

Abstract
Background:
Sevoflurane preconditioning (SP) has been shown to invoke potent myocardial protection in animal studies and clinical trials. However, the mechanisms underlying SP are complex and not yet well understood. We investigated the hypothesis that the cardioprotection afforded by SP is mediated via the Wnt/glycogen synthase kinase 3β (GSK3β)/β-catenin signaling pathway.
Methods:
Two models were established: A Langendorff perfused rat heart model and the H9C2 cell hypoxia/reoxygenation model. Both rats and H9C2 cells were randomly divided into 6 groups as follows: S group, ischemia-reperfusion (I/R) group, DMSO group, IWP group, SP group, and SP + IWP group. Hemodynamic parameters, lactate dehydrogenase (LDH) activity in coronary effluent and cell culture supernatant, and the infarct size were measured to evaluate myocardial ischemia-reperfusion injuries. To determine the activity of Wnt/GSK3β/β-catenin signaling pathway, the expressions of Wnt3a, phospho-GSK3β, and β-catenin were measured by Western blotting.
Results:
SP improved cardiac function recovery, reduced infarct size (18 ± 2% in the SP group compared with 35 ± 4% in the I/R group; P < 0.05), decreased LDH activity in coronary effluent, and culture supernatant. IWP-2, an inhibitor of Wnt, abolished the cardioprotection by SP. In addition, Western blotting analysis demonstrated that the expressions of Wnt3a, phospho-GSK3β, and β-catenin significantly (P < 0.05) increased in the I/R group, compared with the S group; and compared to I/R group, SP significantly (P < 0.05) increased Wnt3a, phospho-GSK3β, and β-catenin expressions. Pretreatment with IWP-2 significantly (P < 0.05) abolished SP-induced Wnt/GSK3β/β-catenin signaling activation.
Conclusions:
The results showed for the first time that cardioprotection afforded by SP may be mediated partly via the Wnt/GSK3β/β-catenin signaling pathway.
Keywords: Cardioprotection, Ischemia-reperfusion Injury, Preconditioning, Sevoflurane, Wnt/Glycogen Synthase Kinase 3β/β-catenin Signaling
INTRODUCTION
Ischemia-reperfusion (I/R) injury occurs due to blood restoration after a critical period of coronary artery obstruction. It is associated with clinical problems, such as thrombolysis, angioplasty, and coronary bypass surgery. However, the abrupt reperfusion of an ischemic myocardium can itself lead to additional myocardial injury, resulting in a spectrum of reperfusion-associated pathologies, which is termed as myocardial ischemia-reperfusion injury (MIRI).
A large number of animal studies and clinical trials have indicated that sevoflurane can protect the myocardium from I/R injuries when applied before ischemic event, and the characteristics of this protection are partly similar to those of the classic ischemic preconditioning (IPC), which was induced by short periods of ischemia and reperfusion and has been the most potent innate protective mechanisms against I/R injury so far.[1,2,3] However, the specific mechanisms underlying sevoflurane preconditioning (SP) are complex and not yet well understood. Exploring mechanisms of cardioprotection afforded by SP is of great significance to the study of MIRI and its treatment. Numerous studies have investigated the mechanisms involved in SP-induced cardioprotection, which may be associated with a variety of intracellular signal transduction pathways.
Wnt/glycogen synthase kinase 3β (GSK3β)/β-catenin signaling pathway, the canonical Wnt pathway, controls a variety of life processes, including the organism growth, development, diseases, aging, and death, as well as cell differentiation and maintenance of form and function, immune, stress, cell carcinogenesis, and cell apoptosis.[4,5,6,7,8,9] Researches have demonstrated that Wnt/GSK3β/β-catenin signaling pathway involved in myocardial remodeling and cardiovascular diseases. GSK3β, as the central substance of Wnt/GSK3β/β-catenin signaling pathway, played an important role in apoptosis and necrosis in MIRI.[10,11] Besides, GSK3β is the common target of the various signaling pathway with myocardial protective function.[12,13] It was reported that GSK3β played an important role in both IPC and postconditioning. Inactivation of GSK3β showed obviously cardioprotective effects.[14,15,16]
However, it is not clear whether Wnt/GSK3β/β-catenin signaling pathway is involved in SP-induced reduction of MIRIs. Here, we hypothesized that the Wnt/GSK3β/β-catenin signaling pathway played an important role in myocardial protection afforded by SP.
METHODS
Animals
Adult, male, and healthy Wistar rats weighing 220–280 g were provided by the Lukang Animal Feed Distribution Center of Jining City, Shandong Province (China) and housed on a 12 h light/dark cycle with free access to food and water. The environment temperature and humidity were maintained between 22°C and 24°C, and 40–60%. All animal care and experimental protocols complied with the guidelines of the Animal Care and Use Committee of Xuzhou Medical College.
