

Abstract
Background:
Najanalgesin, a toxin isolated from the venom of Naja naja atra, has been shown to exert significant analgesic effects in a neuropathic pain model in rats. However, the molecular mechanism underlying this protective effect of najanalgesin is poorly understood. The present study sought to evaluate the intracellular signaling pathways that are involved in the antinociceptive effect of najanalgesin on neuropathic pain.
Methods:
The antinociceptive properties of najanalgesin were tested in hind paw withdrawal thresholds in response to mechanical stimulation. We analyzed the participation of the mitogen-activated protein kinase p38, extracellular-regulated kinase (ERK), and c-Jun N-terminal kinase (JNK) by western blot analysis. This inhibition of JNK was confirmed by immunohistochemistry.
Results:
The phosphorylation levels of JNK (as well as its downstream molecule c-Jun), p38, and ERK were significantly increased after injury. Najanalgesin only inhibited JNK and c-Jun phosphorylation but had no effect on either ERK or p38. This inhibition of JNK was confirmed by immunohistochemistry, which suggested that the antinociceptive effect of najanalgesin on spinal nerve ligation-induced neuropathic pain in rats is associated with JNK activation in the spinal cord.
Conclusion:
The antinociceptive effect of najanalgesin functions by inhibiting the JNK in a neuropathic pain model.
Keywords: c-Jun, c-Jun NH2-terminal Kinase, L5 Spinal Nerve Ligation, Najanalgesin
INTRODUCTION
Neuropathic pain is a devastating condition, and our lack of knowledge regarding its pathogenesis has hampered efforts to find an effective treatment. Increasing evidence has shown that mitogen-activated protein kinase (MAPK) plays important roles in the induction and maintenance of chronic pain. The MAPK family consists of three primary members: Extracellular signal-regulated kinases (ERK), p38 MAPK, and c-Jun N-terminal kinase (JNK); these three kinases correspond to three distinct signaling pathways.[1,2,3,4] Experiments were designed to elucidate the signaling cascades that induce allodynia in a neuropathic pain model. Previously published studies have described the role of MAPKs in the regulation of allodynia and hyperalgesia. MAPKs have, therefore, been suggested as a specific target in neuropathic pain responses. To investigate whether the inhibition of neuropathic pain by najanalgesin is regulated by a MAPK pathway, we examined the effect of najanalgesin on neuropathic pain by measuring the phosphorylation of ERK, JNK, and p38 MAPK.
Najanalgesin is purified from cobra venom and consists of a single polypeptide with a molecular mass of 6714 Da with an LD50 of 2.69 mg/kg. In our laboratory, we found that najanalgesin had an analgesic effect on pain resulting from heat and chemical burns.[5] Additionally, we evaluated the antinociceptive effect of najanalgesin in a rat model of neuropathic pain, which was induced by L5 spinal nerve ligation (SNL). We observed that najanalgesin had antinociceptive effects on the central and peripheral system in a rat neuropathic pain model.[6] However, the effect of najanalgesin on the MAPK pathways after nerve injury remains unclear. In the present study, we investigated the effect of najanalgesin in rats using a neuropathic pain model, focusing on the MAPK family members’ JNK, ERK, and p38 MAPK.
METHODS
Drugs administration
The MAPK kinase (MEK) 1/2 inhibitor (PD98059; 0.1 μg/μl; EMD Biosciences, La Jolla, CA, USA), the p38 inhibitor 4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl) imidazole (SB203580; 0.5 μg/μl; Calbiochem, USA), and the JNK inhibitor anthrax 1,9-cd] pyrazol-6 (2H)-one (SP600125; 2.5 μg/μl; Calbiochem) were prepared in 50% dimethyl sulfoxide (DMSO). Both DMSO (50%) and normal saline were used as vehicle controls.
Rats were allowed to survive for up to 7 days after surgery. The doses used for intrathecal najanalgesin (40 ng/kg) were selected according to our pilot study. JNK, phosphor-JNK (Thr183/Tyr185), phosphor-p38, phosphor-ERK, and phosphor-c-Jun antibodies were purchased from Cell Signaling Technology Inc., USA.
