAACCAGTGCAGTTT- GAAGATG-3′ |
5′-GGCAGAAGATTGTGGTGCAGGGAGAACCAGGGGAT- GAGTTCTTCA-3′ |
132 |
| EXON 10 | 5′-TTGGTGATTTTATTATAGGGGTCAGCTGCTGTGCTA- CAACGTCGGTC-3′ |
5′-AGAAAATGAAGAGTTTGTTGAAGTGGGAA- GATTGGGGCCTTCTGATT-3′ |
136 |
| EXON 11 | 5′-G TGCGTTAAGCTGGACCGACCTAGATTT- GAACGTGTTCTTGGCCCAT-3′ |
5′-GCTCAGACATCCTCAAACGAAACATCCAGCAGTACAA- CAGTTTTGTG-3′ |
136 |
| b) MLPA probesb | |||
| EXON 3 | 5′-GGTTAAAGGTAGGAGGCGACGAGGTGCTA-3′ | 5′-TCAGCGCTGAGGTCTACACGGAGGAAGAT-3′ | 100 |
| IVS2_B | 5′-TTCCTGGAGATCCGGTTTTGAAATGGGTCAC-3′ | 5′-TGTAAGGTGATCCCTCTTCTTTGGACATCAA-3′ | 104 |
| IVS2_C | 5′-GGGGGGTTGTGTTTCTGTTACCTTCAACTACTCCA-3′ | 5′-TAAGAAATGTACCAAAAACTCTTGACAGTTGGTCT-3′ | 112 |
| IVS2_D | 5′-GCTCTGAGTTGTCAGATTAGGTTATATTCAGATTGCT-3′ | 5′-GTTTTTAAAAGACTCACTAGAATCCATGAGACACTGC-3′ | 116 |
| IVS2_E | 5′-CGGGTCAAATGAATGAGAAAGTAGAAGATAACA- CATTTT-3′ |
5′-TATTTTTTCCCTTTTACTTCAGGCACAACAACTAATTCT-3′ | 120 |
| IVS6 | 5′-GTTCTCTAAGTATCTGTGACTGGTATTAGTGCTTAGCT- GACCT-3′ |
5′-TTTTCCCTTACATCCTTACAGAGTGCTTTTTTCTTATCA- GATA-3′ |
128 |
| c) Reference genesc | |||
| SERPINB2 | 5′-CAGAGAACTTTACCAGCTGTGGGTTCATGCA-3′ | 5′-GCAGATCCAGAAGGGTAGTTATCCTGATGCG-3′ | 104 |
| DACH1 | 5′-CTAGACCTGGAAGGCCTCCTAAGAGGACTCAAA-3′ | 5′-GTGTCACCTCCCCAGAGAACTCTCACATCATGCCGCA-3′ | 112 |
| ING1 | 5′-CAGAGATCCTGAAGGAGCTAGACGAGTGCTAC- GAGCGCT-3′ |
5′-TCAGTCGCGAGACAGACGGGGCGCAGAAGCGGCG- GATGC-3′ |
120 |
| SS18 | 5′-GACAGCATTACCAAGGACAGCAGCCACCTATGGGAAT- GATG-3′ |
5′-GGTCAAGTTAACCAAGGCAATCATATGATGGGTCAGAGA- CAGATT-3′ |
128 |
Sequences of the 12 pairs of oligonucleotides designed to analyze the 12 exons of PRKAR1A gene.
Sequences of the oligonucleotides designed specifically to localize the breakpoints of exons 3–6 deletion in one family of CNC.
Sequences of the oligonucleotides designed for the 4 reference genes.
In order to validate the method, series of normal genomic DNA and a set of plasmids containing the PRKAR1A cDNA with or without deletions of exons 3, 4, 7, 9, or 11 were analyzed. As indicated in Supplementary Figure 1, see section on supplementary data given at the end of this article, the exonic deletion was identified for each of these plasmids, in comparison with the WT plasmid.
An additional MLPA assay, using the same protocol described above, was set up specifically for introns 3 and 6 in order to identify the breakpoint regions in one CNC family suspected to present a large deletion (see below). The oligonucleotides used for the MLPA probes are indicated in Table 1b.
PRKAR1A cDNA constructs
The coding sequence for human PRKAR1A gene was obtained by RT-PCR and subcloned as previously described (16). To establish controls for PRKAR1A MLPA, each of the five exons (exons 3, 4, 7, 9, and 11) presenting a purely exonic MLPA probe was deleted in the cDNA sequence by site-directed mutagenesis using the Quick change Site-directed mutagenesis kit (Stratagene, LaJolla, CA, USA). The oligonucleotides used for mutagenesis are described in Supplementary Table 1, see section on supplementary data given at the end of this article.
The deletion mutant observed in one family of CNC was reproduced by site-directed mutagenesis with the oligonucleotides described in Supplementary Table 1. As previously described (16), two plasmids were used as templates: pcDNA3-hPRKAR1A and phPRKAR1A-hRluc. The resulting plasmids were called phPRKAR1A-ΔE3–6 and phPRKAR1A-hRluc-ΔE3–6 respectively.