Isolated, perfused heart preparation
Wistar rats (220–280 g) were anesthetized with sodium pentobarbital (30 mg/kg) and anticoagulated with an intraperitoneal injection of heparin (500 IU/kg). The hearts were rapidly excised by bilateral thoracotomy, placed in ice-cold buffer, and the aorta was cannulated with a 50 ml syringe needle. Isolated hearts were perfused retrogradely at a constant perfusion pressure of 100 cmH2O (≈75 mmHg) with a modified Krebs–Henseleit (K-H) buffer containing 118.5 mmol/L NaCl, 24.8 mmol/L NaHCO3, 11 mmol/L D-glucose, 4.7 mmol/L KCl, 1.2 mmol/L MgSO4·7H2O, 1.2 mmol/L KH2 PO4, 2.25 mmol/L CaCl2·2H2O, pH 7.4. The perfusate buffer was saturated with a 95% O2 and 5% CO2 gas mixture at 37°C before use. A latex balloon was inserted into the left ventricle via the left atrium, inflated with distilled water and then connected to the MacLab/4S ADC attached to the computer. The balloon was inflated with water to adjust the left ventricular end-diastolic pressure (LVEDP) to 7–10 mmHg at the beginning of the experiment, and the volume was kept constant for the duration of the study. The LV function index was monitored continuously with a computer-based system for the acquisition of data regarding LVEDP, left ventricular developed pressure (LVDP), positive and negative LVDP/dt (+dp/dtmax, −dp/dtmax), and heart rate (HR). Before each experimental protocol was initiated, the isolated hearts were allowed to stabilize at 37°C for 10 min. Hearts were excluded if after stabilization they failed to develop steady sinus rhythm or their rate <200 beats/min.
Ischemia/reperfusion injury experimental protocol
The whole procedure lasted 120 min. All animals (except for the rats in the sham groups) were subjected to 30 min of ischemia followed by 60 min of reperfusion. Rats were randomly divided into 6 groups as follows (n = 72, 12 per group): (1) Sham group (Group S), (2) I/R group (Group I/R), (3) I/R + dimethyl sulfoxide group (Group DMSO), (4) I/R + IWP group (Group IWP), (5) SP group (Group SP), and (6) SP + Wnt inhibitor IWP-2 group (Group SP + IWP). The hearts are continuously perfused for 120 min in Group S. After 10 min of equilibration, the isolated hearts were continuously perfused for 20 min, then subjected to 30 min of ischemia followed by 60 min of reperfusion in Group I/R; Groups DMSO, IWP, SP and SP + IWP received 15 min of perfusion with K-H solution containing 0.5 ml/L DMSO, 10 µmol/L IWP (SIGMA-ALDRICH, USA), 2.4 vol% sevoflurane, 2.4 vol% sevoflurane + 10 µmol/L IWP, respectively, followed by 5 min washout before I/R.
H9C2 cell culture and hypoxia/reoxygenation treatment
Once recovery, the H9C2 cells (derived from rat embryonic cardiomyocytes BDlX cell lines, and purchased from Beijing North Carolina Biotechnology Institute Chuanglian) were cultured in Dulbecco's Modified Eagle's medium (DMEM, Invitrogen, USA) with 4500 mg/L glucose supplemented with 10% (v/v) fetal bovine serum (Hangzhou Sijiqing Biological Engineering Materials Co., Ltd., China). Cells were routinely grown in 75 cm2 flasks at 37°C in a humidified atmosphere with 5% CO2 prior to passage. After 3–6 passages, the cells were switched to serum-free 4500 mg/L glucose medium and cultured for 12 h to be synchronized, for subsequent experiments.
Hypoxia/reoxygenation-induced H9C2 cell injury was performed as described previously.[17] After the preincubation, the cell culture medium was replaced with serum-free low glucose DMEM (glucose 1000 mg/L, HyClone, USA) preequilibrated with 95% N2 and 5% CO2 and then, cells were placed into a three gas incubator, set to 5% CO2/95% N2 at 37°C for 12 h and followed by reoxygenation for 2 h. Reoxygenation was accomplished by replacing the serum-free low glucose DMEM with normal cell medium under normoxic conditions. Exposure to sevoflurane was carried out by incubating the cells for 15 min in 2.4 vol% sevoflurane (approximately 1.0 minimum alveolar concentration) saturated normal high-glucose DMEM in a sterile airtight container. The container has two vents, an air inlet that has a constant supply of 97.5% O2-2.4 vol% sevoflurane, the outlet that was connected to anesthetic gas monitor for monitoring sevoflurane concentration continuously. The sevoflurane-containing medium was removed immediately before hypoxic conditions and then the cells were washed with phosphate-buffered saline (PBS) three times.[18] The H9C2 cells were randomly divide into 6 groups as follows: The S group, I/R group, DMSO group, IWP group, SP group and SP + IWP group. The DMSO group, IWP group, SP group, and SP + IWP group received 15 min pretreatment with normal complete medium containing 0.25 ml/L DMSO, 5 µmol/L IWP, 2.4 vol% sevoflurane, 2.4 vol% sevoflurane + 5 µmol/L IWP, respectively, followed by 3 times washout with PBS before I/R.
Measurements of hemodynamics
At the end of equilibration (T0), 30 min of reperfusion (T30) and 60 min of reperfusion (T60), the measured hemodynamic parameters were recorded. The measured hemodynamic parameters were LVDP, LVEDP, the maximum increase in the rate of LVDP (+dP/dt), maximum decrease in the rate of LVDP (−dP/dt), and HR.
Detection of lactate dehydrogenase activity in coronary effluent and cell culture supernatant
Lactate dehydrogenase (LDH) leakage was used as an indicator of cell membrane damage. At the end of equilibration, 30 min of reperfusion, and the end of H9C2 cell reoxygenation, coronary effluent, and cell-culture supernatant were collected. Take the cell culture supernatant 50 µl, coronary effluent 1 ml from each group, and test LDH activity values according to LDH kit instructions (Nanjing Jiancheng Bioengineering Institute, China), respectively.
