

Abstract
Proteolysis Targeting Chimera (PROTAC) technology is a rapidly emerging alternative therapeutic strategy with the potential to address many of the challenges currently faced in modern drug development programs. PROTAC technology employs small molecules that recruit target proteins for ubiquitination and removal by the proteasome. The synthesis of PROTAC compounds that mediate the degradation of c-ABL and BCR-ABL by recruiting either Cereblon or Von Hippel Lindau E3 ligases is reported. During the course of their development, we discovered that the capacity of a PROTAC to induce degradation involves more than just target binding: the identity of the inhibitor warhead and the recruited E3 ligase largely determine the degradation profiles of the compounds; thus, as a starting point for PROTAC development, both the target ligand and the recruited E3 ligase should be varied to rapidly generate a PROTAC with the desired degradation profile.
Keywords: Drug Design, cancer, drug design, E3 ubiquitin ligases, inhibitors, protein degradation
Chronic myelogenous leukemia (CML) is most often caused by the loss of autoinhibitory constraints on the c-ABL kinase domain in the oncogenic fusion protein BCR-ABL. This constitutively active tyrosine kinase drives uncontrolled cellular proliferation through STAT5, MAPK, PI3K/Akt, and CrkL signaling pathways.[1–3] With the advent of tyrosine kinase inhibitors (TKIs) targeting BCR-ABL, CML has become a chronic but manageable disease. Imatinib mesylate, the first TKI developed against BCR-ABL, binds competitively at the ATP-binding site of c-ABL and inhibits both c-ABL and BCR-ABL, leading to inhibition of cell proliferation and apoptosis of non-progenitor leukemic cells.[4,5] Second-generation TKIs (such as dasatinib, bosutinib) were subsequently developed to treat CML patients with acquired resistance to imatinib.[6] Despite the remarkable success of BCR-ABL TKIs, all CML patients must remain on lifelong treatment owing to persistent leukemic stem cells (LSCs) in spite of BCR-ABL inhibition. One hypothesis suggests that BCR-ABL acts as a protein scaffold for compensatory signaling pathways, allowing LSCs to survive kinase inhibition.[7–9] Therefore, knockdown of BCR-ABL has the potential to replace the need for continuous treatment with a cure for CML.
Recently, our lab and other groups have developed a small-molecule drug platform that works by protein degradation and has the potential to address the challenges faced in current drug development programs.[10–13] Proteolysis Targeting Chimera (PROTAC) technology utilizes hetero-bifunctional small molecules whereby one end of the molecule recruits an E3 ubiquitin ligase while the other end engages the target protein.[14] Upon ternary complex formation, the recruited E3 ligase ubiquitinates the target, leading to subsequent degradation by the proteasome (Figure 1A). In contrast to inhibitor-based pharmacology, PROTAC technology requires only transient binding to any surface of the target to catalytically induce ubiquitination and degradation; thus, PROTACs have emerged as a novel therapeutic approach to target so called “undruggable” proteins and have successfully been employed to degrade several proteins such as the estrogen-related receptor alpha,[13] cellular retinoic acid binding proteins,[15] and BRD4.[10–12] Despite these prior success stories, there have been no examples of PROTAC-induced degradation of tyrosine kinases thus far.[13] In this study, we sought to induce degradation of the BCR-ABL fusion protein as an archetypical tyrosine kinase implicated in cancer.
Figure 1.
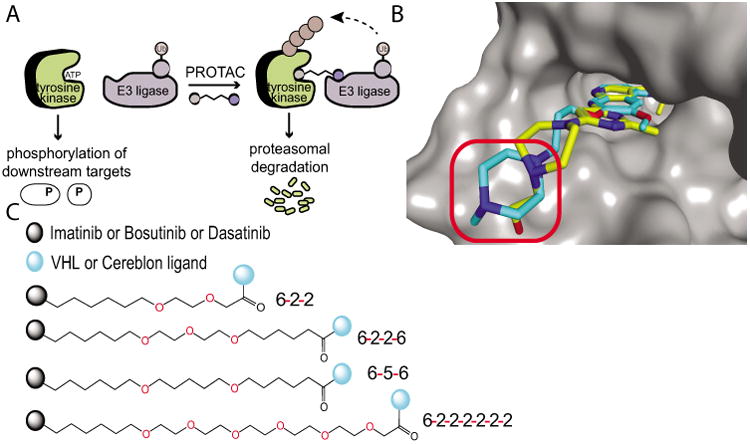
Approach to PROTAC development. A) PROTACs act through proximity-induced ubiquitination, leading to degradation by the proteasome. B) Overlay of bosutinib (blue; PDB: 3UE4) onto c-ABL-dasatinib crystal structure (yellow; PDB: 2GQG). Linkers were attached via the solvent exposed site (red circle). C) Linkers utilized to connect the respective TKI to the E3 recruiting ligand.
