

ABSTRACT
To successfully replicate in an infected host cell, a virus must overcome sophisticated host defense mechanisms. Viruses, therefore, have evolved a multitude of devices designed to circumvent cellular defenses that would lead to abortive infection. Previous studies have identified Nek9, a cellular kinase, as a binding partner of adenovirus E1A, but the biology behind this association remains a mystery. Here we show that Nek9 is a transcriptional repressor that functions together with E1A to silence the expression of p53-inducible GADD45A gene in the infected cell. Depletion of Nek9 in infected cells reduces virus growth but unexpectedly enhances viral gene expression from the E2 transcription unit, whereas the opposite occurs when Nek9 is overexpressed. Nek9 localizes with viral replication centers, and its depletion reduces viral genome replication, while overexpression enhances viral genome numbers in infected cells. Additionally, Nek9 was found to colocalize with the viral E4 orf3 protein, a repressor of cellular stress response. Significantly, Nek9 was also shown to associate with viral and cellular promoters and appears to function as a transcriptional repressor, representing the first instance of Nek9 playing a role in gene regulation. Overall, these results highlight the complexity of virus-host interactions and identify a new role for the cellular protein Nek9 during infection, suggesting a role for Nek9 in regulating p53 target gene expression.
IMPORTANCE In the arms race that exists between a pathogen and its host, each has continually evolved mechanisms to either promote or prevent infection. In order to successfully replicate and spread, a virus must overcome every mechanism that a cell can assemble to block infection. On the other hand, to counter viral spread, cells must have multiple mechanisms to stifle viral replication. In the present study, we add to our understanding of how the human adenovirus is able to circumvent cellular roadblocks to replication. We show that the virus uses a cellular protein, Nek9, in order to block activation of p53-regulated gene GADD45A, which is an important player in stress response and p53-mediated cell cycle arrest. Importantly, our study also identifies Nek9 as a transcriptional repressor.
INTRODUCTION
The NIMA family of kinases is a relatively poorly understood family of serine/threonine kinases. The prototype, NimA (Never in mitosis, gene A), was first identified in Aspergillus as a protein responsible for regulation of mitotic progression (1). There is only one NimA kinase in Aspergillus; however, in humans there are 11 NimA-related kinases (2). The first mammalian NimA-related protein kinase (Nek) to be cloned was Nek1 (3). Subsequently, Nek9 was identified based on its β-casein kinase activity isolated from rabbit lung (4), initially called Nek8. Nek9 was also cloned as a protein associated with Nek6 (5), named Nercc1. Human Nek9 is 979 amino acids long, with an N-terminal kinase domain of the NimA family, a central regulator of chromosome condensation 1 (RCC1)-like domain, and a C-terminal coiled-coil motif (4). The human Nek9 protein interacts with a number of other proteins, including the adenovirus E1A oncoprotein (6), the FACT complex (7), Nek6 and Nek7 (5, 8, 9), Ran GTPase (5), CHK1 (10), the Epstein-Barr virus (EBV) tegument protein BGLF2 (11), and NEDD1 (12). Most of the work on Nek9 has centered on its role in mitosis and cell cycle control. For example, Nek9 is required for proper centrosome separation (9), while Nek9 depletion was found to lead to catastrophic mitosis via impairment of spindle dynamics and mitotic checkpoint control (13). Interestingly, Nek9 has recently been implicated in the DNA replication stress response via interaction and activation of the CHK1 kinase (10). Surprisingly, Nek9 has also been shown to drive cancer cell proliferation in cells lacking functional p53 (also known as TP53) (14). These seemingly opposite functions of the protein are difficult to reconcile but nevertheless highlight the diverse processes that Nek9 participates in.
Human adenovirus (HAdV) is a small, nonenveloped DNA virus with a double-stranded linear genome that infects predominantly terminal differentiated epithelial cells (15). The first viral protein expressed during infection is that encoded by the immediate early gene 1A (E1A). E1A remodels the intracellular environment in order to enable viral genome to be replicated. This involves deregulation of the cell cycle by direct modulation of cellular factors, as well as activation of expression of other viral early genes (for a review of E1A functions, see references 16 and 17). E1A binds to a large variety of cellular proteins, including cell cycle regulators, transcription factors, transcriptional coregulators, chromatin remodeling factors, and a host of other proteins with diverse functions (16). Interestingly, E1A was shown to bind to Nek9 via the highly conserved N terminus of E1A (6). This interaction alters the subcellular localization of a Nek9 mutant lacking the RCC1-like domain, but effects on adenovirus growth remained elusive.
In the present study, we set out to identify the functional significance of the interaction between Nek9 and E1A. We show that knockdown of Nek9 via RNA interference (RNAi) leads to reduced virus growth. Surprisingly, knockdown of Nek9 led to enhanced expression of the viral E2 transcriptional unit responsible for viral genome replication. However, the overall number of viral genomes was significantly reduced under these conditions. Overexpression of Nek9 enhanced viral genome replication despite a modest reduction in E2 expression early in the infection. Nek9 was found to colocalize with viral replication centers in infected cells and viral E4 orf3 protein via transfection assays and during infection. Importantly, reduction in Nek9 levels led to an increase in expression of GADD45A (Growth Arrest and DNA-Damage-inducible, 45 Alpha). Nek9 was also recruited to the p53-regulated, internal GADD45A promoter together with E1A. This represents, as far as we are aware, the first report of Nek9 playing a role in transcriptional regulation. Together, these results highlight a novel function for Nek9 in the innate antiviral response via its role in the downregulation of GADD45A expression and identify a new pathway on which E1A impinges in order to enable a productive viral infection. Importantly, our study highlights the complexity and importance of silencing p53 target genes by HAdV and identifies a cellular factor, Nek9, coopted by the virus for this purpose.
MATERIALS AND METHODS
Antibodies.
Mouse monoclonal anti-E1A M73 and M58 antibodies were previously described (18) and were grown in-house and used as the hybridoma supernatant. For immunoprecipitations (IPs), 25 μl was used, and for Western blot assays a dilution of 1:400 was used. 12CA5 antihemagglutinin (anti-HA) mouse monoclonal antibody was previously described (19); 25 μl of hybridoma supernatant was used in chromatin immunoprecipitation (ChIP) experiments. Mouse monoclonal anti-myc 9E10 antibody was previously described (20) and was grown in-house. For IPs, 50 μl of 9E10 hybridoma supernatant was used, while for Western blots the supernatant was used at a 1:100 dilution. Mouse monoclonal anti-72k DNA-binding protein (DBP) antibody was previously described (21) and was used at a dilution of 1:400 for Western blotting. Anti-adenovirus type 5 (ab6982) and anti-Nek9 (ab138488) antibodies were purchased from Abcam and were used at recommended dilutions. Rabbit polyclonal anti-Nek9 antibody was previously described (6) and was a generous gift from Peter Whyte. Rat monoclonal anti-E4 orf3 antibody was previously described (22) and was a generous gift from Thomas Dobner.
Cell and virus culture.
IMR-90 (ATCC CCL-186), HT1080 (ATCC CCL-121), and MEF/3T3-Nek9V5 cells were grown in Dulbecco's modified Eagle's medium (HyClone) supplemented with 10% fetal bovine serum (Invitrogen) and streptomycin-penicillin (HyClone). All virus infections were carried out in serum-free medium for 1 h, after which saved complete medium was added without removal of the infection medium. MEF/3T3-Nek9V5 cells were a generous gift from Peter Whyte; these cells express a tetracycline (Tet)-regulated murine Nek9 with a C-terminal V5 tag. To induce the expression of Nek9, doxycycline medium was removed from the cells, cells were washed with phosphate-buffered saline (PBS) three times, and Tet-free medium was applied to the cells for 2 h, washed off again, and replaced with Tet-free medium. Cells were then incubated for at least 24 h prior to viral infection in order to overexpress Nek9. Cells were maintained in Tet-free medium for the duration of the experiment in order to maintain Nek9 overexpression.
Chromatin immunoprecipitation.
ChIP was carried out essentially as previously described (23). IMR-90 cells were infected with the indicated adenoviruses at a multiplicity of infection (MOI) of 5 and harvested 24 h after infection for ChIP analysis. For immunoprecipitation of E1A, monoclonal M73 and M58 antibodies were used. For immunoprecipitation of Nek9, the polyclonal anti-Nek9 antibody was used (6). Mouse monoclonal 12CA5 anti-HA antibody was used as a negative IgG control.
PCRs were carried out for HAdV5 early and major late promoters using SYBR Select master mix for CFX (Applied Biosystems) according to the manufacturer's directions, using 3% of total ChIP DNA as the template and a CFX96 Real Time PCR instrument (Bio-Rad). The annealing temperature used was 60°C, and 40 cycles were run. Primers for viral promoters were previously described in reference 24, while primers for cellular promoters were previously described in reference 25.