Animals
Male Sprague-Dawley rats (200–220 g) were purchased from the Laboratory Animal Center at Sun Yat-sen University. All procedures were conducted in accordance with the National Institutes of Health guidelines on animal care. The experimental design resulted in four groups of rats (n = 8 per group): (1) Control group of the sham-operated rats; (2) SNL group of rats with L5 spinal nerve ligation; (3) najanalgesin group of treatment injury rats with 40 ng/kg najanalgesin; (4) inhibitor group of treatment rats with PD98059 or SB203580 or SP600125. All rats were housed in separate cages with free access to food and water and maintained on a 12-h light/dark cycle. Efforts were made to minimize the number of animals used and their suffering.
Surgery
All experimental procedures were performed according to previously established methods.[6] A sterile polyethylene-10 catheter was inserted into the rat's lumbar subarachnoid space, and drugs were administered in a volume of 10 μl. The L5 spinal nerve was tightly ligated with silk suture. In sham-operated rats, the nerve was exposed without ligature. Special care was taken to prevent infection and minimize inflammation.
Behavioral testing
On each testing day, the rats were brought into the behavioral room at least 30 min prior to the test session. Mechanical sensitivity was assessed using the up-down method as previously described using a set of von Frey hairs (Ugo Basile, Italy).[6,7]
Western blotting
Equal amounts of the samples were run on a 12% dodecyl sulfate-polyacrylamide gel and separated using SDS-PAGE. Antibodies against JNK (1:1000; Cell Signaling Technology), ERK (1:1000; Cell Signaling Technology), and p38 (1:1000; Cell Signaling Technology), as well as a monoclonal primary antibody against β-actin (mouse, 1:500; Boshide), were diluted in tris-buffered saline containing 0.1% Triton X-100 and 5% bovine serum albumin and used as primary antibodies.[8]
Immunohistochemistry
Immunohistochemistry was performed on 25-μm free-floating L5 spinal cord sections as previously described.[7] Spinal cord sections were incubated overnight at 4°C with JNK primary antibody (1:1000) and subsequently incubated with a Cyc3-conjugated secondary antibody (1:6000, Boshide, China). To assess nonspecific staining, control sections were incubated in the absence of primary antibodies. The mean value of JNK-IR was obtained within the spinal dorsal horn laminae I–III from six randomly selected sections from each animal.[8]
Statistical analysis
All data were expressed as the mean ± standard error (SE) of the mean. One-way analysis of variance (ANOVA) followed by Tukey's test was used for immunohistochemistry and western blotting data. A P < 0.05 was considered as statistically significant.
RESULTS
The rats subjected to the neuropathic pain model developed robust behavioral allodynia after SNL, which persisted for at least 2 weeks. Sham-operated rats showed no apparent response to mechanical stimuli at any of the investigated time points.
In the mechanical withdrawal threshold testing, groups of rats were injected intrathecally with saline, najanalgesin, or SP600125; the paw withdrawal threshold was measured for 24 h after injection. The injection of najanalgesin resulted in the significantly high threshold compared with saline treatment (P < 0.05) started 1 h and lasted past 24 h. SP600125 produced antinociceptive action than those observed with saline treatment at 1, 3, and 9 h after injection (P < 0.05), [Figure 1]. Our study suggested that najanalgesin has a significant antinociceptive effect in a neuropathic pain model in rats.[6] To determine whether the inhibition of neuropathic pain by najanalgesin is regulated by the MAPK pathways, we examined the effect of najanalgesin on the phosphorylation levels of ERK, JNK, and p38 MAPK by Western blot analysis.
Figure 1.
