

ABSTRACT
The alphaherpesviral envelope protein pUS9 has been shown to play a role in the anterograde axonal transport of herpes simplex virus 1 (HSV-1), yet the molecular mechanism is unknown. To address this, we used an in vitro pulldown assay to define a series of five arginine residues within the conserved pUS9 basic domain that were essential for binding the molecular motor kinesin-1. The mutation of these pUS9 arginine residues to asparagine blocked the binding of both recombinant and native kinesin-1. We next generated HSV-1 with the same pUS9 arginine residues mutated to asparagine (HSV-1pUS9KBDM) and then restored them being to arginine (HSV-1pUS9KBDR). The two mutated viruses were analyzed initially in a zosteriform model of recurrent cutaneous infection. The primary skin lesion scores were identical in severity and kinetics, and there were no differences in viral load at dorsal root ganglionic (DRG) neurons at day 4 postinfection (p.i.) for both viruses. In contrast, HSV-1pUS9KBDM showed a partial reduction in secondary skin lesions at day 8 p.i. compared to the level for HSV-1pUS9KBDR. The use of rat DRG neuronal cultures in a microfluidic chamber system showed both a reduction in anterograde axonal transport and spread from axons to nonneuronal cells for HSV-1pUS9KBDM. Therefore, the basic domain of pUS9 contributes to anterograde axonal transport and spread of HSV-1 from neurons to the skin through recruitment of kinesin-1.
IMPORTANCE Herpes simplex virus 1 and 2 cause genital herpes, blindness, encephalitis, and occasionally neonatal deaths. There is also increasing evidence that sexually transmitted genital herpes increases HIV acquisition, and the reactivation of HSV increases HIV replication and transmission. New antiviral strategies are required to control resistant viruses and to block HSV spread, thereby reducing HIV acquisition and transmission. These aims will be facilitated through understanding how HSV is transported down nerves and into skin. In this study, we have defined how a key viral protein plays a role in both axonal transport and spread of the virus from nerve cells to the skin.
INTRODUCTION
Deletion of the US9 gene, encoding the conserved type II transmembrane viral envelope protein pUS9 (1) (Fig. 1), has been undertaken in a range of Alphaherpesvirinae subfamily members, and neuronal spread has been assessed in several biological models. The relative contribution of pUS9 to neuronal spread within this subfamily was found to differ. Equine herpesvirus type 1 (EHV-1) and bovine herpesvirus type 1 (BHV-1) pUS9 can complement for the loss of pUS9 in swine pathogen pseudorabies virus (PrV) (1). In contrast, varicella-zoster virus (VZV) and herpes simplex virus 1 (HSV-1) pUS9 are unable to complement for a loss of pUS9 in PrV (1). In PrV (2–5), BHV-1 (6), and BHV-5 (7), pUS9 plays a major role in axonal sorting and/or anterograde axonal transport. For PrV there is also evidence for pUS9-independent anterograde axonal transport of some viral components (8, 9). In HSV-1 the contribution of pUS9 to these processes is less clearly defined (10–13), although a cooperative role for HSV-1 pUS9 and the viral glycoproteins gE/gI in axonal sorting and/or anterograde axonal transport has been documented (11, 12, 14).
FIG 1.

Key domains of pUS9 are conserved across the Alphaherpesvirinae subfamily. pUS9 is a type II transmembrane protein consisting of an acidic domain and a basic domain within the cytoplasmic tail, in addition to a transmembrane (TM) domain and a short C-terminal extracellular domain (ED). The encoded pUS9 sequences were obtained from the following herpesviral genome sequence accession numbers: HSV-1 strain 17, X14112; HSV-1 strain F, GU734771; HSV-2, Z86099; simian agent 8 (SA8), AY714813; herpesvirus B (HVB), AF533768; BHV-1, AJ004801; BHV-5, AY261359; PrV, BK001744; EHV-1, AY665713; equine herpesvirus 4 (EHV-4), AF030027; and VZV, X04370. Clustal W alignments were generated using Lasergene (version 11.1) MegAlign from DNAStar. The kinesin-1 binding domain (residues 56 to 60 are underlined for HSV-1) determined in this study maps within the basic domain of pUS9. The consensus residue is shown above the alignments when at least 5 residues of one type only match between aligned sequences; otherwise, residues are indicated by dots or dashes. Consensus strength is shown above the alignments as vertical bars (red, 11 residues match; orange, 9 residues match; green, 7 residues match; light blue, 5 residues match; dark blue, <5 residues match).
In the case of PrV, pUS9 has been shown to interact with the host anterograde molecular motor kinesin-3 (KIF1A) to mediate axonal sorting and anterograde axonal transport (15). The interaction of PrV pUS9 with kinesin-3 depends on the presence of the additional viral glycoproteins gE and gI (16). Phosphorylation of PrV pUS9, although not essential, does enhance the interaction with kinesin-3 (17). To date, no interaction of HSV-1 pUS9 with a kinesin motor has been reported. There appears to be fundamental differences between PrV and HSV-1, as HSV-1 pUS9 was found not to bind kinesin-3 when introduced into a PrV pUS9 deletion background (15).
Members of the pUS9 subfamily share a domain structure comprising both acidic (18) and basic (1) domains within the cytoplasmic tail (Fig. 1). The deletion of the acidic domain in pUS9 reduces anterograde axonal transport for a number of Alphaherpesvirinae subfamily members, including PrV (18), BHV-1 (19), and HSV-1 (20). No studies to date have addressed the role of the pUS9 basic domain in anterograde axonal transport and spread from neurons to skin cells.
Our aim was to determine the mechanism of the pUS9-dependent anterograde axonal transport of HSV-1. We defined a minimal kinesin binding domain (KBD) within the pUS9 basic domain for the host anterograde molecular motor kinesin-1 (KIF5B). A conservative amino acid substitution of the KBD in the whole virus established that this domain contributes to anterograde axonal transport and spread from neurons to skin cells.
MATERIALS AND METHODS
Cell culture and viruses.
Vero cells were maintained in Dulbecco's modified Eagle medium (DMEM) (Invitrogen) supplemented with 10% (vol/vol) fetal bovine serum (FBS) (JRH Bioscience). All cells were grown at 37°C (5% CO2). HSV-1F parental virus (wild-type strain F), HSV-1pUS9KBDM, and rescuant HSV-1pUS9KBDR were propagated in Vero cells. For virus infections, virus inocula were prepared in DMEM supplemented with 2% FBS and 1% penicillin and streptomycin and plated onto cells in monolayers for 1 h at 37°C, unless otherwise specified. After this time, fresh DMEM, supplemented as described for inocula, was added. Plaque assays were performed as previously described (21).
Expression constructs.
The cytoplasmic tail (CT) of HSV-1 pUS9, corresponding to amino acids 1 to 64, was amplified from previously described displayTarget/pUS9 CT (22) and inserted into EcoRI/XhoI-digested pGEX-5X-1. The pUS9 fragments (amino acid numbering) 1-60, 1-55, 1-51, and 1-31 then were amplified from pGEX-5X-1/pUS9 1-64 and inserted into EcoRI/XhoI-digested pGEX-5X-1. Mutagenesis of pUS9 residues 56 to 60 or 57 to 61 from RRRRR to NNNNN in the context of construct pGEX-5X-1/pUS9 1-64 was performed using a QuikChange lightning site-directed mutagenesis kit (Agilent). The dynein light-chain subunit roadblock (DYNLRB) was amplified from a human brain cDNA library (Display Systems Biotech) and inserted into the XhoI site of pET28b (Merck). The construct pET28a/uKHC814-963, encoding the C-terminal tail domain of kinesin-1 (also designated uKHC or KIF5B), has been described previously (23).