Immunofluorescence.
IMR-90 or HT1080 cells were plated at low density (∼40,000 cells per chamber) on chamber slides (Nalgene Nunc) and subsequently infected or transfected as described above. Twenty-four hours after final transfection or infection, cells were fixed in 4% formaldehyde, blocked in blocking buffer (1% normal goat serum, 1% bovine serum albumin [BSA], 0.2% Tween 20 in PBS), and stained with specific primary antibodies. M73 was used neat (hybridoma supernatant), E2 DBP antibody was used at a 1:2 dilution (hybridoma supernatant), Nek9 antibody was used at a dilution of 1:100, 9E10 anti-myc antibody was used neat, rat anti-E4 orf3 monoclonal was used at a dilution of 1:5 (hybridoma supernatant), and Alexa Fluor 488 and 594 secondary antibodies (Jackson ImmunoResearch) were used at a dilution of 1:600. After staining and extensive washing, slides were mounted using Prolong Gold with DAPI (4′,6-diamidino-2-phenylindole; Invitrogen) and imaged using a Zeiss LSM700 confocal laser scanning microscope. Images were analyzed using the Zeiss ZEN software package.
Real-time gene expression analysis.
IMR-90 cells were infected with dl309 at an MOI of 5. Total RNA was extracted using the TRIzol Reagent (Sigma) at the indicated time points according to the manufacturer's instructions. Total RNA (1.25 μg) was used in a reverse transcriptase (RT) reaction using SuperScript VILO reverse transcriptase (Invitrogen) according to the manufacturer's guidelines and random hexanucleotides for priming. The cDNA was subsequently used for real-time expression analysis via the Bio-Rad CFX96 real-time thermocycler. Analysis of expression data was carried out using the Pfaffl method (26), and the data were normalized to GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mRNA levels and compared between siControl- and siNek9-transfected cells. Primers used for E1B, E2, E3, E4, and hexon were previously described (24). Total E1A was detected with primers binding within exon 2: TCCGGTCCTTCTAACACACC and GGCGTTTACAGCTCAAGTCC, as previously described (27). Primers for cellular p53-regulated genes were previously described (28).
siRNA knockdown.
Short interfering RNA (siRNA) knockdown was carried out as previously described (23). Briefly, IMR-90 cells were transfected with Nek9-specific Silencer siRNA (Life Technologies catalog no. 1114) using SilentFect reagent (Bio-Rad) according to the manufacturer's specifications and 10 nM final siRNA concentration. Silencer Select negative-control siRNA number 1 (Life Technologies) was used as the negative siRNA control.
Viral genome quantification.
IMR-90 cells were lysed in lysis buffer (50 mM Tris [pH 8.1], 10 mM EDTA, and 1% SDS) on ice for 10 min. Lysates were sonicated briefly in a Covaris M220 focused ultrasonicator to break up cellular chromatin and subjected to digestion using proteinase K (NEB) according to the manufacturer's specifications. Following digestion, viral DNA was purified using the GeneJET PCR Purification kit (Thermo-Fisher). PCRs were carried out using SYBR Select master mix for CFX (Applied Biosystems) according to the manufacturer's directions with 2% of total purified DNA as the template and a CFX96 Real Time PCR instrument (Bio-Rad). The standard curve for absolute quantification was generated by serially diluting pXC1 plasmid containing the left end of the HAdV5 genome starting with a concentration of 1.0 × 107 copies per reaction mixture down to 1.0 copy per reaction mixture. The primers used were the same as those used for expression analysis of E1B region, the annealing temperature used was 60°C, and 40 cycles were run.
Virus growth assay.
Arrested IMR-90 cells were infected with HAdV5 dl309 (29) at an MOI of 5 or as indicated, in serum-free medium. Virus was adsorbed for 1 h at 37°C under 5% CO2, after which cells were bathed in conditioned medium and were reincubated at 37°C under 5% CO2. Virus titers were determined 24, 48, and 72 h after infection, and plaque assays were performed on 293 cells by serial dilution.
RESULTS
Nek9 knockdown reduces HAdV growth.
In order to identify the role that Nek9 plays in adenovirus infection and the reasons behind it being targeted by E1A, we began our investigation by determining the effect of Nek9 knockdown on adenovirus growth in dl309-infected normal human fibroblasts, IMR-90 (Fig. 1A). IMR-90 cells were transfected with either a negative-control siRNA (siControl) or Nek9-specific siRNA (siNek9) and infected 24 h later with HAdV dl309, which expresses wild-type (wt) E1A. Knockdown of Nek9 modestly reduced virus growth, with approximately a 3-fold reduction in endpoint virus titers at 72 h after infection (Fig. 1A), and slowed down the appearance of the cytopathic effect by approximately 12 h (not shown). To ensure that Nek9 levels were reduced after siRNA transfection, we monitored the protein levels by Western blotting (Fig. 1A, inset). To determine the levels of Nek9 in arrested cells, the natural target of HAdV, we performed Western blotting for the protein in IMR-90 cells that were subconfluent (SC), just confluent (JC), arrested (A), or arrested and infected for 24 h with dl309 (A + dl309) (Fig. 1B). Levels of Nek9 were highest in growing or just confluent cells. Nek9 protein was slightly reduced in abundance in arrested cells and was not altered by infection. Overall, these results demonstrate that reduction in Nek9 levels has a negative effect on HAdV growth and that infection does not alter the level of Nek9 protein expression.
FIG 1.
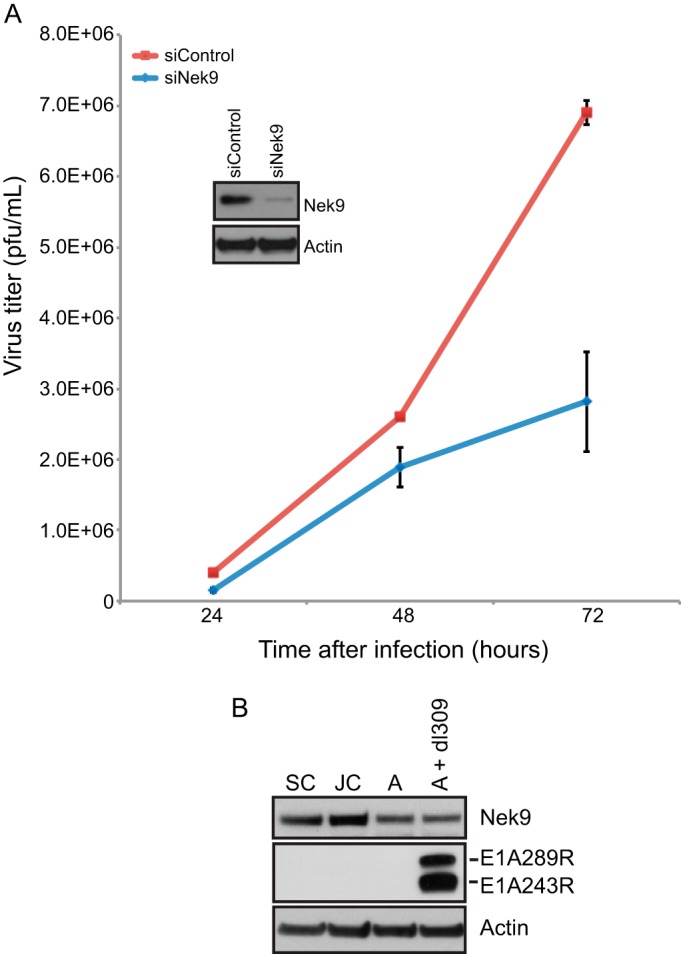
Virus growth in Nek9-depleted IMR-90 cells. (A) IMR-90 cells were transfected with short interfering RNAs, either siControl as a control or siNek9 to deplete Nek9. Twenty-four hours after siRNA transfection, cells were infected with HAdV5 dl309 at an MOI of 5. Virus titers were determined at the indicated time points by plaquing on 293 cells. Nek9 knockdown was monitored by Western blotting as shown in the inset, which shows knockdown at 48 h after infection. n = 4; error bars represent standard deviations (SD). (B) Levels of Nek9, E1A, or actin showing protein expression in IMR-90 cells that are subconfluent and dividing (SC), just confluent cells that have been confluent for less than 24 h (JC), arrested cells that have been confluent for 72 h (A), and arrested cells infected with dl309 at an MOI of 5 and analyzed 24 h after infection (A + dl309). For the Western blot analysis, 20 μg of total cell lysate was resolved on SDS-PAGE.
Nek9 affects viral gene expression and protein levels.