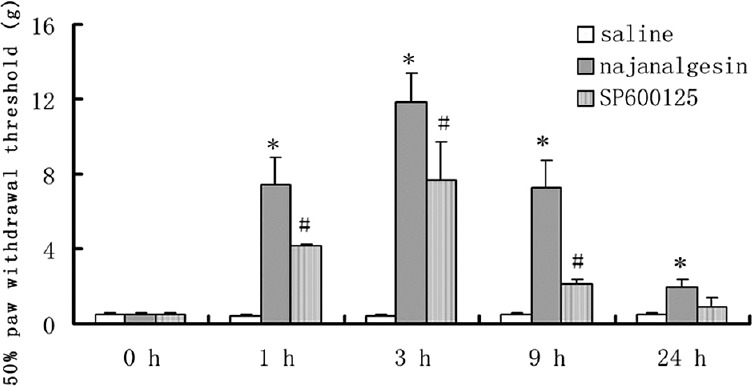
Effects of najanalgesin on mechanical allodynia in the rat spinal nerve ligation model up to 24 h after drug administration. After administration of najanalgesin, saline, or SP600125, the paw withdrawal threshold in response to mechanical stimuli was measured at different time points after the injection (n = 8). *P < 0.05, #P< 0.01 compared with the mean values of saline group.
As shown in Figure 2, p-p38 MAPK was consistently increased in the spinal dorsal cord of injured rats. However, there was no significant change after treatment with najanalgesin. When we investigated the possible role of ERK, we observed a significant increase in ERK phosphorylation levels after nerve injury. ERK activation was increased 10-fold in the SNL group compared with the control group. Interestingly, najanalgesin treatment had no effect on this increase in phosphorylated ERK (p-ERK) [Figure 3].
Figure 2.
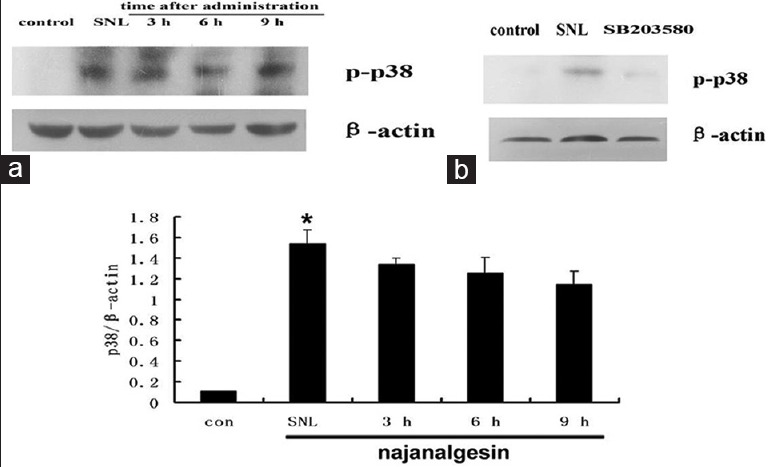
Changes in p38 expression in the spinal dorsal horn after L5 spinal nerve ligation and najanalgesin treatment. (a) Immunoblots against p38 in the spinal dorsal horn of the control, spinal nerve ligation, and najanalgesin treatment groups. The p38 expression at all of the investigated time points was quantified and graphed. (b) Images of protein bands corresponding to p-p38 after intrathecal SB203580 administration. The data are represented as the mean ± standard error of the mean (n = 4). *P < 0.05 versus the control group.
Figure 3.
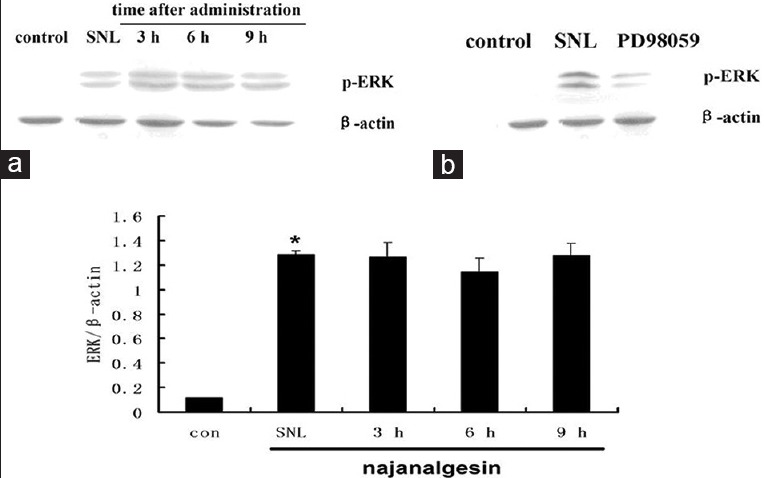
Changes in extracellular-regulated kinase expression in the spinal dorsal horn after L5 spinal nerve ligation and najanalgesin treatment. (a) Immunoblots against extracellular-regulated kinase in the spinal dorsal horn of the control, spinal nerve ligation, and najanalgesin treatment groups. The extracellular-regulated kinase expression at all of the investigated time points was quantified and graphed. (b) Images of protein bands corresponding to p-extracellular-regulated kinase after intrathecal PD98059 administration. The data are represented as the mean ± standard error of the mean (n = 4). *P < 0.05 versus the control group.
