

Abstract
Background:
It has been demonstrated that only 10%–20% cigarette smokers finally suffer chronic obstructive pulmonary disease (COPD). The underlying mechanism of development remains uncertain so far. Nitric oxide (NO) has been found to be closely associated with the pathogenesis of COPD, the alteration of NO synthase (NOS) expression need to be revealed. The study aimed to investigate the alterations of NOS isoforms expressions between smokers with and without COPD, which might be helpful for identifying the susceptibility of smokers developing into COPD.
Methods:
Peripheral lung tissues were obtained from 10 nonsmoker control subjects, 15 non-COPD smokers, and 15 smokers with COPD. Neuronal NOS (nNOS), inducible NOS (iNOS), and endothelial NOS (eNOS) mRNA and protein levels were measured in each sample by using real-time polymerase chain reaction and Western blotting.
Results:
INOS mRNA was significantly increased in patients with COPD compared with nonsmokers and smokers with normal lung function (P < 0.001, P = 0.001, respectively). iNOS protein was also higher in COPD patients than nonsmokers and smokers with normal lung function (P < 0.01 and P = 0.01, respectively). However, expressions of nNOS and eNOS did not differ among nonsmokers, smokers with and without COPD. Furthermore, there was a negative correlation between iNOS protein level and lung function parameters forced expiratory volume in 1 s (FEV1) (% predicted) (r = −0.549, P = 0.001) and FEV1/forced vital capacity (%, r = −0.535, P = 0.001).
Conclusions:
The expression of iNOS significantly increased in smokers with COPD compared with that in nonsmokers or smokers without COPD. The results suggest that iNOS might be involved in the pathogenesis of COPD, and may be a potential marker to identify the smokers who have more liability to suffer COPD.
Keywords: Chronic Obstructive Pulmonary Disease, Nitric Oxide Synthase, Smoking
INTRODUCTION
Chronic obstructive pulmonary disease (COPD) is a major and increasing global health problem, which is predicated to become the third most common cause of death and the fifth most common cause of disability in the world by 2020.[1,2] Cigarette smoking is the most important risk factor for COPD. However, only 10%–20% of the chronic smokers develop into COPD, suggesting that other factors, including genetic factors may play a role in the development of COPD.[3,4]
It has been reported that exhaled nitric oxide (NO) level from the lung peripheral (Calv) is increased in COPD.[5] The biological properties of NO in the airways are two-faced. On one hand, NO regulates physiological function. It controls vascular tone and also acts as a neurotransmitter of the inhibitory nonadrenergic noncholinergic nerves in the airways.[6] On the other hand, NO interacts with superoxide anion (O2 −) to produce the highly cytotoxic and strong nitrosant peroxynitrite (ONOO−), which has been reported to cause lipid peroxidation, DNA damage, and disturb protein function.[7] Moreover, NO has been implicated in the pathophysiology of inflammatory airway diseases such as COPD and asthma.[6]
Nitric oxide is synthesized from L-arginine by the enzyme NO synthase (NOS), of which three isoforms have been identified.[8,9,10] Neuronal NOS (nNOS) and endothelial NOS (eNOS) are constitutively expressed enzymes whose activities are regulated by intracellular calcium and calmodulin. Small amounts of NO are generated by these two isoforms, whereas the activity of inducible NOS (iNOS) is independent of calcium but is transcriptionally regulated by lipopolysaccharide, inflammatory cytokines, and so on.
Nitric oxide synthase gene polymorphisms have been studied in many diseases such as asthma[11,12] and tuberculosis.[13,14] However, few genetic studies have been reported in COPD.[15,16] Thus, we hypothesized that genetic variants of NOS may contribute to the pathogenesis of COPD. In this study, we investigate the protein and mRNA expressions of three NOS isoenzymes in the peripheral lung tissue of patients with COPD. It is aimed to investigate the alteration of NOS expression which might be related to genetic variants of NOS. The study is expected to be helpful for identifying the liability of cigarette smokers suffering COPD.
