

Abstract
Background:
Facioscapulohumeral muscular dystrophy (FSHD), a common autosomal dominant muscular disorder, is caused by contraction of the D4Z4 repeats on 4q35. The complicated genotype-phenotype correlation among different ethnic population remains a controversial subject. We aimed to refine this correlation in order to provide new information for genetic counseling.
Methods:
Here, a cohort of 136 Chinese families including 178 affected individuals and 137 unaffected members were investigated. Genetic analyses were performed using the p13E-11, 4qA and 4qB probes after pulsed field gel electrophoresis separation and southern blotting. A 10-grade FSHD clinical severity scale was adopted for clinical assessment. The genotype-phenotype correlation was established by linear regression analyses.
Results:
We observed a roughly inversed correlation between the short EcoRI fragment size and age-corrected clinical severity score in 154 symptomatic patients (P < 0.05). Compared to male patients, a significant higher proportion of females in both asymptomatic carriers and severe patients showed larger variation in the size of short EcoRI fragment. A high incidence (19/42, 45.2%) of asymptomatic (or minimally affected) carriers was found in familial members.
Conclusions:
Although the number of D4Z4 repeats is known as one of the critical influences on genotype-phenotype correlation, a majority of phenotypic spectrum was still incompatible with their heterozygous contraction of the D4Z4 repeat, especial in female cases. Our results suggest that there are multi-factors synergistically modulating the phenotypic expression.
Keywords: Asymptomatic, Facio-scapulohumeral Muscular Dystrophy, Genetic Counseling, Phenotype
INTRODUCTION
Autosomal dominant facioscapulohumeral muscular dystrophy (FSHD, OMIM 158900) is referred as one of the most prevalent hereditary neuromuscular disorders.[1] Approximately 95% patients presented clinical symptoms before the second decades.[2] The disease is foremost characterized by progressive and asymmetric weakness of muscles in the facial and shoulder-girdle, later involving pelvic and proximal lower limb. The clinical spectrum is widely variable, ranging from asymptomatic gene carriers to patients with wheelchair dependency.[3] No biochemical, histological, or instrumental markers are available to independently confirm a specific FSHD diagnosis except genetic testing.
In most patients (>95%), the molecular defect is linked to a reduced EcoRI fragment (<38 kb) with contraction of the 3.3 kb D4Z4 repeat on the subtelomere of chromosome 4q35, which has been considered as a key diagnostic signature with the p13E-11 probe. The copies of 4q35-located D4Z4 repeat in the normal individuals range from 11 to 150, whereas FSHD patients carry <10 repeats. However, at least one repeat is required for developing FSHD.[4,5] Our previous study emphasized that D4Z4 repeat length analysis alone was not sufficient for the genetic diagnostics, especially when used as an exclusion criterion.[6] The chromosome 10q26 region displays a highly homologous EcoRI fragment including D4Z4 repeats, and ought to be distinguished by double enzymes EcoRI, EcoRI/BlnI (AvrII) digestion.[7] A pair of additional 4q subtelomeric variations (4qA/4qB) locates immediately distal to D4Z4 repeat, and FSHD is uniquely associated with the 4qA variant.[8] Consequently, integral molecular diagnosis is based on the analysis of reduced D4Z4 repeats on the permissive 4qA chromosome by southern blotting of genomic DNA after digestion with a specific set of restriction enzymes.[9]
Although the identity of the specific gene(s) responsible for FSHD is still elusive, the deletion of D4Z4 repeats leading to the chromatin relaxation and the epigenetic overexpression of DUX4 retrogene (MIM *606009) (encoding the double-homeobox 4) from the distal telomeric D4Z4 repeat is proposed as the pathogenic mechanism.[10,11] It is prevalently accepted that there is a reverse correlation between D4Z4 repeats number and clinical severity.[12] However, the wide inter- and intra-familial variability of clinical expression appears to be more pronounced than expected. And several observations including novel large-scale genotype-phenotype analyses and increasing atypical patients have emerged to complicate this relationship.[13,14]
Because the genetic diagnosis for FSHD is laborious and time-consuming, there were only a few reports about the genetically confirmed patients in China.[15] In the present study, we performed the completely genetic testing for a large cohort of FSHD patients according to the guideline of the international consortium. The potential relationship of genotype-phenotype has been examined to assess the impact of the molecular defect on phenotypic manifestations. In all, our investigation offered the available data for the molecular analysis and clinical spectrum of Chinese FSHD patients.