Herein, we describe the successful development of the first PROTACs that induce the degradation of an oncogenic tyrosine kinase, BCR-ABL. In the development process, we evolved a synthetic strategy for PROTAC design that incorporates variations in both warhead and E3 ligase ligands and allows one to rapidly assess the degradation profiles of PROTAC families.
To produce BCR-ABL degrader compounds, we conjugated BCR-ABL TKIs (imatinib, bosutinib, and dasatinib) that bind the c-ABL kinase domain, to a known Von Hippel Lindau (VHL) E3 ubiquitin ligase ligand or to the thalidomide derivative, pomalidomide, to recruit Cereblon (CRBN) E3 ligase.[10,13,16,17] The resulting bifunctional compounds are expected to bind BCR-ABL by the TKI moiety and VHL or CRBN via its recruiting ligand. Using the crystal structures of the c-ABL kinase domain in complex with the TKIs (imatinib, dasatinib, and bosutinib), we were able to predict the best position to attach our linkers to the respective TKIs such that critical binding interactions were not disrupted (Figure 1B).[18–20] Four different linkers of varying composition and length were evaluated as a survey of the potential chemical space (Figure 1C). These linkers contain a mixture of hydrophobic and hydrophilic moieties to balance the hydrophobicity/hydrophilicity of the resulting hybrid compounds. The panel of hybrid compounds was then assayed for retention of binding to c-ABL kinase domain through KinomeScan (Table 1). All of the compounds lost affinity for the phosphorylated and non-phosphorylated form of ABL compared to the parent compound.
Table 1.
Selected PROTAC affinities for the ABL kinase domain.[a]
| Compund | ABL (non-phosphorylated) | ABL (phosphorylated) |
|---|---|---|
| Imatinib | 0.86 | 36 |
| IMA-6-2-2-6-VHL | 4.3 | 93 |
| IMA-6-2-2-6-CRBN | 6.2 | 110 |
| Bosutinib | 0.063 | 0.023 |
| BOS-6-2-2-6-VHL | 1.4 | 0.63 |
| BOS-6-2-2-6-CRBN | 0.91 | 0.55 |
| Dasatinib | 0.03 | 0.02 |
| DAS-6-2-2-6-VHL | 0.92 | 0.47 |
| DAS-6-2-2-6-CRBN | 0.60 | 0.32 |
Affinity values in nm.
Interestingly, within each warhead series, the 1,5-bis(hexyloxy)pentane linker (hereby designated 6-5-6) yielded compounds with the most significant loss (maximum 86-fold) in binding affinity Supporting Information, Table S1). Speculatively, the more hydrophobic nature of 6-5-6 could lead to the compound folding back on itself; therefore, there could be a larger entropic cost to binding. Despite some loss in affinity, all hybrid compounds still bound non-phosphorylated c-ABL in the low nanomolar range (0.28 nm–24 nm). The bosutinib- and dasatinib-based PROTACs bound phosphorylated c-ABL in the high picomolar range (88 pm–1500 pm).
All PROTACs were then tested for c-ABL and BCR-ABL degradation in cell culture. Surprisingly, no degradation of BCR-ABL or c-ABL was observed in K562 CML cells when treated with imatinib-VHL (IMA-VHL) or imatinib-CRBN (IMA-CRBN) PROTACs, despite the fact that the PROTACs bound their targets, as evidenced by the reduced phosphorylation of CrkL and STAT5 at higher concentrations Supporting Information, Figures S1–S4). Since the imatinib warhead did not engage c-ABL or BCR-ABL to the same extent as the parent compound in cell culture, we hypothesized that a more potent inhibitor warhead (bosutinib or dasatinib) would be a better choice for the next PROTAC series.