METHODS
Subjects
Three groups of subjects from Tongji Hospital undergoing lung tumor surgery were enrolled. The COPD diagnosis was established according to the Global Initiative for Chronic Obstructive Lung Disease guidelines.[17] All subjects were in stable condition without history of inhaled or oral corticosteroid at least in 3 months prior to enrollment. Exclusion criteria included chronic respiratory failure, positive bronchodilator to test, bronchial asthma, tuberculosis, cardiovascular diseases, and chronic metabolic diseases. Detailed clinical information about these subjects is listed in Table 1. The study was approved by the Ethics Committee of the Tongji Hospital, and all the subjects signed informed consent.
Table 1.
Characteristics of subjects
| Characteristics | NSC (n = 10) | SC (n = 15) | COPD (n = 15) |
|---|---|---|---|
| Age, year | 54.4 ± 10.7 | 54.3 ± 7.2 | 58.9 ± 10.0 |
| Sex: Male/female | 9/1 | 15/0 | 15/0 |
| Smoking history, pack/year | 0 | 33.9 ± 21.0 | 40.0 ± 22.4 |
| BMI, kg/m2 | 24.2 ± 2.0 | 23.7 ± 3.5 | 21.7 ± 3.7 |
| FEV1 (%predicted) | 95.6 ± 14.6 | 95.6 ± 14.4 | 71.9 ± 9.4*,† |
| FEV1/FVC (%) | 78.2 ± 4.2 | 78.3 ± 6.3 | 61.1 ± 7.5*,† |
Parameter values are mean ± SD for subjects. NSC: Nonsmokers control subjects; SC: Smoker control subjects; COPD: Chronic obstructive pulmonary disease; Pack/year: 1-year smoking 20 cigarettes per day; BMI: Body mass index; FEV1: Forced expiratory volume in 1 s; FVC: Forced vital capacity; SD: Standard deviation. *Significantly different from control nonsmokers: P < 0.001; †Significantly different from control smokers: P < 0.001.
Lung tissue study
The peripheral lung tissues were obtained at the time of the surgical procedure. Just after resection, specimens were taken from an area at least 5 cm from the tumor, and immediately frozen in liquid nitrogen and stored at −80°C. The sample was divided into two parts. One was for real-time polymerase chain reaction (PCR) analysis, and the other was for Western blotting detection.
Real-time quantitative polymerase chain reaction
Total RNA was extracted from lung tissue using TRIzol (Invitrogen, USA) according to the manufacturers’ instructions and quantified by the absorption at 260 nm. 2.5 μg of total RNA was reverse transcribed to cDNA using a PrimeScript® RT Master Mix kit (DRR036A, TaKaRa Bio, Japan). nNOS, iNOS, and eNOS transcript levels were quantified by real-time PCR using the SYBR® Premix Ex Taq™ kit (DRR420A, TaKaRa Bio, Japan). PCR was performed using the ABI PRISM® 7900HT real-Time PCR System (Applied Biosystems, USA) as follows: 95°C for 30 s as pretreatment, 40 cycles of 95°C for 5 s for denaturation, and 60°C for 30 s for annealing/extension, and each sample was run in triple. The cycle threshold value (Ct) obtained for each gene of the target was normalized with that of β-actin, a housekeeping gene. In order to obtain relative fold-change of gene expression, 2−ΔΔCt method was used.[18] ΔCt = Ct (gene of target) − Ct (housekeeping gene), ΔΔCt = ΔCt (study group) − ΔCt (control group), then the regulation of individual genes was calculated using the formula, 2−ΔΔCt. The primers were designed by TaKaRa Bio and are listed in Table 2.
Table 2.