Functional characterization of the deletion mutant
EBV-immortalized B-lymphocytes of one of the CNC patients (II-2) with a deletion of PRKAR1A gene and HEK293 transfected with the phPRKAR1A-ΔE3–6 or the WT phPRKAR1A constructs were solubilized and analyzed by western blot using classical procedures. The PRKAR1A proteins were identified with the antiPKA (R1) antibody (#610166, BD Transduction Laboratories, NJ, USA). The degradation of the WT and mutated PRKAR1A proteins was characterized on the same transfected HEK293 cells after treatment with cycloheximide (20 μg/ml) for 0–4 h, followed by western blot.
Interactions between regulatory and catalytic subunits of the PRKAR1A proteins were analyzed by bioluminescence resonance energy transfer (BRET) assay, as previously described (16, 17).
Results
CNC, PPNAD, and PRKAR1A gene mutations
Between January 2003 and June 2011, 99 kindred were investigated for a loss-of-function mutation of the PRKAR1A gene (Tables 2 and 3). This genetic test was indicated for a suspicion of CNC (46 cases) or isolated PPNAD (53 cases). There were 19 familial cases of CNC. A PRKAR1A point mutation was identified in 33 of the 46 analyzed CNC cases (77%). In 13 cases, no mutation was identified by sequencing. The diagnosis of isolated PPNAD was suspected in 53 cases presenting either hypercortisolism with histologically confirmed PPNAD (39/53 cases) or ACTH-independent hypercortisolism with normal or bilateral micronodular aspect of the adrenal glands at magnetic resonance imaging and/or bilateral hyperfixation of the adrenal at iodocholesterol scintigraphy (14/53 cases). A mutation of PRKAR1A was identified in 14 cases (26%), 13 of them in the group of histological PPNADs (33%) and one of them in the group of clinical PPNADs (7%). Therefore, more than half of the genetic investigations (52/99) were unsuccessful for the PRKAR1A gene and one possible explanation was that large rearrangements of the gene are missed by sequencing.
Table 2.
Clinical and mutational summaries of groups of familial and sporadic CNC.
| n | Age | Sex (M/F) | PPNAD | Myxoma | Lentiginosis |
Other
tumors |
PRKAR1A
mutation |
PRKAR1A
deletion |
PDE11A
mutation |
|
|---|---|---|---|---|---|---|---|---|---|---|
| Familial CNC | 19 | 37±18 | 8/11 | 10 | 11 | 16 | 8 | 14 | 1 | 0 |
| Sporadic CNC | 27 | 40±17 | 7/20 | 18 | 18 | 20 | 7 | 19 | 0 | 1 |
Table 3.
Clinical and mutational summaries of groups of confirmed or suspected PPNAD. Details on the clinical definition of each group are described in the ‘Subjects and methods’ section.
| n | Age | Sex (M/F) |
PRKAR1A
mutation |
PRKAR1A
deletion |
PDE11A
mutation |
|
|---|---|---|---|---|---|---|
| Confirmed PPNAD | 39 | 28±15 | 5/33 | 13 | 0 | 4 |
| Suspected PPNAD | 14 | 34±17 | 2/12 | 1 | 0 | 1 |
PRKAR1A gene deletion in a family with CNC
Using MLPA assay, the DNA of 13 CNC and 32 PPNAD patients, presenting no identified point-mutation by sequencing, were analyzed for exonic insertion/deletion of the PRKAR1A gene. None of the PPNAD patients exhibited a potential large rearrangement of the gene, and only one sample in the patients with CNC was strongly suspected to exhibit a deletion of the gene.
Pedigree of this family is shown in Fig. 1, and clinical presentation of different relatives of this family is detailed in Table 4. The proband (I-2) belongs to a Portuguese family. He underwent two surgeries for cardiac myxoma, the first one at the age of 62 years. He died 2 years ago at 72 years old. His daughter (II-2) is 52 years old and presents with a typical clinical profile of CNC. She underwent several surgeries: bilateral adrenalectomy for PPNAD at age 10, resection of a GH-producing pituitary adenoma in early adulthood, followed by a cardiac myxoma surgery at age 31 and resection of breast and uterine adenoma. Her son (III-2) is 19 years old and underwent cardiac surgery for myxoma at 7 months of age followed by a unilateral adrenalectomy for PPNAD at the age of 7 and a dorsal myxoma surgery at the age of 17 years (Table 4). All three first-degree relatives were presented with multiple skin tags, suggesting lentiginosis. Other studied members of the family had no symptom of CNC, including individuals I-1, II-3, and III-1 of the pedigree (Fig. 1).
Figure 1.