Through a similar synthetic route, we conjugated bosutinib to the VHL recruiting ligand, producing bosutinib-VHL (BOS-VHL) PROTACs. Despite target engagement as determined by inhibition of downstream signaling, the BOS-VHL PROTACs also did not induce degradation of BCR-ABL or c-ABL (Figure 2A). This finding was consistent across several different linkers connecting the bosutinib inhibitor and the VHL recruiting ligand (Supporting Information, Figures S2–S4). The representative blot shown in Figure 2A is of BOS-VHL with the linker 1-{2-[2-(hexyloxy)ethoxy]ethoxy}hexane hereby designated as 6-2-2-6, which refers to the alkyl/ether composition.
Figure 2.
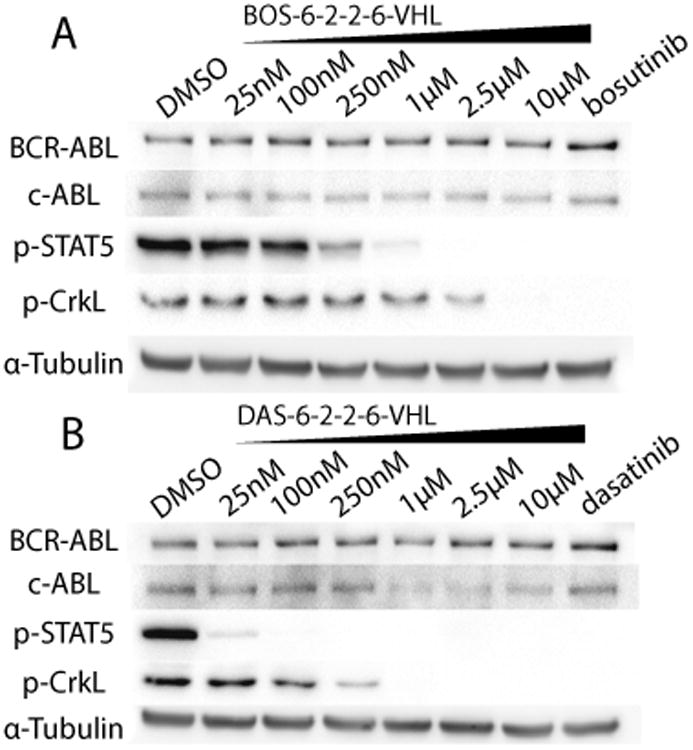
VHL-based PROTACs. A) BOS-6-2-2–6-VHL, and B) DAS-6-2-2-6-VHL were incubated with K562 human chronic myelogenous leukemia cells for 24 h. The concentrations of the parent inhibitors are 1μm. As determined by immunoblot, degradation of c-ABL can be observed with DAS-VHL starting at 1μm; however, no degradation of BCR-ABL was observed in any of the VHL-based PROTACs.
Along with the BOS-VHL series, we also incorporated dasatinib as the ligand binding warhead. In contrast to the IMA-VHL and BOS-VHL PROTACs, dasatinib-based PROTAC (DAS-VHL) induced a clear (>65%) decrease of c-ABL at 1μm PROTAC concentration (Figure 2B). The apparent decrease of protein degradation seen at higher PROTAC concentrations (10μm) has been observed with other PROTACs and is attributed to the formation of separate c-ABL-PROTAC and VHL-PROTAC dimers rather than the c-ABL-PROTAC-VHL trimeric complex required for productive ubiquitination.[13,21] The c-ABL degradation seen with the prototype DAS-VHL PROTAC was consistently observed with PROTACs possessing different linkers Supporting Information, Figures S2–S4). Thus, we have found that, independent of simple target binding, the inhibitor warhead (imatinib, bosutinib, or dasatinib) largely determines the capability of a PROTAC to induce c-ABL degradation.