METHODS
Patients
This study concerned 178 affected individuals (86 females, 92 males; mean age at investigation, 30.3 ± 11.7 years; range, 5–61 years) and 137 unaffected members belonging to 136 unrelated families, in which at least one subject with a contracted 4qA allele. Of these families, 70 had multiple affected individuals, and 66 had only one affected individual. All subjects were of Han ethnic origin. Each patient was given a thorough clinical examination at least twice during the last decade, and the medical history included family history, gender, symptoms of disease onset, age onset, disease duration was well obtained. Other clinical evaluation included creatine kinase levels, electromyography, and muscle biopsy. All patients fulfilled FSHD clinical criteria.[16] Informed consent was obtained from each patient or legal guardian of those under 18 years of age, and the protocol was approved by the local ethics committee for medical research.
Evaluation of clinical severity
In the present study, muscle strength was objectively evaluated by manual muscle testing (MMT) according to the standard procedure. A modified Medical Research Council scale score and a 10-grade FSHD clinical severity scale were adopted to evaluate the phenotypic spectrum, ranking from mildly affected (facial involvement only) at 0.5 to severe weakness (wheelchair-dependent) at 5.[12] These data documented decline in strength relied mainly on neurological examination, which is more objective than the criteria like onset age and extend to include the available affected members of each family. Given variation of onset age, we adopted the Age-corrected clinical severity score (CSS) to assess the relationship of EcoRI fragment and clinical phenotype.[17] Five affected relatives younger than 10 years were a lack of MMT to evaluate the CSS. For statistical analysis, 173 patients were classified into six distinct severity groups (I–VI) according to the CSS (I: 0–1; II: 1.5; III: 2–2.5; IV: 3; V: 3.5; VI: 4–5).
DNA isolation, analysis of D4Z4 repeats, and 4qA/4qB variant determination
High molecular weight DNA was extracted from peripheral lymphocytes derived from freshly collected blood using the standard protocols. Five micrograms of DNA were digested with EcoRI, EcoRI/BlnI, or HindIII (Takara, Japan), and separated by pulsed-field gel electrophoresis on a 1.2% agarose gel (Sangon, China) in × 0.5 TBE for 39 h. Nytran + membranes (GE Healthcare, USA) for allele sizing were hybridized with probe p13E-11, and those for allele typing were hybridized with probe 4qA and 4qB as previously described.[6,15]
Statistical analysis
Data were analyzed by a statistical package (the SPSS program PASW Statistics 17.0, SPSS Inc, Chicago, USA). Continuous data were presented as mean ± standard deviation and categorical variables as percentage. Linear regression analyses were employed to examine the relationship between the age-corrected CSS and fragment size. The Chi-square test was adopted to compare the proportions of patients among different groups. Two sample t-test and one-way analysis of variance were used to compare the continuous variables among groups for normally distributed values (age, EcoRI fragment size). Other data for nonnormally distributed values were analyzed by Kruskall–Wallis test (median CSS).
RESULTS
Clinical findings and genetic characteristics
Molecular analysis revealed that 178 individuals from total 136 families carried 4q35 reduced EcoRI fragments in the range from 10 kb to 35 kb (1–9 D4Z4 repeats) with p13E-11 probe. The mean fragment size of the probands was 19.2 ± 4.9 kb. Importantly, all the polymorphic D4Z4 repeat contractions possessed the 4qA allele with two allelic variant genotypes on chromosome 4q35, including 4qA/4qA homozygote and 4qA/4qB heterozygote. Among them, 151 patients carried standard configuration distribution without translocation or mosaicism and the remaining 27 patients displayed a nonstandard configuration of the chromosome 4q35.
The mean onset age in this cohort was 18.4 ± 9.0 years, with a wide range from childhood to adulthood. As to analysis of clinical manifestation [Table 1], 59.1% (91/154) of the patients displayed asymmetrical weakness at the first time of neurological examination. The disturbances related symptoms to facial and scapulo-humeral weakness are quite typical onset pattern to the FSHD diagnostic criteria, which were reported to be the presenting symptoms by 137 of the 154 patients (89.0%). When facial weakness was considered separately from scapulo-humeral invasion, the onset of facial weakness was predominant in 12 patients at the mean age of 12.5 ± 5.4 years. By contrast, 125 patients began with scapulo-humeral weakness were at the mean age of 18.9 ± 5.4 years. There was significant difference for age of onset between facial weakness and scapula-humeral weakness (t = 3.258, P < 0.05). Nontypically initial symptoms were involved in various body regions. Onset with atypical proximal weakness in lower limbs was collected in 11 patients, including 7 unilateral, and 4 bilateral cases. Abnormal walking posture and back pain were the predominated initial manifestation in 2 patients. Unilateral foot drop was the major presentation in 4 patients. Meanwhile, associated signs of facial or scapulo-humeral region were also demonstrated at the first neurological examination in these patients with atypical onset. The significant higher proportion of female than male was observed in atypical onset pattern (χ2 = 5.41, P < 0.05).
Table 1.