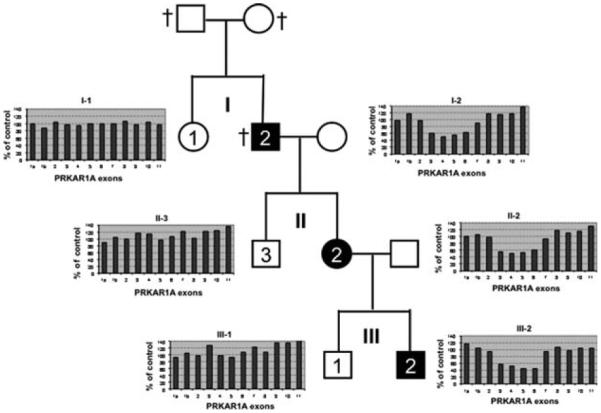
Genealogical tree of a Portuguese family analyzed for PRKAR1A gene deletion at three generations (labeled in roman numerical: I, II, and III). Three patients (I-2, II-2, and III-2) presented with a typical CNC and are represented as black squares (male) or circle (female) with number in white. Three siblings without any CNC clinical signs were also studied (I-1, II-3, and III-1, white squares or circle with black numbers). Deceased persons are indicated by †. MLPA results for each of the studied patients are represented as a box containing a histogram (x-axis, number of the studied exon; y-axis, value (%) of the area of the peak compared with a panel of controls). A value of 100% indicates the presence of two alleles: 50% one allele and 0% the absence of exon detection.
Table 4.
Clinical presentation of a family with Carney complex (CNC). Three individuals with CNC and three individuals without any clinical symptom of CNC were analyzed for point mutation and large rearrangement of the PRKAR1A gene.
| Patients | Sex (M/F) | Age (years) |
Adrenal disease
(age at diagnosis) |
Myxoma
(age at diagnosis) |
Lentiginosis | Other |
|---|---|---|---|---|---|---|
| I-1 | F | 71 | – | – | – | |
| I-2 | M | 72 | – | Cardiac myxoma (62) Second cardiac myxoma – surgery |
Yes | Died at 72 years old |
| II-2 | F | 52 | ACTH independent Cushing’s syndrome Bilateral adrenalectomy (10) |
Cardiac myxoma (31) Multiple cutaneous myxoma |
Yes | GH pituitary adenoma Breast and uterine adenoma |
| II-3 | M | 43 | – | – | – | |
| III-1 | M | 26 | – | – | – | |
| III-2 | M | 19 | PPNAD Unilateral adrenalectomy (7) |
Cardiac myxoma (30 months) |
Yes | Dorsal myxoma, surgery at 17 years old, probable GH pituitary adenoma currently under evaluation |
Genomic DNA of patients I-2 and II-2 was extensively sequenced for the identification of point-mutation in the coding sequence of the PRKAR1A gene. No mutation was observed in the 12 exons of the PRKAR1A gene. As for the other studied cases of typical CNC or PPNAD with no identified mutation, the PRKAR1A gene was investigated for large rearrangements. As shown in Fig. 1, the DNA of the two patients (I-2 and II-2) presented a deletion of exons 3–6. Four other members of the family were investigated for this deletion and only patient III-2, but not relatives I-1, II-3, and III-1, presented the same deletion. This result was confirmed on a second independent DNA sample for patients I-2, II-2, and III-2.
In order to confirm this deletion and identify its breakpoints, we performed several strategies. We first designed primers every 500 bp across the 10 kb sequence between the end of exon 2 and the beginning of exon 7 to amplify the fragments to be sequenced (data not shown). Unfortunately, no PCR fragment was identified. Second, we analyzed the 10 kb sequence between exons 2 and 7 and identified two Alu sequences at the end of PRKAR1A intron 2 and one in intron 6, with respectively 67 and 77% identity with the two intron 2 Alu sequences. This suggested a potential recombination event between two of these sequences. Oligonucleotides designed before the first (intron 2) Alu sequence and after the last (intron 6) Alu sequence failed to amplify a PCR fragment in the CNC patient II-2. Finally, we designed five MLPA probes (Table 1b) in the 10 kb potentially deleted for the patients in order to narrow the search of the breakpoints (Fig. 2A and B). The four MLPA probes located in intron 2 gave a deletion profile, indicating that intron 2 was almost totally deleted. However, the MLPA probe located in intron 6 had a normal profile, whereas a second probe to exon 6 confirmed its deletion. Altogether, these data approximately located the breakpoints and allowed us to design appropriate oligonucleotides to amplify and sequence the affected region. Figure 2C shows the chromatogram of the sequence around the breakpoints. The 5′ breakpoint is at position 66 512 656 on chromosome 17 and the 3′ breakpoint is at position 66 521 194, resulting in a deletion of 8538 bp. No homology between the two regions around the 5′ and 3′ breakpoints was identified, with the exception of a short 5 bp sequence (TCATT). This analysis confirms the results observed by MLPA and validates this assay to identify large rearrangements of the PRKAR1A gene.
Figure 2.
