

ABSTRACT
We recently showed that the interaction between Kaposi's sarcoma-associated herpesvirus (KSHV) tegument proteins ORF33 and ORF45 is crucial for progeny virion production, but the exact functions of KSHV ORF33 during lytic replication were unknown (J. Gillen, W. Li, Q. Liang, D. Avey, J. Wu, F. Wu, J. Myoung, and F. Zhu, J Virol 89:4918–4931, 2015, http://dx.doi.org/10.1128/JVI.02925-14). Therefore, here we investigated the relationship between ORF33 and ORF38, whose counterparts in both alpha- and betaherpesviruses interact with each other. Using specific monoclonal antibodies, we found that both proteins are expressed during the late lytic cycle with similar kinetics and that both are present in mature virions as components of the tegument. Furthermore, we confirmed that ORF33 interacts with ORF38. Interestingly, we observed that ORF33 tightly associates with the capsid, whereas ORF38 associates with the envelope. We generated ORF33-null, ORF38-null, and double-null mutants and found that these mutants apparently have identical phenotypes: the mutations caused no apparent effect on viral gene expression but reduced the yield of progeny virion by about 10-fold. The progeny virions also lack certain virion component proteins, including ORF45. During viral lytic replication, the virions associate with cytoplasmic vesicles. We also observed that ORF38 associates with the membranes of vesicles and colocalizes with the Golgi membrane or early endosome membrane. Further analyses of ORF33/ORF38 mutants revealed the reduced production of virion-containing vesicles, suggesting that ORF33 and ORF38 are involved in the transport of newly assembled viral particles into cytoplasmic vesicles, a process important for viral maturation and egress.
IMPORTANCE Herpesvirus assembly is an essential step in virus propagation that leads to the generation of progeny virions. It is a complicated process that depends on the delicate regulation of interactions among virion proteins. We previously revealed an essential role of ORF45-ORF33 binding for virus assembly. Here, we report that ORF33 and its binding partner, ORF38, are required for infectious virus production due to their important role in the tegumentation process. Moreover, we found that both ORF33 and ORF38 are involved in the transportation of virions through vesicles during maturation and egress. Our results provide new insights into the important roles of ORF33 and ORF38 during viral assembly, a process critical for virus propagation that is intimately linked to KSHV pathobiology.
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV) is etiologically associated with Kaposi's sarcoma (KS) as well as primary effusion lymphoma and multicentric Castleman's disease (1–3). As a herpesvirus, KSHV alternates between two life cycles, latency and lytic replication. Latency is a dormant state during which only a few viral genes are expressed, whereas the lytic cycle leads to the expression of the full panel of viral genes, ultimately resulting in the production of progeny virions (4, 5).
Herpesvirus virions consist of four morphologically distinct structures: genome, capsid, tegument, and envelope. Among these, the tegument is the most complex in composition. While capsid proteins are well conserved among all herpesviruses, some tegument proteins are unique to each subfamily (6). Tegument proteins can have structural roles in the assembly of mature virions and/or regulatory roles important for establishing latency during primary infection (7–10). Our laboratory has been interested in ORF45, a multifunctional tegument protein that is unique to gammaherpesviruses. Although ORF45 is conserved in gammaherpesviruses, the overall sequence homology is low, except for a few short discrete regions. Among these, the extreme C terminus has the highest homology, implying an important functional role of this region. This was first established when it was discovered that deleting the conserved C terminus of mouse hepatitis virus 68 (MHV-68) ORF45 abolished the production of progeny virions, but the exact role of this region remained unknown (11). We recently found that the C terminus of KSHV ORF45 binds to and thereby stabilizes ORF33. This interaction is critical for the accumulation of ORF33 protein in cells and the production of progeny virions (12).
Unlike ORF45, ORF33 is conserved among all herpesviruses (13–15). Its homologues, herpes simplex virus 1 (HSV-1) UL16, Epstein-Barr virus (EBV) BGLF2, and human cytomegalovirus (HCMV) UL94, all are present in the tegument layer of mature virions (13, 16–23), but the exact roles of the ORF33 homologues in herpesviral replication remain elusive. Although the deletion of UL16 reduces the viral yield of HSV-1 (alphaherpesvirus) only moderately (24), the deletion of UL94 abolishes progeny virion production of HCMV (betaherpesvirus) (15, 25). In gammaherpesviruses, ORF33 of MHV-68 initially was found to be essential for viral replication by genome-wide signature-tagged transposon mutagenesis studies (26). Guo et al. further showed that ORF33-null mutation does not affect viral DNA replication, viral gene expression, or capsid assembly but abolishes the release of infectious virions, resulting in the accumulation of partially tegumented viral particles in the cytoplasm (13).
Because ORF45-null mutation or deletion of the C-terminal 19 amino acids (aa) abolished the accumulation of ORF33 protein in KSHV-infected cells (12), the extent to which the phenotypes of these ORF45 mutants can be attributed to the loss of ORF33 protein was unclear. Furthermore, although the exact roles of ORF33 homologues have remained unclear, a unifying feature seems to be their interaction with ORF38, another herpesviral core protein that is also present in mature virions and appears to be required for their optimal production (27–31). Therefore, we characterized ORF33 and ORF38 during KSHV lytic replication. Using our newly developed monoclonal antibodies, we found that both proteins are expressed late during the lytic cycle with similar kinetics. We also confirmed that both proteins are present in mature virions as tegument proteins. Interestingly, we found that upon treating virions with detergent, ORF38 is easily solubilized, whereas ORF33 remains stably associated with the capsid. Furthermore, we confirmed that ORF33 interacts with ORF38. We generated ORF33-null, ORF38-null, and double null mutants and found that these mutants appear to have identical phenotypes: the mutations caused no apparent effect on viral gene expression but reduced the yield of progeny virions by about 10-fold. The progeny virions apparently lack certain virion component proteins. We also observed that ORF38 associates with cytoplasmic membranes and colocalizes with the Golgi membrane or early endosome membrane. Further analyses revealed a reduced association of virion proteins with vesicles in cells, suggesting that ORF33 and ORF38 are involved in the transportation of newly assembled viral particles through cytoplasmic vesicles, a process important for viral maturation and egress.