Despite the success of DAS-VHL with the c-ABL degradation, no degradation of BCR-ABL was seen with any of the VHL-based PROTACs. This lack of degradation cannot be attributed to loss of binding affinity, as these VHL-based PROTACs still bind and inhibit both c-ABL and BCR-ABL in cell culture (Figure 2). Since E3 ligase presentation to the target is important for ubiquitination of an available lysine residue, we speculated that a differently oriented E3 ligase is required for adequate ubiquitination and degradation of BCR-ABL. Given the recent success of induced protein degradation through the recruitment of the Cereblon (CRBN) E3 ligase, we hypothesized that a switch to the CRBN E3 ligase may allow us to access BCR-ABL for degradation as well as provide the first head-to-head comparison between the two E3 ligases.[10,11]
When dasatinib was conjugated to pomalidomide to recruit CRBN, the dasatinib-CRBN (DAS-CRBN) PROTAC not only retained its ability to induce degradation of c-ABL (>85% at 1μm) but also induced BCR-ABL degradation (>60% at 1μm), demonstrating the first PROTAC-induced degradation of an oncogenic tyrosine kinase (Figure 3). This result was consistent across the several different linkers used previously in the series of VHL-based PROTACs Supporting Information, Figures S1–S4). Even more strikingly, when the VHL recruiting ligand in the bosutinib PROTAC series was exchanged for the CRBN ligand, we observed c-ABL (>90%) and BCR-ABL (>80%) degradation at 2.5μm (Figure 3A). The accessibility of BCR-ABL and c-ABL for degradation with the BOS-CRBN series stands in contrast with the BOS-VHL series where, despite target engagement, no degradation of c-ABL or BCR-ABL was observed. Thus, the inactive BOS-VHL compounds were converted to active BCR-ABL and c-ABL degrader compounds by switching to the CRBN E3 ligase. As demonstrated by these two inhibitor warhead series, the oncogenic fusion protein BCR-ABL is differentially susceptible to PROTAC-mediated degradation, depending on the E3 ligase (VHL or CRBN) recruited to the target.
Figure 3.
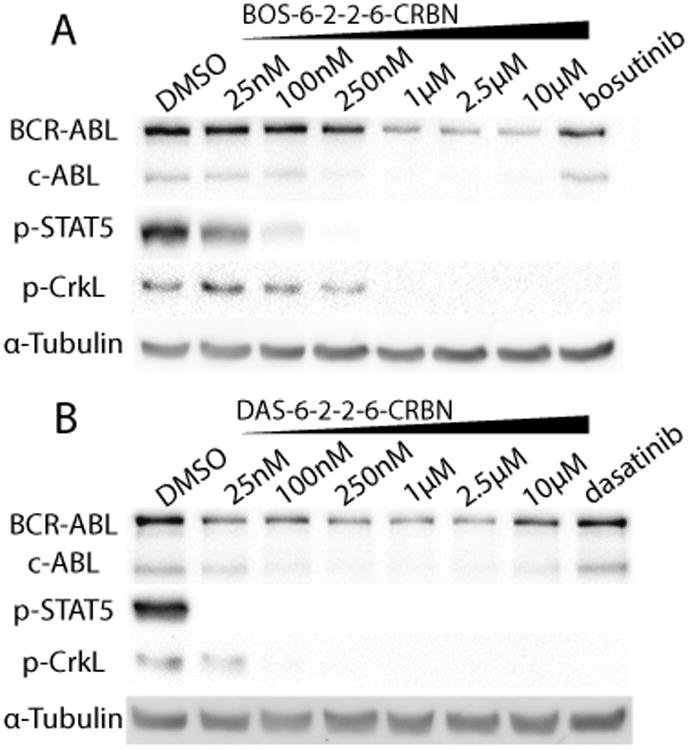
CRBN-based PROTACs. A) BOS-6-2-2–6-CRBN and B) DAS-6-2-2-6-CRBN were incubated with K562 cells for 24 h. The concentrations of the parent inhibitors are 1μm. As determined by immunoblot, degradation of BCR-ABL and c-ABL can be observed in the DAS-CRBN and BOS-CRBN series.
As BCR-ABL degradation can be observed at 25 nm with the DAS-6-2-2-6-CRBN PROTAC, we next sought to determine the cellular effects of the PROTAC (Figure 3B). In a cell viability assay, we determined that DAS-6-2-2-6-CRBN is active against BCR-ABL driven K562 with a half-maximal response concentration (EC50) of 4.4±2.1 nm (Figure 4). Furthermore, the PROTAC compound is more than 103-fold less active against the non-BCR-ABL driven cell lines, HEK293T and SK-BR-3 breast carcinoma. Thus, this PROTAC compound retains selective activity against the BCR-ABL driven cell line K562 and will be an excellent tool for future investigations into kinase-independent roles of BCR-ABL in LSCs.
Figure 4.