In vitro pulldown assay.
Glutathione S-transferase (GST)-tagged and His6-tagged proteins were expressed and harvested as previously described (23, 24). In each case, the lysis buffer consisted of PBS and 0.1% (vol/vol) Triton X-100 plus 1% (vol/vol) bacterial protease inhibitor cocktail (Sigma). Rat brain nerve terminal synaptosomes were prepared as previously described (25) and lysed in 5 mM Tris, pH 7.5, 150 mM NaCl, 1% (vol/vol) Triton X-100, 1 mM EDTA, 1 mM EGTA, leupeptin (10 μg/ml), 1 mM phenylmethylsulfonyl fluoride (PMSF), and EDTA-free complete protease inhibitors (Roche). Conditions for binding of GST-tagged proteins to glutathione-Sepharose beads (Merck) and washing differed according to whether subsequent pulldowns were performed with bacterial lysates (24) or rat brain nerve terminal synaptosomes (26). Immobilized GST fusion proteins then were incubated with mixing at 4°C for 3 h with 1 ml of bacterial lysate containing His6-tagged protein or 1 h with 1 ml of lysate of rat brain synaptosomes. Beads were pelleted by spinning at 500 × g for 5 min and then washed with PBS plus 0.1% (vol/vol) Triton X-100 for His6-tagged protein pulldown or PBS for rat brain synaptosome pulldown to remove unbound proteins. Bound protein complexes then were eluted by heating in 50 to 100 μl of 2× reducing Laemmli buffer for 4 min at 95°C.
Construction of recombinant viruses.
The two recombinant HSV-1 viruses, HSV-1pUS9KBDM and HSV-1pUS9KBDR, were generated by homologous recombination and galK selection/counterselection (27) using a bacterial artificial chromosome (BAC)-cloned HSV-1 parental strain F genome (YEbac102; kindly provided by Yasushi Kawaguchi [28]) as previously described (29). The first-stage targeting cassette was generated using oligonucleotide primers US9galK-Forward and US9galK-Reverse (Table 1), which replaces the region of US9 encoding amino acid residues 57 to 61 in YEbac102 with galK. For HSV-1pUS9KBDM, the second-stage targeting cassette was generated using oligonucleotide primers US9KBDM-Forward and US9KBDM-Reverse (Table 1). This results in the removal of the galK insert in US9 and mutation of the US9 region encoding amino acid residues 57 to 61 from the sequence RRRRR to NNNNN. The same approach was used to generate the rescuant HSV-1pUS9KBDR using the HSV-1pUS9KBDM BAC construct as the starting template for replacement of US9, encoding amino acid residues 57 to 61, with galK. The second-stage targeting cassette was generated using oligonucleotide primers US9KBDR-Forward and US9KBDR-Reverse (Table 1). This results in the removal of the galK insert in US9 and restoration of the US9 region encoding amino acid residues 57 to 61 back to the wild-type RRRRR sequence. Clones which had successfully replaced galK with the desired modification initially were identified by restriction enzyme analysis using Bsp119I digestion and then subsequently confirmed by direct sequencing of the entire pUS9 open reading frame (ORF). To release the viral genome from the BAC backbone and produce infectious virus, modified HSV-1 BAC DNA was cotransfected with a Cre recombinase-expressing plasmid into Vero cells.
TABLE 1.
Oligonucleotides used for construction of recombinant viruses and for qPCR of DNA from ganglia
| Name | Sequencea |
|---|---|
| US9galK-Forward | CGGCCAACGACTTCCTCGTACGCATGGGCCGCCAACAGTCGGTATTAAGGCCTGTTGACAATTAATCATCGGCA |
| US9galK-Reverse | GACAGAACGGCCACGAGGAGACAGGCGATCACCATGCCGACGCAGCGGGTTCAGCACTGTCCTGCTCCTT |
| US9KBDM-Forward | CGGCCAACGACTTCCTCGTACGCATGGGCCGCCAACAGTCGGTATTAAGGAATAACAACAATAACACCCGCTGCGTCGGCATGGTGATCGCCTGTCTCCTCGTGGCCGTTCTGTC |
| US9KBDM-Reverse | GACAGAACGGCCACGAGGAGACAGGCGATCACCATGCCGACGCAGCGGGTGTTATTGTTGTTATTCCTTAATACCGACTGTTGGCGGCCCATGCGTACGAGGAAGTCGTTGGCCG |
| US9KBDR-Forward | CGGCCAACGACTTCCTCGTACGCATGGGCCGCCAACAGTCGGTATTAAGGCGTCGACGCAGACGCACCCGCTGCGTCGGCATGGTGATCGCCTGTCTCCTCGTGGCCGTTCTGTC |
| US9KBDR-Reverse | GACAGAACGGCCACGAGGAGACAGGCGATCACCATGCCGACGCAGCGGGTGCGTCTGCGTCGACGCCTTAATACCGACTGTTGGCGGCCCATGCGTACGAGGAAGTCGTTGGCCG |
| UL35-Forward | GTCTTGGCCACCAATAACTC |
| UL35-Reverse | GGGTAAACGTGTTGTTTGCG |
| mGAPDH-Forward | GATGGGTGTGAACCACGAG |
| mGAPDH-Reverse | GTGATGGCATGGACTGTGG |
Homology arms for BAC recombination are underlined.
Infected cell lysates.
Vero cells in 6-well plates were infected with HSV-1 at an MOI of 5 PFU/ml and then incubated at 37°C for 26 h. At the time of harvest, cells were washed twice with PBS before the addition of 200 μl per well of lysis buffer (PBS, 0.1% [vol/vol] Triton X-100, 1% [vol/vol] mammalian protease inhibitors [Sigma]) and incubated at 4°C for 10 min with gentle rocking. After incubation, cells were scraped into lysis buffer and transferred to prechilled Eppendorf tubes. Cells were lysed by incubation on ice for 30 min, with vortexing every 10 min, before pelleting of cell debris at 8,000 × g for 20 min at 4°C.
Analysis of protein complexes.
Proteins were separated by SDS-PAGE, and the gels were further processed for either total protein staining using Coomassie brilliant blue R-250 or immunoblotting as previously described (29, 30). Detection was performed with an Odyssey infrared imaging system (LiCor Biosciences).
Antibodies.
Primary antibodies for immunoblotting included the following: mouse monoclonal antibodies against the His6 tag (1:200 dilution; Santa Cruz), HSV-1 pUL19 (VP5; DM165) (1:5,000 dilution; provided by Frazer Rixon [31]), HSV-1 gE (1:1,000 dilution; Abcam), HSV-1 pUL48 (VP16) (1:1,000 dilution; Santa Cruz), and α-actin (1:1,000 dilution; Abcam); goat polyclonal antibody against kinesin-1 (1:300 dilution; Everest Biotech); and rabbit polyclonal antibodies against HSV-1 pUL37 (1:5,000 dilution; provided by Thomas Mettenleiter [32]), HSV-1 gD (1:4,000 dilution; Abcam), and HSV-1 pUS9 (1:1,000 dilution; provided by David Johnson [12]). Secondary antibodies for the Odyssey system included goat anti-mouse IRDye 680-conjugated IgG (1:5,000 dilution; LiCor), donkey anti-goat IRDye 680-conjugated IgG (1:5,000 dilution; LiCor), and goat anti-rabbit IRDye 800-conjugated IgG (1:5,000 dilution; LiCor).
Primary antibodies for confocal microscopy included rabbit polyclonal against purified HSV-1 nuclear C capsids (PTNC) (1:5,500 dilution; provided by Frazer Rixon [33]) and mouse monoclonal antibody against HSV-1 ICP27 (1:100 dilution; Abcam). Secondary antibodies for confocal microscopy included anti-rabbit Alexa Fluor 543 (1:400 dilution; Life Technologies) and anti-mouse Alexa Fluor 633 (1:100 dilution; Life Technologies).