Peripheral nerve injury significantly induced the activation of JNK without causing any significant changes in total JNK protein levels. JNK activation was increased by eight-fold in the SNL group compared with the sham group (P < 0.01). Interestingly, we observed a significant decrease in the phosphorylation of JNK at 3 and 9 h after treatment with 40 ng/kg najanalgesin [Figure 4]. The phosphorylation of c-Jun followed a similar pattern to JNK after treatment with najanalgesin. The levels of phosphorylated c-Jun decreased progressively from 3 h to 9 h [Figure 5]. Based on these results, we used three known pharmacological inhibitors of various MEKs to confirm the specificity of najanalgesin activity. We used inhibitors against ERK (PD98059),[9] p38 (SB203580),[10] and JNK (SP600125).[11] All of these inhibitors decreased the phosphorylation of p38 [Figure 1], ERK [Figure 2], and JNK [Figure 3]. Western blotting revealed that rats subjected to nerve injury had increased levels of p-JNK, p38, and p-ERK. Treatment with najanalgesin inhibited the injury-induced increase in p-JNK, whereas p-ERK and p38 were unchanged. This result suggests that JNK may have a key role in the antinociceptive effect of najanalgesin on neuropathic pain.
Figure 4.
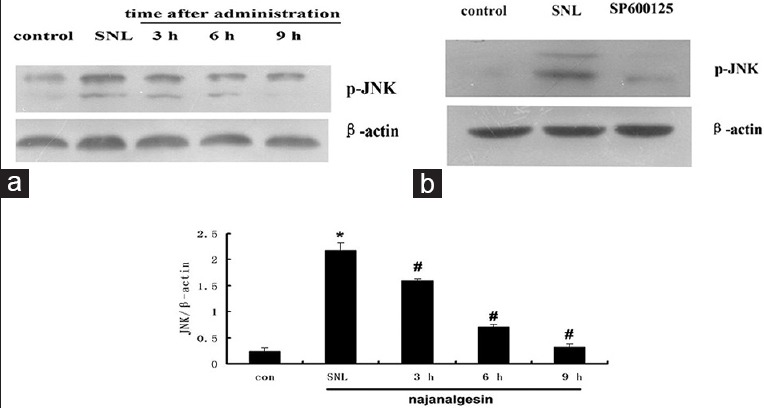
Changes in c-Jun N-terminal kinase expression in the spinal dorsal horn after L5 spinal nerve ligation and najanalgesin treatment. (a) Immunoblots against c-Jun N-terminal kinase in the spinal dorsal horn of the control, spinal nerve ligation, and najanalgesin treatment groups. The c-Jun N-terminal kinase expression at all of the investigated time points was quantified and graphed. (b) Images of the protein bands corresponding to p-JNK after intrathecal SP600125 administration. The data are represented as the mean ± standard error of the mean (n = 4). *P < 0.05 versus the control group; #P < 0.05 versus the spinal nerve ligation group.
Figure 5.