Oligonucleotide primers for real-time PCR used in the study
| Primers | Prime sequences (5’-3’) | Product length (bp) |
|---|---|---|
| nNOS | ||
| Forward | TGCGAACGTACGAAGTGACCA | 149 |
| Reverse | GCAGTGTCTGTCCGCGCTTA | |
| iNOS | ||
| Forward | ACGTGCGTTACTCCACCAACAA | 109 |
| Reverse | CGGATGAGCTGAGCATTCCA | |
| eNOS | ||
| Forward | GCTGTCTGCATGGACCTGGA | 119 |
| Reverse | TCCACGATGGTGACTTTGGCTA | |
| β-actin | ||
| Forward | TGGCACCCAGCACAATGAA | 186 |
| Reverse | CTAAGTCATAGTCCGCCTAGAAGCA |
nNOS: Neuronal nitric oxide synthase; iNOS: Inducible nitric oxide synthase; eNOS: Endothelial NOS; PCR: Polymerase chain reaction.
Western blotting
Frozen samples were weighed (about 100 mg) and the tissue was homogenized (1:7 w/v) with hand-made glass pestles in cold RIPA buffer and the protease inhibitors phenylmethylsulfonyl fluoride (1 mmol/L) (G2002, Guge Bio, China). Samples were then centrifuged at 12,000 ×g for 5 min at 4°C. The supernatant was immediately transferred into fresh cold microcentrifuge tubes, and the protein concentration was assessed using the BCA protein assay kit (P0010S, Beyotime Bio, China). The resulting supernatant was mixed with four-fold volume of loading buffer (G2013, Guge Bio, China) and then was boiled for 5 min. 60 μg/sample was separated on 8% sodium dodecyl sulfate (SDS)-polyacrylamide gel (Guge Bio, China) and transferred to 0.45 μm polyvinylidene difluoride membrane using a transblotting apparatus (Bio-Rad). The transfer buffer contained 25 mmol/L Tris, 190 mmol/L glycine, 0.1% SDS, and 20% methanol and was carried out at 300 mA for 2.5 h. The membranes were then blocked by shaking in a blocking buffer containing 5% nonfat dry milk and 0.1% Tween 20 at room temperature for 1 h. Then membranes were incubated overnight at 4°C with primary antibody, anti-nNOS mouse monoclonal antibody at 1:500 dilution (610308, BD Biosciences, USA), anti-iNOS rabbit polyclonal antibody at 1:2000 dilution (ab3523, Abcam, UK), anti-eNOS mouse monoclonal antibody at 1:250 dilution (610296, BD Biosciences, USA), and anti-β-actin rabbit polyclonal antibody at 1:1000 (1616-R, Santa Cruz, USA). After washing with TBST 3 times, 10 min/time, the membranes were then incubated with secondary antibodies. The secondary antibodies, a horseradish peroxidase-linked sheep anti-rabbit immunoglobulin G (IgG) for iNOS, β-actin (KPL, USA) and a horseradish peroxidase-linked sheep anti-mouse IgG for nNOS, eNOS (KPL, USA), were both incubated at 1:3000 dilution in blocking solution at room temperature for 1.5 h. The immunoreactive bands were detected by chemiluminescence according to the specifications provided by the manufacturer (ECL kit, Beyotime Bio, China). Films were scanned, and the protein content was assessed by densitometry using Quantity One software (Bio-Rad, USA). The results were normalized to the β-actin content.
Statistical analysis
Data were presented as mean ± standard division or median ± interquartile (IQR) for not normally distributed data. Differences characteristics data among three groups were analyzed using analysis of variance (ANOVA) and LSD for multiple comparison test. The Kruskal–Wallis test was used to compare nonnormal distribution data, when significant, by the Mann–Whitney U-test for comparison between groups. Relationships between various parameters were studied by calculating the Spearman's correlation coefficient. A P < 0.05 was considered significant. SPSS version 18.0 (SPSS Inc., IL, USA) was used for all statistical analyses.