Facioscapulohumeral muscular dystrophy clinical data of typical and nontypical onset (91/154 asymmetrical weakness)*
| Iterms | Initial symptoms (n) | ||
|---|---|---|---|
| Typical onset | Nontypical onset | ||
| Facial (12) | Scapulo-humeral (125) | Lower limbs proximal weakness (11)/foot drop (4)/others (2) | |
| Male/ | 9/3 | 72/53 | 5/12 |
| female (n/n) | |||
| Age at the | 12.5 ± 5.4 | 18.9 ± 5.4 | 22.3 ± 13.1 |
| onset (years)† | |||
| EcoRI | 20.2 ± 7.4 | 19.5 ± 4.9 | 19.7 ± 4.2 |
| fragment (kb)‡ | |||
*Five affected children younger than 10 years were considered as preclinical cases and excluded; †,‡EcoRI fragment size and the onset of age was given as means ± SD. SD: Standard deviation.
Analysis of the 4q35-EcoRI fragment and size distribution
About 51.4% (70/136) of the probands were familial cases and their 42 relatives were detected to carry the consistently same EcoRI fragments. In the total affected relatives, 19 were asymptomatic (or minimally affected) carriers with EcoRI fragments ranging from 13 kb to 30 kb (mean size 19.6 ± 4.3 kb). All asymptomatic carriers were more than 20 years old (mean age 36.4 ± 10.5 years), which were not considered in the preclinical condition. Among the 66 isolated cases, 35 (23 males and 12 females) were de novo since both parents were available to confirm their normal EcoRI fragments. The remaining 31 patients were uncertain because it was unable to get molecular analysis from either of their parents.
Size distribution of the short EcoRI fragments from136 probands showed statistically significant difference between the sporadic and familial cases (χ2 = 5.69, P < 0.05, Figure 1). 67.1% (47/70) of the familial cases carried 4q35 EcoRI fragment ranging from16 kb to 24 kb, whereas a lower proportion (42.9%, 15/35) of sporadic patients was detected with the same range.
Figure 1.
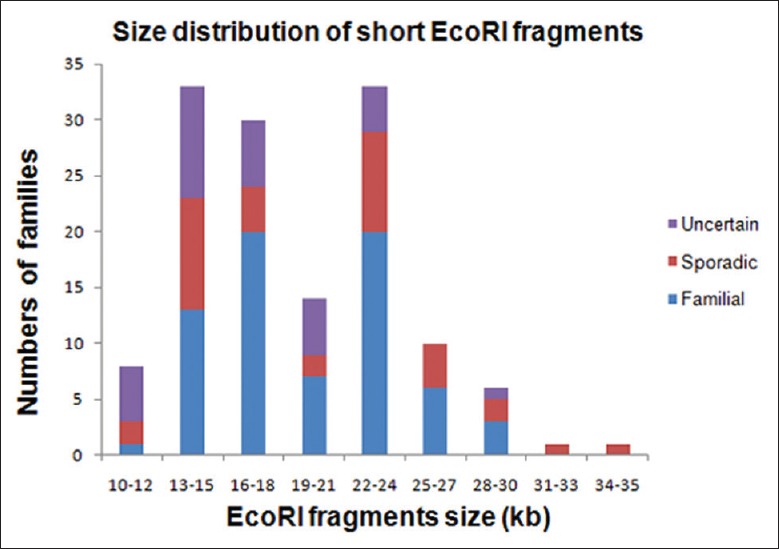
Size distribution of the p13E-11 EcoRI short fragments in 136 unrelated facioscapulohumeral muscular dystrophy probands.
Correlation between the size of short EcoRI fragment and age-corrected clinical severity score
Correlation in total symptomatic patients
It showed the significantly reverse correlation between the EcoRI fragment size and age-corrected CSS (F = 12.803, P < 0.01, Figure 2a). When male and female were observed respectively, only significant correlation for males (F = 9.257, P < 0.01). There was no correlation between the age of onset and the EcoRI fragment size in both genders (P > 0.05).
Figure 2.