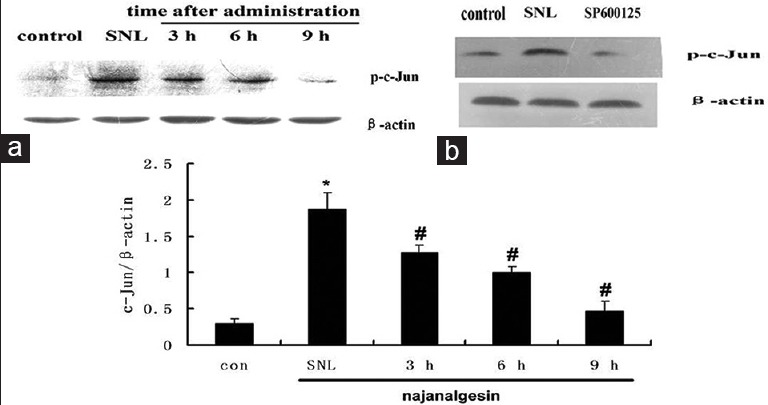
Changes in c-Jun expression in the spinal dorsal horn after L5 spinal nerve ligation and najanalgesin treatment. (a) Immunoblots against p-c-Jun in the spinal dorsal horn of the control, spinal nerve ligation, and najanalgesin treatment groups. (b) Images of the protein bands corresponding to p-c-Jun after intrathecal SP600125 administration. The c-Jun expression at all of the investigated time points was quantified and graphed. The data are represented as the mean ± standard error of the mean. (n = 4). *P < 0.05 versus the control group; #P < 0.05 versus the spinal nerve ligation group.
The changes in JNK expression were confirmed by immunohistochemistry analysis. The robust increase of JNK expression in JNK-IR cells was observed in the superficial region (laminae I–III) of the dorsal horn. Low basal levels of constitutive JNK expression were observed in control group. However, intrathecal administration of najanalgesin effectively attenuated the increase in JNK expression. Consistent with the results obtained by western blot analysis, najanalgesin treatment of rats subjected to L5 SNL led to a decrease in ipsilateral JNK immunoreactivity at 3, 6, and 9 h compared with the control group [Figure 6]. These findings suggest that the antinociceptive effect of najanalgesin inhibits JNK in a rat model of neuropathic pain.
Figure 6.
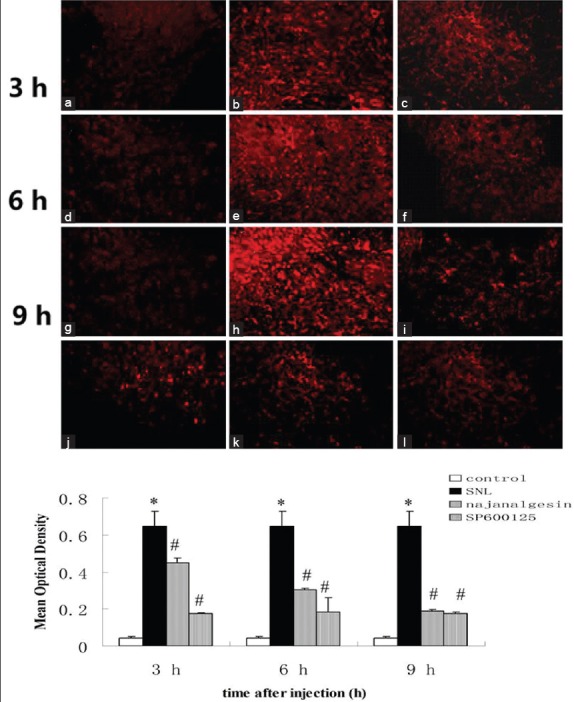
Changes in c-Jun N-terminal kinase protein expression in the spinal dorsal horn after L5 spinal nerve ligation, transection-induced neuropathic pain, and najanalgesin treatment. Images show c-Jun N-terminal kinase immunostaining in the spinal dorsal horn in the control, spinal nerve ligation, najanalgesin treatment, and SP600125 treatment groups. Lower magnification images showing the area of the dorsal horn that was analyzed. c-Jun N-terminal kinase-IR activation was not observed in the control group (a, d, g). Prominent astrocyte activation was observed in rats treated with intrathecal saline after spinal nerve ligation (b, e, h). These responses were markedly inhibited in rats treated with either intrathecal najanalgesin (c, f, i) or intrathecal SP600125 (j, k, l) for 3, 6, and 9 h. The c-Jun N-terminal kinase protein expression at all of the investigated time points was quantified and graphed. (a-i) ×200. The data are represented as the mean ± standard error of the mean. (n = 6). *P < 0.05 versus the control group; #P < 0.05 versus the spinal nerve ligation group.