MATERIALS AND METHODS
Cell culture and transfection.
HEK293 and HEK293T cells were cultured under 5% CO2 at 37°C in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and antibiotics. iSLK-puro cells carrying wild-type or mutant bacterial artificial chromosome 16 (BAC16) were cultured in DMEM containing 10% FBS, 450 μg/ml G418, 1 μg/ml puromycin, and 500 μg/ml hygromycin. The transfection of BAC16 into the iSLK-puro cells was performed in 24-well plates with Effectene transfection reagent (Qiagen). Transfected cells were selected with 500 μg/ml hygromycin (Invitrogen, Carlsbad, CA) to establish stable cells as previously described (32).
Antibodies and reagents.
Anti-ORF38 and anti-ORF33 monoclonal antibodies were generated by the FSU hybridoma facility. The detailed procedures of antibody production were described in our previous study (12). All other antibodies used in this study were described previously (32, 33). Doxycycline, sodium butyrate, isopropyl β-d-1-thiogalactopyranoside (IPTG), and N-ethylmaleimide (NEM) were purchased from Sigma-Aldrich (St. Louis, MO). MBP-ORF33 proteins were obtained from the National Cancer Institute.
Expression and preparation of GST fusion proteins.
Glutathione S-transferase (GST)-tagged protein purification was described previously (33). Briefly, a GST-tagged ORF38 expression construct was generated by cloning the ORF38 coding sequence into the pGEX-5X-1 vector. The resultant plasmid was transformed into Escherichia coli BL21-CodonPlus-RP competent cells. After induction with 0.5 mM IPTG at 18°C overnight, the bacteria were harvested by centrifugation. The pellets were resuspended, sonicated in lysis buffer (50 mM Tris-HCl, pH 7.4, 500 mM NaCl, 10% glycerol, 1% Triton X-100, 0.2 mM phenylmethylsulfonyl fluoride [PMSF]), and centrifuged at 20,000 × g for 10 min. The supernatant was mixed with glutathione Sepharose (Sigma) and washed extensively with lysis buffer. Bound proteins were eluted with 50 mM reduced glutathione in 50 mM Tris-HCl, pH 8.0, and the eluates were dialyzed against buffer (20 mM Tris-HCl, pH 7.4, 500 mM NaCl, 10% glycerol).
GST pulldown assays.
GST pulldown assays were performed as described previously (33). Briefly, GST-ORF38- or GST-bound glutathione agarose beads were incubated with lysates of HEK293T cells transiently expressing Flag-ORF33 for 3 h at 4°C. The beads then were washed three times each with whole-cell lysis buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% NP-40, 1 mM sodium orthovanadate [Na3VO4], 40 mM-glycerophosphate, 1 mM sodium fluoride, 10% glycerol, 5 mM EDTA, 5 μg/ml of aprotinin, 5 μg/ml of leupeptin, 5 mM benzamidine, and 1 mM PMSF) and 1× phosphate-buffered saline (PBS), mixed with an equal volume of 2× Laemmli loading buffer, and boiled for 10 min. The input/eluates were resolved by SDS-PAGE and analyzed by Coomassie staining and/or Western blotting. For NEM crystalline treatment, HEK293T cells transiently expressing Flag-ORF33 were pretreated with 20 mM NEM in PBS for 30 min before cell lysis for GST pulldown. For hydrogen peroxide (H2O2) treatment, the cells were pretreated with 500 μM H2O2 in DMEM for 30 min before cell lysis for GST pulldown. For dithiothreitol (DTT) and N-acetylcysteine amide (NAC) treatment, the cells were pretreated with 10 mM DTT or 10 mM NAC in DMEM for 3 h before cell lysis for GST pulldown.
Western blot analysis.
Proteins were resolved by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were stained with Ponceau S as described previously (34), blocked in 5% dried milk in 1× PBS plus 0.2% Tween 20, and then incubated with diluted primary antibodies for 2 h at room temperature or overnight at 4°C. Anti-rabbit and anti-mouse IgG antibodies conjugated to horseradish peroxidase (Pierce) were used as the secondary antibodies. SuperSignal chemiluminescence reagents (Pierce) were used for detection.
Genetic manipulation of KSHV BAC genome.
The mutagenesis of BAC16 was described previously (32). In brief, the Kan/I-SceI cassettes were amplified from plasmid pEPKan-S by PCR with the following primers: ΔORF33-S (5′-TATGGCTAGCCGGAGGCGCAAACTTCGGAATTTCCTAAACTAGTAATGCATATGGACTGTTAACAGGATGACGACGATAAG TAGGG-3′), ΔORF33-A (5′-ATATGGTCCCCTGACATTGGGTTAACAGTCCATATGCATTACTAGTTTAGGAAATTCCGAAGTTGCCAGTGTTACAACCAATTA ACC-3′), ΔORF38-S (5′-TTTGGTCAAGAAGTGCGAAGGACACCTTTCCATATATCAAGTGGGATTTCTCCTATCTATCAGGATGACGACGATAAGTAGGG-3′), and ΔORF38-A (5′-GGCTGTGAGGGACGTTTGCAGATAGATAGGAGAAATCCCACTTGATATATGGAAAGGTGTCGC CAGTGTTACAACCAATTAACC-3′). The purified PCR fragment was electroporated into BAC16-containing E. coli GS1783 cells that had been induced at 42°C for 15 min. The recombinant clones were selected at 32°C on LB plates containing 34 μg/ml chloramphenicol and 50 μg/ml kanamycin and then characterized by RFLP (restriction fragment length polymorphism). Positive clones were induced at 42°C again and plated on LB plates containing 1% l-arabinose for secondary recombination. We then picked replicas of the clones from l-arabinose plates onto plates with 34 μg/ml chloramphenicol alone or plates with 34 μg/ml chloramphenicol plus 50 μg/ml kanamycin. The kanamycin-sensitive clones were second-recombinant clones and confirmed by RFLP and sequencing.