RESULTS
Subjects
Subject characteristics are reported in Table 1. There are no significant differences in age, sex, BMI. Smoking history was similar in patients with COPD and smoker control (SC) subjects. Forced expiratory volume in 1 s (FEV1) (% predicted) and FEV1/forced vital capacity (FVC) (%) were significantly lower in patients with COPD as compared with both nonsmokers control (NSC) and SC subjects (P < 0.001).
Expressions of three NOS isoforms mRNA in three groups
Figure 1 shows the fold changes of mRNA of NOS isoforms expressions in peripheral lung tissue of NSC subjects, SC subjects, and COPD patients. There were significant increases in iNOS mRNA expression in COPD patients compared to NSC and SC subjects (P < 0.001, P = 0.001, respectively). However, no significant differences were found in nNOS and eNOS mRNA levels among the three groups.
Figure 1.

mRNA expression of (a) nNOS; (b) iNOS; and (c) eNOS in human peripheral lung tissue from NSC subjects, SC subjects, and COPD patients by real-time polymerase chain reaction. The data are presented as mean ± standard division. #P < 0.001 versus patients with NSC. ΔP = 0.001 versus patients with SC. nNOS: Neuronal nitric oxide synthase; iNOS: Inducible nitric oxide synthase; eNOS: Endothelial nitric oxide synthase; NSC: Nonsmokers control; SC: Smokers control; COPD: Chronic obstructive pulmonary disease.
Expressions of three NOS isoforms protein in three groups
NOS protein expressions in peripheral lung tissue are shown in Figures 2–4. iNOS protein levels were significantly higher in COPD patients (1.7, IQR: 0.6–2.6) compared with NSC subjects (0.4, IQR: 0.2–0.9) and SC subjects (0.5, IQR: 0.3–1.0) (P < 0.01, P = 0.01, respectively) [Figure 3]. However, there were no significant differences in nNOS and eNOS protein levels among the three groups [Figures 2 and 4]. These results are consistent with mRNA of NOS isoforms expressions. In the three groups, iNOS optical densities were significantly negatively correlated with FEV1 (% predicted) (r = −0.549, P = 0.001) and FEV1/FVC (%, r = −0.535, P = 0.001).
Figure 2.
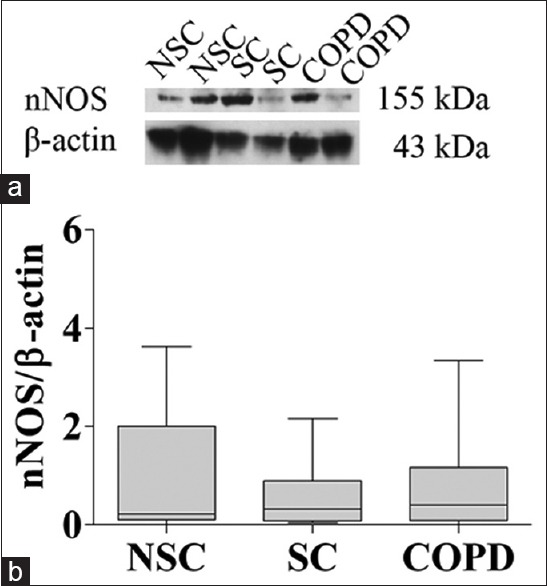
nNOS protein expression in peripheral lung tissue from NSC subjects, SC subjects, and COPD patients. (a) Representative Western blot analysis of nNOS and β-actin; (b) Densitometric analysis of nNOS and β-actin bands. Each bar represents the median ± interquartile of each group. nNOS: Neuronal nitric oxide synthase; NSC: Nonsmokers control; SC: Smokers control; COPD: Chronic obstructive pulmonary disease.
Figure 4.