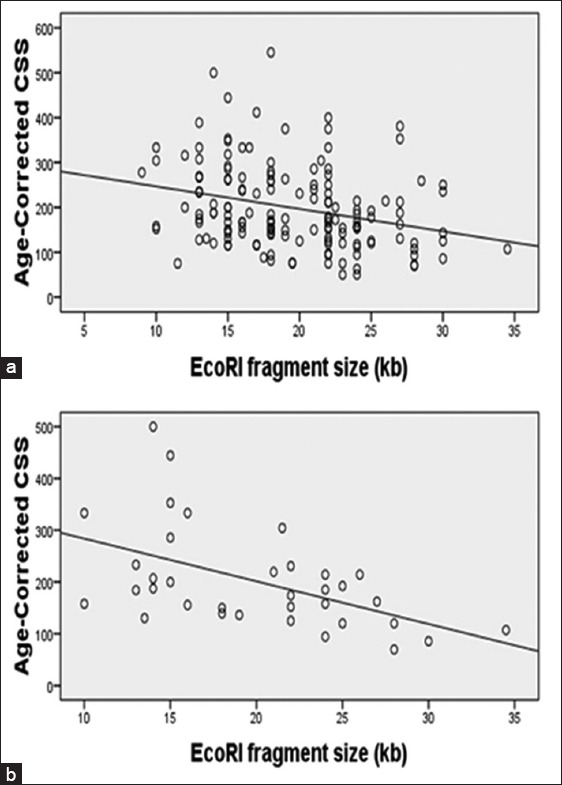
Correlation between the size of short EcoRI fragment and Age-corrected clinical severity score. (a) The diagram refers to the correlation in 154 symptomatic patients (r = −0.279, P < 0.005); (b) The diagram refers to the correlation only in 35 sporadic patients (r = −0.512, P < 0.005).
Sporadic and familial cases
There was significant correlation between the EcoRI fragment size and age-corrected CSS in 35 sporadic cases (F = 11.711, P < 0.01, Figure 2b). Compared to the correlation in total, the coefficient r raised to 0.512. However, the correlation in the familial cases and clinically affected relatives showed closely matched the coefficient of total (F = 5.297, r = −0.241, P < 0.05).
The proportion of males versus females in different clinical severity score groups
In Group I, the proportion of females was significantly higher than that of males among asymptomatic carriers (16 females, 3 males; χ2 = 10.86, P < 0.01) [Table 2]. If the symptomatic carriers were considered as nonpenetrants, the penetrance for FSHD-associated allele was estimated to 89.0% (154/173). There was significantly lower penetrance in females (68/84, 81.0%) than that in males (86/89, 96.6%).
Table 2.
The classification of clinical severity score, mean age and EcoRI fragment in 173 facioscapulohumeral muscular dystrophy patients
| Groups* | Male | Female | ||||||
|---|---|---|---|---|---|---|---|---|
| n | Age (years)† | EcoRI fragment (kb) | Range (kb) | n | Age (years)† | EcoRI fragment (kb) | Range (kb) | |
| I | 3 | 40.0 ± 12.5 | 19.3 ± 3.8 | 15–22 | 16 | 35.2 ± 10.2 | 19.7 ± 5.8 | 13–30 |
| II | 22 | 28.2 ± 10.1 | 21.7 ± 6.1 | 12–35 | 14 | 29.7 ± 12.0 | 20.1 ± 4.5 | 10–28 |
| III | 18 | 25.9 ± 5.9 | 20.0 ± 5.6 | 13–30 | 9 | 30.0 ± 13.4 | 19.3 ± 4.1 | 13–24 |
| IV | 27 | 30.0 ± 10.7 | 19.2 ± 4.4 | 12–26 | 20 | 30.3 ± 12.1 | 21.2 ± 5.2 | 13–30 |
| V | 15 | 34.3 ± 13.5 | 18.6 ± 3.9 | 13–29 | 13 | 36.3 ± 13.2 | 16.5 ± 5.8 | 10–27 |
| VI | 4 | 28.5 ± 7.6 | 13.5 ± 2.7 | 10–15 | 12 | 33.4 ± 11.1 | 18.6 ± 4.3 | 14–27 |
| Total | 89 | 29.7 ± 10.5 | 19.6 ± 5.2 | 10–35 | 84 | 32.2 ± 11.9 | 19.4 ± 5.2 | 10–30 |
*Groups according to clinical severity score. I: Asymptomatic individuals; II: 1.5; III: 2–2.5; IV: 3; V: 3.5; VI: 4–5; †Age: The age on examination.
Herein, we classified severely affected patients into Group VI (CS4–5), in which the proportion of females showed higher than males (χ2 = 4.93, P < 0.05). All male patients in this group presented severe phenotype with very small EcoRI fragment ranging from 10 kb to 15 kb. However, a large heterozygosity of EcoRI fragment ranging from 14 kb to 27 kb was observed in severe female patients. Only one early wheelchair-dependent male was evaluated as CS5 in our study, who presented marked lumbar lordosis and joint contracture with the smallest fragment size of 10 kb (Only one D4Z4 repeat left, Figure 3). On the other hand, the proportion of males was significantly different from females in sum Group II to Group V (χ2 = 17.37, P < 0.01). However, there was no significant difference between males and females in each group separately (II–V, P > 0.05).
Figure 3.