To make a revertant mutant, we replaced ORF33 and ORF38 mutants with wild-type sequence by using a homologous recombination strategy similar to that described above. The Kan/I-SceI cassettes were amplified from plasmid pEPKan-S by PCR with the following primers: ORF33Rev-S (5′-TATGGCTAGCCGGAGGCGCAAACTTCGGAATTTCCTAAACAAGGAATGCATATGGACTGTTAACAGGATGACGACGATAAGTAGGG-3′), ORF33Rev-A (5′-ATATGGTCCCCTGACATTGGGTTAACAGTCCATATGCATTCCTTGTTTAGGAAATTCCGAAGTTGCCAGTGTTACAACCAATTAACC-3′), ORF38Rev-S (5′-TTTGGTCAAGAAGTGCGAAGGACACCTTTCCATATATCAAATGGGATTTCTCCTATCTATCAGGAT GAC GACGATAAGTAGGG-3′), and ORF38Rev-A (5′-GGCTGTGAGGGACGTTTGCAGATAGATAGGAGAAATCCCATTTGATATATGGAAAGGTGTCGCCAGTGTTACAACCAATTA ACC-3′).
Membrane flotation assay.
The membrane flotation assay was performed as described previously (14). Briefly, HEK293T cells transiently expressing Flag-ORF33 and/or ORF38 or iSLK.BAC16 cells induced by doxycycline and sodium butyrate were harvested, washed twice with PBS, and then resuspended in 300 ml of hypotonic lysis buffer (10 mM Tris-HCl, pH 7.4, 0.2 mM MgCl2) on ice for 20 min. Swollen cells were lysed on ice by 50 strokes with a Dounce homogenizer and then centrifuged at 1,500 × g for 5 min to remove unbroken cells and nuclei. The supernatants were mixed with 1.2 ml of 65% (wt/wt) sucrose, placed at the bottom of a Beckman SW55Ti tube, and sequentially overlaid with 1.75 ml of 45% and 0.35 ml of 25% sucrose. The sucrose solutions used were made in TNE buffer (100 mM NaCl, 10 mM Tris-HCl pH 7.4, 1 mM EDTA). The samples were spun for 20 h at 200,000 × g at 4°C in a Beckman ultracentrifuge, and six equal-volume fractions were collected from the top. Samples were separated by 12% SDS-PAGE, followed by Western blot analysis using the indicated antibodies.
Indirect immunofluorescence staining.
HEK293T cells cultured on coverslips in 12-well plates were cotransfected with plasmids expressing ORF38 or different red fluorescent protein (RFP)-tagged endomembrane markers, including endoplasmic reticulum (Sec61), Golgi (eNOS 1-33), early endosome (Rab5), late endosome (Rab7), and lysosome (Lamp1) markers, using Fugene 6 (Promega). After 24 h, the transfected cells were fixed with 3% paraformaldehyde in PBS for 10 min at room temperature, permeabilized with 0.2% Triton X-100 for 15 min on ice, and then incubated with ORF38 antibodies for 1 h at room temperature. After four washes with PBS with 0.1% Tween 20, the cells were incubated with Alexa 488-labeled secondary antibodies (Invitrogen) for 1 h, stained with 4′,6-diamidino-2-phenylindole (DAPI) (1 μg/ml) for 10 min, and mounted in 50% glycerol solution. The images were acquired with a Nikon confocal microscope A1R using a 63× objective. The colocalization was quantified with Pearson's correlation coefficient using ImageJ software.
Real-time quantitative PCR (qPCR) analysis of virion DNA.
The virion DNAs were prepared as previously described (32). The medium from induced iSLK.BAC16 cells was collected, centrifuged, and passed through a 1-μm filter to clear cell debris. The cleared supernatants (200 μl) were incubated with 10 U of Turbo DNase (Ambion, Austin, TX) at 37°C for 1 h to degrade extravirion DNAs. The reaction was stopped by the addition of EDTA, followed by heat inactivation at 70°C. Twenty microliters of proteinase K solution and 200 μl of buffer AL from a DNeasy kit (Qiagen, Valencia, CA) then were added. The mixture was kept at 70°C for 15 min and then extracted with phenol-chloroform. The DNA was precipitated by the addition of 2 volumes of isopropanol with glycogen as a carrier, and the DNA pellet was dissolved in 40 μl of Tris-EDTA buffer. Two microliters of DNA was used in SYBR green real-time PCRs using KSHV-specific primers, ORF73-LCN (5′-CGCGAATACCGCTATGTACTCA-3′) and OFF73-LCC (5′-GGAACGCGCCTCATACGA-3′), with a Bio-Rad C1000 thermal cycler and CFX96 real-time detection system. Viral DNA copy numbers were calculated with external standards of known concentrations of serially diluted BAC16 DNA ranging from 1 to 107 genome copies per reaction.