Reduced virus growth in Nek9 knockdown cells suggested that Nek9 either enhances virus growth by directly participating in viral replication or that it may be involved in inhibition of the innate antiviral response. To elucidate which was the case, we initially analyzed viral gene expression in IMR-90 cells in which Nek9 was knocked down by RNAi and compared it to the expression in cells treated with a negative-control siRNA (Fig. 2A) by reverse transcriptase quantitative PCR (RT-qPCR). Unexpectedly, we observed an enhancement in the expression of the viral E2A transcriptional unit (Fig. 2A, orange bar) as early as 20 h after infection. Expression of other viral genes was largely unaltered, with the exception of hexon being reduced 48 h after infection (Fig. 2A, blue bar) and the E3A transcriptional unit showing a small elevation in expression at 48 h after infection in Nek9-depleted cells. These observations were unexpected as we did not anticipate an increase in expression of viral genes after knockdown due to the observed reduction in viral growth; nevertheless, the reduced hexon mRNA level corroborates the reduction in observed viral titers.
FIG 2.

Nek9 affects viral gene expression. (A) IMR-90 cells that were transfected with siRNAs, either siControl as a control or siNek9 to deplete Nek9, were infected with dl309 24 h after knockdown at an MOI of 5. Viral gene expression was monitored by RT-qPCR at the indicated time. GAPDH mRNA was used as the normalization control. Insets show cellular and viral protein expression at 16 and 48 h after viral infection in control or Nek9-depleted cells after resolving 20 μg of total protein from infected IMR-90 cells. n = 4; error bars represent SD. (B) MEF/3T3-Nek9V5 cells were induced to express Nek9 by removal of doxycycline as described in Materials and Methods. Cells were infected with dl309 at an MOI of 5 24 h after Nek9 induction. Viral gene expression was monitored by RT-qPCR as described for panel A with mouse GAPDH mRNA as the normalization control. The inset shows Nek9 induction 24 h after doxycycline removal from the cell culture medium. n = 3; error bars represent SD.
To verify that the altered levels of mRNA translated into changes in protein levels, we performed Western blotting for E1A, E2 DBP, and the late proteins hexon, penton, and protein V in Nek9 knockdown and control cells (Fig. 2A, inset). We observed an increase in the levels of the E2 DBP protein only very early in the infection (16 h), while a minimal reduction in Nek9-depleted cells was observed 48 h after infection. Unexpectedly, the observed increase in E2 DBP levels at 16 h following Nek9 knockdown did not correlate with changes in mRNA levels, suggesting that either translation or protein stability may be affected.
Interestingly, we observed a considerable reduction in E1A and protein V at 48 h after infection. The reduced E1A levels were not correlated with mRNA levels, which remained relatively stable. Similarly, there was a small but noticeable reduction in penton protein levels, but hexon was largely unchanged following Nek9 knockdown. This was unexpected considering the significant reduction in hexon mRNA observed after Nek9 knockdown. Likely, the reduced hexon mRNA level is still at a saturation concentration for the cell to be able to translate into protein. Indeed, levels of hexon mRNA at 48 h under both siControl and siNek9 conditions were more than 1,000 times higher than at 16 h and more than 30 times higher than at 24 h after infection (not shown).
Our observation that knockdown of Nek9 led to enhanced expression of the E2 transcriptional unit suggested that this protein may negatively regulate some aspects of viral gene expression. To test whether overexpression of Nek9 reduced viral gene expression, in particular the E2 transcriptional unit, we performed RT-qPCR on RNA from dl309-infected mouse embryo fibroblast 3T3 (MEF/3T3) cells, which express an inducible murine Nek9 tagged with a C-terminal V5-tag. Removal of doxycycline induces Nek9 expression in less than 24 h in these cells. Although murine cells do not lead to production of human adenovirus particles, all early events of infection are similar to what occurs in human cells, and it therefore makes for a useful model. MEF/3T3-Nek9V5 cells were induced to overexpress Nek9 by replacing medium with tetracycline-doxycycline-free medium for 24 h prior to infection with dl309 virus, and cells were maintained in the Nek9-induced state for the duration of the experiment. Expression of viral genes was analyzed at the indicated time points and shows that most viral genes are unaffected by Nek9 overexpression. Interestingly, there was a reduction in E2 mRNA levels 16 h after infection and a small enhancement in hexon mRNA levels 48 h after infection. Surprisingly, E2 mRNA levels were significantly higher at 48 h after infection in Nek9-overexpressing cells than in the uninduced control cells.
Overall, these results demonstrate that Nek9 plays a role in regulating the expression of certain viral genes and suggest that Nek9 may function as a transcriptional coregulator.
Nek9 is recruited to viral promoters during infection.
The observed alteration in expression of the viral E2 gene under various levels of Nek9 protein suggested that Nek9 may play a role in transcriptional regulation. This is unsurprising since Nek9 possesses an RCC1-like domain that is present in RCC1 for the purposes of docking with chromatin and associates with the FACT transcriptional complex (7, 30, 31). Furthermore, the association of Nek9 with E1A may enable it to indirectly associate with promoter regions via E1A. The kinase activity of Nek9 may therefore be brought to bear on histones or other factors within the vicinity of the promoter region and alter their activities. To test whether Nek9 is recruited to viral promoters, we performed ChIP of dl309-infected IMR-90 cells (Fig. 3). Nek9 was found to occupy the E2 early promoter and, to a lesser extent, the E3 promoter, but not E4 nor the major late promoter (MLP). E1A was found on all promoters tested, as previously observed (24). Recruitment of Nek9 to the E2 early promoter correlated with alteration in expression of E2A (Fig. 2). Although the E3 promoter was also occupied by Nek9 during infection, the occupancy was ∼20-fold lower than at the E2 early promoter, which correlated with much lower effects of Nek9 depletion on E3 expression. The negative enrichment versus IgG control observed for the E4 and the MLP promoters indicated lack of occupancy by Nek9 at these sites, and this correlated with RT-qPCR results for expression of E4 orf6/7 and hexon early in the infection. The altered expression of hexon late in the infection is therefore unlikely to be directly caused by Nek9 but rather is likely a consequence of other factors. Together, these results demonstrate, for the first time, that Nek9 can be recruited to viral promoters and alter viral gene expression.
FIG 3.
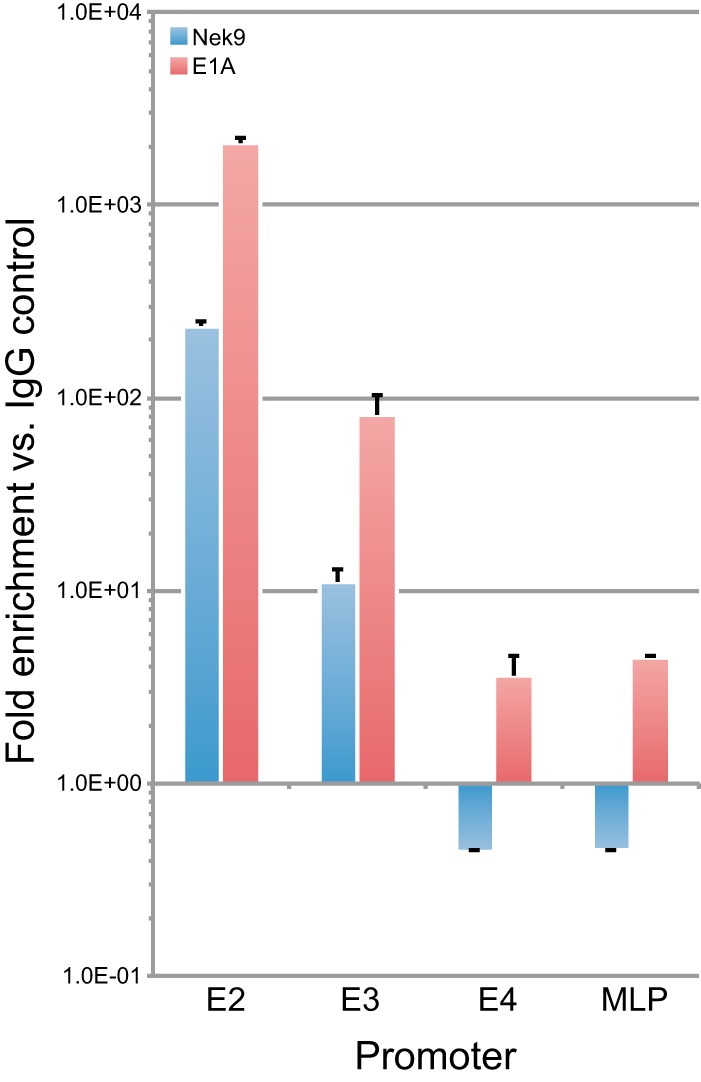
Nek9 associates with the viral E2 early and E3 promoters. IMR-90 cells were infected with dl309 at an MOI of 5, and ChIP was performed 24 h after infection as described in Materials and Methods. Data are represented as fold enrichment versus IgG negative control (12CA5 monoclonal antibody). n = 3; error bars represent SD.