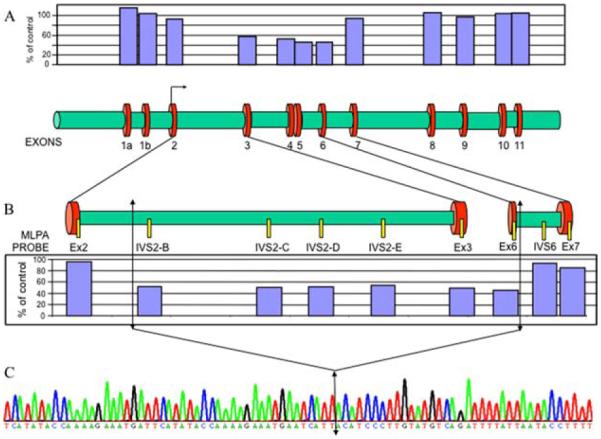
Breakpoints localization of the deletion of exons 3–6. (A, lower part) Structure of the PRKAR1A gene at scale with the 12 exons and the introns. (A, upper part) MLPA histogram for patient II-2 showing a heterozygous deletion of exons 3–6. The breakpoints are therefore located in introns 2 and 6. (B, upper part) Schematic representation of introns 2 and 6 with the positions of the different MLPA probes designed to locate the breakpoints. (B, lower part) Histograms of this specific MLPA analysis identify the breakpoints at the proximal sequences of introns 2 and 6. Arrows locate these potential breakpoints. (C) Sequence analysis around the breakpoints, identifying precisely the size of the deletion. The double arrow indicates the fusion point between introns 2 and 6.
Functional consequences of exons 3–6 deletion
Deletion of exons 3–6 should correspond to a shorter mRNA and a truncated protein if expressed, resulting in haploinsufficiency. A careful analysis of the deleted sequence indicates that deletion of exons 3–6 was an in-frame deletion, potentially resulting in a new protein missing aminoacids 60–183, corresponding to the four last aminoacids of the dimerization domain, the inhibitory site, which interacts with the catalytic subunit and is the beginning of the cAMP-binding domain A (Fig. 3). This truncated protein was analyzed by western blot in HEK293 cells transfected with the mutated cDNA or in lymphocytes of patient II-2. As shown in Fig. 4A, the truncated PRKAR1A protein is expressed as a 31 kDa band, in addition to the WT protein (49 kDa) expressed by the other allele (patient lymphocytes) or the endogenous PRKAR1A gene of HEK293. The intensity of the band for the truncated protein was weaker than that of the WT protein expressed in the lymphocytes, or equal to the endogenous protein expressed in HEK293 despite overexpression of the mutated one. This observation suggests a lower expression of the truncated protein either by reduction of its biosynthesis or by increase of its degradation. We further investigate the latter hypothesis by measuring the half lives of the WT and mutated proteins after blocking their biosynthesis with cycloheximide (Fig. 4B). As shown on the quantification in Fig. 4C, the half-life of the truncated protein is very short (<2 h), whereas that of the WT protein is between 4 and 5 h.
Figure 3.
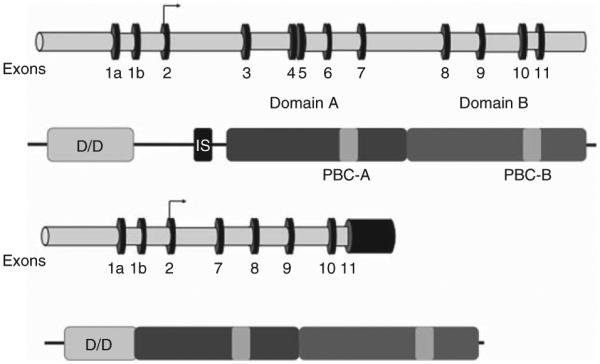
Schematic representation of the WT and Δ exons 3–6 mutant PRKAR1A genes and proteins. The genes are represented with exonic sequences (black and numbered) and intronic sequences (grey). The proteins are represented with their functional domains: dimerization domain (D/D); inhibitory sequence which binds to the catalytic subunit (IS); and the two cAMP-binding domains (A in dark grey and B in light grey) with their phosphobinding cassettes (PBC). The deleted mutant protein lacks the last four amino acids of the dimerization domain, the inhibitory sequence and the first 41 aminoacids of cAMP-binding domain A.
Figure 4.