Zosteriform abrasion model of HSV-1 infection.
Female adult C57BL/6 mice, 6 to 8 weeks of age, were purchased from the Animal Resource Centre (Perth, Australia) and acclimatized, and experiments were conducted with the approval of the Children's Medical Research Institute/Children's Hospital at Westmead animal ethics committee. To mimic the route of HSV infection in humans, we used an epicutaneous flank scarification skin infection model as previously described (34). Briefly, mice were anesthetized, the left flank was shaved and depilated (Nair), and then the mice were infected topically by applying 10 μl of virus (1 × 106 PFU) or PBS within a 2-mm2 area on the flank skin, which was scarified 20 times using an 18-guage needle, and a dressing was applied after 1 h for 24 h to facilitate infection. Mice were scored for skin lesion severity at primary skin site P (site of infection) and secondary skin site S (site of spread of virus from the ganglia) on a scale from 0 to 5: 0, no lesion; 1, redness or swelling; 2, individual vesicle formation; 3, coalesced and/or ulcerated vesicles; 4, severe ulceration; and 5, necrosis.
For the determination of the copy number of HSV-1 DNA genome in the ganglia, total DNA was extracted from DRG (T8-T12) tissue using a DNeasy blood and tissue kit (Qiagen). Viral DNA was quantified by real-time PCR using primers specific for the HSV-1 UL35 gene (Table 1). PCR was performed with 10 μl of SsoAdvanced Universal Sybr green Supermix (Bio-Rad) with primers at a concentration of 200 nM and approximately 200 ng of HSV-1-infected mouse DRG genomic DNA in a total reaction volume of 20 μl. PCR was performed under the following conditions: after 3 min at 98°C, the samples were subjected to 50 cycles of 98°C for 15 s and 60°C for 30 s for both UL35 and mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers (Table 1). A plasmid (pGEX-5X-1) containing the UL35 target sequence was used to create a standard curve of the cycle threshold (CT) values from serial 10-fold dilutions of the plasmid (2.5 × 108 to 2.5 × 104 copies/well). The CT values of the unknown samples were plotted on the standard curve, and copy numbers were determined and normalized to those of mouse GAPDH. Each sample and standard were analyzed in duplicate.
Preparation of dissociated rat neuronal cultures.
DRG neurons were prepared from 4-day-old Wistar rat neonates as previously described (35). Briefly, DRG neurons were dissociated in Hanks calcium- and magnesium-free solution (Invitrogen) plus 0.25% (wt/vol) trypsin (Sigma) and 0.05% (wt/vol) collagenase (Worthington Biomedical Co.) for 30 min at 37°C followed by DNase (10 mg/ml; Sigma) for 5 min at 37°C, washed two times by centrifugation at 80 × g, and passed through a 35% Percoll gradient (Sigma). The cell pellet was resuspended in 500 μl of neurobasal medium (Invitrogen), plated onto Matrigel-coated plastic coverslips, and cultured at 37°C with 5% CO2 in neurobasal medium supplemented with 4 mM l-glutamine (Invitrogen), 2% (vol/vol) B-27 (Invitrogen), and 7S nerve growth factor (100 ng/ml) (Sigma) for 3 days prior to HSV-1 infection. The use of rat neonates was approved by the Western Sydney Local Health District and the University of Sydney animal research ethics committees.
Neuronal cultures in three-chamber microfluidic devices.
Standard neuron devices (SND450; obtained from Xona Microfluidics) were used to study virus anterograde axonal transport and spread from axons to Vero cells. Cover glasses (24 mm by 40 mm) were plasma bonded to the neuron devices by exposing the surfaces of both cover glass and device to glow discharge treatment for 10 s (under 0.39 mBar) with air using a Pelco easiGlow glow discharge cleaning system (Ted Pella). After plasma bonding, the devices were coated by overnight incubation in 0.5 mg/ml poly–d-lysine in borate buffer (pH 8.5) at 37°C with 5% CO2. The devices then were washed three times with water, followed by a second overnight incubation in laminin (10 μg/ml) (Sigma) at 37°C with 5% CO2. The devices were washed three times with neurobasal medium prior to seeding of the neuronal cell suspension. Rat DRG dissociated neurons were prepared as described above, and 10 μl of suspension containing approximately 40,000 neurons was added to the somal compartments of the devices. Neurons were grown in neurobasal medium supplemented with 4 mM l-glutamine, 2% (vol/vol) B-27, and 7S nerve growth factor (100 ng/ml) for 4 to 5 days at 37°C with 5% CO2 to allow the axons to grow into the axonal compartment of the device. A total of 50,000 Vero cells then were added to the axonal compartment of the devices 24 h prior to infection. Ten microliters of virus suspension (5 PFU/cell) was added in the somal (neuronal cell body) compartment, the virus inoculum was removed at 2 h postinfection (p.i.), and cells were washed with neurobasal medium. The medium in the axonal compartment was removed at 8 h p.i. and replaced with medium containing foscarnet (100 μg/ml) (Sigma) to prevent secondary virus spread in Vero cells.
Immunofluorescence and confocal microscopy.
Neuronal cultures in three-chamber microfluidic devices were fixed in situ at 22 h p.i. by addition of 3% (vol/vol) formaldehyde to both somal and axonal compartments and incubation for 30 min at room temperature, followed by addition of 0.1% Triton X-100 (vol/vol) in PBS for 5 min. The cultures were immunostained by the addition of primary antibodies to both somal and axonal compartments and incubated overnight at 4°C, followed by incubation with secondary antibodies for 1 h at room temperature. The cultures were washed six times after each antibody incubation step in PBS. PBS from the last wash then was removed and replaced with Fluoromount G (SouthernBiotech) prior to examination using a Leica SP5 II confocal microscope. For axonal transport studies, axons in close proximity to microgrooves from 10 randomly selected fields of view (1,024 by 1,024 pixels) were manually counted for the presence of HSV-1 capsids. For studies involving virus spread from axons to Vero cells, the total number of HSV-1 ICP27-positive Vero cells in the axonal chambers was manually counted.
Statistical analysis.
For studies of HSV-1 capsid transport along axons in the axonal compartment of the microfluidic devices, the number of capsid-positive axons and the total number of axons observed were recorded in each of 3 separate experiments performed for the HSV-1pUS9KBDM and HSV-1pUS9KBDR virus pair. Statistical analysis was performed using a generalized linear mixed-effect model (GLME) with a logit link function to investigate the effects of HSV-1pUS9KBDM versus HSV-1pUS9KBDR status on the odds of an axon being positive for capsid. In this model, the grouping variable was each experiment, which was considered a random effect. Condition (HSV-1pUS9KBDM versus HSV-1pUS9KBDR) was a fixed effect. Estimates of the ln(odds ratio) and associated 95% confidence intervals were back transformed to produce estimated odds ratios of observed capsid-positive axons under the HSV-1pUS9KBDM versus HSV-1pUS9KBDR condition.
For studies of HSV-1 spread from axons to nonneuronal Vero cells in the axonal compartment, the total number of HSV-1 ICP27-positive Vero cells in the axonal compartment was considered a Poisson variable and log transformed in order to stabilize the variance prior to analysis. The number of ICP27-positive final cells (from 40,000) never exceeded 1,400 in any experiment under either the HSV-1pUS9KBDM or HSV-1pUS9KBDR condition. A linear mixed-effect model (LME) was used to investigate the effects of HSV-1pUS9KBDM versus those of HSV-1pUS9KBDR. In this model, the grouping variable was each experiment and was considered a random effect, whereas the HSV-1pUS9KBDM versus HSV-1pUS9KBDR condition was considered a fixed effect. Estimates and their 95% confidence intervals were back transformed to produce estimated relative rates of ICP27-positive Vero cells under the HSV-1pUS9KBDM versus HSV-1pUS9KBDR condition.