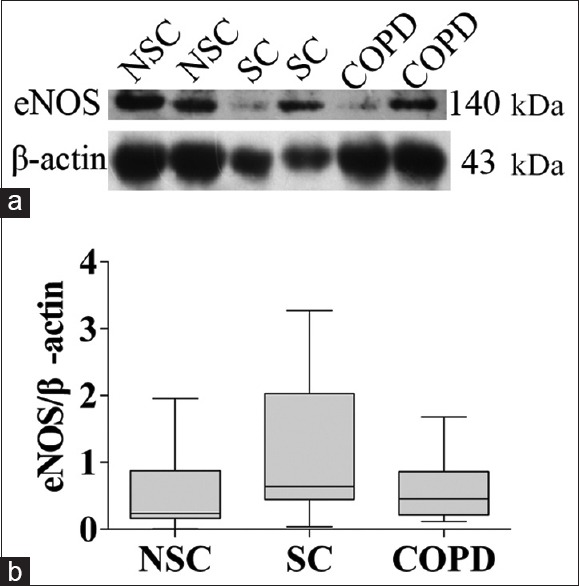
eNOS protein expression in peripheral lung tissue from NSC subjects, SC subjects, and COPD patients. (a) Representative Western blot analysis of eNOS and β-actin; (b) Densitometric analysis of eNOS and β-actin bands. Each bar represents the median ± interquartile of each group. eNOS: Endothelial nitric oxide synthase; NSC: Nonsmokers control; SC: Smokers control; COPD: Chronic obstructive pulmonary disease.
Figure 3.
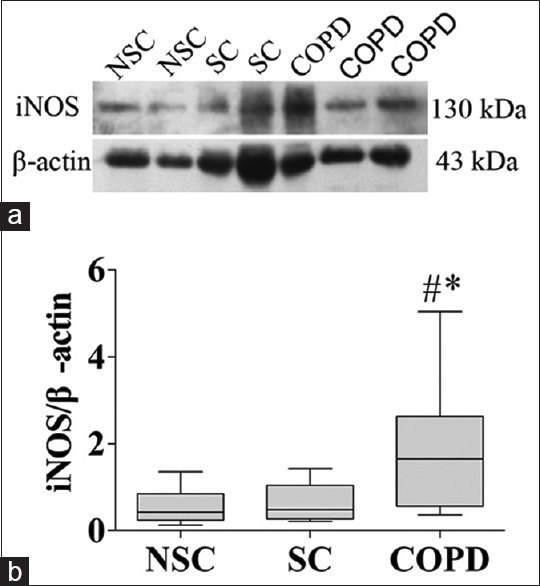
iNOS protein expression in peripheral lung tissue from NSC subjects, SC subjects, and COPD patients. (a) Representative Western blot analysis of iNOS and β-actin; (b) Densitometric analysis of iNOS and β-actin bands. Each bar represents the median ± interquartile of each group. *P < 0.01 versus group NSC, #P = 0.01 versus group SC. iNOS: inducible nitric oxide synthase; NSC: Nonsmokers control; SC: Smokers control; COPD: Chronic obstructive pulmonary disease.
DISCUSSION
The present study suggested that iNOS mRNA and protein levels were significantly increased in the peripheral lung tissues from COPD patients compared with NSC and SC subjects. Furthermore, there is a significant negative correlation between the iNOS expression and the level of airflow limitation as measured by FEV1 (% predicted) and FEV1/FVC (%). These results indicate that iNOS might be involved in the progression of COPD, and the alteration of NOS may play a role in the susceptibility of smokers suffering COPD.
Our results confirmed previous findings on increased iNOS expression in COPD patients.[19,20,21,22] We extended these findings to peripheral lung tissues via real-time PCR and Western blot analysis. The association between iNOS expression and airflow limitation is also consistent with the results in the bronchial tissue of COPD patients.[22] However, Brindicci et al.[23] found that nNOS protein expression negatively correlated with FEV1 (% predicted) and FEV1/FVC (%). This discrepancy is uncertain and may be explained by the fact that the differences in the severity of the airflow obstruction in COPD patients. The enrolled patients in their study were mostly severe COPD (FEV1 46.0 ± 0.1, 20.3 ± 1.8, and FEV1/FVC 53 ± 5.5, 35.9 ± 3), whereas our study only included patients with less severe degree of obstruction of COPD (FEV1 71.9 ± 9.4, and FEV1/FVC 61.1 ± 7.5).