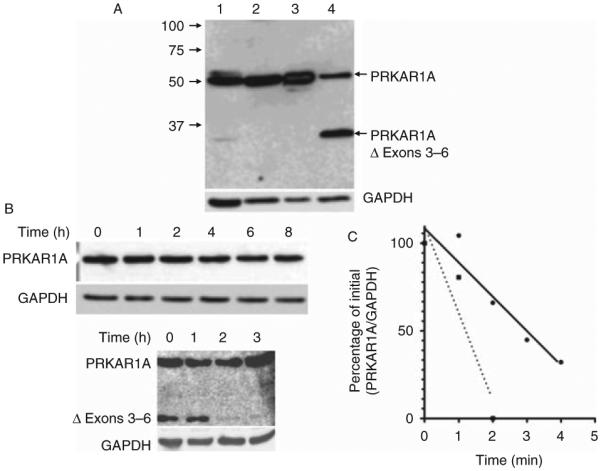
Expression and degradation of the WT and Δ3–6 deletion mutant. (A) Western blot analysis of PRKAR1A. Lane 1, lymphocytes of patient II-2 (60 μg protein extract); lane 2, HEK293 cells (60 mg protein extract); lane 3, HEK293 cells transfected with the WT PRKAR1A construct (10 μg protein extract); and lane 4, HEK293 cells transfected with the deletion mutant construct (20 μg protein extract). Western blot of the same membrane for GAPDH is shown in the lower part of the figure as control. (B) Degradation of PRKAR1A in transfected cells. Western blot of PRKAR1A and GAPDH was performed on HEK293 cells transfected with WT (upper part) or deletion mutant (lower part) PRKAR1A constructs and after incubation with cycloheximide (20 μg/ml) for the indicated time. (C) Typical degradation curves for WT (black dots and solid line) or deletion mutant (black squares and dotted line).
Since the degradation of PRKAR1A is dependent on its association to the catalytic subunit, we investigated whether this truncated regulatory subunit is still able to bind to the catalytic subunit. The deletion of the interacting site and the rapid degradation of the truncated protein suggested an abnormal binding to the catalytic subunit. This was confirmed experimentally using BRET: with increasing concentrations of PRKAC1A-YFP, no BRET signal was detected for Δ1-6-PRKAR1A-luciferase, whereas a strong and saturable signal was observed for the WT PRKAR1A-luciferase (Fig. 5).
Figure 5.
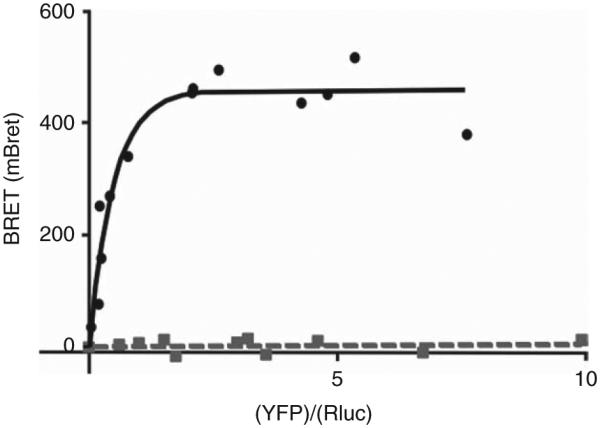
Interaction of the WT and truncated mutant PRKAR1A with the catalytic PKA subunit analyzed with BRET technique. Fusion proteins were designed corresponding to WT and deletion mutant of PRKAR1A-R-Luciferase and EYFP-PRKAC1A. These constructions were expressed in HEK293 cells with a constant expression of the luciferase constructs and increasing expression of the EYFP construct. BRET was measured for each condition and expressed as the arbitrary unit (mBRET; ordinate of the graph). In abscissa is represented as the ratio between the amounts of EYFP fusion protein and of the R-luciferase fusion protein. For the WT PRKAR1A (black dots and solid line) there is a saturable and important interaction between the two subunits, whereas the deletion mutant (grey squares and dotted line) is unable to interact with the catalytic subunit.
Altogether, these data indicate that CNC in the three patients of this Portuguese family is the consequence of a deletion of exons 3–6 of the PRKAR1A gene. This gene deletion results in the transcription and translation of a truncated protein with a shorter half-life probably in part due to its absence of association with the catalytic subunit. The resulting consequence remains haploinsufficiency.
Discussion
In this study, large rearrangements of PRKAR1A gene were investigated in CNC and sporadic PPNAD using a MLPA technique. For 8 years, our laboratory was able to analyze the PRKAR1A sequence in order to detect point mutations in 46 kindred of CNC and 53 cases of sporadic PPNAD. The CNC kindred were selected for their typical clinical presentation, i.e. familial cases of CNC (19 kindred) with at least two of the three cardinal lesions (PPNAD, myxoma, and lentiginosis) associated or not with other tumors found classically in CNC. Sporadic cases of CNC (27 kindreds) met the same criteria of diagnosis, but such a diagnosis was identified in only one member of the kindred at the time of the genetic test. Considering the clinical information obtained by the oncogenetic laboratory at the time of the genetic test, the observed ratio between familial vs sporadic CNC (40 vs 60%) is unusual compared with the ratio reported in the literature (70 vs 30%) from the clinician point of view (6). Our cohort excluded isolated myxoma or lentiginosis. As already described, probands of CNC are more often women (67%) than men (33%) and the mean age at diagnosis is around 40 years old (6). In this selected population, high frequencies of PPNAD (61%), myxoma (63%), and lentiginosis (78%) were observed. As expected, the frequency of PRKAR1A point mutation in this classical CNC cohort is high (72%). It is higher than the 62% described recently in a cohort of 185 families of CNC and/or PPNAD (6).