The statistical software SPSS, version 22, and SPLUS 8.2 were used to analyze the data. Two-tailed tests with a significance level of 5% were used throughout. For the zosteriform abrasion model, the Wilcoxon signed-rank test was used to calculate the statistically significant differences between samples. P < 0.05 was considered significant.
RESULTS
HSV-1 pUS9 contains a kinesin-1 binding domain.
We have previously shown that the molecular motor kinesin-1 colocalizes with HSV-1 during anterograde axonal transport (36). Our first aim was to determine if HSV-1 pUS9 directly interacts with kinesin-1 in the absence of other viral proteins.
An in vitro pulldown assay was employed using the cytoplasmic tail of HSV-1 pUS9, which contains conserved acidic and basic domains (Fig. 1). GST-tagged pUS9 1-64 specifically bound the His6-tagged kinesin-1 C terminus, which contains the known cargo binding domain (24) (Fig. 2). Initial C-terminal truncations of the pUS9 cytoplasmic tail revealed that the basic domain, and not the acidic domain, is responsible for kinesin-1 binding (Fig. 2).
FIG 2.

Basic domain of HSV-1 pUS9 contains a kinesin-1 binding domain. (A) Summary of C-terminal truncations of the cytoplasmic tail (corresponds to residues 1 to 64) of HSV-1 pUS9 and their ability to bind kinesin-1. The kinesin-1 binding domain maps within the basic domain of pUS9. (B) In vitro pulldown assay showing His6-tagged kinesin-1 C-terminal domain and not His6-tagged dynein light-chain roadblock (DYNLRB) binds preferentially to GST-tagged pUS9 1-64. (Upper) Immunoblots (anti-His6 tag) to detect His6-tagged proteins eluted from boiled GST Bind beads. (Lower) Total protein stains with Coomassie blue to confirm the presence of GST-tagged proteins eluted from boiled GST Bind beads.
Further C-terminal truncations within the basic domain of pUS9 then were tested using the same in vitro pulldown assay (Fig. 3). pUS9 fragment 1-60 and not 1-55 binds kinesin-1, which indicates that the kinesin-1 binding domain of pUS9 comprises residues 56 to 60 (Fig. 3). This region in the basic domain of HSV-1 pUS9 is a sequence of 5 consecutive positively charged arginine residues followed by another arginine at position 61 (Fig. 1). This positively charged region is conserved across the Alphaherpesvirinae subfamily (Fig. 1).
FIG 3.

Further definition of the kinesin-1 binding domain within the basic domain of HSV-1 pUS9. (A) Summary of C-terminal truncations of the cytoplasmic tail (corresponds to residues 1 to 64) of HSV-1 pUS9 and their ability to bind kinesin-1. The kinesin-1 binding domain maps to pUS9 residues 56 to 60 within the basic domain. (B) In vitro pulldown assay showing His6-tagged kinesin-1 C-terminal domain and not His6-tagged dynein light-chain roadblock (DYNLRB) binds preferentially to GST-tagged pUS9 1-64 and 1-60. (Upper) Immunoblots (anti-His6 tag) to detect His6-tagged proteins eluted from boiled GST Bind beads. (Lower) Total protein stains with Coomassie blue to confirm the presence of GST-tagged proteins eluted from boiled GST Bind beads.
To confirm whether the sequence of positively charged arginines in HSV-1 pUS9 is essential for binding kinesin-1, we replaced them with polar asparagine residues. We made two mutants in the context of GST-pUS9 1-64, including R56-60N, based on the initial mapping results, and R57-61N, which targets the additional arginine at position 61 in HSV-1 pUS9 (Fig. 1). Using the in vitro pulldown experiments, they both abolished His6-tagged kinesin-1 binding. For simplicity, the results are shown only for pUS9 R57-61N, which we have subsequently termed the kinesin binding domain mutant (KBDM) (Fig. 4A). Therefore, a cluster of positive charges is the most important kinesin-1 binding determinant.
FIG 4.

Confirmation that the pUS9 kinesin binding domain (KBD) is essential for binding kinesin-1. (A and B) In vitro pulldown assay was undertaken with a GST-tagged HSV-1 pUS9 cytoplasmic tail (residues 1 to 64) containing a substitution mutation. This mutation, KBDM, consisted of pUS9 residues 57 to 61 with the sequence RRRRR mutated to NNNNN. Binding of either the His6-tagged kinesin-1 C-terminal domain (A) or native kinesin-1 (derived from rat brain synaptosomes) (B) was abolished for the GST-tagged pUS9 KBDM but not the GST-tagged pUS9 WT. This also illustrates that HSV-1 pUS9 binds native kinesin-1. The upper images in each case are immunoblots (anti-His6 tag [A] or anti-kinesin-1 [B]) to detect either His6-tagged proteins or kinesin-1 eluted from boiled GST Bind beads. The lower images in panels A and B are total protein stains with Coomassie blue to confirm the presence of GST-tagged proteins eluted from Bind GST Bind beads.
To determine whether pUS9 can bind endogenous kinesin-1, we again used GST-pUS9 1-64 WT and confirmed the specific binding of full-length kinesin-1 (110 kDa) from rat brain nerve terminals (synaptosomes) (Fig. 4B). This kinesin-1 binding also was abolished with GST pUS9 1-64 KBDM (Fig. 4B).
Characterization of HSV-1 containing pUS9KBDM.
To establish the role of the pUS9 KBD in HSV-1 replication, we engineered the pUS9 KBD mutation into HSV-1 strain F. The amino acid sequence of pUS9 is identical between HSV-1 strain 17 (the source of recombinant pUS9 used in binding assays) and HSV-1 strain F (Fig. 1). In addition, we also repaired the introduced pUS9 mutation back to the wild-type sequence. These recombinant viruses, designated HSV-1pUS9KBDM and HSV-1pUS9KBDR, respectively, grew to titers similar to those of the HSV-1 parental strain F in Vero cells. This is not surprising, given HSV-1 pUS9 is nonessential for viral growth in cultured nonneuronal cells (37). Analysis of the expression of a number of viral structural proteins from infected Vero cells highlights that capsid (pUL19), tegument (pUL37 and pUL48), and glycoprotein (gD and gE) all are expressed at equivalent levels for both HSV-1pUS9KBDM and HSV-1pUS9KBDR relative to that of HSV-1Fparental (Fig. 5). Only in the case of pUS9 itself was there an observed change in expression for HSV-1pUS9KBDM relative to that of HSV-1Fparental (Fig. 5). For HSV-1pUS9KBDR, the expression of pUS9 was similar to that of HSV-1Fparental (Fig. 5). The fact that expression levels of pUS9 have increased in HSV-1pUS9KBDM suggests that mutation of arginines at positions 57 to 61 to asparagines results in a more stable protein.
FIG 5.