Recently, Seimetz et al.[24] reported that smoke exposure caused emphysema and pulmonary hypertension in wild-type mice and in eNOS−/− mice, but not in iNOS−/− mice. Moreover, treatment of wild-type mice with an iNOS inhibitor prevented structural and functional alterations of both the lung vasculature and alveoli and also reversed established disease. These findings supported the important role of iNOS in the pathogenesis of COPD. We infer that up-regulation of iNOS may be the major source of exhaled NO in COPD[5] and the alteration of iNOS levels may be as a result of genetic variants of iNOS which may be helpful for identifying the liability of cigarette smokers suffering COPD.
Inducible NOS gene polymorphism has been studied in many diseases such as asthma[11,12] and tuberculosis.[13,14] However, few genetic studies have been reported in COPD.[15,16] Arif et al.[16] reported that the association of eNOS polymorphism (−786T/C, −922A/G, and 4B/4A) with the susceptibility to COPD. And the combination of genotypes containing −786C and 4A alleles correlated with reduced FEV1 values and nitrite levels. However, we did not find any differences of eNOS expression among three groups. Furthermore, Brindicci et al.[23] also demonstrated that there were no differences among patients with mild to moderate COPD, NSC subjects, and SC subjects. And no correlation was detected between eNOS expression and FEV1 (% predicted). This discrepancy may be explained by the fact that a relatively small number of patients and with less severe degree of obstruction of COPD in our study. There may also be different genetic background in population. However, the possibility cannot be excluded that both eNOS and iNOS gene polymorphism are involved in susceptibility to COPD. Further studies are needed to explore eNOS and iNOS gene polymorphism in large study subjects.
The underlying mechanism of iNOS taking part in the pathogenesis of COPD is still unknown. A possible mechanism is that an increase in lung iNOS expression in patients with COPD increases NO synthesis. NO interacts with O2 − to produce the formation of the strong ONOO−, which can lead to nitration (addition of - NO2) of most classes of biological molecules. ONOO− may thus provoke inhibition of mitochondrial respiration, protein dysfunction, and damage to cell membranes and DNA.[7] Seimetz et al.[24] have also found that ONOO− indeed caused nitration in primary isolated alveolar epithelial cells, pulmonary vascular endothelial cells, and pulmonary arterial smooth muscle cells from resistance vessels. Furthermore, elevated levels of ONOO− have been found in human patients with COPD.[25] Hence, further studies are required to confirm and explore the molecular mechanism.
We must acknowledge that our study has several limitations. First, we obtained all specimens of peripheral lung tissue during lung tumor surgery. The specimens were only from less severe COPD. Although we can also obtain peripheral lung tissue specimens through lung volume reduction surgery, only a few patients with severe COPD could undergo that type surgery. Second, although we took specimens from an area at least 5 cm from the tumor, we cannot exclude that a possible cancer microenvironment may influence the results. Third, the relatively small number of subjects is a potential weakness of our study. Finally, we did not measure NOS activity, all specimens were obtained after surgery resection, which might cause NOS activity decrease and influence the results.
The current study showed that iNOS expression was significantly increased in the peripheral lung tissue of patients with COPD and associated with the degree of airflow limitation. These results suggest that iNOS might be involved in the pathogenesis of COPD and may be a potential marker to identify the smokers who have more liability to suffer COPD. Future studies are needed to explore iNOS gene polymorphisms in the development of COPD.
ACKNOWLEDGMENTS
We would like to thank Ran Wang and Bo Zhang for excellent technical support.