Virus purification and infection.
iSLK cells carrying wild-type or mutant BAC16 were induced for 5 days, and then medium was collected and centrifuged to remove cell debris. After passing through a 1-μm filter, the clarified medium was concentrated using ultrafiltration disks (300-kDa nominal molecular mass limit; PBMK06210; Millipore) and a Millipore Amicon stirred cell (model 8200) according to the manufacturer's instructions. The concentrated virus was further purified at 10,0000 × g for 1 h on a 12% to 32% step dextran gradient according to a previous study (35). The virion fractions were collected and then concentrated to the desired volume by Amicon ultra centrifugal filters (UFC510008) and stored at −80°C. The viral genome copy number of stock virus then was quantified by real-time quantitative PCR. Infection was carried out as previously described (32, 33). Briefly, HEK293 cells plated in 24-well plates were incubated with 2-fold serial dilutions of concentrated virus plus Polybrene (4 μg/ml) and spun at 800 × g for 1 h at room temperature. The plates then were incubated at 37°C for another 2 h, and the inocula were removed and replaced with fresh medium with 5% FBS. The next day, medium was replaced with fresh medium containing 1% FBS. Green fluorescent protein (GFP) expression was assessed 48 h after infection using an inverted fluorescence microscope or flow cytometry. For flow cytometry analysis (by fluorescence-activated cell sorting [FACS]), cells were washed twice and resuspended in PBS, followed by analysis by a BD FACSCanto analyzer to measure GFP expression.
Sucrose gradient.
A sucrose gradient was performed according to a previously study (36). Briefly, 20% to 60% linear sucrose gradients were made with the Biocomp gradient master. Equal amounts of wild-type and mutant virions were added to the top of each gradient, which then were centrifuged at 10,0000 × g for 1 h with an SW41 Beckman rotor. Twenty continuous fractions were collected, and the level of ORF26 was detected in each fraction by Western blotting.
RESULTS
Both ORF33 and ORF38 are expressed during the late phase of lytic replication and are present in the tegument layer of virions.
To characterize ORF33 and ORF38 proteins, we generated monoclonal antibodies against them. When purified virions or lysates of induced but not uninduced iSLK.BAC16 cells were analyzed by Western blotting, the anti-ORF33 antibody detected a protein of ∼38 kDa (Fig. 1A), confirming that ORF33 is a virion component as reported previously (21). Similarly, the anti-ORF38 antibody detected a protein of ∼10 kDa in the lysates of induced cells or virions (Fig. 1B), suggesting that ORF38 is also a virion protein. Their apparent sizes match the expected sizes of ORF33 (334 aa) and ORF38 (61 aa). Using these antibodies, we next examined the expression kinetics of ORF33 and ORF38 in iSLK.BAC16 cells. Unlike the immediate-early gene products RTA and ORF59/PF8, which were readily detectable 1 day postinduction (dpi), ORF33 and ORF38 were not detectable until 2 dpi, exhibiting kinetics similar to that of the late gene and capsid protein ORF26 (Fig. 1C). Furthermore, expression of ORF33 and ORF38 was sensitive to treatment with phosphonoacetic acid (PAA), an inhibitor of viral DNA replication, indicating that both ORF33 and ORF38 are true late proteins (Fig. 1C).
FIG 1.

Characterization of KSHV ORF33 and ORF38. (A and B) Generation of anti-ORF33 and anti-ORF38 monoclonal antibodies. iSLK carrying BAC16 was left untreated or was induced with doxycycline and sodium butyrate for 3 days. The uninduced (UI) or induced (IN) cell lysates, as well as extracellular virions, were resolved by SDS-PAGE and then analyzed by Western blotting using anti-ORF33 (A) or anti-ORF38 (B) antibody. The black arrows indicate ORF33 (A) or ORF38 (B). IB, immunoblot. (C) ORF33 and ORF38 express with true late kinetics. iSLK cells carrying BAC16 were induced with doxycycline and sodium butyrate in the presence or absence of phosphonoacetic acid (PAA), and the cell lysates were harvested at the indicated day postinduction (dpi). Total proteins were resolved by SDS-PAGE and detected with the indicated antibodies. M, molecular size marker. (D) Differential sensitivity of ORF33 and ORF38 to trypsin digestion. The extracellular virions were concentrated and purified as outlined in Materials and Methods. The purified virions were treated with Triton X-100, trypsin, or both. Virion envelope, tegument, and capsid proteins were analyzed by Western blotting with the indicated antibodies. (E) Sensitivities of ORF33 and ORF38 to detergent treatment. Purified virions were treated with increasing concentrations of NP-40, after which the virions were pelleted down through a 25% sucrose cushion. The virion proteins in the supernatant (s) or pellet (p) were detected with the indicated antibodies. (F and G) The molecular numbers of ORF33 and ORF38 are comparable in the viral particle. Purified virions (108) and serially diluted GST-ORF38 and MBP-ORF33 proteins were resolved by SDS-PAGE and then analyzed by Western blotting with the indicated antibodies. The blot signals were plotted with ImageJ, and ORF33 and ORF38 copy numbers were determined by using GST-ORF38 or MBP-ORF33 as standards. Values stated in the text are representative of technical duplicates.