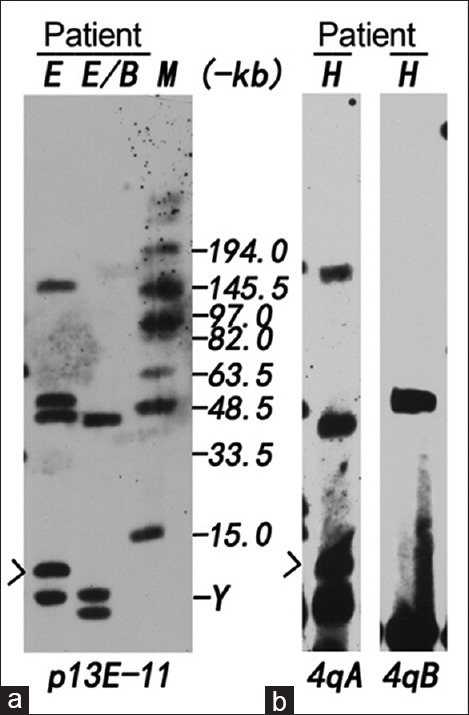
Analysis of the patient with the smallest EcoRI fragment of 10 kb. (a) DNA was digested with EcoRI (E) and EcoRI/BlnI (B), separated by pulsed-field gel electrophoresis and hybridized with probe p13E-11. 4q35-located alleles are resistant to BlnI, and 10q26-located alleles are sensitive to BlnI. Two 4q-type alleles of 10 kb (arrow) and 50 kb; two 10q-type alleles of 42 kb and 156 kb; (b) The same DNA was digested with Hind III (H) and hybridized with probes 4qA and 4qB. 4q-type allele of 10 kb (arrow) was identified as 4qA variant and 50 kb-allele was 4qB variant. M: MidRange PFG marker Y: Y chromosome.
Clinical severity score comparing in groups with different D4Z4 repeats
If the difference of age at examination was not considered, no significant correlation of CSS with short fragment size was observed in all symptomatic patients. In order to distinctly investigate the correlation between D4Z4 repeats and clinical severity, our patients were classified into three groups (A, B, C) according to different D4Z4 repeats [Figure 4]. Although the median CSS of these three groups showed no different (H = 3.020, P > 0.05), the relative proportion of patients with clinical severity classification was quite different. As mentioned above, only one patient with the smallest EcoRI fragment of CS 5 belonged to Group A. The patients of group A showed larger variation of CS ranging from 0.5 to 5. Compared to CS3.5–5 in Group A (32.7% + 2.0%), the proportion of patients with CS3.5-4.5 was remarkably decreased in Group B (22.5%) and Group C (13.6%). However, there was no significant correlation between Group A and group (B + C) with CS 3.5–4.5(χ2 = 3.54, P > 0.05). Conversely, 45.5% of the patients in Group C presented mild involvement of facial and scapulohumeral regions compared with 29.0% of the patients in groups (A + B), although it was showed no significant difference between Group C and groups (A + B) with CS0.5–1.5 (χ2 = 2.17, P > 0.05). Among 16 patients with severe phenotype (CS4-5) (16/173, 9.24%), 9 patients (9/49, 18.4%) in group A compared with 6 (6/102, 5.89%) in Group B and 1 (1/22, 4.54%) in Group C. These data indicated that significant difference between Group A and groups (B + C) for developing severe phenotype (χ2 = 5.34, P < 0.05).
Figure 4.
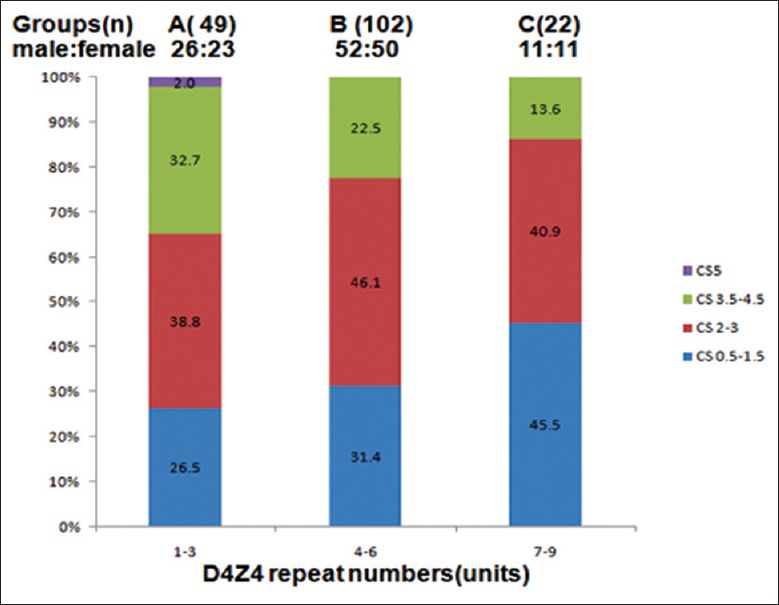
Relative proportion of patients with reduced D4Z4 repeats in each class of CCS.