Western blotting analysis
Once completion of the experimental period, the myocardium and H9C2 cells were lysed in ice-cold radioimmunoprecipitation assay lysis buffer containing 1% phenylmethylsulfonyl fluoride, at 4°C for 30 min and then the homogenate was incubated and centrifuged. The supernatant was collected, and the protein concentration was determined using the bicinchoninic acid protein assay kit according to the manufacturer's protocol (Beyotime, USA). The supernatant was mixed with ×4 loading buffer and heated for 15 min at 100°C. The extracts were injected into each sample hole and separated by SDS-polyacrylamide gel electrophoresis. After electrophoresis, proteins were electrophoretically transferred to a polyvinylidene difluoride filter membrane (0.45 mm, Millipore, USA). The membrane was blocked in washing buffer with 5% nonfat milk for 2 h and incubated overnight with the corresponding primary antibodies at 4°C. The membrane was placed at room temperature for 0.5 h, and then incubated with secondary antibody. The signals of detected proteins were visualized by BCIP/NBT alkaline phosphatase chromogenic color kit (Promega Corporation, Madison, WI, USA). The staining was quantified by scanning the films, and the band density was determined with Image-J software (Open source, http://rsb.info.nih.gov/ij/index.html).
Determination of infarct size
At the end of the reperfusion period, hearts were frozen at − 80°C for 30 min. The frozen hearts were cut transversely into 5 pieces, each 1 mm thick, and stained with 1% TTC for 30 min in a 37°C water bath. Then the slices were fixed in 4% formaldehyde solution for 15 min. At last, each slice was photographed by Epson Perfection V300 Photo scanner. The viable myocardium stained brick red, and infarct tissues appeared pale white. Infarct and LV area were measured using Image-J software, with the infarct size expressed as a percentage of the total LV area.
Statistical analysis
Data were analyzed using GraphPad Prism version 5.00 software (SanDiego, California, USA). Data are reported as means ± standard deviation, and n refers to the number of experiments. The group was compared using repeated measures analysis of variance data, comparisons between groups were performed by one-way analysis of variance followed by a post-hoc testing (Newman–Keuls test). Differences with P < 0.05 were considered as statistically significant.
RESULTS
Hemodynamic parameters
The hemodynamic data are shown in Figure 1. There were no differences of baseline hemodynamics among the experimental groups (P > 0.05). Compared with S group, the other groups had a significant decrease in HR, LVDP and dp/dtmax, an great increase (P < 0.05) in LVEDP at 30 min after reperfusion (T30), 60 min after reperfusion (T60). Compared to I/R group, the SP group showed an obvious increase (P < 0.05) in ± dp/dtmax and LVDP, and a significant decrease (P < 0.05) in LVEDP, the IWP group had a decrease in ± dp/dtmax and LVDP, and a significant increase (P < 0.05) in LVEDP at T30 and T60. The ± dp/dtmax and LVDP significantly decreased (P < 0.05) and the LVEDP significantly increased (P < 0.05) in SP + IWP group compared with the SP group at T30 and T60
Figure 1.
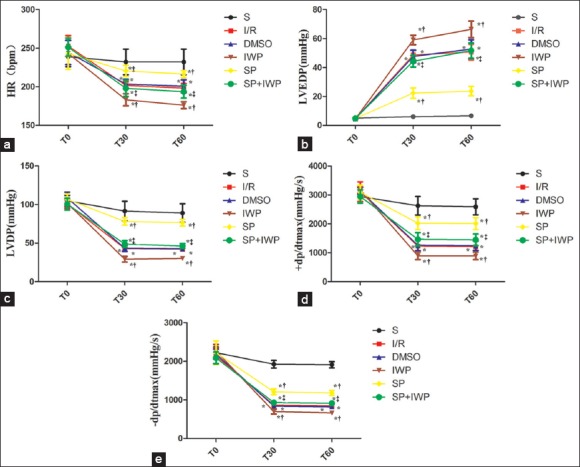
(a) Data are reported as means ± standard deviation (n = 12 for each group). *P < 0.05 versus S; †P < 0.05 versus I/R; ‡P < 0.05 versus sevoflurane preconditioning (two-way ANOVA). S: Sham group; I/R: Ischemia/reperfusion; DMSO: Dimethyl sulfoxide; IWP: Ischemia/reperfusion + inhibitor IWP-2; SP: Sevoflurane preconditioning; SP + IWP: Sevoflurane preconditioning + inhibitor IWP-2. T0: The end of equilibration; T30: 30 min after reperfusion; T60: 60 min after reperfusion; HR: Heart rate; (b) Data are reported as means ± standard deviation (n = 12 for each group). *P < 0.05 versus S; †P < 0.05 versus I/R; ‡P < 0.05 versus sevoflurane preconditioning (two-way ANOVA). T0: The end of equilibration; T30: 30 min after reperfusion; T60: 60 min after reperfusion; LVEDP: Left ventricular end-diastolic pressure; I/R: Ischemia-reperfusion; (c) Data are reported as means ± standard deviation (n = 12 for each group). *P < 0.05 versus S; †P < 0.05 versus I/R; ‡P < 0.05 versus sevoflurane preconditioning (two-way ANOVA). T0: The end of equilibration; T30: 30 min after reperfusion; T60: 60 min after reperfusion; LVDP: Left ventricular developed pressure; I/R: Ischemia-reperfusion; (d) Data are reported as means ± standard deviation (n = 12 for each group). *P < 0.05 versus S; †P < 0.05 versus I/R; ‡P < 0.05 versus sevoflurane preconditioning (two-way ANOVA). T0: The end of equilibration, T30: 30 min after reperfusion, T60: 60 min after reperfusion; +dp/dt = Max rate of left ventricular development pressure; I/R: Ischemia-reperfusion; (e) Data are reported as means ± standard deviation (n = 12 for each group). *P < 0.05 versus S; †P < 0.05 versus I/R; ‡P < 0.05 versus sevoflurane preconditioning (two-way ANOVA). T0: The end of equilibration, T30: 30 min after reperfusion, T60: 60 min after reperfusion; −dp/dt: Max rate of left ventricular fall pressure; I/R: Ischemia-reperfusion.