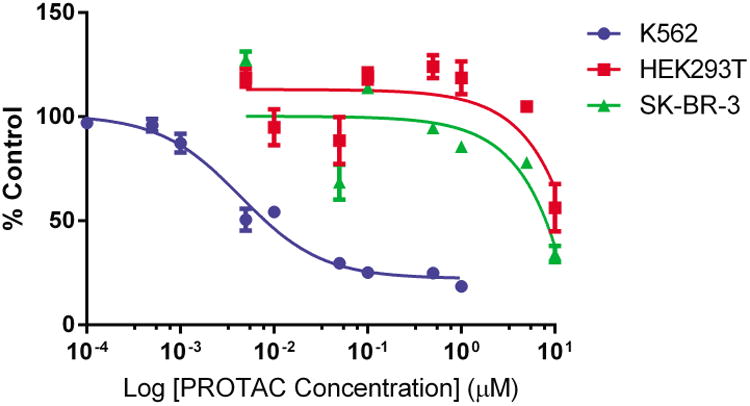
Cell viability with DAS-6-2-2-6-CRBN. This PROTAC is >103-fold more effective against the BCR-ABL driven cell line K562 over non-BCR-ABL driven cell lines as determined by CellTiter-Glo Luminescent Cell Viability assay after a 48 h treatment. Error bars displayed are the standard error of the median (n=3). Data was normalized to DMSO-treated controls.
In summary, we report the design and synthesis of bifunctional small molecules based on two potent TKIs (bosutinib and dasatinib) that mediate the degradation of c-ABL and BCR-ABL by hijacking either CRBN or VHL E3 ubiquitin ligase. Furthermore, these novel PROTACs are selective against the BCR-ABL driven cell line K562. In the course of their development, we have found that changing the inhibitor warhead and the recruited E3 ligase influences which protein targets are susceptible to PROTAC-induced degradation (Figure 5). By varying the recruited E3 ligase, we have found that the substrate spectrum of PROTACs can be significantly altered. This last observation hints at the potential to narrow the selectivity of a promiscuous inhibitor by creating a more selective degrader via the coupling to different E3 ligase recruiting ligands. In conclusion, for those seeking to utilize PROTAC technology for post-translational protein removal, a starting point for synthesis should focus on exchanging the identity of the target ligand and the recruited E3 ligase in order to quickly survey the degradation potential of small-molecule PROTACs.
Figure 5.
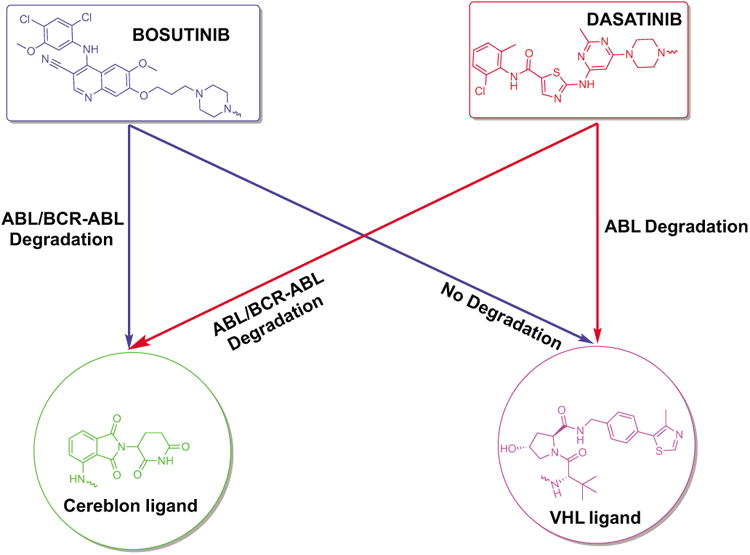
Summary. Varying the inhibitor warhead and the recruited E3 ligase permits targets to be accessed for degradation. IMA-based PROTACs did not induce the degradation of c-ABL or BCR-ABL, despite target engagement.
Supplementary Material
Figure S1. IMA-based PROTACs with linker 6-2-2-6. A) IMA-6-2-2-6-VHL and B) IMA-6-2-2-6-CRBN were incubated with K562 human chronic yelogenous leukemia cell line for 24 hrs. The concentration of the parent inhibitors are at 1μM. As determined by immunoblot, no degradation of c-ABL or BCR-ABL was observed in any of the IMA-based PROTACs.