To confirm that virion-associated ORF33 and ORF38 reside interior to the virion envelope, we treated the purified virions with trypsin in the presence and absence of the detergent Triton X-100. In the absence of Triton X-100, the envelopment protein K8.1 was readily degraded by trypsin, while ORF33, ORF38, and ORF45, as well as the capsid protein ORF26, were not, suggesting that both ORF33 and ORF38 are protected from trypsin digestion by an intact viral envelope (Fig. 1D). When the viral envelope was dissolved by Triton X-100, ORF38 and ORF45 were degraded by trypsin, while ORF33 remained partially resistant to trypsin digestion (Fig. 1D). These results suggested that both ORF33 and ORF38 proteins reside inside the virion envelope and that, compared to the other tegument proteins assessed, ORF33 is more stably associated with the capsid. To further confirm this observation, we treated the purified virions with increasing concentrations of the detergent NP-40 and then separated the soluble proteins (in the supernatant; s) from the capsid (in the pellet; p) by ultracentrifugation. As shown in Fig. 1E, ORF33 behaved like capsid proteins ORF26 and ORF65, remaining insoluble at the highest concentration of NP-40. These results provide further evidence that ORF33 tightly associates with the capsid. Interestingly, ORF38 behaved like the envelope protein K8.1 and became soluble even at the lowest concentration of NP-40, suggesting that it associates with the envelope. ORF45 was partially soluble under these conditions, as previously reported (21). We next attempted to determine the abundance and relative ratios of these proteins in extracellular KSHV virions. Using known concentrations of GST-ORF38 and MBP-ORF33 purified proteins as standards, we estimated that there are ∼2,000 ± 565 copies of ORF33 and ∼1,659 ± 465 copies of ORF38 per viral particle based on viral genome copy number (Fig. 1F and G). There results suggested that ORF33 and ORF38 are abundant and present in a relatively equimolar ratio in extracellular virions.
The interactions between ORF33 and ORF38.
Because homologues of ORF33 and ORF38 in alpha- and betaherpesviruses are known to interact with each other, we next assessed the extent of interaction between ORF33 and ORF38. Indeed, ORF33 was efficiently pulled down by GST-ORF38 but not GST alone (Fig. 2A). To determine the specificity of ORF33-ORF38 binding, we tested whether ORF33 interacts with ORF38 homologues of other gammaherpesviruses and vice versa. Despite the fact that ORF33 and ORF38 are highly conserved among gammaherpesviruses, we found that KSHV ORF38 and Ross River virus (RRV) ORF38 interacted with their own version of ORF33 but not with other ORF33 homologues (Fig. 2B). Interestingly, EBV BBLF1 interacted with both KSHV ORF33 and its own ORF33 (Fig. 2B). None of these ORF38 homologues interacted with ORF33 homologues of alpha- or betaherpesviruses under the same conditions (data not shown). These results suggest that the interactions between ORF38 and ORF33 are highly specific and conserved among all three subfamilies of herpesviruses.
FIG 2.

Interaction between ORF33 and ORF38. (A) GST-ORF38 interacts with ORF33. GST or GST-ORF38 was used to pull down cell lysates expressing Flag-ORF33, and the input/eluates were probed with anti-FLAG antibody. (B) Interactions between ORF33 and ORF38 homologues. GST-ORF38 proteins were used to pull down (PD) lysates of HEK293T cells expressing different Flag-ORF33 homologs. The inputs/eluates were probed with the indicated antibodies. (C) ORF33 and ORF38 interact in cells. Lysates of HEK293T cells transiently expressing Flag-ORF33 and/or ORF38 were subjected to a membrane flotation assay as described in Materials and Methods. Six fractions were collected and analyzed by Western blotting with the indicated antibodies. Representative blots of our three replicates are shown. (D) The effect of N-ethylmaleimide (NEM) on GST-ORF38–ORF33 interaction. GST-ORF38 was used to pull down lysates of HEK293T cells expressing Flag-ORF33 that were pretreated with NEM or not pretreated. The input/eluates were analyzed by Western blotting with the indicated antibodies. (E) The interaction between ORF33 and ORF38 occurred preferentially in oxidized environments. The pulldown was performed as described for panel D, except that cells were pretreated with H2O2, NAC, or DTT as described in Materials and Methods. (F) The interaction between GST-ORF38 and ORF33 is not mediated by disulfide bond formation. The complex of GST-ORF38 with Flag-ORF33 was treated with increasing concentrations of DTT. The proteins released into the eluate or retained on the beads were analyzed by Western blotting.
We next used coimmunoprecipitation to determine whether the two interact in cells but failed to detect positive interaction. This is not surprising, since the interaction between HSV UL16 and UL11 appeared to be dynamic and was destroyed upon cell lysis (14). Because HSV UL11 is associated with the membrane but UL16 is not, membrane flotation assays have been used to demonstrate their interactions (14). We adapted this assay to examine the interaction between ORF33 and ORF38. As shown in Fig. 2C, ORF38 was detected in the top fractions, which contain the cytoplasmic membrane and associated proteins, whereas ORF33 was detected only in the bottom fractions when expressed alone. However, ORF33 signal was detected in the top fraction when coexpressed with ORF38 (Fig. 2C). These results are indicative of interaction between ORF38 and ORF33 in cells.
Because previous studies using HSV and HCMV implicated a role of cysteines in the interaction between ORF33 and ORF38 homologues (14, 28), we asked whether these cysteine residues are involved in the interaction between KSHV ORF33 and ORF38. We performed GST pulldown experiments in the presence of NEM, a small membrane-permeable thiol blocker that modifies free cysteines. NEM treatment caused a noticeable mobility shift of ORF33 on PAGE and abolished its binding to GST-ORF38, suggesting roles of cysteines of ORF33 in its interaction with ORF38 (Fig. 2D). Because cysteine is sensitive to the redox conditions, we treated Flag-ORF33-transfected cell lysates with dithiothreitol (DTT), N-acetyl-l-cysteine (NAC), or hydrogen peroxide (H2O2) before the pulldown assay. We found that the ORF33-ORF38 interaction occurred preferably in oxidized environments (Fig. 2E), further implying the importance of cysteine residues for ORF33-ORF38 interaction. We postulated that ORF33 and ORF38 interact through the formation of intermolecular disulfide bonds. However, the fact that their interaction survives treatment with up to 100 mM DTT suggests otherwise (Fig. 2F). Collectively, these results indicate that the cysteines of ORF33 are involved in its interactions with ORF38 but not through disulfide bond formation with ORF38. These results also suggest that their interaction can be regulated by the redox environment.