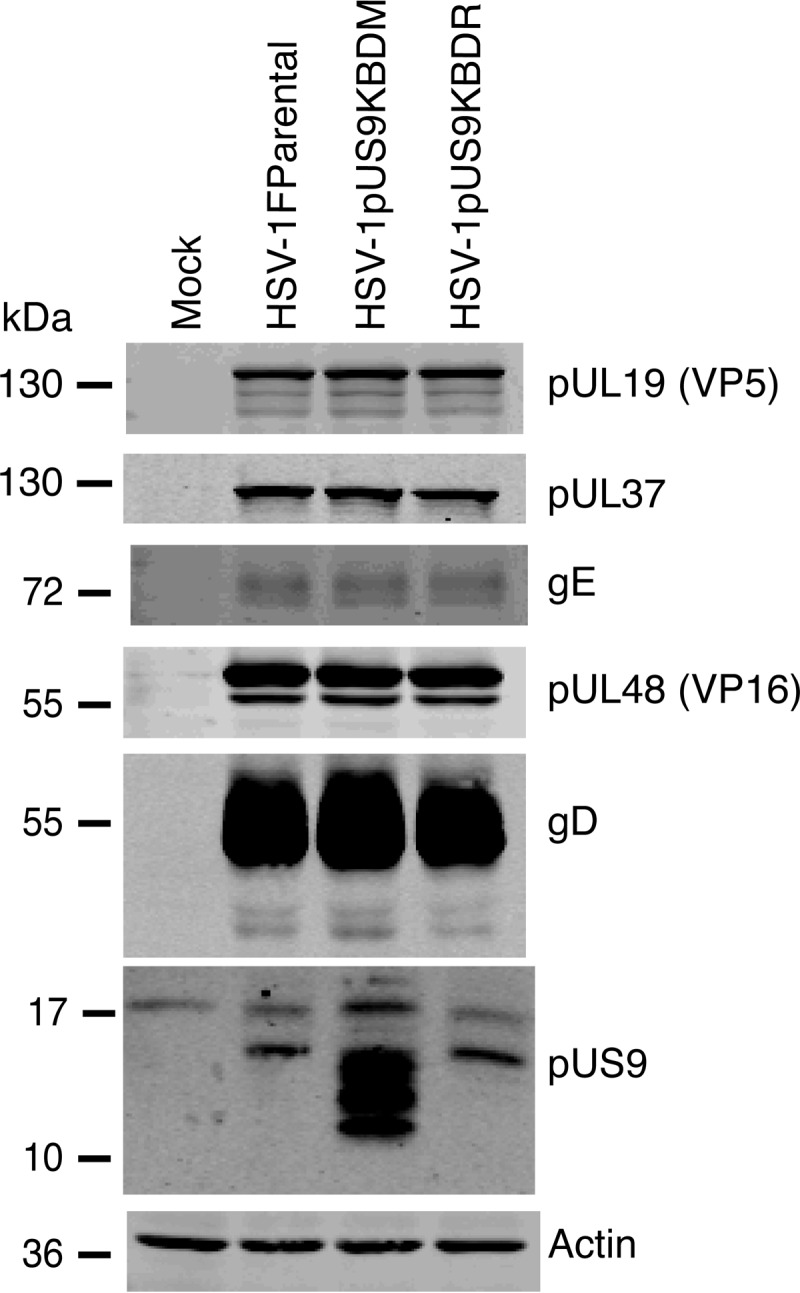
Characterization of HSV-1pUS9KBDM and HSV-1pUS9KBDR (rescuant), generated by BAC recombination. Vero cell lysates harvested at 24 h p.i. and immunoblotted with the indicated antibodies are shown. Relative to those of HSV-1Fparental (F strain), HSV-1pUS9KBDM and HSV-1pUS9KBDR showed similar expression of major viral structural proteins. Only in the case of HSV-1pUS9KBDM was there enhanced expression of pUS9.
Multiple bands observed for HSV-1 pUS9 (Fig. 5) have been noted on SDS-PAGE previously for wild-type untagged (38) and GFP-tagged (15) PrV pUS9 as well as wild-type HSV-1 pUS9 (39). These multiple bands were attributed to either the phosphorylation state of pUS9 (presumably via the acidic domain) (17) or a common result of membrane protein chemistry (40) or possibly lysine-free ubiquitination (39). Therefore, for the HSV-1 pUS9 wild type and mutant, one would expect the same number of multiple equivalent bands, but they are more evident in the mutant form of pUS9, which appears to be more stable, resulting in higher expression levels (Fig. 5). It is unlikely these additional bands are competing for pUS9 binding partners, as they also usually are present in wild-type forms of pUS9.
We next analyzed HSV-1pUS9KBDM with respect to HSV-1pUS9KBDR in two biological models of neuronal spread. In the first instance, we used the mouse flank zosteriform abrasion model of HSV-1 infection. The scoring of lesion formation at the primary site of infection illustrated no differences between HSV-1pUS9KBDM and HSV-1pUS9KBDR in initial replication in skin (Fig. 6A and B). In contrast, lesion formation at the secondary skin site showed a significant decrease for HSV-1pUS9KBDM relative to that of HSV-1pUS9KBDR that peaked at day 8 p.i., indicative of a defect in axonal transport/neuronal spread (Fig. 6A and B). The determination of viral DNA genome copy number by qPCR in DRG tissue at day 4 p.i. showed no significant difference between HSV-1pUS9KBDM and HSV-1pUS9KBDR (Fig. 6C). This indicates that there was no defect in retrograde axonal transport for HSV-1pUS9KBDM, and the difference in lesion formation at the secondary site is in fact due to decreased anterograde axonal transport and/or spread from neurons to skin cells of HSV-1pUS9KBDM.
FIG 6.

HSV zosteriform disease in the mouse flank infection model as a means to assess viral neuronal spread. Anesthetized, 6-week-old female adult C57BL/6 mice were scarified on the flank within a 2-mm2 area using an 18-gauge needle and then topically infected with 1 × 106 PFU/mouse of HSV-1pUS9KBDR or HSV-1pUS9KBDM. Mice were scored for HSV disease at the primary skin site (measure of primary infection) and the secondary skin site (measure of ganglionic neuronal spread) per the lesion scoring scheme given in Materials and Methods. (A) Mean skin lesion scores (± SEM) at selected times (n = 5 mice/time point) from days 1 to 28 p.i. for HSV-1pUS9KBDM and HSV-1pUS9KBDR. Similar primary skin lesion scores were observed. A significant decrease in secondary skin lesion score (*, P < 0.05 by Wilcoxon signed-rank test), which peaked at day 8 p.i., was observed for HSV-1pUS9KBDM compared to that of HSV-1pUS9KBDR. In each case HSV-1pUS9KBDR behaved the same as parental strain F (not shown). (B) Illustration of the progression of zosteriform disease at both primary and secondary skin sites in the mouse flank model using HSV-1pUS9KBDM and HSV-1pUS9KBDR. Similar disease progression was observed at the primary (P) site for both viruses with a peak decrease at the secondary (S) site at day 8 p.i. for HSV-1pUS9KBDM. (C) qPCR analysis of total DNA extracted from mouse DRG tissue (n = 5 mice/virus) at day 4 p.i. Levels of total viral genome copy number present in DRG tissue from infected mice were not significantly different between HSV-1pUS9KBDM and HSV-1pUS9KBDR. This confirms that there were no defects in the retrograde transport of HSV-1pUS9KBDM. The standard curve was based on a plasmid containing the HSV-1 UL35 gene. Samples were normalized to mGAPDH.