Isolated PPNAD is another clinical situation, for which investigation of the PRKAR1A sequence is necessary (5). In this study, we have distinguished the histologically confirmed PPNAD from the PPNAD suspected on clinical and imaging criteria. We confirm previous studies indicating that PRKAR1A gene point mutations are frequent in confirmed isolated PPNAD (33%), but clearly less frequent than in classical CNC. Interestingly, the frequency of identified mutations is very low (7%) in PPNAD suspected on fluctuant ACTH-independent Cushing’s syndrome with no alteration of the adrenal CT-scan and/or bilateral fixation at iodocholesterol scintigraphy. This observation indicates that these clinical and imaging criteria are insufficient for the diagnosis of PPNAD, and in rare cases genetic testing may help with the diagnosis. Taken altogether, 30–40% of the index cases of CNC and at least 66% of the isolated PPNAD had no point mutation of the PRKAR1A gene, and investigation of other genetic causes (PDE11A and PDE8B mutations) for these remaining families estimated their frequency at 12% of the patients with PPNAD (7).
Another possibility never investigated on a large scale in absence of an appropriate methodology is the presence of large rearrangements of the PRKAR1A gene. The presented MLPA technique offers the possibility of such an investigation. In the CNC cohort presented in this study, there was a large rearrangement in one out of 13 families of CNC without any point mutation of the gene. In addition, none of the 39 suspected or even confirmed PPNAD without PRKAR1A gene mutation presented a large rearrangement of the gene. From this study and a previous one (11), it is clear that the frequency of large deletions affecting the PRKAR1A gene is low. In the present cohort, it represents around 2% of the families, a number to compare with the 6% observed by Horvath et al. (11) in a series of 36 CNC without small intragenic mutations.
The first reported large rearrangement of PRKAR1A gene corresponded to a complete deletion of not only this gene but also 13 adjacent genes, resulting in a severe phenotype of mental retardation and microcephaly in a young boy (10). Two other shorter deletions were described in apparently sporadic cases of CNC using Southern blot analysis (11). The first one is a 3876 bp deletion including part of the promoter and the untranslated first exon of the PRKAR1A gene. This deletion should logically result in a quantitative defect in PRKAR1A expression and haploinsufficiency. The second is a 5149 bp deletion including the third exon and parts of the flanking introns. Such a deletion should result in a shorter RNA. The present paper reports another deletion of 8538 bp (66 512 656–66 521 194 bp) of chromosome 17 in the human genome sequence, which encompasses exons 3–6 and parts of the flanking introns. The recombination event resulting in this deletion did not involve the Alu sequences located in introns 2 and 6, since the identified breakpoints are located either 4.5 kb above the exon 2 Alu sequences or 130 bp above the exon 6 homologous Alu sequence. The breakpoint includes a very short sequence of 5 bp (TCATT) homologous in the two introns. Similar results were observed for the two other analyzed deletions (11). There is no doubt that systematic analysis of PRKAR1A gene large rearrangements using MLPA or other techniques in all CNC without PRKAR1A mutations will allow the identification of more CNC families with this type of alteration.
Finally, we found that the deletion of exons 3–6 results in an in-frame deletion with the potentially translated protein missing the last four aminoacids of the dimerization domain, the inhibitory sequence, and the first 41 aminoacids of cAMP-binding domain A. This observation is reminiscent of the previously reported deletion of exon 3, which is also an in-frame deletion (11), and a splice site mutation (c.708+1G>T) resulting in an exon 7 (former exon 6) skipping event (18). The deletion of exons 3–6 results in an expressed protein, which was detected both in the immortalized lymphocytes of the patients and in HEK293 cells transfected with the corresponding vector containing the mutant cDNA sequence. The apparent molecular weight (MW) was around 31 kDa, close to expected theoretical MW of this truncated protein (29.15 kDa). This truncated protein is rapidly degraded as assessed by its half-life <2 h, leading to partial haploinsufficiency, whereas the WT protein has a longer half-life above 4 h, as already described for the mouse PRKAR1A (19). The remaining undegraded truncated protein is unable to bind to the catalytic subunit as demonstrated by BRET techniques. Taken together, these functional alterations of PRKAR1A-Δ3–6 result in inappropriate activation of the enzyme and CNC phenotypes. Similar observations were made for the exons 3 and 7 deletion mutants described previously (20, 21). Δ3-PKAR1A mutant does not interact with the catalytic subunit and its overexpression results in increased activation of the PKA enzyme in response to cAMP. In addition, it presents a reduced binding to cAMP (20). The Δ7-PRKAR1A mutant results in a significantly higher cAMP-induced kinase activity compared with the WT, in both CRE-luciferase reporter assay and CREB phosphorylation (5, 18, 21). Taken together, these data confirm that in-frame deletions of exon 3, exons 3–6, or exon 7 result in expressed proteins with defective properties of inhibitory binding to the catalytic subunits and therefore increased activity of the enzyme. Whether some of these deleted proteins are still able to form dimers is not known; however in this case, they may act as dominant negative by trapping the WT PRKAR1A subunits for example. Some of these dominant negative properties may explain the severe phenotype of CNC in these patients, which includes early onset of the disease, frequent occurrence of the three major localizations of the disease (PPNAD, myxoma, and lentiginosis), and also multiple other tumoral localizations. Interestingly, the presented family does not fit in this category, suggesting that the molecular consequences of the exons 3–6 deletion are not similar.