DISCUSSION
Increasing evidence has shown that MAPKs play important roles in the induction and maintenance of chronic pain. ERK, p38, and JNK represent three different signal transduction pathways that contribute to pain sensitization after tissue and nerve injury.[12] ERK, p38, and JNK are downstream of many kinases and are activated in primary sensory neurons, dorsal horn neurons, and spinal glial cells, and contribute to neuropathic pain via transcriptional, translational, and posttranslational regulation.[13] The inhibition of all three MAPK pathways has been shown to attenuate inflammatory and neuropathic pain in different animal models.[14]
In the present study, we observed that peripheral nerve injury resulted in a marked increase in the activation of p38, ERK, and JNK (P < 0.01). The increased activation of both ERK and p38 MAPK was not attenuated by najanalgesin administration. However, the increase in JNK was attenuated with najanalgesin. The transcription c-Jun is the best-known substrate of JNK, and it is activated mainly through double phosphorylation by the JNK and also has a phosphorylation-independent function.[11] Our study showed that the levels of phosphorylated c-Jun were markedly increased in the SNL group and that this increase was prevented by najanalgesin treatment, which implies the involvement of JNK in the antinociceptive effect of najanalgesin. Consistent with these results, we also demonstrated by immunohistochemistry that the activation of JNK is involved in the antinociceptive effect of najanalgesin.
The MAPK pathways are complex and not yet fully understood, as different cell types respond to the same nerve injury by activating different signaling pathways. It has been suggested that the ERK pathway, which is primarily triggered in response to growth factors and mitogen stimulation, is involved in cellular proliferation and differentiation.[8] Specifically, ERK activation in spinal cord dorsal horn neurons occurs by nociceptive activity by regulating the activity of glutamate receptors and potassium channels, as well as inducing gene transcription.[15,16] Based on our results, ERK may be not involved in the analgesic effect of najanalgesin treatment, suggesting that najanalgesin may act through another signaling pathway. p38 was shown to be rapidly and robustly activated in the superficial spinal dorsal horn after nerve injury and localized primarily to microglia.[17,18] Peripheral nerve injury leads to the activation of both microglia and astrocytes in the spinal cord but with different time courses.[12] Microglia are involved in the early response to neuropathic pain, but the persistent expression of astrocyte markers correlates with the persistence of neuropathic pain symptoms. Activated glial cells release proinflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and IL-6, as well as other substances that enhance neuronal central sensitization and nerve injury-induced persistent pain.[19,20] In this study, najanalgesin treatment was administered after the initial pain was inflicted and during the maintenance of the pain. Therefore, p38 may be not involved in antinociception of najanalgesin on the maintenance of neuropathic pain. Whether p38 would be involved in the antinociception with najanalgesin on the early response to neuropathic pain, we need to investigate it further.
The previous study showed that JNK is persistently activated in the spinal dorsal horn after nerve injury.[12] JNK has three isoforms: JNK1, JNK2, and JNK3.[21] JNK3 is primarily expressed in the brain and has different roles than JNK1 and JNK2.[22] However, JNK1 and JNK2 are heavily expressed in the spinal cord and are the predominant isoforms after nerve injury.[13] pJNK1/2 is persistently increased in astrocytes on 1, 3, 10, and 21 days in the spinal cord after partial sciatic nerve injury.[23] Additionally, c-Jun, the major transcription factor downstream of JNK, is also activated in the spinal cord after nerve injury.[11] The JNK/c-Jun pathway is activated in the spinal astrocytes, which is also critical for the maintenance of neuropathic pain. In this study, there were marked increases in the JNK and c-Jun levels after SNL, and both of these protein levels decreased after najanalgesin treatment. In addition, we have observed that astrocytes were involved in the antinociception of najanalgesin during the maintenance of pain in the previous study.[7] Therefore, the effect of najanalgesin may act through the JNK signaling pathway in the spinal cord on the maintenance of neuropathic pain.