A three-chamber microfluidic system then was utilized to directly assess anterograde axonal transport and spread from distal axons to nonneuronal cells of HSV-1pUS9KBDM and HSV-1pUS9KBDR. Rat DRG neurons were seeded in the somal compartment to produce axons which extend into the axonal compartment via microgrooves in the middle compartment. Indicator Vero cells then were plated in the axonal compartment prior to the infection of neurons with HSV-1 in the somal compartment (and with the addition of the viral DNA replication inhibitor foscarnet in the axonal compartment to prevent secondary virus spread in Vero cells). Staining of infected neurons in the somal compartment at 22 h p.i. showed similar levels of infection for HSV-1pUS9KBDM and HSV-1pUS9KBDR (not shown). Staining of distal axons in the axonal compartment for viral capsid at 22 h p.i. illustrated a significant reduction in the proportion of axons stained for capsids, highlighting a reduction in axonal anterograde transport for HSV-1pUS9KBDM compared to that for HSV-1pUS9KBDR (Fig. 7). In our companion study (53), we have shown no defect in a pUS9 full deletion mutant virus in egress from the cell body, so the defect in the current study for a partial pUS9 mutant must be axon specific. The odds ratio for observing HSV-1pUS9KBDR versus HSV-1pUS9KBDM capsid-positive axons was estimated to be 4.1 (95% confidence interval, 2.8 to 6.1; P < 0.001). A reduction in spread from distal axons to Vero cells for HSV-1pUS9KBDM compared to that for HSV-1pUS9KBDR also was observed (Fig. 8). The relative rate of final HSV-1-positive Vero cells for HSV-1pUS9KBDR versus HSV-1pUS9KBDM was estimated to be 8.9 (95% confidence interval, 5.6 to 14.1; P = 0.002).
FIG 7.

Analysis of anterograde axonal transport of HSV-1 capsids. Neonatal rat DRG neurons were dissociated, pelleted through a 35% Percoll gradient, and plated into the somal compartment of three-chamber microfluidic devices. Cultures were incubated for 5 to 6 days to allow axons to grow into the axonal compartment via the middle grooves. Vero cells were added to the axonal compartment 24 h prior to the addition of either HSV-1pUS9KBDM or HSV-1pUS9KBDR (5 PFU/cell) to the somal compartment. Foscarnet (100 μg/ml) was added to the axonal compartment 8 h p.i. to prevent secondary virus spread in Vero cells. The cultures were fixed at 22 h p.i., immunostained for C capsids (PTNC), and examined using a Leica SP5 confocal microscope. (A) Representative confocal micrographs of axons (indicated by arrows) emerging from the middle grooves (visible on the left side of the lower images) into the axonal compartment. These axons were counted from 10 randomly selected fields of view and scored as positive or negative for the presence of HSV-1 capsid (red in the upper images). Scale bars, 25 μm. (B) The percentage of capsid-positive (+ve) axons versus capsid-negative (−ve) axons in either case is shown for three separate experiments. A significant reduction in capsid transport along axons was observed for HSV-1pUS9KBDM compared to that of HSV-1pUS9KBDR.
FIG 8.

HSV-1 spread from axons to nonneuronal Vero cells. Neonatal rat DRG neurons were dissociated, pelleted through a 35% Percoll gradient, and plated into the somal compartment of three-chamber microfluidic devices. Cultures were incubated for 5 to 6 days to allow axons to grow in the axonal compartment. Vero cells were added to the axonal compartment 24 h prior to addition of either HSV-1pUS9KBDM or HSV-1pUS9KBDR (5 PFU/cell) to the somal compartment. Foscarnet (100 μg/ml) was added to the axonal compartment 8 h p.i. to prevent secondary virus spread in Vero cells. The cultures were fixed at 22 h p.i., immunostained for HSV-1 immediate-early protein ICP27, and examined using a Leica SP5 confocal microscope. (A) Representative confocal micrographs of Vero cells in the axonal compartment. (Lower) The presence of axons innervating the Vero cells was confirmed for each virus. (Upper) Vero cells were counted from 10 randomly selected fields of view and scored as positive or negative for the presence of HSV-1 ICP27 (blue). Scale bars, 25 μm. (B) The total number of ICP27-positive Vero cells in the axonal compartment was counted for each virus in three separate experiments (Exp). A significant reduction in virus spread from axons to Vero cells was observed for HSV-1pUS9KBDM compared to that of HSV-1pUS9KBDR.
DISCUSSION
In this study, we have defined a kinesin-1 binding site within the basic domain of HSV-1 pUS9. Recombinant HSV-1 containing a conservative substitution of this domain was tested in two biological models. This revealed an overall decrease in anterograde axonal transport and spread from distal axons to nonneuronal cells in an in vitro three-chamber microfluidic system employing rat DRG neurons as well as a decrease in spread in an in vivo zosteriform mouse model.
Two possible scenarios have been proposed to explain the reduction in anterograde axonal transport observed with gE, gI, and pUS9 deletion mutants in HSV-1. First, the viral protein facilitates sorting into the proximal axon by promoting the loading of viral components onto kinesin motors in the neuronal cell body. Second, the viral protein binds kinesin directly or via an adaptor to mediate anterograde transport of viral components toward axon tips (11). Our identified interaction of kinesin-1 and the pUS9 basic domain most likely plays a functional role in the context of the motor-adaptor scenario to directly mediate axonal anterograde transport. In support of this, a recent study has shown that HSV-1 pUS9 still undergoes microtubule-dependent axonal transport in neurons in a process independent of other viral proteins (41). This axonal transport also did not depend on the acidic domain of pUS9 (41). It is also of note that the pUS9 basic domain contains a consensus recognition sequence for HSV-1 pUS3 kinase (42). Mutation of this region could disrupt phosphorylation of pUS9 by pUS3, but the in vivo role of such phosphorylation has not been determined (42).
The fact that the kinesin-1 binding site in pUS9 maps to a series of arginine residues centered around residue 58 in the basic domain is of particular note. Analysis of the genome of two HSV-1 strains, KOS and McIntyre, have identified the introduction of a stop codon at position 58 in pUS9 (43–45). Whether the R58stop mutation of pUS9 directly affects viral neuronal spread as a result of an inability to recruit kinesin-1 is an intriguing possibility. In the case of KOS, the resulting truncated pUS9 has been shown to be unstable, resulting in little or no protein expression, making it effectively equivalent to a pUS9 deletion mutant (44). The fact there is no obvious axonal transport phenotype reported for KOS in animal models (46) may be due to the presence of compensating mutations, such as an observed extension of the C-terminal end of pUS8A (44, 45). However, we have observed a neuronal spread defect for KOS in vitro using dissociated rat DRG neurons (53). In the case of McIntyre, another mutation of note is a frameshift mutation which results in an extension of the C-terminal tail of gI (43), which, along with truncated pUS9, may contribute directly to the observed anterograde spread defect of this strain (47, 48).
Our study suggests that HSV-1 pUS9, via the basic domain, also contributes to viral spread from neurons. Similar observations have been reported for an HSV-1 pUS9 full deletion mutant (11). Our current findings with the three-chamber system using an HSV-1 pUS9 basic domain mutant also are similar to our observations with a complete deletion of pUS9 (53). A role for HSV-1 gE/gI extracellular domains in contributing to both anterograde axonal transport and possibly also in the spread from distal axons to nonneuronal cells has been reported recently (14) and adds further support to the notion of redundancy in transport processes essential for viral replication (49).
As observed in cell culture, HSV-1 assembly and egress from the cell body of neurons appears not to require intact pUS9, as strain McIntyre, encoding truncated pUS9 (43), still is able to move to presynaptic neurons (47). However, from our observations it appears that HSV-1 pUS9 via the KDB plays a major role in anterograde axonal transport and neuronal spread from the distal axon. These processes would require interactions with cytoskeletal transport proteins, such as kinesin-1, as well as vesicular trafficking proteins or proteins involved in exocytosis which are enriched in growth cones (50–52). We have previously identified such classes of proteins, including Rab3A, SNAP-25, GAP-43, and kinesin-1, colocalizing with HSV-1 virions in growth cones (36).