Generation of ORF33- and ORF38-null mutants in BAC16.
To investigate the roles of ORF33 and ORF38 in the context of KSHV lytic replication, we introduced premature stop codons or a mutation at the start codons of ORF33 or ORF38, or both, in BAC16, an infectious bacterial artificial clone of KSHV (37), using two-step RED recombineering technology (32, 38, 39). For the ORF33 mutant, two consecutive stop codons were introduced after the 13th codon. For the ORF38 mutant, we changed the start codon ATG to GTG (37). The double mutant included both of these mutations in BAC16 (Fig. 3A). These alterations were designed so as not to affect the overlapping amino acid sequences of ORF32 and ORF37 (37).
FIG 3.

Construction of ORF33-null, ORF38-null, and ORF33/ORF38-null mutants in BAC16. (A) Schematic diagram of ORF33-null or ORF38-null mutant constructions. For the ORF33 mutant, the 14th and 15th codons (AAG and GAA) were changed to two consecutive stop codons (TAG and TAA), thereby introducing a new restriction enzyme site (SpeI). For the ORF38 mutant, the start codon ATG was mutated to GTG. This modification resulted in the replacement of ORF37 codon AAA with AAG, both of which code for lysine. The blue box indicates overlap sequences. The indicated base pair number is derived from the previously annotated BAC16-GFP sequence (37). (B) RFLP analysis of wild-type and mutant BACs. Electrophoresis of KpnI- or SpeI-digested wild-type, mutant, and revertant BAC16 DNAs is shown. The arrow indicates the wild-type pattern, while the arrowheads indicate the predicted changes as a consequence of the newly introduced SpeI site in the ORF33 mutant. (C) Sequences of the wild type, mutants, and revertants in the ORF33 or ORF38 loci. The sequence chromatographs are shown, and the predicted mutations are boxed.
To ensure the phenotypic changes were a consequence of the designed mutation rather than unintentional random mutations, we restored the sequence to the original wild-type sequence, producing the revertants stop33-Rev and stop38-Rev. The wild-type and mutant BACs were analyzed by restriction fragment length polymorphism (RFLP), using the restriction enzyme KpnI or SpeI (Fig. 3B). Because a new SpeI site was created in the stop33 mutant, digestion of stop33 mutants with SpeI yielded fragments of 4.8 kb and 2.6 kb (arrowheads) instead of the 7.4-kb fragment (arrow) seen following digestion of the wild-type or revertant BACs. As expected, digestion with KpnI revealed no discernible difference among these constructs, suggesting that unintentional recombination was not a concern. These constructs were further validated by sequencing the PCR-amplified fragments from the designed loci (Fig. 3C).
ORF33 and ORF38 are not essential for viral gene expression but are required for production of progeny infectious virions.
The DNAs of stop33, stop38, the stop33/38 double mutant, and their revertants were transfected into iSLK cells, which express RTA under a doxycycline-inducible promoter (40). After hygromycin selection, stable iSLK cells harboring different KSHV BACs and expressing comparable levels of GFP were obtained. Upon induction with doxycycline and sodium butyrate, these cells express RTA, which in turn initiates the cascade of lytic gene expression, ultimately resulting in the release of progeny viral particles. Besides the expected loss of ORF33 and/or ORF38 signal in the respective iSLK.BAC16 mutants, we observed no dramatic difference in the expression of early (RTA, PF8, and ORF45) or late (ORF33, ORF38, ORF52, and ORF26) genes (Fig. 4), suggesting that ORF33 and ORF38 are not required for viral gene expression.
FIG 4.

Loss of ORF33 and/or ORF38 does not affect viral gene expression. iSLK.BAC16 wild-type or mutant cells were mock treated (M) or induced with doxycycline and sodium butyrate. The cell lysates were harvested at the indicated dpi and analyzed by Western blotting with the indicated antibodies.
To determine whether the loss of ORF33 and/or ORF38 affects the production of progeny viruses, we measured extracellular viral genomic copies by qPCR (Fig. 5A). We detected an ∼10-fold reduction of viral yield between the stop33 or stop38 mutant and the wild type or their revertants. We also analyzed the virion composition of the mutant viruses and found a corresponding ∼10-fold reduction in the level of ORF26 (Fig. 5B). Importantly, we noticed that the double mutation caused no additional defect in virion production (Fig. 5A and B), suggesting that ORF33 and ORF38 play roles in the same related pathways during the late lytic cycle. We infected HEK293 cells with 2-fold serial dilutions of progeny viruses (normalized by viral genome copy numbers). Because successfully infected cells express GFP, infected cells can be counted by flow cytometry, and the percentage of GFP-positive cells represents the infectivity. At an equivalent dilution, the infectivity of stop33, stop38, or the double mutant were at least an order of magnitude lower than those of the wild-type or revertant viruses (Fig. 5C). These results indicated that ORF33 and ORF38 are required for the production of infectious progeny virions.
FIG 5.