Lactate dehydrogenase activity in coronary effluent and cell culture supernatant
As shown in Figure 2a, there were no differences of baseline LDH release among all the experimental groups (P > 0.05). Thirty minutes later after reperfusion, except for the S group, all other groups had a significant increase (P < 0.05) in LDH release. However, compared to those in I/R group, LDH significantly decreased (P < 0.05) in SP and increased (P < 0.05) in IWP group, there was no difference between DMSO group and the SP + IWP group. LDH release had a great increase in SP + IWP group compared with the SP group [Figure 2b]. The change trend of LDH release in cell culture supernatant at the end of reoxygenation are the same as those in coronary effluent at 30 min after reperfusion [Figure 2c–d].
Figure 2.
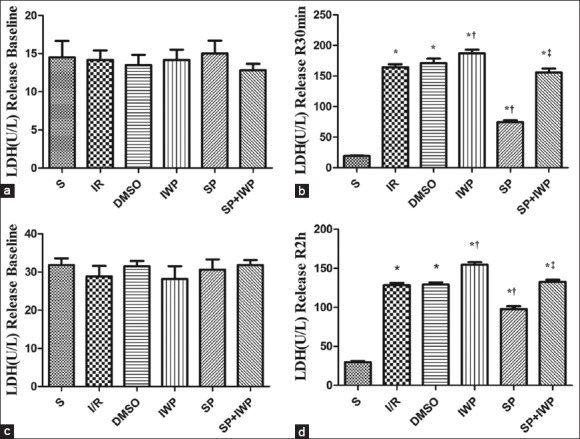
(a) Data are reported as means ± standard deviation. *P < 0.05 versus S; †P < 0.05 versus ischemia-reperfusion; ‡P < 0.05 versus sevoflurane preconditioning (one-way ANOVA). Coronary effluent was collected for lactate dehydrogenase activity measurement after equilibration in isolated rat heart. n = 12; (b) Data are reported as means ± standard deviation. *P < 0.05 versus S; †P < 0.05 versus ischemia-reperfusion; ‡P < 0.05 versus sevoflurane preconditioning (one-way ANOVA). Coronary effluent was collected for lactate dehydrogenase activity measurement at reperfusion 30 min in isolated rat heart. n = 12; (c) Data are reported as means ± standard deviation. *P < 0.05 versus S; †P < 0.05 versus ischemia-reperfusion; ‡P < 0.05 versus sevoflurane preconditioning (one-way ANOVA). Cell supernatant was collected for lactate dehydrogenase activity measurement at the end of equilibration. n = 6; (d) Data are reported as means ± standard deviation. *P < 0.05 versus S; †P < 0.05 versus ischemia-reperfusion; ‡P < 0.05 versus sevoflurane preconditioning (one-way ANOVA). Cell supernatant was collected for lactate dehydrogenase activity measurement at the end of cell reoxygenation. n = 6.
Sevoflurane preconditioning activates the Wnt/glycogen synthase kinase 3β/β-catenin signaling pathway in the isolated rat hearts
As shown in Figure 3, compared with the S group, the expressions of Wnt3a, p-GSK3β and β-catenin significantly (P < 0.05) increased in I/R group, DMSO group, SP group and the SP + IWP group; SP significantly (P < 0.05) increased levels of myocardial Wnt3a [Figure 3a], p-GSK3β [Figure 3b] and β-catenin [Figure 3c], and the IWP-2 decreased the levels, compared with those in I/R group. Pretreatment with IWP-2 abolished SP-induced increase of Wnt3a, p-GSK3β, and β-catenin.
Figure 3.
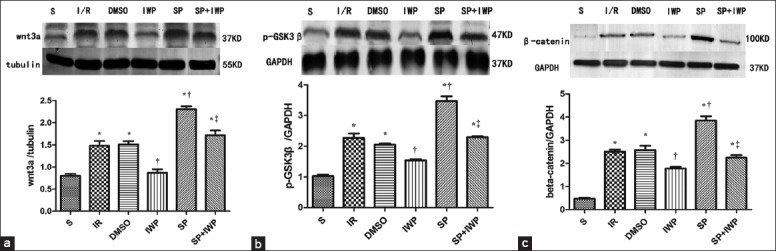
Western blotting analysis of myocardium Wnt3a. The Wnt3a (37 kDa) (a), was analyzed by Western blot with specific Wnt3a antibody at the end of reperfusion. Three hearts were used in each group. Data are reported as means ± standard deviation. *P < 0.05 versus S; †P < 0.05 versus ischemia-reperfusion; ‡P < 0.05 versus sevoflurane preconditioning (one-way ANOVA); Western blot analysis of myocardium p-GSK3β. The p-GSK3β (47 kDa) (b) was analyzed by Western blot with specific p-GSK3β antibody at the end of reperfusion. Three hearts were used in each group. Data are reported as means ± standard deviation. *P < 0.05 versus S; †P < 0.05 versus ischemia-reperfusion; ‡P < 0.05 versus sevoflurane preconditioning (one-way ANOVA); Western blot analysis of myocardium β-catenin. The β-catenin (c) was analyzed by Western blot with specific β-catenin antibody at the end of reperfusion. Three hearts were used in each group. Data are reported as means ± standard deviation. *P < 0.05 versus S; †P < 0.05 versus ischemia-reperfusion; ‡P < 0.05 versus sevoflurane preconditioning (one-way ANOVA).