Footnotes
Edited by: Xiu-Yuan Hao
Source of Support: This work was supported by the grants from Natural Science Foundation of China (No. 81270106), Ministry of Science and Technology of China (No. 2012BAI05B00) and Ministry of Public Health (No. 201002008).
Conflict of Interest: None declared.
REFERENCES
- 1.Murray CJ, Lopez AD. Alternative projections of mortality and disability by cause 1990-2020: Global Burden of Disease Study. Lancet. 1997;349:1498–504. doi: 10.1016/S0140-6736(96)07492-2. [DOI] [PubMed] [Google Scholar]
- 2.Lopez AD, Murray CC. The global burden of disease, 1990-2020. Nat Med. 1998;4:1241–3. doi: 10.1038/3218. [DOI] [PubMed] [Google Scholar]
- 3.Snider GL. Chronic obstructive pulmonary disease: Risk factors, pathophysiology and pathogenesis. Annu Rev Med. 1989;40:411–29. doi: 10.1146/annurev.me.40.020189.002211. [DOI] [PubMed] [Google Scholar]
- 4.Barnes PJ. Chronic obstructive pulmonary disease. N Engl J Med. 2000;343:269–80. doi: 10.1056/NEJM200007273430407. [DOI] [PubMed] [Google Scholar]
- 5.Brindicci C, Ito K, Resta O, Pride NB, Barnes PJ, Kharitonov SA. Exhaled nitric oxide from lung periphery is increased in COPD. Eur Respir J. 2005;26:52–9. doi: 10.1183/09031936.04.00125304. [DOI] [PubMed] [Google Scholar]
- 6.Ricciardolo FL, Sterk PJ, Gaston B, Folkerts G. Nitric oxide in health and disease of the respiratory system. Physiol Rev. 2004;84:731–65. doi: 10.1152/physrev.00034.2003. [DOI] [PubMed] [Google Scholar]
- 7.van der Vliet A, Eiserich JP, Shigenaga MK, Cross CE. Reactive nitrogen species and tyrosine nitration in the respiratory tract: Epiphenomena or a pathobiologic mechanism of disease? Am J Respir Crit Care Med. 1999;160:1–9. doi: 10.1164/ajrccm.160.1.9807044. [DOI] [PubMed] [Google Scholar]
- 8.Charles IG, Palmer RM, Hickery MS, Bayliss MT, Chubb AP, Hall VS, et al. Cloning, characterization, and expression of a cDNA encoding an inducible nitric oxide synthase from the human chondrocyte. Proc Natl Acad Sci U S A. 1993;90:11419–23. doi: 10.1073/pnas.90.23.11419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marsden PA, Schappert KT, Chen HS, Flowers M, Sundell CL, Wilcox JN, et al. Molecular cloning and characterization of human endothelial nitric oxide synthase. FEBS Lett. 1992;307:287–93. doi: 10.1016/0014-5793(92)80697-f. [DOI] [PubMed] [Google Scholar]
- 10.Nakane M, Schmidt HH, Pollock JS, Förstermann U, Murad F. Cloned human brain nitric oxide synthase is highly expressed in skeletal muscle. FEBS Lett. 1993;316:175–80. doi: 10.1016/0014-5793(93)81210-q. [DOI] [PubMed] [Google Scholar]
- 11.Batra J, Pratap Singh T, Mabalirajan U, Sinha A, Prasad R, Ghosh B. Association of inducible nitric oxide synthase with asthma severity, total serum immunoglobulin E and blood eosinophil levels. Thorax. 2007;62:16–22. doi: 10.1136/thx.2006.057935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leung TF, Liu EK, Tang NL, Ko FW, Li CY, Lam CW, et al. Nitric oxide synthase polymorphisms and asthma phenotypes in Chinese children. Clin Exp Allergy. 2005;35:1288–94. doi: 10.1111/j.1365-2222.2005.02342.x. [DOI] [PubMed] [Google Scholar]