Nek9 depletion reduces viral genome replication.
In order to explicate the mechanism leading to reduced virus growth in Nek9-depleted cells, we analyzed viral genome copies in IMR-90 cells in which Nek9 was knocked down by siRNA and in MEF/3T3-Nek9V5 cells in which Nek9 was overexpressed (Fig. 4). Reduction in cellular Nek9 levels resulted in reduced viral genome copies per cell (Fig. 4A), despite an elevation in viral E2 transcripts and viral E2 DBP (Fig. 2A). The reduction in genome copies was approximately 3-fold, which parallels the reduced virus titers 72 h after infection. Significantly, overexpression of Nek9 led to a 4-fold increase in viral genomes in MEF/3T3-Nek9V5 cells (Fig. 4B). The enhancement in viral genome copies occurred despite a reduction in E2 mRNA levels early in viral infection. Overall, these results demonstrate that Nek9 depletion negatively affects viral genome replication and likely contributes to reduced viral titers.
FIG 4.
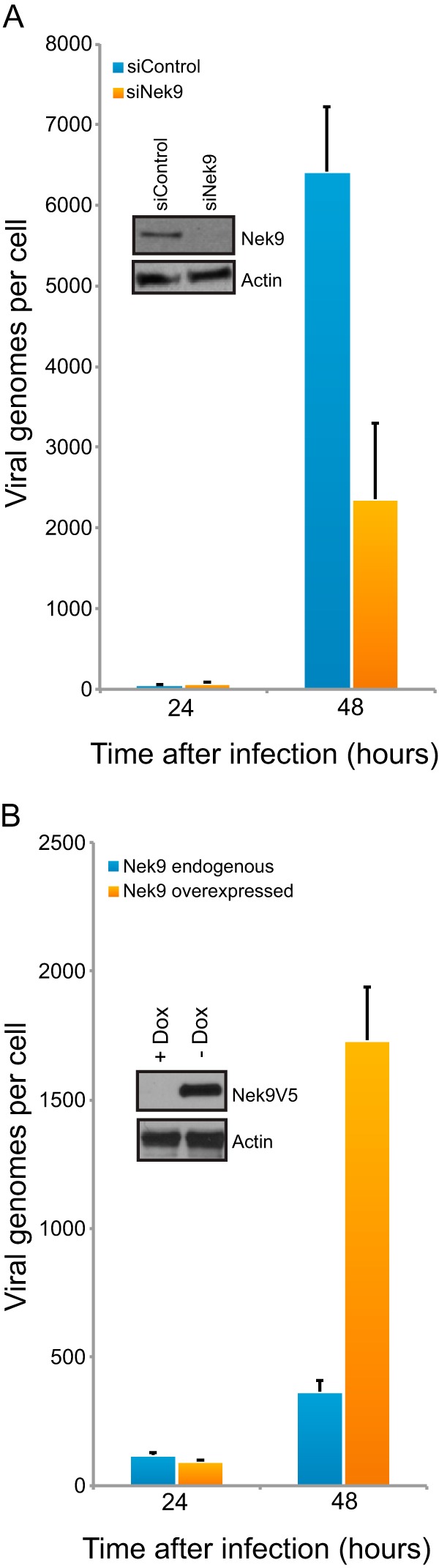
Nek9 affects viral DNA replication. (A) IMR-90 cells that were transfected with siRNAs, either siControl as a control or siNek9 to deplete Nek9, were infected with dl309 24 h after knockdown at an MOI of 5. Viral genomes were quantified by qPCR at the indicated times as described in Materials and Methods. Nek9 depletion was monitored by Western blotting (inset). n = 3; error bars represent SD. (B) MEF/3T3-Nek9V5 cells were induced to express Nek9 by removal of doxycycline as described in Materials and Methods. Cells were infected with dl309 at an MOI of 5 24 h after Nek9 induction. Viral genomes were quantified by qPCR at the indicated times as described in Materials and Methods. Nek9 induction was monitored by Western blotting (inset) (24 h time point shown). n = 3; error bars represent SD.
Nek9 localizes to viral replication centers and colocalizes with the viral E4 orf3 protein.
Since we observed that Nek9 affects viral genome replication and virus growth, we investigated the localization of the protein in infected IMR-90 cells (Fig. 5). To visualize viral replication centers, cells were stained for the viral E2 DBP and cellular Nek9 protein. In uninfected cells, Nek9 showed a predominantly cytoplasmic localization (Fig. 5A, top row), which is consistent with previous reports (6). However, in dl309-infected IMR-90 cells, the subcellular localization of Nek9 was altered, with the protein colocalizing with viral replication centers. We observed two distinct localizations during infection, as shown in Fig. 5, which depended on the extent of viral replication centers. The localization to distinct viral replication centers was the more commonly observed phenotype, occurring in approximately 60% of infected cells based on analysis of three random fields of view. Nonetheless, the localization of Nek9 during infection was altered substantially. To ensure that the nuclear staining observed with the Nek9-specific antibody in the infected cells was caused by Nek9, we stained infected cells in which Nek9 was depleted (Fig. 5B). In these cells, Nek9 was undetectable in the nucleus.
FIG 5.
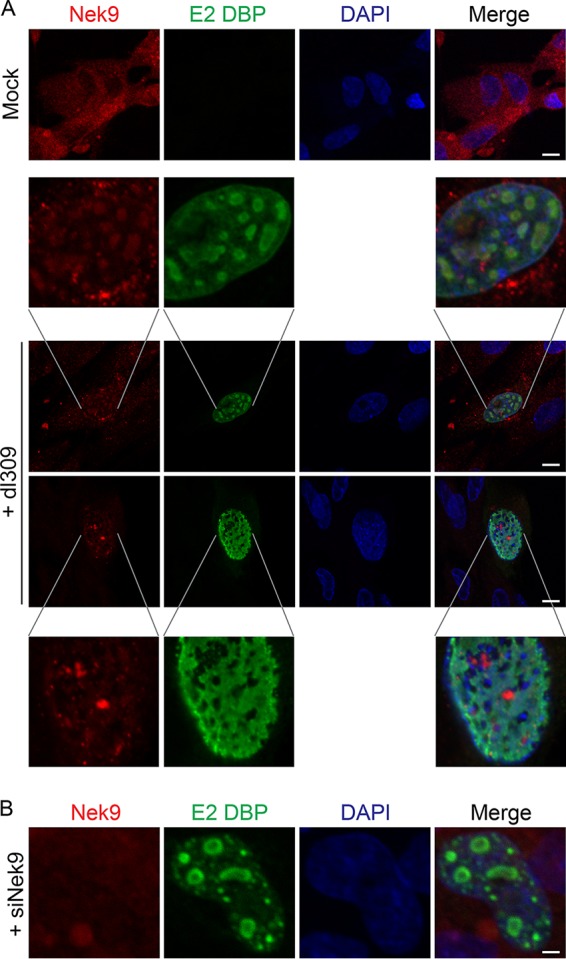
Nek9 localizes to viral replication centers in infected cells. (A) IMR-90 cells were infected with dl309 at an MOI of 10, fixed, and stained for E2 DBP and Nek9 24 h after infection. Uninfected, control IMR-90 cells were also imaged, showing normal distribution of Nek9 (top panel). DAPI was used as a nuclear counterstain. Bar, 7.5 μm. (B) IMR-90 cells were initially transfected with siNek9 to deplete Nek9, infected with dl309 at an MOI of 5, and stained as described for panel A. Bar, 2 μm.
The nuclear localization of Nek9 following infection weakly resembled the previously reported subcellular localization of E4 orf3 (28, 32) and required a productive viral replication, as we did not observe altered Nek9 distribution in mouse fibroblasts (data not shown). To investigate whether E4 orf3 was sufficient to alter the subcellular distribution of Nek9, we expressed myc-tagged E4 orf3 in HT1080 human fibrosarcoma cells and stained them for E4 orf3 using myc-specific antibody and for Nek9 using a rabbit polyclonal antibody (Fig. 6). We used HT1080 cells instead of IMR-90 cells due to cell death following transfection. E4 orf3 was found to localize to the nucleus in four distinct patterns (Fig. 6), likely dependent on the level of E4 orf3 protein. We observed E4 orf3 protein as nuclear foci, large nuclear spots similar to the nucleolus, distinct nuclear tracks, or a combination of tracks and large spots. The focus phenotype was least frequently observed, occurring in <10% of E4 orf3-expressing cells, while the other phenotypes had similar distributions, occurring in about 30% of E4 orf3-expressing cells each, based on analysis of three random fields of view. Regardless of the observed localization of E4 orf3, Nek9 distribution was consistently altered in the presence of E4 orf3, where it was observed either to be localizing with the large nuclear spots and/or nuclear tracks or to be overall more nuclear than cells not transfected with E4 orf3 (Fig. 6). These results suggest that Nek9 is recruited to the nuclear structures formed by E4 orf3. We attempted to determine whether Nek9 and E4 orf3 interact via coimmunoprecipitation assays, but we were unable to detect a stable interaction.