In conclusion, a routine-based assay by MLPA has been developed for the detection of PRKAR1A gene large rearrangements. This assay was validated and was used to detect such gene modifications in a series of CNC and PPNAD negative for a point-mutation of the PRKAR1A gene. This test was able to detect a deletion of exons 3–6 in a family of CNC, a diagnosis confirmed by direct sequencing of the breakpoints. The corresponding truncated protein is expressed, but is rapidly degraded and does not bind to the catalytic subunit.
Supplementary Material
Acknowledgements
Many thanks to Dr R Vargas Poussou for valuable discussions about Alu sequences.
Funding
This work was supported by recurrent funding from INSERM and Assistance Publique – Hôpitaux de Paris.
Footnotes
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Supplementary data
This is linked to the online version of the paper at http://dx.doi.org/10.1530/EJE-13-0740.
References
- 1.Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS, Cho-Chung YS, Stratakis CA. Mutations of the gene encoding the protein kinase A type I-α regulatory subunit in patients with the Carney complex. Nature Genetics. 2000;26:89–92. doi: 10.1038/79238. doi:10.1038/79238. [DOI] [PubMed] [Google Scholar]
- 2.Kirschner LS, Sandrini F, Monbo J, Lin JP, Carney JA, Stratakis CA. Genetic heterogeneity and spectrum of mutations of the PRKAR1A gene in patients with the Carney complex. Human Molecular Genetics. 2000;9:3037–3046. doi: 10.1093/hmg/9.20.3037. doi:10.1093/hmg/9.20.3037. [DOI] [PubMed] [Google Scholar]
- 3.Carney JA, Gordon H, Carpenter PC, Shenoy BV, Go VL. The complex of myxomas, spotty pigmentation, and endocrine overactivity. Medicine. 1985;64:270–283. doi: 10.1097/00005792-198507000-00007. doi:10.1097/00005792-198507000-00007. [DOI] [PubMed] [Google Scholar]
- 4.Groussin L, Horvath A, Jullian E, Boikos S, Rene-Corail F, Lefebvre H, Cephise-Velayoudom FL, Vantyghem MC, Chanson P, Conte-Devolx B, et al. A PRKAR1A mutation associated with primary pigmented nodular adrenocortical disease in 12 kindreds. Journal of Clinical Endocrinology and Metabolism. 2006;91:1943–1949. doi: 10.1210/jc.2005-2708. doi:10.1210/jc.2005-2708. [DOI] [PubMed] [Google Scholar]
- 5.Groussin L, Jullian E, Perlemoine K, Louvel A, Leheup B, Luton JP, Bertagna X, Bertherat J. Mutations of the PRKAR1A gene in Cushing’s syndrome due to sporadic primary pigmented nodular adrenocortical disease. Journal of Clinical Endocrinology and Metabolism. 2002;87:4324–4329. doi: 10.1210/jc.2002-020592. doi:10.1210/jc.2002-020592. [DOI] [PubMed] [Google Scholar]
- 6.Bertherat J, Horvath A, Groussin L, Grabar S, Boikos S, Cazabat L, Libe R, Rene-Corail F, Stergiopoulos S, Bourdeau I, et al. Mutations in regulatory subunit type 1A of cyclic adenosine 5′-monophosphate-dependent protein kinase (PRKAR1A): phenotype analysis in 353 patients and 80 different genotypes. Journal of Clinical Endocrinology and Metabolism. 2009;94:2085–2091. doi: 10.1210/jc.2008-2333. doi:10.1210/jc.2008-2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horvath A, Bertherat J, Groussin L, Guillaud-Bataille M, Tsang K, Cazabat L, Libe R, Remmers E, Rene-Corail F, Faucz FR, et al. Mutations and polymorphisms in the gene encoding regulatory subunit type 1-α of protein kinase A (PRKAR1A): an update. Human Mutation. 2010;31:369–379. doi: 10.1002/humu.21178. doi:10.1002/humu.21178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horvath A, Boikos S, Giatzakis C, Robinson-White A, Groussin L, Griffin KJ, Stein E, Levine E, Delimpasi G, Hsiao HP, et al. A genome-wide scan identifies mutations in the gene encoding phosphodiesterase 11A4 (PDE11A) in individuals with adrenocortical hyperplasia. Nature Genetics. 2006;38:794–800. doi: 10.1038/ng1809. doi:10.1038/ng1809. [DOI] [PubMed] [Google Scholar]
- 9.Horvath A, Mericq V, Stratakis CA. Mutation in PDE8B, a cyclic AMP-specific phosphodiesterase in adrenal hyperplasia. New England Journal of Medicine. 2008;358:750–752. doi: 10.1056/NEJMc0706182. doi:10.1056/NEJMc0706182. [DOI] [PubMed] [Google Scholar]