Sevoflurane preconditioning activates the Wnt/glycogen synthase kinase 3β/β-catenin signaling pathway in H9C2 cell
To further prove the reliability of the experiment, H9C2 cell line was used. As shown in Figure 4, compared with the S group, the expressions of Wnt3a, p-GSK3β, and β-catenin significantly (P < 0.05) increased in groups I/R, DMSO, SP, SP + IWP; Compared with the I/R group, expressions of Wnt3a, p-GSK3β, and β-catenin significantly (P < 0.05) increased in SP group, decreased in IWP group. Pretreatment with IWP-2 significantly (P < 0.05) abolished SP-induced increase of Wnt3a, p-GSK3β, and β-catenin expressions.
Figure 4.
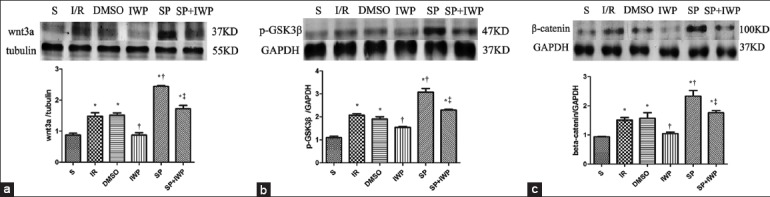
Western blotting analysis of H9C2 cells. The Wnt3a (37 kDa) (a), was analyzed by Western blot with specific Wnt3a antibody at the end of reperfusion. Three hearts were used in each group. Data are reported as means ± standard deviation. *P < 0.05 versus S; †P < 0.05 versus ischemia-reperfusion; ‡P < 0.05 versus sevoflurane preconditioning (one-way ANOVA); GSK3β: Glycogen synthase kinase 3β; Western blot analysis of H9C2 cells. The p-GSK3β (47 kDa) (b) was analyzed by Western blot with specific p-GSK3β antibody at the end of reperfusion. Three hearts were used in each group. Data are reported as means ± standard deviation. *P < 0.05 versus S; †P < 0.05 versus ischemia-reperfusion; ‡P < 0.05 versus sevoflurane preconditioning (one-way ANOVA); GSK3β: Glycogen synthase kinase 3β; Western blot analysis of H9C2 cells. The β-catenin (c) was analyzed by Western blot with specific β-catenin antibody at the end of reperfusion. Three hearts were used in each group. Data are reported as means ± standard deviation. *P < 0.05 versus S; †P < 0.05 versus ischemia-reperfusion; ‡P < 0.05 versus Sevoflurane preconditioning (one-way ANOVA).
Effects of myocardial infarct size
As shown in Figure 5, infarct sizes significantly (P < 0.05) increased at the end of reperfusion compared to S group. Compared to those in I/R group, infarct size significantly decreased (P < 0.05) in SP and increased (P < 0.05) in IWP group, there was no difference (P > 0.05) between DMSO group and the SP + IWP group. Infarct size had a great increase (P < 0.05) in SP + IWP group compared with the SP group.
Figure 5.
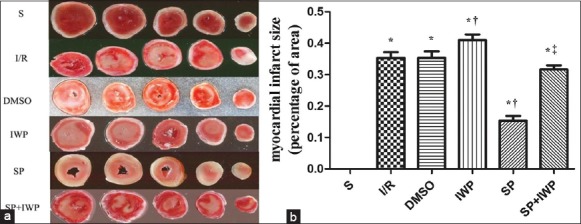
Effects of myocardial infarct size of isolated hearts in 6 groups. (a) Representative cross-sections of rat hearts from 6 groups after ischemia-reperfusion and staining with TTC to visualize the infarcted area; Effects of myocardial infarct size of isolated hearts in 6 groups; (b) Infarct size expressed as percentage of left ventricular area for each group. Values are means ± standard deviation (n = 6 for each group), *P < 0.05 versus S; †P < 0.05 versus ischemia-reperfusion; ‡P < 0.05 versus sevoflurane preconditioning (one-way ANOVA).
DISCUSSION
Our present study reveals a novel mechanism underlying sevoflurane-induced cardioprotection. We found that SP could activate Wnt/GSK3β/β-catenin signaling pathway in myocardium. Besides decreasing the infarct size and the LDH release, SP also significantly up-regulated the expression of Wnt3a, p-GSK3β, and β-catenin in isolated rat heart after I/R and the supematant of H9C2 cell line after hypoxia/reoxygenation. Pretreatment with Wnt inhibitor IWP-2 significantly abolished SP-induced cardioprotection and Wnt/GSK3β/β-catenin signaling activation. These results suggest that sevoflurane-induced cardioprotection may be mediated by Wnt/GSK3β/β-catenin signaling pathway.
IPC induced by short periods of ischemia and reperfusion is well recognized as the most potent innate protective mechanisms against IR injury.[19,20] However, despite large of experimental evidence confirming the benefits, it is difficult to carry out IPC in clinic considering its safety and ethics. Pharmacological preconditioning has been proposed. Volatile anesthetics have a long history in clinical anesthesia.