FIG 6.
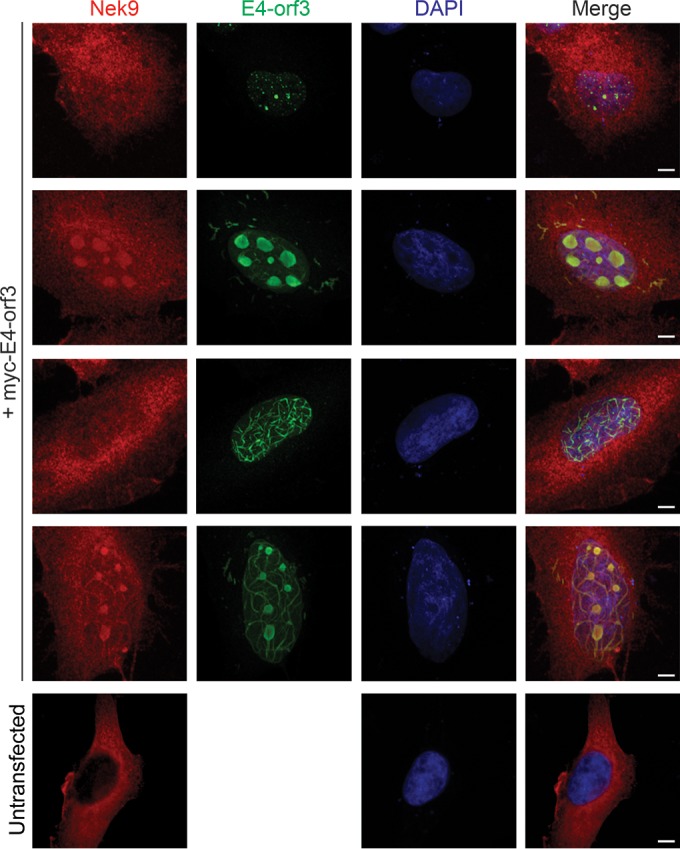
Nek9 colocalizes with E4 orf3 in HT1080 cells. HT1080 cells were transfected with pCAN-myc-E4-orf3 plasmid to express E4-orf3, and 24 h after transfection the cells were fixed and stained for Nek9 and myc. DAPI was used as a nuclear counterstain. Four different morphologies of E4-orf3 and Nek9 are shown. Bar, 5 μm.
In order to determine Nek9 and E4 orf3 localization in the infected cell, we infected IMR-90 cells with dl309, dl1101 (33), or Ad5.inORF3 (34). In dl309-infected IMR-90 cells, Nek9 colocalized with E4 orf3 nuclear tracks and viral replication centers (Fig. 7). To ensure that the observed staining for Nek9 was specific, we also labeled infected cells for Nek9 but did not use primary antibodies for E4 orf3 or E2 DBP (labeled “Not stained for DBP or E4-orf3”). In this case, Nek9 staining was retained. Furthermore, we observed nuclear tracks in DBP-stained cells only when DBP levels were very low (not shown), likely early in infection. Likewise, we have observed E2 DBP-like Nek9 localization in a subset of E4 orf3-labeled cells, likely representing a late point in the infection (not shown). Infection with dl1101, which expresses an E1A mutant with residues 4 to 25 (33) deleted and removes the Nek9-binding site (6), formed E4 orf3 nuclear tracks. However, Nek9 did not localize to these and was instead found to localize to the viral replication centers and the nucleus. Deletion of E4 orf3 prevented the formation of nuclear tracks and resulted in Nek9 localizing only to viral replication centers and the nucleus. Lastly, siRNA-mediated depletion of Nek9 resulted in no nuclear Nek9 staining or colocalization with E4 orf3 (Fig. 7B), similarly to what we observed with E2 DBP (Fig. 5B).
FIG 7.

Nek9 colocalizes with E4-orf3 during viral infection of IMR-90 cells. (A) IMR-90 cells were infected with dl309, Ad5.inORF3, or dl1101 at an MOI of 10, fixed, and stained for E2 DBP, E4-orf3, or Nek9 as indicated 24 h after infection. DAPI was used as a nuclear counterstain. Cells labeled “Not stained for DBP or E4-orf3” were labeled for Nek9 only. Bar, 5 μm. (B) IMR-90 cells in which Nek9 was depleted by siRNA were stained for Nek9 and E4-orf3 24 h after infection with dl309 at an MOI of 10. Bar, 5 μm.
Overall, these results demonstrate that Nek9 is recruited to viral replication centers during productive viral infection independently of E4 orf3 protein and that colocalization to E4 orf3 nuclear tracks appears to depend on the expression of E4 orf3 and, partly at least, on the interaction of E1A and Nek9.
Nek9 depletion in infected cells leads to upregulation of GADD45A.
Colocalization of Nek9 with E4 orf3 suggested that Nek9 may play a role in E4 orf3-mediated silencing of cellular genes that would negatively impact virus growth (28). In particular, E4 orf3 was found to silence p53-responsive genes during viral infection (28). To investigate whether cellular p53-responsive genes were affected by Nek9 knockdown in infected IMR-90 cells, we carried out RT-qPCR on p21, PIG3, MDM2, and GADD45A genes after Nek9 knockdown and infection (Fig. 8A). These genes were previously reported to be affected by E4 orf3 during infection (28) and, considering the observed colocalization of Nek9 and E4 orf3, made for a logical target of investigation. Most of the genes analyzed were not altered in expression after Nek9 depletion and dl309 infection compared to control knockdown and infection. The only exception was GADD45A, which was upregulated approximately 3-fold at 16 h after infection and increased up to 8-fold 48 h after infection. This result was specific to GADD45A, as we did not see alteration in GADD45B expression in the same assay (data not shown). This result suggested that Nek9 may be a negative regulator of transcriptional activation and may be an essential cofactor for virus-mediated transcriptional silencing of the GADD45A gene. To investigate this possibility, we performed ChIP on promoters of the genes analyzed (MDM2, PIG3, p21, and GADD45A), looking at occupancy of Nek9 in uninfected and infected cells (Fig. 8B and C, respectively) and E1A in infected cells (Fig. 8C). For GADD45A promoter regions, we analyzed occupancy at the proximal promoter upstream of the transcriptional start site (p1 in Fig. 8) and the intragenic p53-regulated promoter (p53 in Fig. 8). In uninfected cells, Nek9 was found to occupy predominantly the GADD45A promoters at a low level (approximately 10-fold enrichment over IgG negative control) and was not found on MDM2, PIG3, or p21 promoters. In infected cells, the occupancy was altered, such that Nek9 was found on all promoters. However, occupancy on GADD45Ap1 did not increase and was similar to that observed in uninfected cells. Interestingly, Nek9 and E1A were found to occupy only the p53-regulated GADD45A promoter after infection of IMR-90 cells with dl309 (Fig. 8C). We observed minimal enrichment of E1A at other promoters analyzed, suggesting that E1A and Nek9 target the p53-regulated promoter together in order to subvert p53-mediated activation of GADD45A expression following infection. Together, these results demonstrate that Nek9 and E1A likely collaborate to inhibit p53-mediated activation of the GADD45A gene after infection, since depletion of Nek9 inhibited the ability of HAdV to silence GADD45A expression. Furthermore, these data show that Nek9 is recruited to cellular p53-regulated promoters during viral infection.
FIG 8.

Nek9 affects GADD45A expression during viral infection and is recruited to the intronic, p53-regulated GADD45A promoter. (A) IMR-90 cells that were transfected with siRNAs, either siControl as a control or siNek9 to deplete Nek9, were infected with dl309 24 h after knockdown at an MOI of 5. Cellular gene expression was monitored by RT-qPCR at the indicated time. GAPDH mRNA was used as the normalization control. Note that these are the same samples as in Fig. 2A. n = 4; error bars represent SD. (B) ChIP was carried out on uninfected and subconfluent IMR-90 cells using the Nek9 antibody as described in Materials and Methods. Data are represented as fold enrichment versus IgG negative control (12CA5 monoclonal antibody). Occupancy was analyzed for Nek9 at the MDM2, PIG3, p21, GADD45Ap1 (not bound by p53), and GADD45Ap53 (bound by p53) promoters. n = 3; error bars represent SD. (C) IMR-90 cells were infected with dl309 at an MOI of 5, and ChIP was performed 24 h after infection using E1A or Nek9 antibodies as indicated and as described in Materials and Methods. Data are represented as fold enrichment versus IgG negative control (12CA5 monoclonal antibody). Occupancy was analyzed for Nek9 and E1A at the MDM2, PIG3, p21, GADD45Ap1, and GADD45Ap53 promoters. n = 3; error bars represent SD.