DISCUSSION
The contraction of D4Z4 repeat in 4q35 is responsible for FSHD, so the 4q-derived EcoRI fragment is considered as one major factor for determining clinical phenotype, including age of onset, clinical course, and age at loss of ambulation.[18] Our study represents new insights into genotype-phenotype correlations in Chinese FSHD, and supports that other epigenetic factors appear to involve in phenotypic regulation.[19,20]
In agreement with the clinical description of FSHD, the characteristic presentation at onset associated with facial or scapulohumeral weakness was observed in most of our patients. The major clinical problem with typical onset was characterized by difficulty in arm abduction, often with asymmetry. Facial weakness was one common associated sign on the neurological examination, while only a few of our patients were aware of facial involvement as initial presentation at early age. Although the obvious appearance of facial weakness is helpful for FSHD diagnosis, the facial-sparing phenotype at late-onset was often associated with very small 4q35 deletion, which alerts the clinician for the disease heterogeneity. As a clinically heterogeneous myopathy, the atypical patterns of FSHD were usually characterized by the focal presentation and have been reported on the increasing basis.[21,22,23] Foot drop, which was observed in 2.6% (4/154) of our series, has been known as an uncommon feature of disease onset when carefully investigated.[24] Although all showed the associated sign of facial or scapulohumeral region, these atypical cases were often delayed diagnosis by the highly focal presentation. It is worth noting that the distinct foot drop as initial manifestation ought to be distinguishing from other mimic neuromuscular disorder, such as distal myopathy with rimmed vacuoles. Another atypical presentation characterized by proximal lower limb weakness was found in 7.1% of our patients, in greatly accordance with the report in Italy.[24] The clinical course in these cases seemed to different from the common occurrence, which involvement of lower limbs is expected to occur in the later stage. In our patients with atypical onset, the 4q35 EcoRI fragment size was similar to that one found in majority with typical onset. Therefore, the genetic and epigenetic factors contributing to these disease manifestations need further investigation. Because other unclassified myopathies similar to atypical forms of FSHD may carry nonpathogenic short EcoRI fragment on 4q35, analysis of the D4Z4 repeat length alone easily leads to misdiagnosis, and its distinguishing requires to determinate 4qA/4qB variant.
The remarkable intra- and inter-familial clinical heterogeneity is one well-known characteristic of FSHD. In our survey, the most severe patient was detected with the smallest 10 kb fragment (one D4Z4 repeat), and obviously clinical variability was noticed in patients carrying contracted fragments above 13 kb, for there is no significant correlation of mutation spectrum associated with phenotypic manifestation. However, we indicated a roughly inversed correlation between the EcoRI fragment size and age-corrected CSS. This result supports that the various age at examination could influence the genotype-phenotype correlation and deteriorating synergistic factors could affect the disease progression.[25] Further analysis revealed that inverse correlation was only significantly observed in male, inconsistent with a similar survey.[26] On the other hand, females (16/19) predominantly occupied in our asymptomatic carriers and half of them (8/16) were probands’ mother. In addition, both asymptomatic gene carriers and patients with severe phenotype (CS4-5) were predominately occupied by females, which showed large variation of EcoRI fragment incompatible with clinical phenotype. Hence, the clinicians should catch more attention to perform genetic test in the proband's female relatives. Now, increasing evidences support that the (epi) genetic or unknown modifying genes/factors contribute to phenotypic heterogeneity in FSHD, especial in females. Accordingly, it is difficult to provide accurate clinical prognosis and genetic counseling for the asymptomatic gene carriers or late-onset patients.
The intra-familial variability is another genetic influential factor in analysis of genotype-phenotype correlation. The correlation between the EcoRI fragment size and age-corrected CSS in our sporadic cases displayed much stronger supporting that intra-familial variability is higher than inter-familial one.[18] The familial patients with EcoRI fragment ranging from 16 kb to 24 kb presented significantly higher proportion, indicating that this length size of EcoRI fragment is more stably co-segregated. We speculated those familial patients with very small EcoRI fragment often cause severe phenotype and affect fertility. In contrast to the variable proportion of asymptomatic carriers in other studies (3.5–50%),[25,27] our results revealed that high incidence (14/42, 45.2%) of asymptomatic (or minimally affected) gene carriers in some pedigrees with at least one symptom-free individual. Inconsistent to asymptomatic carrier with relatively large fragment size from 21 kb to 27 kb in previous study,[12] we detected more various fragment size from 13 kb to 30 kb in nonpenetrant carriers. Therefore, it is difficult to offer genetic counseling for the families, especially female cases, upon determining whether their offsprings with segregated short EcoRI fragment is in preclinical phase or asymptomatic carrier.