Sevoflurane, a new type of volatile anesthetic which has been widely used in clinical anesthesia, provides more controllable depth of anesthesia and more rapid recovery than other volatile anesthetics. It was reported SP could reduce MIRI.[2,3,21] However, the specific mechanisms underlying SP remained unclear. Previous studies showed that the underlying mechanisms of SP may be involved in modulation of the phosphatidylinositol-3-kinase (PI3K)/Akt[21] and its downstream component GSK3β,[22] activation of extracellular signal-regulated kinase 1/2[23] pathway and so on. However, the role of the Wnt/GSK3β/β-catenin signaling pathway in SP is still unknown.
The first detail of the Wnt/β-catenin network was reported in 1982 with the identification of the proto-oncogene int-1 in mice.[4] Wnt proteins are a class of highly conserved secretory proteins rich in cysteine and expressed in a variety of tissues and cells. Once Wnt ligand binds to a Frizzled family receptor and a coreceptor of the LRP-5/6/arrow family, the disheveled signal transduction molecules are activated, resulting in the phosphorylation of GSK3β at position Ser9 and subsequent inactivation of GSK3β. Since the active GSK3β can form APC/Axin/GSK3β complex and phosphorylate serine and threonine residues of the β-catenin, and then phosphorylation of beta-catenin degrades via the proteasome pathway. Thus, activation of the Wnt/GSK3β/β-catenin signaling pathway can phosphorylate and subsequently inhibit GSK3β activity, leading to the stabilization of β-catenin and its translocation from the cytosol to the nucleus where it interacts with TCF/LEF family transcription factors and start Wnt target gene transcription.[8,24,25,26] It participates in cell differentiation, proliferation, maturity, embryonic development, adult tissue homeostasis and so on.[27]
In this experiment, we attempted to study the role of the Wnt/GSK3β/β-catenin pathway in SP-induced cardioprotection. To increase the reliability of the experiment, the present study used two reliable models, one is the isolated rat heart I/R model, another one is the H9C2 cell H/R model, and both have been widely recognized in studies about cardioprotection. The simulated H9C2 cell hypoxia/reoxygenation model was built according to previous studies.[17,28] H9C2 cells have the morphology and function of myocardial cells. They are cultured more easily than primary neonatal rat cardiomyocytes, and also have passages, characteristics different from primary cardiomyocytes. The results showed that Wnt/GSK3β/β-catenin signaling pathway was activated in I/R group, compared with the S group, and it might be a self-protection mechanism. SP played an important role in cardioprotection by further activating Wnt/GSK3β/β-catenin signaling pathway. Furthermore, the Wnt inhibitor IWP-2 not only inhibited SP-induced Wnt/GSK3β/β-catenin signaling activation, but also abolished cardioprotection afforded by SP. Thus, the results also demonstrated that Wnt ligands were the direct targets of SP. The previous study that Barandon using transgenic mice overexpressing Wnt receptor FrzA also showed that the Wnt pathway contributed to cardioprotection afforded by IPC.[29,30] Above all, The Wnt/GSK3β/β-catenin signaling pathway played an important role in cardioprotection afforded by not only IPC but also sevoflurane pretreatment.
Wnt proteins not only constitute members of the Wnt/GSK3β/β-catenin signaling pathway, but also play an important part in noncanonical Wnt pathway: Wnt/Ca2 + signaling pathway, planar cell polarity (PCP) pathway.[31,32] The PCP pathway includes Wnt/JNK pathway, Wnt/ROCK pathway, and Wnt/PKC pathway.[33] We all know that JNK, ROCK, and PKC are important parts of cell survival signaling pathways, they play important roles in myocardial protection.[34,35,36,37,38,39,40,41] Since then, whether Wnt/JNK pathway, Wnt/ROCK pathway, and Wnt/PKC pathway appear to be of great importance for myocardial protection? Future studies about this are needed.
Provided it is indicated, Wnt, as upstream of various myocardial protective signaling pathways, appears to be a molecular target for I/R treatment and plays an extremely important role in cardioprotection and cardiovascular disease treatment. Through intervention in expression of Wnt ligands or ligands binding to its receptors, it would be likely to regulate various pathways cardioprotection involved, including the canonical Wnt pathway and noncanonical Wnt pathway. Thus, we can achieve an optimal cardioprotection effect. Therefore, further studies about the characteristics and mechanism of the Wnt signaling pathway in cardioprotection are particularly needed, and it is likely to provide new insights into cardiovascular disease treatment for humans in the future.