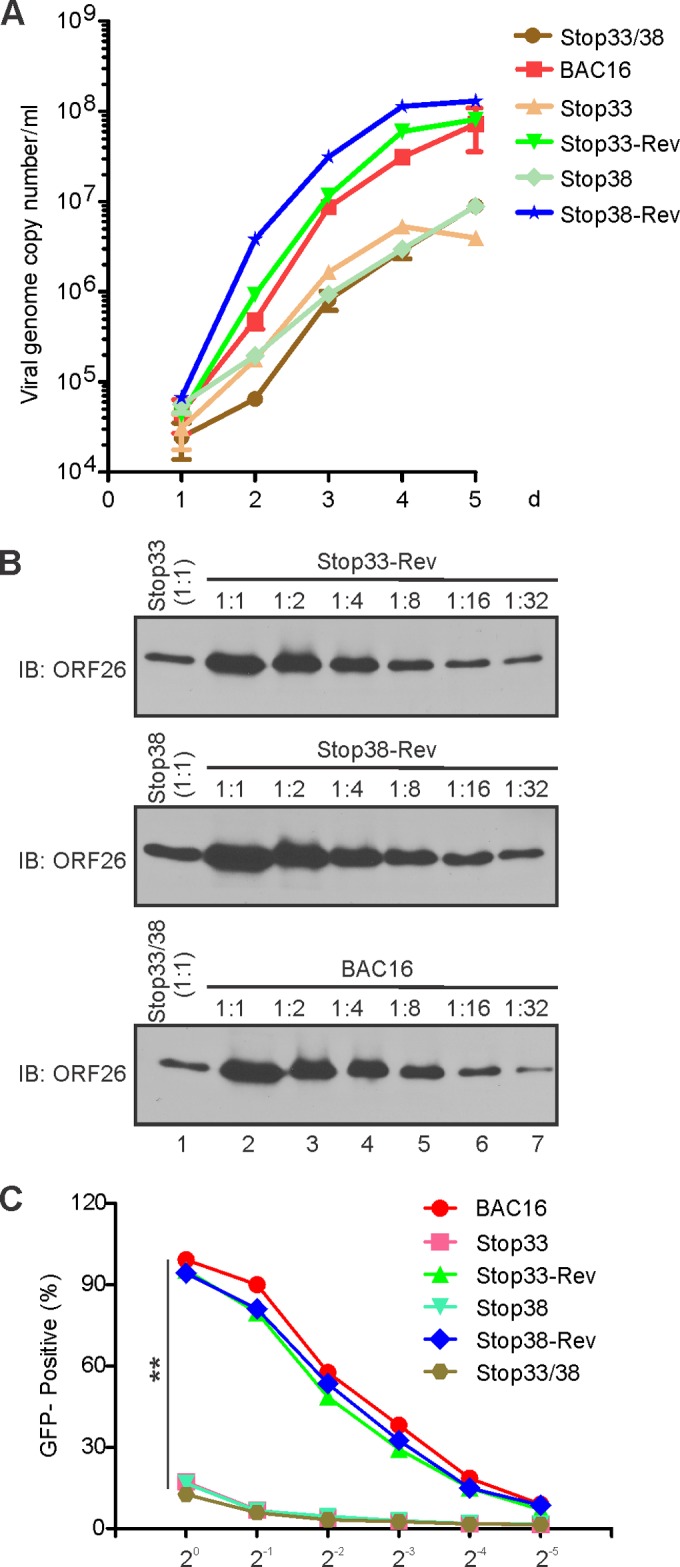
Loss of ORF33 and/or ORF38 compromised the production of infectious virions. (A) Growth kinetics of wild-type, mutant, and revertant KSHV. iSLK.BAC16 cells were mock treated or induced with doxycycline and sodium butyrate. The medium was collected at the indicated dpi, and the extracellular viral genome copy number was calculated by qPCR. (B) ORF33 and ORF38 mutation reduced progeny virion production. The extracellular virions were harvested at 5 dpi and concentrated as outlined in Materials and Methods. Mutant virions and 2-fold serial dilutions of wild-type or revertant virions were analyzed for the level of capsid protein (ORF26) by Western blotting. (C) Loss of ORF33 and/or ORF38 compromised virion infectivity. The virions of the wild type, mutants, and revertants were harvested and concentrated. 293 cells were infected with 2-fold serial dilutions of virions (after normalization by qPCR). Twenty-four hours postinduction, GFP-positive cells were counted by fluorescence-activated cell sorting (FACS). The percentages of GFP-positive cells were calculated for each dilution. **, P < 0.01 by Student's t test.
Loss of ORF33 and ORF38 resulted in change of virion components (affects assembly of other tegument proteins into virions).
We next asked whether the loss of ORF33 and/or ORF38 affects packaging of other proteins into mature virions. Again, we normalized samples by viral genome copy number, and equal loading was confirmed by similar levels of capsid protein ORF26 (Fig. 6A). Strikingly, the loss of ORF33 and/or ORF38 not only affected their own packaging into virions but also abolished that of ORF45. Conceivably, the loss of virion components would alter the buoyant density of the virion. Therefore, we analyzed the virions by 20% to 60% linear sucrose gradient ultracentrifugation. As shown in Fig. 6B, the wild-type virions were detected primarily in fractions 7 to 12, whereas virions of the ORF33/ORF38 double mutant were detected exclusively in fractions 6 to 8 (Fig. 6B). This virion peak shift to lighter fractions indicates a reduced buoyant density of the mutant virions, which supports the notion that they lack virion protein components. The abnormal virion composition likely explains why the mutant virions were less infectious (Fig. 5C).
FIG 6.