On the whole, we only observed rough correlation between the short EcoRI fragment and age-corrected CS, implying that genetic testing of short EcoRI fragment is insufficient for predicting clinical progression. High proportions of family members carrying the same short fragment were asymptomatic or minimally affected, which alerts the clinicians to offer molecular detection for the probands’ relatives in genetic counseling.
ACKNOWLEDGMENTS
The authors thank Dr. Silvère Van der Maarel of Leiden University for his kindly providing the p13E-11, 4qA and 4qB plasmids; Dr. Rossella Tupler of Miogen lab department di Scienze della Vita for technological help. Also, we sincerely thank the participants for their help and willingness to participate in this study, and the anonymous reviewers for improving this manuscript.
Footnotes
Edited by: Ya-Lin Bao
Source of Support: This work was supported by grants from the National Natural Science Foundation of China (81100937, Beijing), National key clinical specialty discipline construction program, and key clinical specialty discipline construction program of Fujian, P.R. China.
Conflict of Interest: None declared.
REFERENCES
- 1.Emery AE. Population frequencies of inherited neuromuscular diseases — A world survey. Neuromuscul Disord. 1991;1:19–29. doi: 10.1016/0960-8966(91)90039-u. [DOI] [PubMed] [Google Scholar]
- 2.Lunt PW, Compston DA, Harper PS. Estimation of age dependent penetrance in facioscapulohumeral muscular dystrophy by minimising ascertainment bias. J Med Genet. 1989;26:755–60. doi: 10.1136/jmg.26.12.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lunt PW, Harper PS. Genetic counselling in facioscapulohumeral muscular dystrophy. J Med Genet. 1991;28:655–64. doi: 10.1136/jmg.28.10.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wijmenga C, Hewitt JE, Sandkuijl LA, Moerer P, Wright TJ, Williamson R, et al. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat Genet. 1992;2:26–30. doi: 10.1038/ng0992-26. [DOI] [PubMed] [Google Scholar]
- 5.van Deutekom JC, Wijmenga C, van Tienhoven EA, Gruter AM, Hewitt JE, Padberg GW, et al. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum Mol Genet. 1993;2:2037–42. doi: 10.1093/hmg/2.12.2037. [DOI] [PubMed] [Google Scholar]
- 6.Wang ZQ, Wang N, van der Maarel S, Murong SX, Wu ZY. Distinguishing the 4qA and 4qB variants is essential for the diagnosis of facioscapulohumeral muscular dystrophy in the Chinese population. Eur J Hum Genet. 2011;19:64–9. doi: 10.1038/ejhg.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deidda G, Cacurri S, Piazzo N, Felicetti L. Direct detection of 4q35 rearrangements implicated in facioscapulohumeral muscular dystrophy (FSHD) J Med Genet. 1996;33:361–5. doi: 10.1136/jmg.33.5.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lemmers RJ, de Kievit P, Sandkuijl L, Padberg GW, van Ommen GJ, Frants RR, et al. Facioscapulohumeral muscular dystrophy is uniquely associated with one of the two variants of the 4q subtelomere. Nat Genet. 2002;32:235–6. doi: 10.1038/ng999. [DOI] [PubMed] [Google Scholar]
- 9.Lemmers RJ, O’Shea S, Padberg GW, Lunt PW, van der Maarel SM. Best practice guidelines on genetic diagnostics of facioscapulohumeral muscular dystrophy: Workshop 9 th June 2010, LUMC, Leiden, The Netherlands. Neuromuscul Disord. 2012;22:463–70. doi: 10.1016/j.nmd.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 10.Lemmers RJ, van der Vliet PJ, Klooster R, Sacconi S, Camaño P, Dauwerse JG, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science. 2010;329:1650–3. doi: 10.1126/science.1189044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wallace LM, Garwick SE, Mei W, Belayew A, Coppee F, Ladner KJ, et al. DUX4, a candidate gene for facioscapulohumeral muscular dystrophy, causes p53-dependent myopathy in vivo. Ann Neurol. 2011;69:540–52. doi: 10.1002/ana.22275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ricci E, Galluzzi G, Deidda G, Cacurri S, Colantoni L, Merico B, et al. Progress in the molecular diagnosis of facioscapulohumeral muscular dystrophy and correlation between the number of KpnI repeats at the 4q35 locus and clinical phenotype. Ann Neurol. 1999;45:751–7. doi: 10.1002/1531-8249(199906)45:6<751::aid-ana9>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 13.Krasnianski M, Eger K, Neudecker S, Jakubiczka S, Zierz S. Atypical phenotypes in patients with facioscapulohumeral muscular dystrophy 4q35 deletion. Arch Neurol. 2003;60:1421–5. doi: 10.1001/archneur.60.10.1421. [DOI] [PubMed] [Google Scholar]