E1A interaction with Nek9 is required for GADD45A transcriptional repression.
To investigate whether suppression of GADD45A transcription by E1A requires an interaction with Nek9, we utilized a mutant virus, dl1101, which removes residues 4 to 25 of E1A (33) and deletes the Nek9 binding site (6). We have also used mutants dl1102 and dl1103, which delete adjacent residues 26 to 35 and 30 to 49 (33, 35), respectively, but retain the Nek9-binding site. Infection of IMR-90 cells with the dl1101 mutant resulted in approximately 2-fold activation of GADD45A expression (Fig. 9A), whereas wild-type virus and the other mutants tested induced repression of GADD45A transcription 24 h after infection, although dl1103 was less efficient at suppression of GADD45A expression than dl309 or dl1102. The activation of GADD45A transcription with dl1101 was comparable to the level of activation of GADD45A transcription observed in Nek9-depleted cells. Similarly, deletion of E4 orf3 impaired the ability of the virus to suppress activation of GADD45A expression following infection.
FIG 9.

Infection-mediated suppression of GADD45A activation relies on Nek9 association with E1A and, to a lesser degree, E4 orf3. (A) IMR-90 cells were infected with dl309, dl1101, dl1102, dl1103, or Ad5.inORF3 at an MOI of 5. Twenty-four hours after infection, total cellular RNA was isolated and expression of GADD45A was analyzed by RT-qPCR as described in Materials and Methods. n = 4; error bars represent SD. (B) IMR-90 cells were infected with dl1101, dl1102, dl1103, or Ad5.inORF3 at an MOI of 5, and ChIP was performed 24 h after infection using E1A or Nek9 antibodies as indicated and as described in Materials and Methods. Data are represented as fold enrichment versus IgG negative control (12CA5 monoclonal antibody). n = 3; error bars represent SD.
In order to elucidate the contribution that E1A binding and E4 orf3 make to the Nek9-mediated suppression of GADD45A expression, we investigated promoter occupancy of E1A and Nek9 in infected IMR-90 cells with mutants that abolish E1A binding to Nek9 (dl1101) or expression of E4 orf3 (Fig. 9B). Nek9 and E1A were recruited to the GADD45A p53-regulated promoter to similar levels after infection with dl1102 or dl1103, both of which express E4 orf3 and bind to Nek9. However, loss of E1A binding to Nek9 prevented recruitment of E1A to the GADD45A promoter but had no effect on Nek9 presence on the promoter. Similarly, loss of E4 orf3 expression resulted in a reduced level of E1A recruitment observed, which was about one-half of that observed with viruses expressing E1A that bound to Nek9 (Fig. 9B and 8C).
Together, these results suggest that an interaction between Nek9 and E1A is necessary for proper transcriptional silencing of GADD45A during viral infection. The results also demonstrate that silencing is dependent on E1A recruitment to the GADD45A promoter and, partly, on the expression of E4 orf3.
DISCUSSION
In the present study, we describe a novel mechanism employed by adenovirus E1A proteins to suppress transcriptional activation of GADD45A. Previous work has shown that HAdV E1A interacts with the NimA-related kinase Nek9, but the consequences of this interaction were unclear (6). Here we show that depletion of Nek9 resulted in a modest reduction in virus growth (Fig. 1A) and enhancement of transcription from the viral E2 transcriptional unit (Fig. 2A). Overexpression of Nek9, on the other hand, resulted in early reduction in E2 transcripts (Fig. 2B). Importantly, we show, for the first time, that Nek9 is targeted to the viral E2 early promoter and, to a lesser degree, to the E3 promoter and a subset of cellular p53-regulated promoters (Fig. 3 and 8), leading to transcriptional repression of some of these genes. Furthermore, we show that Nek9 is a component of viral replication centers and also colocalizes with the viral E4 orf3 protein. Notably, depletion of Nek9 inhibited viral genome replication, whereas overexpression of the protein resulted in enhancement of viral genome replication in infected cells (Fig. 4). Lastly, we show that an interaction between Nek9 and E1A is important for transcriptional suppression of GADD45A and recruitment of E1A to the p53-regulated GADD45A promoter (Fig. 9), which was also partly supported by the viral E4 orf3 protein.
The role of Nek9 in cell division has been well established (9, 12, 13, 36). However, studies have also shown that the protein has other functions. Nek9 is targeted by adenovirus E1A and EBV BGLF2 during viral infection (6, 11), supports cell growth in p53-null cancer cells (14), has recently been shown to play a role in the DNA replication stress response (10), and has been implicated in interphase progression through interaction with the FACT complex (7). These studies provide a picture of a protein with multiple activities. This pleiotropy of function is unsurprising considering the structure of Nek9, which consists of a NimA-like kinase domain and a central RCC1-like motif with largely unknown function (4, 5). Our results suggest that Nek9 interacts with chromatin (Fig. 3, 8, and 9) and can affect transcription likely as a repressor. These results are highly novel and suggest a potential function for the RCC1-like domain within Nek9. It is plausible that this region is what allows Nek9 to be recruited to chromatin, although further experimentation is necessary to verify this hypothesis. This is unsurprising considering that RCC1 itself binds to DNA via a seven-propeller motif that is intact in Nek9 (5, 31).
Nek9 depletion was shown to induce p21 expression in Epstein-Barr virus (EBV)-infected cells, suggesting that, similarly to what we report here, Nek9 may function as a negative regulator of transcription. Unlike what was seen in that study (11), we did not observe upregulation of p21 following adenovirus infection in Nek9-depleted cells (Fig. 8). It is likely that the observed difference is due to the mechanism of replication of these two different viruses. Whereas EBV and other herpesviruses will induce cell cycle arrest at G1/S via activation of genes like p21, adenovirus will drive cells into S phase in order to allow for viral DNA replication to occur.
Suppression of activation of GADD45A via Nek9 is an important contributor to virus growth. This is highlighted by sacrificial reduction in expression of some viral genes as a likely side effect of GADD45A suppression. This is exemplified by the reduced viral titers in spite of enhanced expression of E2 proteins early in infection. Ultimately, it appears that suppression of GADD45A expression is more important to productive viral infection than reduction of certain viral proteins. Likely, the initial deficit in viral proteins will be compensated for later in the infection once viral genomes begin to replicate, not unlike what we have previously observed with DREF (24). Our results suggest that inhibition of GADD45A is an important mechanism that enhances viral DNA replication. Indeed, GADD45A has been shown to play an important role in DNA damage-induced cell cycle arrest (for a review, see reference 37) via its interaction with p21 and Cdc2 (38, 39). Furthermore, GADD45A binds to PCNA and leads to changes in PCNA function, transforming it into a regulator of cell division (40). These roles of GADD45A paint a clear picture as to why E1A would drive repression of GADD45A transcription. High levels of GADD45A may be deleterious to virus growth via inhibition of cell cycle progression essential for viral DNA replication. Furthermore, GADD45A may directly affect viral genome replication by interfering with PCNA function. On the other hand, efficient silencing of GADD45A leads to more efficient replication of the viral genome and likely higher viral titers. However, we cannot exclude a direct role for Nek9 in viral genome replication as suggested by its colocalization to viral replication centers during infection (Fig. 5 and 7). It would be of interest to establish whether Nek9 is a bona fide regulator of GADD45A or whether it is recruited for this role by E1A and whether the kinase or the RCC1-like domains are essential for this function. Observation of Nek9 at the GADD45A promoter in the absence of infection (Fig. 8B) suggests that it may play a role in the regulation of GADD45A expression under normal conditions.
Suppression of p53 activity is of overriding significance to the virus. HAdVs have evolved multiple proteins and mechanisms in order to deal with this powerful suppressor of the cell cycle (41). Among these are the degradation and suppression of p53 through the activities of the E1B 55k and E4 orf6 proteins (42, 43). Binding of E1B 55k to the transactivation domain of p53 inhibits transcriptional activation by p53 (44, 45). Recently, it has also been shown that E4 orf3 can remodel chromatin in order to silence p53-regulated genes, as well as other genes that could interfere with viral replication (28, 32, 46). Here we add to our understanding of the various mechanisms whereby HAdV suppresses p53-responsive genes and show that Nek9 colocalizes with the viral E4 orf3 protein previously implicated in p53 target gene silencing (28). Our assays focused on the differences observed between normal and Nek9-depleted cells, and of the several potential p53-regulated genes only GADD45A stood out as affected. Although this does not exclude the possibility that other p53-regulated genes are affected, it still shows that Nek9 plays a role in regulation of at least a subset of p53-responsive genes and, for the first time, shows that Nek9 functions in transcription. Interestingly, although Nek9 was found to occupy all analyzed p53-regulated promoters after infection, E1A was found to be enriched only at the intronic p53-regulated promoter of GADD45A (Fig. 8C). Since GADD45A was the only gene affected after Nek9 depletion and viral infection, this suggests that E1A is required for silencing of GADD45A during infection and that upon Nek9 knockdown this repression is impaired. Indeed, a mutant of E1A that is unable to bind to Nek9 was not recruited to the GADD45A promoter and did not suppress GADD45A expression during infection. Interestingly, genes not activated following Nek9 knockdown and infection did not have E1A recruited to their promoters, further implicating E1A in Nek9-mediated transcriptional silencing of a subset of p53-regulated genes. Suppression of GADD45A activation also relied, in part, on the E4 orf3 protein. A virus lacking this protein was deficient in GADD45A transcriptional repression and recruitment of E1A to the p53-regulated GADD45A promoter (Fig. 9). It is plausible that the nuclear structures formed by E4 orf3 enhance recruitment of E1A to certain Nek9-occupied promoters. Alternatively, this may be an indirect effect caused by inhibition of E1A by other E4 proteins, which may be elevated in expression in the absence of E4 orf3 (47–49). Since we do not observe silencing of all promoters that are occupied by Nek9, it is likely that a specific set of conditions must be met in order to silence a promoter. What these conditions are remains uncertain.