Further studies are required to dissect the functional relevance of the kinesin-1–pUS9 interaction in anterograde axonal transport and spread from distal axons to nonneuronal cells. In addition, the role of the pUS9 acidic domain remains to be established precisely, given that this domain has been shown to be important for anterograde spread in a number of alphaherpesviruses (18–20). Finally, whether, and to what degree, the interaction of kinesin-3 and pUS9 identified in PrV (15, 16) also is important for anterograde spread of HSV-1 requires further elucidation.
ACKNOWLEDGMENTS
This work was supported by bridging grants from the Westmead Millennium Foundation (to R.J.D., B.K., and M.M.S.) and the Westmead Charitable Trust (to R.J.D.).
We thank Karen Byth, Westmead Hospital, Westmead, Australia, for assistance in statistical analysis and Phillip Robinson, Children's Medical Research Institute, Westmead, Australia, for helpful discussions.
REFERENCES
- 1.Lyman MG, Kemp CD, Taylor MP, Enquist LW. 2009. Comparison of the pseudorabies virus Us9 protein with homologs from other veterinary and human alphaherpesviruses. J Virol 83:6978–6986. doi: 10.1128/JVI.00598-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brideau AD, Card JP, Enquist LW. 2000. Role of pseudorabies virus Us9, a type II membrane protein, in infection of tissue culture cells and the rat nervous system. J Virol 74:834–845. doi: 10.1128/JVI.74.2.834-845.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ch'ng TH, Enquist LW. 2005. Neuron-to-cell spread of pseudorabies virus in a compartmented neuronal culture system. J Virol 79:10875–10889. doi: 10.1128/JVI.79.17.10875-10889.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lyman MG, Curanovic D, Enquist LW. 2008. Targeting of pseudorabies virus structural proteins to axons requires association of the viral Us9 protein with lipid rafts. PLoS Pathog 4:e1000065. doi: 10.1371/journal.ppat.1000065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lyman MG, Feierbach B, Curanovic D, Bisher M, Enquist LW. 2007. Pseudorabies virus Us9 directs axonal sorting of viral capsids. J Virol 81:11363–11371. doi: 10.1128/JVI.01281-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Butchi NB, Jones C, Perez S, Doster A, Chowdhury SI. 2007. Envelope protein Us9 is required for the anterograde transport of bovine herpesvirus type 1 from trigeminal ganglia to nose and eye upon reactivation. J Neurovirol 13:384–388. doi: 10.1080/13550280701375433. [DOI] [PubMed] [Google Scholar]
- 7.Chowdhury SI, Onderci M, Bhattacharjee PS, Al-Mubarak A, Weiss ML, Zhou Y. 2002. Bovine herpesvirus 5 (BHV-5) Us9 is essential for BHV-5 neuropathogenesis. J Virol 76:3839–3851. doi: 10.1128/JVI.76.8.3839-3851.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daniel GR, Sollars PJ, Pickard GE, Smith GA. 2015. Pseudorabies virus fast-axonal transport occurs by a pUS9-independent mechanism. J Virol 89:8088–8091. doi: 10.1128/JVI.00771-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kratchmarov R, Enquist LW, Taylor MP. 2015. Us9-independent axonal sorting and transport of the pseudorabies virus glycoprotein gM. J Virol 89:6511–6514. doi: 10.1128/JVI.00625-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.LaVail JH, Tauscher AN, Sucher A, Harrabi O, Brandimarti R. 2007. Viral regulation of the long distance axonal transport of herpes simplex virus nucleocapsid. Neuroscience 146:974–985. doi: 10.1016/j.neuroscience.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Howard PW, Howard TL, Johnson DC. 2013. Herpes simplex virus membrane proteins gE/gI and US9 act cooperatively to promote transport of capsids and glycoproteins from neuron cell bodies into initial axon segments. J Virol 87:403–414. doi: 10.1128/JVI.02465-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Snyder A, Polcicova K, Johnson DC. 2008. Herpes simplex virus gE/gI and US9 proteins promote transport of both capsids and virion glycoproteins in neuronal axons. J Virol 82:10613–10624. doi: 10.1128/JVI.01241-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McGraw HM, Awasthi S, Wojcechowskyj JA, Friedman HM. 2009. Anterograde spread of herpes simplex virus type 1 requires glycoprotein E and glycoprotein I but not Us9. J Virol 83:8315–8326. doi: 10.1128/JVI.00633-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Howard PW, Wright CC, Howard T, Johnson DC. 2014. Herpes simplex virus gE/gI extracellular domains promote axonal transport and spread from neurons to epithelial cells. J Virol 88:11178–11186. doi: 10.1128/JVI.01627-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kramer T, Greco TM, Taylor MP, Ambrosini AE, Cristea IM, Enquist LW. 2012. Kinesin-3 mediates axonal sorting and directional transport of alphaherpesvirus particles in neurons. Cell Host Microbe 12:806–814. doi: 10.1016/j.chom.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kratchmarov R, Kramer T, Greco TM, Taylor MP, Ch'ng TH, Cristea IM, Enquist LW. 2013. Glycoproteins gE and gI are required for efficient KIF1A-dependent anterograde axonal transport of alphaherpesvirus particles in neurons. J Virol 87:9431–9440. doi: 10.1128/JVI.01317-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kratchmarov R, Taylor MP, Enquist LW. 2013. Role of Us9 phosphorylation in axonal sorting and anterograde transport of pseudorabies virus. PLoS One 8:e58776. doi: 10.1371/journal.pone.0058776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brideau AD, Eldridge MG, Enquist LW. 2000. Directional transneuronal infection by pseudorabies virus is dependent on an acidic internalization motif in the Us9 cytoplasmic tail. J Virol 74:4549–4561. doi: 10.1128/JVI.74.10.4549-4561.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chowdhury SI, Brum MC, Coats C, Doster A, Wei H, Jones C. 2011. The bovine herpesvirus type 1 envelope protein Us9 acidic domain is crucial for anterograde axonal transport. Vet Microbiol 152:270–279. doi: 10.1016/j.vetmic.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 20.Draper JM, Huang G, Stephenson GS, Bertke AS, Cortez DA, LaVail JH. 2013. Delivery of herpes simplex virus to retinal ganglion cell axon is dependent on viral protein Us9. Investig Ophthalmol Vis Sci 54:962–967. doi: 10.1167/iovs.12-11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ko DH, Cunningham AL, Diefenbach RJ. 2010. The major determinant for addition of tegument protein pUL48 (VP16) to capsids in herpes simplex virus type 1 is the presence of the major tegument protein pUL36 (VP1/2). J Virol 84:1397–1405. doi: 10.1128/JVI.01721-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee JH, Vittone V, Diefenbach E, Cunningham AL, Diefenbach RJ. 2008. Identification of structural protein-protein interactions of herpes simplex virus type 1. Virology 378:347–354. doi: 10.1016/j.virol.2008.05.035. [DOI] [PubMed] [Google Scholar]
- 23.Diefenbach RJ, Mackay JP, Armati PJ, Cunningham AL. 1998. The C-terminal region of the stalk domain of ubiquitous human kinesin heavy chain contains the binding site for kinesin light chain. Biochemistry 37:16663–16670. doi: 10.1021/bi981163r. [DOI] [PubMed] [Google Scholar]
- 24.Diefenbach RJ, Diefenbach E, Douglas MW, Cunningham AL. 2002. The heavy chain of conventional kinesin interacts with the SNARE proteins SNAP25 and SNAP23. Biochemistry 41:14906–14915. doi: 10.1021/bi026417u. [DOI] [PubMed] [Google Scholar]