- 13.Gómez LM, Anaya JM, Vilchez JR, Cadena J, Hinojosa R, Vélez L, et al. A polymorphism in the inducible nitric oxide synthase gene is associated with tuberculosis. Tuberculosis (Edinb) 2007;87:288–94. doi: 10.1016/j.tube.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 14.Qidwai T, Jamal F. Inducible nitric oxide synthase (iNOS) gene polymorphism and disease prevalence. Scand J Immunol. 2010;72:375–87. doi: 10.1111/j.1365-3083.2010.02458.x. [DOI] [PubMed] [Google Scholar]
- 15.Ahsan A, Ram R, Baig MA, Pasha MA. ACE I allele and eNOS G allele crosstalk may have a role in chronic obstructive pulmonary disease. Clin Biochem. 2004;37:1037–40. doi: 10.1016/j.clinbiochem.2004.05.026. [DOI] [PubMed] [Google Scholar]
- 16.Arif E, Ahsan A, Vibhuti A, Rajput C, Deepak D, Athar M, et al. Endothelial nitric oxide synthase gene variants contribute to oxidative stress in COPD. Biochem Biophys Res Commun. 2007;361:182–8. doi: 10.1016/j.bbrc.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 17.Vestbo J, Agusti A, Anzueto A, Bames P, Calverley P, Fabbri L, et al. Global Strategy for Diagnosis, Management, and Prevention of COPD. 2010. Available from: http://www.goldcopd.com .
- 18.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 19.Ichinose M, Sugiura H, Yamagata S, Koarai A, Shirato K. Increase in reactive nitrogen species production in chronic obstructive pulmonary disease airways. Am J Respir Crit Care Med. 2000;162:701–6. doi: 10.1164/ajrccm.162.2.9908132. [DOI] [PubMed] [Google Scholar]
- 20.Maestrelli P, Páska C, Saetta M, Turato G, Nowicki Y, Monti S, et al. Decreased haem oxygenase-1 and increased inducible nitric oxide synthase in the lung of severe COPD patients. Eur Respir J. 2003;21:971–6. doi: 10.1183/09031936.03.00098203. [DOI] [PubMed] [Google Scholar]
- 21.Ricciardolo FL, Caramori G, Ito K, Capelli A, Brun P, Abatangelo G, et al. Nitrosative stress in the bronchial mucosa of severe chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2005;116:1028–35. doi: 10.1016/j.jaci.2005.06.034. [DOI] [PubMed] [Google Scholar]
- 22.Tadié JM, Henno P, Leroy I, Danel C, Naline E, Faisy C, et al. Role of nitric oxide synthase/arginase balance in bronchial reactivity in patients with chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol. 2008;294:L489–97. doi: 10.1152/ajplung.00109.2007. [DOI] [PubMed] [Google Scholar]
- 23.Brindicci C, Kharitonov SA, Ito M, Elliott MW, Hogg JC, Barnes PJ, et al. Nitric oxide synthase isoenzyme expression and activity in peripheral lung tissue of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181:21–30. doi: 10.1164/rccm.200904-0493OC. [DOI] [PubMed] [Google Scholar]
- 24.Seimetz M, Parajuli N, Pichl A, Veit F, Kwapiszewska G, Weisel FC, et al. Inducible NOS inhibition reverses tobacco-smoke-induced emphysema and pulmonary hypertension in mice. Cell. 2011;147:293–305. doi: 10.1016/j.cell.2011.08.035. [DOI] [PubMed] [Google Scholar]
- 25.Osoata GO, Hanazawa T, Brindicci C, Ito M, Barnes PJ, Kharitonov S, et al. Peroxynitrite elevation in exhaled breath condensate of COPD and its inhibition by fudosteine. Chest. 2009;135:1513–20. doi: 10.1378/chest.08-2105. [DOI] [PubMed] [Google Scholar]