In conclusion, the present study has identified a link between HAdV E1A, Nek9, and selective suppression of p53-responsive gene GADD45A. We show that depletion of Nek9 in infected cells results in reduced viral growth but paradoxically enhances expression from the viral E2 transcription unit. Importantly, we show that Nek9 is recruited to viral promoters and at least one cellular promoter, the p53-regulated third intron promoter of GADD45A, leading to transcriptional suppression of GADD45A. Notably, our study identified Nek9 as a factor recruited by the virus in order to silence certain p53-regulated genes, highlighting the complexity and importance of p53 control during viral infection. Much remains to be discovered about the mechanisms behind the Nek9-mediated suppression of E2 and GADD45A. What is the role, if any, of the kinase domain or the RCC1-like domain? Does the closely related Nek8 function similarly? Nevertheless, this study has highlighted some of the reasons behind the interaction of Nek9 and E1A, summarized in Fig. 10, and has identified Nek9 as a novel transcriptional regulator.
FIG 10.

Model describing the effects that HAdV has on Nek9 and the mechanism of induction of GADD45A. Infection of cells with HAdV will lead to production of E1A, which will subsequently associate with Nek9, which is either in the cytoplasm or in the nucleus potentially associated with the GADD45A promoter. This complex is then targeted to the GADD45A promoter and induces transcriptional silencing, enhancing virus growth. Note that although Nek9 is shown to associate with the promoter, other factors may mediate DNA binding. Under conditions where Nek9 is not present (such as siRNA-mediated depletion), E1A is unable to repress the GADD45A promoter and GADD45A protein will be made, inhibiting virus growth. The role of E4 orf3 is uncertain, but it may assist in recruitment of E1A to p53-regulated promoters.
ACKNOWLEDGMENTS
We are indebted to Joe Mymryk for countless reagents and invaluable discussions. We are particularly grateful to Patrick Hearing for his generosity in gifting us Ad5.inORF3 and U2OS-Tet-HA-ORF3 cells. We thank Thomas Dobner for the E4-orf3 antibody, Peter Whyte for the MEF/3T3-Nek9V5 cells and Nek9 antibody, and Phil Branton for E2 DBP hybridoma supernatant. We thank Karen Brassinga for access to the Zeiss LSM700 suite. We also thank Jasmine Frost, Olanubi Oladunni, Braden Arbuckle, and Michael Wu for technical assistance. We are grateful to Michael Golding and Sean McKenna for invaluable comments on the manuscript. P.P. also thanks Stanislawa Pelka for invaluable support and assistance.
Funding Statement
This work was supported by grants from the Natural Sciences and Engineering Research Council (NSERC; grant RGPIN/435375-2013), Manitoba Medical Service Foundation (grant MMSF 8-2013-04), and Research Manitoba (MHRC Establishment Grant) to P.P. R.J. was supported by an NSERC Studentship, and S.R. was supported by a University of Manitoba Graduate Student Scholarship. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Morris NR. 1975. Mitotic mutants of Aspergillus nidulans. Genet Res 26:237–254. doi: 10.1017/S0016672300016049. [DOI] [PubMed] [Google Scholar]
- 2.O'Connell MJ, Krien MJ, Hunter T. 2003. Never say never. The NIMA-related protein kinases in mitotic control. Trends Cell Biol 13:221–228. [DOI] [PubMed] [Google Scholar]
- 3.Letwin K, Mizzen L, Motro B, Ben-David Y, Bernstein A, Pawson T. 1992. A mammalian dual specificity protein kinase, Nek1, is related to the NIMA cell cycle regulator and highly expressed in meiotic germ cells. EMBO J 11:3521–3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holland PM, Milne A, Garka K, Johnson RS, Willis C, Sims JE, Rauch CT, Bird TA, Virca GD. 2002. Purification, cloning, and characterization of Nek8, a novel NIMA-related kinase, and its candidate substrate Bicd2. J Biol Chem 277:16229–16240. doi: 10.1074/jbc.M108662200. [DOI] [PubMed] [Google Scholar]
- 5.Roig J, Mikhailov A, Belham C, Avruch J. 2002. Nercc1, a mammalian NIMA-family kinase, binds the Ran GTPase and regulates mitotic progression. Genes Dev 16:1640–1658. doi: 10.1101/gad.972202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pelka P, Scime A, Mandalfino C, Joch M, Abdulla P, Whyte P. 2007. Adenovirus E1A proteins direct subcellular redistribution of Nek9, a NimA-related kinase. J Cell Physiol 212:13–25. doi: 10.1002/jcp.20983. [DOI] [PubMed] [Google Scholar]
- 7.Tan BC, Lee SC. 2004. Nek9, a novel FACT-associated protein, modulates interphase progression. J Biol Chem 279:9321–9330. doi: 10.1074/jbc.M311477200. [DOI] [PubMed] [Google Scholar]
- 8.Regue L, Sdelci S, Bertran MT, Caelles C, Reverter D, Roig J. 2011. DYNLL/LC8 protein controls signal transduction through the Nek9/Nek6 signaling module by regulating Nek6 binding to Nek9. J Biol Chem 286:18118–18129. doi: 10.1074/jbc.M110.209080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bertran MT, Sdelci S, Regue L, Avruch J, Caelles C, Roig J. 2011. Nek9 is a Plk1-activated kinase that controls early centrosome separation through Nek6/7 and Eg5. EMBO J 30:2634–2647. doi: 10.1038/emboj.2011.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith SC, Petrova AV, Madden MZ, Wang H, Pan Y, Warren MD, Hardy CW, Liang D, Liu EA, Robinson MH, Rudra S, Wang J, Ehdaivand S, Torres MA, Wang Y, Yu DS. 2014. A gemcitabine sensitivity screen identifies a role for NEK9 in the replication stress response. Nucleic Acids Res 42:11517–11527. doi: 10.1093/nar/gku840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paladino P, Marcon E, Greenblatt J, Frappier L. 2014. Identification of herpesvirus proteins that contribute to G1/S arrest. J Virol 88:4480–4492. doi: 10.1128/JVI.00059-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sdelci S, Schutz M, Pinyol R, Bertran MT, Regue L, Caelles C, Vernos I, Roig J. 2012. Nek9 phosphorylation of NEDD1/GCP-WD contributes to Plk1 control of gamma-tubulin recruitment to the mitotic centrosome. Curr Biol 22:1516–1523. doi: 10.1016/j.cub.2012.06.027. [DOI] [PubMed] [Google Scholar]
- 13.Kaneta Y, Ullrich A. 2013. NEK9 depletion induces catastrophic mitosis by impairment of mitotic checkpoint control and spindle dynamics. Biochem Biophys Res Commun 442:139–146. doi: 10.1016/j.bbrc.2013.04.105. [DOI] [PubMed] [Google Scholar]
- 14.Kurioka D, Takeshita F, Tsuta K, Sakamoto H, Watanabe S, Matsumoto K, Watanabe M, Nakagama H, Ochiya T, Yokota J, Kohno T, Tsuchiya N. 2014. NEK9-dependent proliferation of cancer cells lacking functional p53. Sci Rep 4:6111. doi: 10.1038/srep06111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed). 2013. Fields virology, 6th ed Wolters Kluwer/Lippincott Williams & Wilkins Health, Philadelphia, PA. [Google Scholar]
- 16.Pelka P, Ablack JN, Fonseca GJ, Yousef AF, Mymryk JS. 2008. Intrinsic structural disorder in adenovirus E1A: a viral molecular hub linking multiple diverse processes. J Virol 82:7252–7263. doi: 10.1128/JVI.00104-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frisch SM, Mymryk JS. 2002. Adenovirus-5 E1A: paradox and paradigm. Nat Rev Mol Cell Biol 3:441–452. doi: 10.1038/nrm827. [DOI] [PubMed] [Google Scholar]