Figure S2. PROTACs with linker 6-5-6. A) IMA-6-5-6-VHL, B) BOS-6-5-6-VHL, C) DAS-6-5-6-VHL, D) IMA-6-5-6-CRBN, E) BOS-6-5-6-CRBN and F) DAS-6-5-6-CRBN were incubated with K562 human chronic myelogenous leukemia cell line for 24 hrs. The concentration of the parent inhibitors are at 1μM. As determined by immunoblot, degradation of c-ABL can be observed with DAS-6-5-6-VHL starting at 2.5μM; however, no degradation of BCR-ABl was observed in any of the VHL-based PROTACs.
Figure S3. PROTACs with linker 6-2-2. A) IMA-6-2-2-VHL, B) BOS-6-2-2-VHL, C) DAS-6-2-2-VHL, D) IMA-6-2-2-CRBN, E) BOS-6-2-2-CRBN and F) DAS-6-2-2-CRBN were incubated with K562 human chronic myelogenous leukemia cell line for 24 hrs. The concentration of the parent inhibitors are at 1μM. As determined by immunoblot, degradation of c-ABL can be observed with DAS-6-2-2-VHL starting at 100nM; however, no degradation of BCR-ABl was observed in any of the VHL-based PROTACs.
Figure S4. PROTACs with linker 6-(2-)5-2. A) IMA-6-(2-)5-2-VHL, B) BOS-6-(2-)5-2-VHL, C) DAS-6-(2-)5-2-VHL, D) IMA-6-(2-)5-2-CRBN, E) BOS-6-(2-)5-2-CRBN, and F) DAS-6-(2-)5-2-CRBN were incubated with K562 human chronic myelogenous leukemia cell line for 24 hrs. The concentration of the parent inhibitors are at 1μM. As determined by immunoblot, degradation of c-ABL can be observed with DAS-6-(2-)5-2-VHL starting at 250nM; however, no degradation of BCR-ABl was observed in any of the VHL-based PROTACs.
Acknowledgments
This work was partially supported by the Leukemia & Lymphoma Society and the NIH (T32GM067543, R35CA197589, R01AI084140, and MTSP TG-T32GM007205). D.H. is supported by the EMBO Long-Term Fellowship ALTF-1102-2014.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201507634.
References
- 1.Hantschel O, Superti-Furga G. Nat Rev Mol Cell Biol. 2004;5:33–44. doi: 10.1038/nrm1280. [DOI] [PubMed] [Google Scholar]
- 2.Hantschel O, Warsch W, Eckelhart E, Kaupe I, Grebien F, Wagner KU, Superti-Furga G, Sexl V. Nat Chem Biol. 2012;8:285–293. doi: 10.1038/nchembio.775. [DOI] [PubMed] [Google Scholar]
- 3.Quintás-Cardama A. J Cortes, Blood. 2008;113:1619–1630. doi: 10.1182/blood-2008-03-144790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Druker B, Tamura S, Buchdunger E. Nat Med. 1996;2:561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 5.Okada M, Adachi S, Imai T, Watanabe K, Toyokuni S, Ueno M, Zervos AS, Kroemer G, Nakahata T. Blood. 2003;103:2299–2307. doi: 10.1182/blood-2003-05-1605. [DOI] [PubMed] [Google Scholar]
- 6.An X, Tiwari AK, Sun Y, Ding PR, Ashby CR, Jr, Chen ZS. Leuk Res. 2010;34:1255–1268. doi: 10.1016/j.leukres.2010.04.016. [DOI] [PubMed] [Google Scholar]
- 7.Wertheim J, Forsythe K, Druker BJ, Hammer D, Boettiger D, Pear W. Blood. 2002;99:4122–4130. doi: 10.1182/blood.v99.11.4122. [DOI] [PubMed] [Google Scholar]
- 8.Ichim CV. Stem Cells Transl Med. 2014;3:405–415. doi: 10.5966/sctm.2012-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamilton A, Helgason GV, Schemionek M, Zhang B, Myssina S, Allan EK, Nicolini FE, Müller-Tidow C, Bhatia R, Brunton VG, et al. Blood. 2011;119:1501–1510. doi: 10.1182/blood-2010-12-326843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, Hines J, Winkler JD, Crew AP, Coleman K, et al. Chem Biol. 2015;22:755–763. doi: 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, Bradner JE. Science. 