In conclusion, our study shows that the myocardial protection afforded by SP may be mediated partly via Wnt/GSK3β/β-catenin signaling pathway.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Xiu -Yuan Hao
REFERENCES
- 1.Feng J, Zhu M, Schaub MC, Gehrig P, Roschitzki B, Lucchinetti E, et al. Phosphoproteome analysis of isoflurane-protected heart mitochondria: Phosphorylation of adenine nucleotide translocator-1 on Tyr194 regulates mitochondrial function. Cardiovasc Res. 2008;80:20–9. doi: 10.1093/cvr/cvn161. [DOI] [PubMed] [Google Scholar]
- 2.Dai AL, Fan LH, Zhang FJ, Yang MJ, Yu J, Wang JK, et al. Effects of sevoflurane preconditioning and postconditioning on rat myocardial stunning in ischemic reperfusion injury. J Zhejiang Univ Sci B. 2010;11:267–74. doi: 10.1631/jzus.B0900390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weber NC, Schlack W. Inhalational anaesthetics and cardioprotection. Handb Exp Pharmacol. 2008;182:187–207. doi: 10.1007/978-3-540-74806-9_9. [DOI] [PubMed] [Google Scholar]
- 4.Nusse R, Varmus HE. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell. 1982;31:99–109. doi: 10.1016/0092-8674(82)90409-3. [DOI] [PubMed] [Google Scholar]
- 5.Melkonyan HS, Chang WC, Shapiro JP, Mahadevappa M, Fitzpatrick PA, Kiefer MC, et al. SARPs: A family of secreted apoptosis-related proteins. Proc Natl Acad Sci U S A. 1997;94:13636–41. doi: 10.1073/pnas.94.25.13636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wodarz A, Nusse R. Mechanisms of Wnt signaling in development. Annu Rev Cell Dev Biol. 1998;14:59–88. doi: 10.1146/annurev.cellbio.14.1.59. [DOI] [PubMed] [Google Scholar]
- 7.Smalley MJ, Dale TC. Wnt signalling in mammalian development and cancer. Cancer Metastasis Rev. 1999;18:215–30. doi: 10.1023/a:1006369223282. [DOI] [PubMed] [Google Scholar]
- 8.Patapoutian A, Reichardt LF. Roles of Wnt proteins in neural development and maintenance. Curr Opin Neurobiol. 2000;10:392–9. doi: 10.1016/s0959-4388(00)00100-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chong ZZ, Maiese K. Targeting WNT, protein kinase B, and mitochondrial membrane integrity to foster cellular survival in the nervous system. Histol Histopathol. 2004;19:495–504. doi: 10.14670/hh-19.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Das A, Xi L, Kukreja RC. Protein kinase G-dependent cardioprotective mechanism of phosphodiesterase-5 inhibition involves phosphorylation of ERK and GSK3beta. J Biol Chem. 2008;283:29572–85. doi: 10.1074/jbc.M801547200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao HK, Yin Z, Zhou N, Feng XY, Gao F, Wang HC. Glycogen synthase kinase 3 inhibition protects the heart from acute ischemia-reperfusion injury via inhibition of inflammation and apoptosis. J Cardiovasc Pharmacol. 2008;52:286–92. doi: 10.1097/FJC.0b013e318186a84d. [DOI] [PubMed] [Google Scholar]
- 12.Yin H, Chao L, Chao J. Adrenomedullin protects against myocardial apoptosis after ischemia/reperfusion through activation of Akt-GSK signaling. Hypertension. 2004;43:109–16. doi: 10.1161/01.HYP.0000103696.60047.55. [DOI] [PubMed] [Google Scholar]
- 13.Murphy E, Steenbergen C. Preconditioning: The mitochondrial connection. Annu Rev Physiol. 2007;69:51–67. doi: 10.1146/annurev.physiol.69.031905.163645. [DOI] [PubMed] [Google Scholar]
- 14.Tong H, Imahashi K, Steenbergen C, Murphy E. Phosphorylation of glycogen synthase kinase-3beta during preconditioning through a phosphatidylinositol-3-kinase – Dependent pathway is cardioprotective. Circ Res. 2002;90:377–9. doi: 10.1161/01.res.0000012567.95445.55. [DOI] [PubMed] [Google Scholar]
- 15.Gomez L, Paillard M, Thibault H, Derumeaux G, Ovize M. Inhibition of GSK3beta by postconditioning is required to prevent opening of the mitochondrial permeability transition pore during reperfusion. Circulation. 2008;117:2761–8. doi: 10.1161/CIRCULATIONAHA.107.755066. [DOI] [PubMed] [Google Scholar]
- 16.Wagner C, Kloeting I, Strasser RH, Weinbrenner C. Cardioprotection by postconditioning is lost in WOKW rats with metabolic syndrome: Role of glycogen synthase kinase 3beta. J Cardiovasc Pharmacol. 2008;52:430–7. doi: 10.1097/FJC.0b013e31818c12a7. [DOI] [PubMed] [Google Scholar]
- 17.Shin EJ, Schram K, Zheng XL, Sweeney G. Leptin attenuates hypoxia/reoxygenation-induced activation of the intrinsic pathway of apoptosis in rat H9c2 cells. J Cell Physiol. 2009;221:490–7. doi: 10.1002/jcp.21883. [DOI] [PubMed] [Google Scholar]
- 18.Conacci-Sorrell M, Zhurinsky J, Ben-Ze’ev A. The cadherin-catenin adhesion system in signaling and cancer. J Clin Invest. 2002;109:987–91. doi: 10.1172/JCI15429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–36. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 20.Kloner RA, Bolli R, Marban E, Reinlib L, Braunwald E. Medical and cellular implications of stunning, hibernation, and preconditioning: An NHLBI workshop. Circulation. 1998;97:1848–67. doi: 10.1161/01.cir.97.18.1848. [DOI] [PubMed] [Google Scholar]
- 21.Chiari PC, Bienengraeber MW, Pagel PS, Krolikowski JG, Kersten JR, Warltier DC. Isoflurane protects against myocardial infarction during early reperfusion by activation of phosphatidylinositol-3-kinase signal transduction: Evidence for anesthetic-induced postconditioning in rabbits. Anesthesiology. 2005;102:102–9. doi: 10.1097/00000542-200501000-00018. [DOI] [PubMed] [Google Scholar]