- 10.Blyth M, Huang S, Maloney V, Crolla JA, Karen Temple I A 2.3Mb deletion of 17q24.2–q24.3 associated with ‘Carney complex plus’. European Journal of Medical Genetics. 2008;51:672–678. doi: 10.1016/j.ejmg.2008.09.002. doi:10.1016/j.ejmg.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 11.Horvath A, Bossis I, Giatzakis C, Levine E, Weinberg F, Meoli E, Robinson-White A, Siegel J, Soni P, Groussin L, et al. Large deletions of the PRKAR1A gene in Carney complex. Clinical Cancer Research. 2008;14:388–395. doi: 10.1158/1078-0432.CCR-07-1155. doi:10.1158/1078-0432.CCR-07-1155. [DOI] [PubMed] [Google Scholar]
- 12.Stratakis CA, Kirschner LS, Carney JA. Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. Journal of Clinical Endocrinology and Metabolism. 2001;86:4041–4046. doi: 10.1210/jcem.86.9.7903. doi:10.1210/jc.86.9.4041. [DOI] [PubMed] [Google Scholar]
- 13.Solberg R, Sandberg M, Spurkland A, Jahnsen T. Isolation and characterization of a human pseudogene for the regulatory subunit RIα of cAMP-dependent protein kinases and its sublocalization on chromosome 1. Genomics. 1993;15:591–597. doi: 10.1006/geno.1993.1112. doi:10.1006/geno.1993.1112. [DOI] [PubMed] [Google Scholar]
- 14.Caceres A, Armengol L, Villatoro S, Gonzalez JR. MLPAstats: an R GUI package for the integrated analysis of copy number alterations using MLPA data. BMC Bioinformatics. 2011;12:147. doi: 10.1186/1471-2105-12-147. doi:10.1186/1471-2105-12-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonzalez JR, Carrasco JL, Armengol L, Villatoro S, Jover L, Yasui Y, Estivill X. Probe-specific mixed-model approach to detect copy number differences using multiplex ligation-dependent probe amplification (MLPA) BMC Bioinformatics. 2008;9:261. doi: 10.1186/1471-2105-9-261. doi:10.1186/1471-2105-9-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Linglart A, Menguy C, Couvineau A, Auzan C, Gunes Y, Cancel M, Motte E, Pinto G, Chanson P, Bougneres P, et al. Recurrent PRKAR1A mutation in acrodysostosis with hormone resistance. New England Journal of Medicine. 2011;364:2218–2226. doi: 10.1056/NEJMoa1012717. doi:10.1056/NEJMoa1012717. [DOI] [PubMed] [Google Scholar]
- 17.Diskar M, Zenn HM, Kaupisch A, Prinz A, Herberg FW. Molecular basis for isoform-specific autoregulation of protein kinase A. Cellular Signalling. 2007;19:2024–2034. doi: 10.1016/j.cellsig.2007.05.012. doi:10.1016/j.cellsig.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 18.Groussin L, Kirschner LS, Vincent-Dejean C, Perlemoine K, Jullian E, Delemer B, Zacharieva S, Pignatelli D, Carney JA, Luton JP, et al. Molecular analysis of the cyclic AMP-dependent protein kinase A (PKA) regulatory subunit 1A (PRKAR1A) gene in patients with Carney complex and primary pigmented nodular adrenocortical disease (PPNAD) reveals novel mutations and clues for pathophysiology: augmented PKA signaling is associated with adrenal tumorigenesis in PPNAD. American Journal of Human Genetics. 2002;71:1433–1442. doi: 10.1086/344579. doi:10.1086/344579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amieux PS, Cummings DE, Motamed K, Brandon EP, Wailes LA, Le K, Idzerda RL, McKnight GS. Compensatory regulation of RIα protein levels in protein kinase A mutant mice. Journal of Biological Chemistry. 1997;272:3993–3998. doi: 10.1074/jbc.272.7.3993. doi:10.1074/jbc.272.7.3993. [DOI] [PubMed] [Google Scholar]
- 20.Greene EL, Horvath AD, Nesterova M, Giatzakis C, Bossis I, Stratakis CA. In vitro functional studies of naturally occurring pathogenic PRKAR1A mutations that are not subject to nonsense mRNA decay. Human Mutation. 2008;29:633–639. doi: 10.1002/humu.20688. doi:10.1002/humu.20688. [DOI] [PubMed] [Google Scholar]
- 21.Meoli E, Bossis I, Cazabat L, Mavrakis M, Horvath A, Stergiopoulos S, Shiferaw ML, Fumey G, Perlemoine K, Muchow M, et al. Protein kinase A effects of an expressed PRKAR1A mutation associated with aggressive tumors. Cancer Research. 2008;68:3133–3141. doi: 10.1158/0008-5472.CAN-08-0064. doi:10.1158/0008-5472.CAN-08-0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.