2015;348:1376–1381. doi: 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zengerle M, Chan KH, Ciulli A. ACS Chem Biol. 2015;10:1770–1777. doi: 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bondeson DP, Mares A, Smith IED, Ko E, Campos S, Miah AH, Mulholland KE, Routly N, Buckley DL, Gustafson JL, et al. Nat Chem Biol. 2015;11:611–617. doi: 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Proc Natl Acad Sci USA. 2001;98:8554–8559. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Itoh Y, Ishikawa M, Kitaguchi R, Sato S, Naito M, Hashimoto Y. Bioorg Med Chem. 2011;19:3229–3241. doi: 10.1016/j.bmc.2011.03.057. [DOI] [PubMed] [Google Scholar]
- 16.Buckley DL, Gustafson JL, Van Molle I, Roth AG, Tae HS, Gareiss PC, Jorgensen WL, Ciulli A, Crews CM. Angew Chem Int Ed. 2012;51:11463–11467. doi: 10.1002/anie.201206231. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2012;124:11630–11634. [Google Scholar]
- 17.Buckley DL, Van Molle I, Gareiss PC, Tae HS, Michel J, Noblin DJ, Jorgensen WL, Ciulli A, Crews CM. J Am Chem Soc. 2012;134:4465–4468. doi: 10.1021/ja209924v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nagar B, Bornmann W, Pellicena P. Cancer Res. 2002;62:4236–4243. [PubMed] [Google Scholar]
- 19.Tokarski JS, Newitt J, Chang CYJ, Cheng JD, Wittekind M, Kiefer SE, Kish K, Lee FYF, Borzillerri R, Lombardo LJ, et al. Cancer Res. 2006;66:5790–5797. doi: 10.1158/0008-5472.CAN-05-4187. [DOI] [PubMed] [Google Scholar]
- 20.Levinson NM, Boxer SG. PLoS One. 2012;7:e29828. doi: 10.1371/journal.pone.0029828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Douglass EF, Miller CJ, Sparer G, Shapiro H, Spiegel DA. J Am Chem Soc. 2013;135:6092–6099. doi: 10.1021/ja311795d. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. IMA-based PROTACs with linker 6-2-2-6. A) IMA-6-2-2-6-VHL and B) IMA-6-2-2-6-CRBN were incubated with K562 human chronic yelogenous leukemia cell line for 24 hrs. The concentration of the parent inhibitors are at 1μM. As determined by immunoblot, no degradation of c-ABL or BCR-ABL was observed in any of the IMA-based PROTACs.
Figure S2. PROTACs with linker 6-5-6. A) IMA-6-5-6-VHL, B) BOS-6-5-6-VHL, C) DAS-6-5-6-VHL, D) IMA-6-5-6-CRBN, E) BOS-6-5-6-CRBN and F) DAS-6-5-6-CRBN were incubated with K562 human chronic myelogenous leukemia cell line for 24 hrs. The concentration of the parent inhibitors are at 1μM. As determined by immunoblot, degradation of c-ABL can be observed with DAS-6-5-6-VHL starting at 2.5μM; however, no degradation of BCR-ABl was observed in any of the VHL-based PROTACs.
Figure S3. PROTACs with linker 6-2-2. A) IMA-6-2-2-VHL, B) BOS-6-2-2-VHL, C) DAS-6-2-2-VHL, D) IMA-6-2-2-CRBN, E) BOS-6-2-2-CRBN and F) DAS-6-2-2-CRBN were incubated with K562 human chronic myelogenous leukemia cell line for 24 hrs. The concentration of the parent inhibitors are at 1μM. As determined by immunoblot, degradation of c-ABL can be observed with DAS-6-2-2-VHL starting at 100nM; however, no degradation of BCR-ABl was observed in any of the VHL-based PROTACs.
Figure S4. PROTACs with linker 6-(2-)5-2. A) IMA-6-(2-)5-2-VHL, B) BOS-6-(2-)5-2-VHL, C) DAS-6-(2-)5-2-VHL, D) IMA-6-(2-)5-2-CRBN, E) BOS-6-(2-)5-2-CRBN, and F) DAS-6-(2-)5-2-CRBN were incubated with K562 human chronic myelogenous leukemia cell line for 24 hrs. The concentration of the parent inhibitors are at 1μM. As determined by immunoblot, degradation of c-ABL can be observed with DAS-6-(2-)5-2-VHL starting at 250nM; however, no degradation of BCR-ABl was observed in any of the VHL-based PROTACs.