- 18.Harlow E, Franza BR Jr, Schley C. 1985. Monoclonal antibodies specific for adenovirus early region 1A proteins: extensive heterogeneity in early region 1A products. J Virol 55:533–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Niman HL, Houghten RA, Walker LE, Reisfeld RA, Wilson IA, Hogle JM, Lerner RA. 1983. Generation of protein-reactive antibodies by short peptides is an event of high frequency: implications for the structural basis of immune recognition. Proc Natl Acad Sci U S A 80:4949–4953. doi: 10.1073/pnas.80.16.4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Evan GI, Lewis GK, Ramsay G, Bishop JM. 1985. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol Cell Biol 5:3610–3616. doi: 10.1128/MCB.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reich NC, Sarnow P, Duprey E, Levine AJ. 1983. Monoclonal antibodies which recognize native and denatured forms of the adenovirus DNA-binding protein. Virology 128:480–484. doi: 10.1016/0042-6822(83)90274-X. [DOI] [PubMed] [Google Scholar]
- 22.Nevels M, Tauber B, Kremmer E, Spruss T, Wolf H, Dobner T. 1999. Transforming potential of the adenovirus type 5 E4orf3 protein. J Virol 73:1591–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pelka P, Ablack JN, Torchia J, Turnell AS, Grand RJ, Mymryk JS. 2009. Transcriptional control by adenovirus E1A conserved region 3 via p300/CBP. Nucleic Acids Res 37:1095–1106. doi: 10.1093/nar/gkn1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Radko S, Koleva M, James KM, Jung R, Mymryk JS, Pelka P. 2014. Adenovirus E1A targets the DREF nuclear factor to regulate virus gene expression, DNA replication, and growth. J Virol 88:13469–13481. doi: 10.1128/JVI.02538-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaeser MD, Iggo RD. 2004. Promoter-specific p53-dependent histone acetylation following DNA damage. Oncogene 23:4007–4013. doi: 10.1038/sj.onc.1207536. [DOI] [PubMed] [Google Scholar]
- 26.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Radko S, Jung R, Olanubi O, Pelka P. 2015. Effects of adenovirus type 5 E1A isoforms on viral replication in arrested human cells. PLoS One 10:e0140124. doi: 10.1371/journal.pone.0140124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soria C, Estermann FE, Espantman KC, O'Shea CC. 2010. Heterochromatin silencing of p53 target genes by a small viral protein. Nature 466:1076–1081. doi: 10.1038/nature09307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones N, Shenk T. 1979. Isolation of adenovirus type 5 host range deletion mutants defective for transformation of rat embryo cells. Cell 17:683–689. doi: 10.1016/0092-8674(79)90275-7. [DOI] [PubMed] [Google Scholar]
- 30.Ohtsubo M, Okazaki H, Nishimoto T. 1989. The RCC1 protein, a regulator for the onset of chromosome condensation locates in the nucleus and binds to DNA. J Cell Biol 109:1389–1397. doi: 10.1083/jcb.109.4.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seino H, Hisamoto N, Uzawa S, Sekiguchi T, Nishimoto T. 1992. DNA-binding domain of RCC1 protein is not essential for coupling mitosis with DNA replication. J Cell Sci 102(Part 3):393–400. [DOI] [PubMed] [Google Scholar]
- 32.Ou HD, Kwiatkowski W, Deerinck TJ, Noske A, Blain KY, Land HS, Soria C, Powers CJ, May AP, Shu X, Tsien RY, Fitzpatrick JA, Long JA, Ellisman MH, Choe S, O'Shea CC. 2012. A structural basis for the assembly and functions of a viral polymer that inactivates multiple tumor suppressors. Cell 151:304–319. doi: 10.1016/j.cell.2012.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Egan C, Jelsma TN, Howe JA, Bayley ST, Ferguson B, Branton PE. 1988. Mapping of cellular protein-binding sites on the products of early-region 1A of human adenovirus type 5. Mol Cell Biol 8:3955–3959. doi: 10.1128/MCB.8.9.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang MM, Hearing P. 1989. Adenovirus early region 4 encodes two gene products with redundant effects in lytic infection. J Virol 63:2605–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jelsma TN, Howe JA, Evelegh CM, Cunniff NF, Skiadopoulos MH, Floroff MR, Denman JE, Bayley ST. 1988. Use of deletion and point mutants spanning the coding region of the adenovirus 5 E1A gene to define a domain that is essential for transcriptional activation. Virology 163:494–502. doi: 10.1016/0042-6822(88)90290-5. [DOI] [PubMed] [Google Scholar]
- 36.Belham C, Roig J, Caldwell JA, Aoyama Y, Kemp BE, Comb M, Avruch J. 2003. A mitotic cascade of NIMA family kinases. Nercc1/Nek9 activates the Nek6 and Nek7 kinases. J Biol Chem 278:34897–34909. [DOI] [PubMed] [Google Scholar]
- 37.Yang Z, Song L, Huang C. 2009. Gadd45 proteins as critical signal transducers linking NF-kappaB to MAPK cascades. Curr Cancer Drug Targets 9:915–930. doi: 10.2174/156800909790192383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jin S, Antinore MJ, Lung FD, Dong X, Zhao H, Fan F, Colchagie AB, Blanck P, Roller PP, Fornace AJ Jr, Zhan Q. 2000. The GADD45 inhibition of Cdc2 kinase correlates with GADD45-mediated growth suppression. J Biol Chem 275:16602–16608. doi: 10.1074/jbc.M000284200. [DOI] [PubMed] [Google Scholar]
- 39.Zhao H, Jin S, Antinore MJ, Lung FD, Fan F, Blanck P, Roller P, Fornace AJ Jr, Zhan Q. 2000. The central region of Gadd45 is required for its interaction with p21/WAF1. Exp Cell Res 258:92–100. doi: 10.1006/excr.2000.4906. [DOI] [PubMed] [Google Scholar]
- 40.Paunesku T, Mittal S, Protic M, Oryhon J, Korolev SV, Joachimiak A, Woloschak GE. 2001. Proliferating cell nuclear antigen (PCNA): ringmaster of the genome. Int J Radiat Biol 77:1007–1021. doi: 10.1080/09553000110069335. [DOI] [PubMed] [Google Scholar]
- 41.Berk AJ. 2005. Recent lessons in gene expression, cell cycle control, and cell biology from adenovirus. Oncogene 24:7673–7685. doi: 10.1038/sj.onc.1209040. [DOI] [PubMed] [Google Scholar]
- 42.Querido E, Blanchette P, Yan Q, Kamura T, Morrison M, Boivin D, Kaelin WG, Conaway RC, Conaway JW, Branton PE. 2001. Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes Dev 15:3104–3117. doi: 10.1101/gad.926401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harada JN, Shevchenko A, Shevchenko A, Pallas DC, Berk AJ. 2002. Analysis of the adenovirus E1B-55K-anchored proteome reveals its link to ubiquitination machinery. J Virol 76:9194–9206. doi: 10.1128/JVI.76.18.9194-9206.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yew PR, Berk AJ. 1992. Inhibition of p53 transactivation required for transformation by adenovirus early 1B protein. Nature 357:82–85. doi: 10.1038/357082a0. [DOI] [PubMed] [Google Scholar]
- 45.Yew PR, Liu X, Berk AJ. 1994. Adenovirus E1B oncoprotein tethers a transcriptional repression domain to p53. Genes Dev 8:190–202. doi: 10.1101/gad.8.2.190. [DOI] [PubMed] [Google Scholar]
- 46.DeHart CJ, Perlman DH, Flint SJ. 2015. Impact of the adenoviral E4 Orf3 protein on the activity and posttranslational modification of p53. J Virol 89:3209–3220. doi: 10.1128/JVI.03072-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bondesson M, Ohman K, Manervik M, Fan S, Akusjarvi G. 1996. Adenovirus E4 open reading frame 4 protein autoregulates E4 transcription by inhibiting E1A transactivation of the E4 promoter. J Virol 70:3844–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mannervik M, Fan S, Strom AC, Helin K, Akusjarvi G. 1999. Adenovirus E4 open reading frame 4-induced dephosphorylation inhibits E1A activation of the E2 promoter and E2F-1-mediated transactivation independently of the retinoblastoma tumor suppressor protein. Virology 256:313–321. doi: 10.1006/viro.1999.9663. [DOI] [PubMed] [Google Scholar]
- 49.Leppard KN. 1997. E4 gene function in adenovirus, adenovirus vector and adeno-associated virus infections. J Gen Virol 78(Part 9):2131–2138. [DOI] [PubMed] [Google Scholar]