CTX is the primary neurotoxic component of Crotalus durissus terrificus snake venom, which modulates inflammatory responses. This toxin inhibits cytokine release (e.g., IL-2, IL-4, and IL-10) in human serum albumin-immunized mice.[24] Our previous study showed that najanalgesin attenuated the activation of spinal astrocytes and reduced the release of proinflammatory cytokines (TNF-α, IL-1β) in the spinal cord.[7] Based on the results of our previous and current studies, najanalgesin produces an antinociceptive effect on the maintenance of neuropathic pain, by attenuating the activation of JNK, and reducing the release of proinflammatory cytokines. And then, we should investigate the mRNA of JNK and give JNK inhibitor to observe the expression of proinflammatory cytokines, in order to make the molecule mechanism of najanalgesin clear in the future.
The present study evaluates the intracellular signaling pathways implicated in the antinociceptive effect of najanalgesin, a toxin isolated from Naja naja atra. Najanalgesin inhibited JNK without affecting the activity of ERK and p38. This inhibition could be one of the mechanisms of najanalgesin that protects against peripheral nerve injury. The results suggest that the antinociceptive effect of najanalgesin is due to inhibition of JNK based on the neuropathic pain model.
Financial support and sponsorship
This study was supported by the grants from the National Natural Science Foundation of China (No. 81300969), the Shandong Province National Natural Science Foundation (No. ZR2012HL27), the project from Weifang Science and Technology Bureau (No. 20121373), and the Weifang Medical Technology Innovation Project (No. K11QC1001).
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Xiu-Yuan Hao
REFERENCES
- 1.Ji RR, Gereau RW 4th, Malcangio M, Strichartz GR. MAP kinase and pain. Brain Res Rev. 2009;60:135–48. doi: 10.1016/j.brainresrev.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ji RR, Suter MR. p38 MAPK, microglial signaling, and neuropathic pain. Mol Pain. 2007;3:33. doi: 10.1186/1744-8069-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–2. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 4.Wang W, Mei XP, Wei YY, Zhang MM, Zhang T, Wang W, et al. Neuronal NR2B-containing NMDA receptor mediates spinal astrocytic c-Jun N-terminal kinase activation in a rat model of neuropathic pain. Brain Behav Immun. 2011;25:1355–66. doi: 10.1016/j.bbi.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 5.Jiang WJ, Liang YX, Han LP, Qiu PX, Yuan J, Zhao SJ. Purification and characterization of a novel antinociceptive toxin from Cobra venom (Naja naja atra) Toxicon. 2008;52:638–46. doi: 10.1016/j.toxicon.2008.06.030. [DOI] [PubMed] [Google Scholar]
- 6.Liang YX, Jiang WJ, Han LP, Zhao SJ. Peripheral and spinal antihyperalgesic activity of najanalgesin isolated from Naja naja atra in a rat experimental model of neuropathic pain. Neurosci Lett. 2009;460:191–5. doi: 10.1016/j.neulet.2009.04.066. [DOI] [PubMed] [Google Scholar]
- 7.Liang Y, Jiang W, Zhang Z, Yu J, Tao L, Zhao S. Behavioral and morphological evidence for the involvement of glial cells in the antinociceptive effect of najanalgesin in a rat neuropathic pain model. Biol Pharm Bull. 2012;35:850–4. doi: 10.1248/bpb.35.850. [DOI] [PubMed] [Google Scholar]
- 8.Lee JY, Choi DC, Oh TH, Yune TY. Analgesic effect of acupuncture is mediated via inhibition of JNK activation in astrocytes after spinal cord injury. PLoS One. 2013;8:e73948. doi: 10.1371/journal.pone.0073948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–32. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 10.Choi DC, Lee JY, Lim EJ, Baik HH, Oh TH, Yune TY. Inhibition of ROS-induced p38MAPK and ERK activation in microglia by acupuncture relieves neuropathic pain after spinal cord injury in rats. Exp Neurol. 2012;236:268–82. doi: 10.1016/j.expneurol.2012.05.014. [DOI] [PubMed] [Google Scholar]