- 14.Ricci G, Scionti I, Sera F, Govi M, D’Amico R, Frambolli I, et al. Large scale genotype-phenotype analyses indicate that novel prognostic tools are required for families with facioscapulohumeral muscular dystrophy. Brain. 2013;136:3408–17. doi: 10.1093/brain/awt226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu ZY, Wang ZQ, Murong SX, Wang N. FSHD in Chinese population: Characteristics of translocation and genotype-phenotype correlation. Neurology. 2004;63:581–3. doi: 10.1212/01.wnl.0000133210.93075.81. [DOI] [PubMed] [Google Scholar]
- 16.Padberg GW, Lunt PW, Koch M, Fardeau M. Diagnostic criteria for facioscapulohumeral muscular dystrophy. Neuromuscul Disord. 1991;1:231–4. doi: 10.1016/0960-8966(91)90094-9. [DOI] [PubMed] [Google Scholar]
- 17.Van Overveld PG, Enthoven L, Ricci E, Rossi M, Felicetti L, Jeanpierre M, et al. Variable hypomethylation of D4Z4 in facioscapulohumeral muscular dystrophy. Ann Neurol. 2005;58:569–76. doi: 10.1002/ana.20625. [DOI] [PubMed] [Google Scholar]
- 18.Lunt PW, Jardine PE, Koch MC, Maynard J, Osborn M, Williams M, et al. Correlation between fragment size at D4F104S1 and age at onset or at wheelchair use, with a possible generational effect, accounts for much phenotypic variation in 4q35-facioscapulohumeral muscular dystrophy (FSHD) Hum Mol Genet. 1995;4:951–8. doi: 10.1093/hmg/4.5.951. [DOI] [PubMed] [Google Scholar]
- 19.Sacconi S, Lemmers RJ, Balog J, van der Vliet PJ, Lahaut P, van Nieuwenhuizen MP, et al. The FSHD2 gene SMCHD1 is a modifier of disease severity in families affected by FSHD1. Am J Hum Genet. 2013;93:744–51. doi: 10.1016/j.ajhg.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lemmers RJ, Goeman JJ, Tawil R, Tapscott SJ, Bakker B, van der Maarel SM, et al. Inter-individual differences in CpG methylation at D4Z4 correlate with clinical variability in FSHD1 and FSHD2. Hum Mol Genet. 2014;486:1–11. doi: 10.1093/hmg/ddu486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uncini A, Galluzzi G, Di Muzio A, De Angelis MV, Ricci E, Scoppetta C, et al. Facioscapulohumeral muscular dystrophy presenting isolated monomelic lower limb atrophy. Report of two patients with and without 4q35 rearrangement. Neuromuscul Disord. 2002;12:874–7. doi: 10.1016/s0960-8966(02)00027-5. [DOI] [PubMed] [Google Scholar]
- 22.Kottlors M, Kress W, Meng G, Glocker FX. Facioscapulohumeral muscular dystrophy presenting with isolated axial myopathy and bent spine syndrome. Muscle Nerve. 2010;42:273–5. doi: 10.1002/mus.21722. [DOI] [PubMed] [Google Scholar]
- 23.Hassan A, Jones LK, Jr, Milone M, Kumar N. Focal and other unusual presentations of facioscapulohumeral muscular dystrophy. Muscle Nerve. 2012;46:421–5. doi: 10.1002/mus.23358. [DOI] [PubMed] [Google Scholar]
- 24.Pastorello E, Cao M, Trevisan CP. Atypical onset in a series of 122 cases with FacioScapuloHumeral Muscular Dystrophy. Clin Neurol Neurosurg. 2012;114:230–4. doi: 10.1016/j.clineuro.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakellariou P, Kekou K, Fryssira H, Sofocleous C, Manta P, Panousopoulou A, et al. Mutation spectrum and phenotypic manifestation in FSHD Greek patients. Neuromuscul Disord. 2012;22:339–49. doi: 10.1016/j.nmd.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 26.Tonini MM, Passos-Bueno MR, Cerqueira A, Matioli SR, Pavanello R, Zatz M. Asymptomatic carriers and gender differences in facioscapulohumeral muscular dystrophy (FSHD) Neuromuscul Disord. 2004;14:33–8. doi: 10.1016/j.nmd.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 27.Goto K, Nishino I, Hayashi YK. Very low penetrance in 85 Japanese families with facioscapulohumeral muscular dystrophy 1A. J Med Genet. 2004;41:e12. doi: 10.1136/jmg.2003.008755. [DOI] [PMC free article] [PubMed] [Google Scholar]