- 22.Pagel PS, Krolikowski JG, Neff DA, Weihrauch D, Bienengraeber M, Kersten JR, et al. Inhibition of glycogen synthase kinase enhances isoflurane-induced protection against myocardial infarction during early reperfusion in vivo. Anesth Analg. 2006;102:1348–54. doi: 10.1213/01.ane.0000202379.61338.37. [DOI] [PubMed] [Google Scholar]
- 23.Krolikowski JG, Weihrauch D, Bienengraeber M, Kersten JR, Warltier DC, Pagel PS. Role of Erk1/2, p70s6K, and eNOS in isoflurane-induced cardioprotection during early reperfusion in vivo. Can J Anaesth. 2006;53:174–82. doi: 10.1007/BF03021824. [DOI] [PubMed] [Google Scholar]
- 24.Cavallo RA, Cox RT, Moline MM, Roose J, Polevoy GA, Clevers H, et al. Drosophila Tcf and Groucho interact to repress Wingless signalling activity. Nature. 1998;395:604–8. doi: 10.1038/26982. [DOI] [PubMed] [Google Scholar]
- 25.Roose J, Molenaar M, Peterson J, Hurenkamp J, Brantjes H, Moerer P, et al. The Xenopus Wnt effector XTcf-3 interacts with Groucho-related transcriptional repressors. Nature. 1998;395:608–12. doi: 10.1038/26989. [DOI] [PubMed] [Google Scholar]
- 26.Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. Beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997;16:3797–804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–80. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 28.Park M, Youn B, Zheng XL, Wu D, Xu A, Sweeney G. Globular adiponectin, acting via AdipoR1/APPL1, protects H9c2 cells from hypoxia/reoxygenation-induced apoptosis. PLoS One. 2011;6:e19143. doi: 10.1371/journal.pone.0019143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barandon L, Dufourcq P, Costet P, Moreau C, Allières C, Daret D, et al. Involvement of FrzA/sFRP-1 and the Wnt/frizzled pathway in ischemic preconditioning. Circ Res. 2005;96:1299–306. doi: 10.1161/01.RES.0000171895.06914.2c. [DOI] [PubMed] [Google Scholar]
- 30.Mirotsou M, Zhang Z, Deb A, Zhang L, Gnecchi M, Noiseux N, et al. Secreted frizzled related protein 2 (Sfrp2) is the key Akt-mesenchymal stem cell-released paracrine factor mediating myocardial survival and repair. Proc Natl Acad Sci U S A. 2007;104:1643–8. doi: 10.1073/pnas.0610024104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li F, Chong ZZ, Maiese K. Winding through the WNT pathway during cellular development and demise. Histol Histopathol. 2006;21:103–24. doi: 10.14670/hh-21.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loh YN, Hedditch EL, Baker LA, Jary E, Ward RL, Ford CE. The Wnt signalling pathway is upregulated in an in vitro model of acquired tamoxifen resistant breast cancer. BMC Cancer. 2013;13:174. doi: 10.1186/1471-2407-13-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knabb MT, Danielsen CA, McShane-Kay K, Mbuy GK, Woodruff RI. Herpes simplex virus-type 2 infectivity and agents that block gap junctional intercellular communication. Virus Res. 2007;124:212–9. doi: 10.1016/j.virusres.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hopper RA, Forrest CR, Xu H, Zhong A, He W, Rutka J, et al. Role and mechanism of PKC in ischemic preconditioning of pig skeletal muscle against infarction. Am J Physiol Regul Integr Comp Physiol. 2000;279:R666–76. doi: 10.1152/ajpregu.2000.279.2.R666. [DOI] [PubMed] [Google Scholar]
- 35.Inagaki K, Begley R, Ikeno F, Mochly-Rosen D. Cardioprotection by epsilon-protein kinase C activation from ischemia: Continuous delivery and antiarrhythmic effect of an epsilon-protein kinase C-activating peptide. Circulation. 2005;111:44–50. doi: 10.1161/01.CIR.0000151614.22282.F1. [DOI] [PubMed] [Google Scholar]
- 36.Minatoguchi S, Uno Y, Kariya T, Arai M, Wang N, Hashimoto K, et al. Cross-talk among noradrenaline, adenosine and protein kinase C in the mechanisms of ischemic preconditioning in rabbits. J Cardiovasc Pharmacol. 2003;41(Suppl 1):S39–47. [PubMed] [Google Scholar]
- 37.Milano G, Morel S, Bonny C, Samaja M, von Segesser LK, Nicod P, et al. A peptide inhibitor of c-Jun NH2-terminal kinase reduces myocardial ischemia-reperfusion injury and infarct size in vivo. Am J Physiol Heart Circ Physiol. 2007;292:H1828–35. doi: 10.1152/ajpheart.01117.2006. [DOI] [PubMed] [Google Scholar]
- 38.Wang J, Yang L, Rezaie AR, Li J. Activated protein C protects against myocardial ischemic/reperfusion injury through AMP-activated protein kinase signaling. J Thromb Haemost. 2011;9:1308–17. doi: 10.1111/j.1538-7836.2011.04331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bao W, Hu E, Tao L, Boyce R, Mirabile R, Thudium DT, et al. Inhibition of Rho-kinase protects the heart against ischemia/reperfusion injury. Cardiovasc Res. 2004;61:548–58. doi: 10.1016/j.cardiores.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 40.Zhang J, Li XX, Bian HJ, Liu XB, Ji XP, Zhang Y. Inhibition of the activity of Rho-kinase reduces cardiomyocyte apoptosis in heart ischemia/reperfusion via suppressing JNK-mediated AIF translocation. Clin Chim Acta. 2009;401:76–80. doi: 10.1016/j.cca.2008.11.016. [DOI] [PubMed] [Google Scholar]
- 41.Lipton SA, Bossy-Wetzel E. Dueling activities of AIF in cell death versus survival: DNA binding and redox activity. Cell. 2002;111:147–50. doi: 10.1016/s0092-8674(02)01046-2. [DOI] [PubMed] [Google Scholar]