- 25.Dunkley PR, Jarvie PE, Robinson PJ. 2008. A rapid Percoll gradient procedure for preparation of synaptosomes. Nat Protoc 3:1718–1728. doi: 10.1038/nprot.2008.171. [DOI] [PubMed] [Google Scholar]
- 26.Brymora A, Valova VA, Robinson PJ. 2004. Protein-protein interactions identified by pull-down experiments and mass spectrometry. Curr Protoc Cell Biol Chapter 17:5.1–5.51. [DOI] [PubMed] [Google Scholar]
- 27.Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33:e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanaka M, Kagawa H, Yamanashi Y, Sata T, Kawaguchi Y. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J Virol 77:1382–1391. doi: 10.1128/JVI.77.2.1382-1391.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kelly BJ, Bauerfeind R, Binz A, Sodeik B, Laimbacher AS, Fraefel C, Diefenbach RJ. 2014. The interaction of the HSV-1 tegument proteins pUL36 and pUL37 is essential for secondary envelopment during viral egress. Virology 454–455:67–77. [DOI] [PubMed] [Google Scholar]
- 30.Vittone V, Diefenbach E, Triffett D, Douglas MW, Cunningham AL, Diefenbach RJ. 2005. Determination of interactions between tegument proteins of herpes simplex virus type 1. J Virol 79:9566–9571. doi: 10.1128/JVI.79.15.9566-9571.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McClelland DA, Aitken JD, Bhella D, McNab D, Mitchell J, Kelly SM, Price NC, Rixon FJ. 2002. pH reduction as a trigger for dissociation of herpes simplex virus type 1 scaffolds. J Virol 76:7407–7417. doi: 10.1128/JVI.76.15.7407-7417.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leege T, Granzow H, Fuchs W, Klupp BG, Mettenleiter TC. 2009. Phenotypic similarities and differences between UL37-deleted pseudorabies virus and herpes simplex virus type 1. J Gen Virol 90:1560–1568. doi: 10.1099/vir.0.010322-0. [DOI] [PubMed] [Google Scholar]
- 33.Pasdeloup D, Blondel D, Isidro AL, Rixon FJ. 2009. Herpesvirus capsid association with the nuclear pore complex and viral DNA release involve the nucleoporin CAN/Nup214 and the capsid protein pUL25. J Virol 83:6610–6623. doi: 10.1128/JVI.02655-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fernandez MA, Yu U, Zhang G, White R, Sparwasser T, Alexander SI, Jones CA. 2013. Treg depletion attenuates the severity of skin disease from ganglionic spread after HSV-2 flank infection. Virology 447:9–20. doi: 10.1016/j.virol.2013.08.027. [DOI] [PubMed] [Google Scholar]
- 35.Miranda-Saksena M, Boadle RA, Armati P, Cunningham AL. 2002. In rat dorsal root ganglion neurons, herpes simplex virus type 1 tegument forms in the cytoplasm of the cell body. J Virol 76:9934–9951. doi: 10.1128/JVI.76.19.9934-9951.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miranda-Saksena M, Boadle RA, Aggarwal A, Tijono B, Rixon FJ, Diefenbach RJ, Cunningham AL. 2009. Herpes simplex virus utilizes the large secretory vesicle pathway for anterograde transport of tegument and envelope proteins and for viral exocytosis from growth cones of human fetal axons. J Virol 83:3187–3199. doi: 10.1128/JVI.01579-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Umene K. 1986. Conversion of a fraction of the unique sequence to part of the inverted repeats in the S component of the herpes simplex virus type 1 genome. J Gen Virol 67:1035–1048. doi: 10.1099/0022-1317-67-6-1035. [DOI] [PubMed] [Google Scholar]
- 38.Brideau AD, Banfield BW, Enquist LW. 1998. The Us9 gene product of pseudorabies virus, an alphaherpesvirus, is a phosphorylated, tail-anchored type II membrane protein. J Virol 72:4560–4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brandimarti R, Roizman B. 1997. Us9, a stable lysine-less herpes simplex virus 1 protein, is ubiquitinated before packaging into virions and associates with proteasomes. Proc Natl Acad Sci U S A 94:13973–13978. doi: 10.1073/pnas.94.25.13973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rath A, Glibowicka M, Nadeau VG, Chen G, Deber CM. 2009. Detergent binding explains anomalous SDS-PAGE migration of membrane proteins. Proc Natl Acad Sci U S A 106:1760–1765. doi: 10.1073/pnas.0813167106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pedrazzi M, Nash B, Meucci O, Brandimarti R. 2014. Molecular features contributing to virus-independent intracellular localization and dynamic behavior of the herpesvirus transport protein US9. PLoS One 9:e104634. doi: 10.1371/journal.pone.0104634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kato A, Yamamoto M, Ohno T, Kodaira H, Nishiyama Y, Kawaguchi Y. 2005. Identification of proteins phosphorylated directly by the Us3 protein kinase encoded by herpes simplex virus 1. J Virol 79:9325–9331. doi: 10.1128/JVI.79.14.9325-9331.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Szpara ML, Tafuri YR, Parsons L, Shreve JT, Engel EA, Enquist LW. 2014. Genome sequence of the anterograde-spread-defective herpes simplex virus 1 strain MacIntyre. Genome Announc 2(6):e01161-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Negatsch A, Mettenleiter TC, Fuchs W. 2011. Herpes simplex virus type 1 strain KOS carries a defective US9 and a mutated US8A gene. J Gen Virol 92:167–172. doi: 10.1099/vir.0.026484-0. [DOI] [PubMed] [Google Scholar]
- 45.Macdonald SJ, Mostafa HH, Morrison LA, Davido DJ. 2012. Genome sequence of herpes simplex virus 1 strain KOS. J Virol 86:6371–6372. doi: 10.1128/JVI.00646-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dix RD, McKendall RR, Baringer JR. 1983. Comparative neurovirulence of herpes simplex virus type 1 strains after peripheral or intracerebral inoculation of BALB/c mice. Infect Immun 40:103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zemanick MC, Strick PL, Dix RD. 1991. Direction of transneuronal transport of herpes simplex virus 1 in the primate motor system is strain-dependent. Proc Natl Acad Sci U S A 88:8048–8051. doi: 10.1073/pnas.88.18.8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garner JA, LaVail JH. 1999. Differential anterograde transport of HSV type 1 viral strains in the murine optic pathway. J Neurovirol 5:140–150. doi: 10.3109/13550289909021996. [DOI] [PubMed] [Google Scholar]
- 49.Diefenbach RJ, Miranda-Saksena M, Douglas MW, Cunningham AL. 2008. Transport and egress of herpes simplex virus in neurons. Rev Med Virol 18:35–51. doi: 10.1002/rmv.560. [DOI] [PubMed] [Google Scholar]
- 50.Igarashi M. 2014. Proteomic identification of the molecular basis of mammalian CNS growth cones. Neurosci Res 88:1–15. doi: 10.1016/j.neures.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 51.Estrada-Bernal A, Sanford SD, Sosa LJ, Simon GC, Hansen KC, Pfenninger KH. 2012. Functional complexity of the axonal growth cone: a proteomic analysis. PLoS One 7:e31858. doi: 10.1371/journal.pone.0031858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nozumi M, Togano T, Takahashi-Niki K, Lu J, Honda A, Taoka M, Shinkawa T, Koga H, Takeuchi K, Isobe T, Igarashi M. 2009. Identification of functional marker proteins in the mammalian growth cone. Proc Natl Acad Sci U S A 106:17211–17216. doi: 10.1073/pnas.0904092106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miranda-Saksena M, Boadle RA, Diefenbach RJ, Cunningham AL. Dual role of herpes simplex virus 1 pUS9 in virus anterograde axonal transport and final assembly in growth cones in distal axons. J Virol, in press. doi: 10.1128/JVI.03023-15. [DOI] [PMC free article] [PubMed] [Google Scholar]