- 11.Zhuang ZY, Wen YR, Zhang DR, Borsello T, Bonny C, Strichartz GR, et al. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: Respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J Neurosci. 2006;26:3551–60. doi: 10.1523/JNEUROSCI.5290-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ji RR, Strichartz G. Cell signaling and the genesis of neuropathic pain. Sci STKE 2004. 2004 doi: 10.1126/stke.2522004re14. reE14. [DOI] [PubMed] [Google Scholar]
- 13.Gao YJ, Ji RR. Activation of JNK pathway in persistent pain. Neurosci Lett. 2008;437:180–3. doi: 10.1016/j.neulet.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ji RR, Kawasaki Y, Zhuang ZY, Wen YR, Zhang YQ. Protein kinases as potential targets for the treatment of pathological pain. Handb Exp Pharmacol. 2007;177:359–89. doi: 10.1007/978-3-540-33823-9_13. [DOI] [PubMed] [Google Scholar]
- 15.Hu HJ, Alter BJ, Carrasquillo Y, Qiu CS, Gereau RW 4th. Metabotropic glutamate receptor 5 modulates nociceptive plasticity via extracellular signal-regulated kinase-Kv4.2 signaling in spinal cord dorsal horn neurons. J Neurosci. 2007;27:13181–91. doi: 10.1523/JNEUROSCI.0269-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu HJ, Carrasquillo Y, Karim F, Jung WE, Nerbonne JM, Schwarz TL, et al. The kv4.2 potassium channel subunit is required for pain plasticity. Neuron. 2006;50:89–100. doi: 10.1016/j.neuron.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 17.Katsura H, Obata K, Mizushima T, Sakurai J, Kobayashi K, Yamanaka H, et al. Activation of Src-family kinases in spinal microglia contributes to mechanical hypersensitivity after nerve injury. J Neurosci. 2006;26:8680–90. doi: 10.1523/JNEUROSCI.1771-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wen YR, Suter MR, Kawasaki Y, Huang J, Pertin M, Kohno T, et al. Nerve conduction blocked in the sciatic nerve prevents but does not reverse the activation of p38 mitogen-activated protein kinase in spinal microglia in the rat spared nerve injury model. Anesthesiology. 2007;107:312–21. doi: 10.1097/01.anes.0000270759.11086.e7. [DOI] [PubMed] [Google Scholar]
- 19.Clark AK, D’Aquisto F, Gentry C, Marchand F, McMahon SB, Malcangio M. Rapid co-release of interleukin 1beta and caspase 1 in spinal cord inflammation. J Neurochem. 2006;99:868–80. doi: 10.1111/j.1471-4159.2006.04126.x. [DOI] [PubMed] [Google Scholar]
- 20.Tsuda M, Mizokoshi A, Shigemoto-Mogami Y, Koizumi S, Inoue K. Activation of p38 mitogen-activated protein kinase in spinal hyperactive microglia contributes to pain hypersensitivity following peripheral nerve injury. Glia. 2004;45:89–95. doi: 10.1002/glia.10308. [DOI] [PubMed] [Google Scholar]
- 21.Manassero G, Repetto IE, Cobianchi S, Valsecchi V, Bonny C, Rossi F, et al. Role of JNK isoforms in the development of neuropathic pain following sciatic nerve transection in the mouse. Mol Pain. 2012;8:39. doi: 10.1186/1744-8069-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuan CY, Whitmarsh AJ, Yang DD, Liao G, Schloemer AJ, Dong C, et al. A critical role of neural-specific JNK3 for ischemic apoptosis. Proc Natl Acad Sci U S A. 2003;100:15184–9. doi: 10.1073/pnas.2336254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mei XP, Zhang H, Wang W, Wei YY, Zhai MZ, Wang W, et al. Inhibition of spinal astrocytic c-Jun N-terminal kinase (JNK) activation correlates with the analgesic effects of ketamine in neuropathic pain. J Neuroinflammation. 2011;8:6. doi: 10.1186/1742-2094-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cardoso DF, Mota I. Effect of Crotalus venom on the humoral and cellular immune response. Toxicon. 1997;35:607–12. doi: 10.1016/s0041-0101(96)00134-1. [DOI] [PubMed] [Google Scholar]