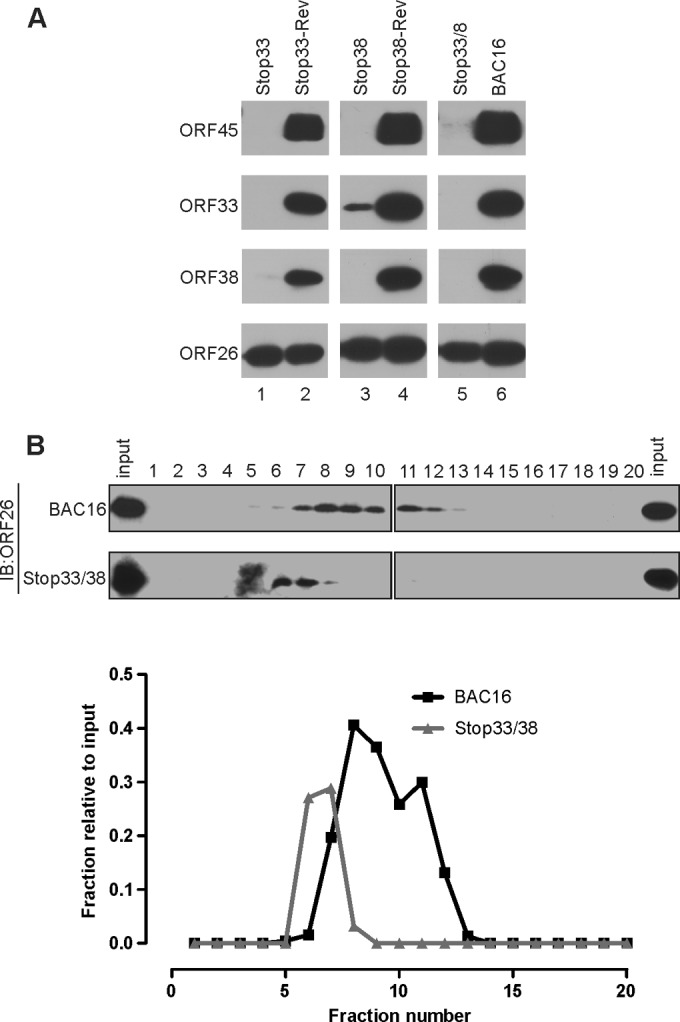
Loss of ORF33 and/or ORF38 affected virion composition. (A) KSHV wild-type and mutant virions were purified and concentrated. After normalization by qPCR, the virion components were analyzed by Western blotting with the indicated antibodies. (B) ORF33/ORF38-null virions have a reduced buoyant density. The same amount of purified wild-type or stop33/38 virions was applied to a 20% to 60% linear sucrose gradient. Twenty continuous fractions were collected and the level of ORF26 protein was assessed. The signal intensity of each fraction relative to the input was calculated and plotted.
Formation of virion-containing vesicles during KSHV lytic replication.
Previous reports show that alpha- and betaherpesviruses take advantage of vesicle release pathways (41–43). To determine if KSHV does the same, we adapted the well-established membrane flotation assay (14, 44). When lysates of KSHV-infected cells were analyzed, we found that most ORF38 protein was detected in fractions 1 and 2, which are known to be enriched for membrane-bound vesicles. This finding supports our conclusion that ORF38 associates with membranes. Interestingly, several virion proteins, including capsid protein ORF26 and tegument proteins ORF33 and ORF45, were present in fraction 1 (Fig. 7A). To ascertain whether this vesicle-rich fraction indeed contained viral particles, we quantified virion DNA from each fraction. As shown in Fig. 7B, about 25.6% of viral DNA was detected in fraction 1, whereas 27.7% of viral DNA was in the bottom fraction, which likely represents the free viral particles (Fig. 7B). We reasoned that the association of ORF38 with cytoplasmic membranes or the virion-containing vesicles might be sensitive to the detergent. As expected, the association of ORF38 with membranes was dramatically disrupted by treatment with Triton X-100 or NP-40, whereas it was completely abolished by SDS treatment. Interestingly, virion component proteins disappeared from the top fraction upon detergent treatment (Fig. 7A). We also quantified the virion DNA in each fraction. We consistently found that treatment with detergents dramatically reduced the presence of virion-containing vesicles in the top fraction (Fig. 7B). Because mild detergent, such as Triton X-100 or NP-40, only disrupts vesicle or virion envelopment, the amount of total virion DNA remained comparable to that of the no-treatment control (Fig. 7B). However, SDS treatment dramatically reduced the total viral DNA level (Fig. 7B), indicating that the capsid structure had been disrupted under this condition. Collectively, our results suggest that virions have been packaged into cytoplasmic vesicles during lytic replication.
FIG 7.

Formation of virion-containing vesicles during KSHV lytic replication. (A and B) iSLK.BAC16 cells were induced with doxycycline and sodium butyrate. Cells were harvested at 60 h postinduction and osmotically disrupted, left untreated (nontreated; NT), or treated with 0.5% Triton X-100, NP-40, or SDS for 30 min on ice and then subjected to a membrane flotation assay. Six continuous fractions were collected. The viral components in each fraction were analyzed by Western blotting using the indicated antibodies (A), and the viral DNA in each fraction was determined by qPCR (B). The numbers in the top left of each graph (Fr1% and Total) indicate the percentages of viral DNA detected in the first fraction relative to the total viral DNA and the total viral DNA of all fractions, respectively.
Loss of ORF33 and ORF38 resulted in reduction of virion-containing vesicles.
We next sought to further characterize the association between ORF38 and cytoplasmic membranes. To do so, we cotransfected cells with plasmids expressing ORF38 or different endomembrane markers and examined their subcellular localization. As revealed in Fig. 8, we found that ORF38 predominantly colocalizes with Golgi or early endosome markers, suggesting that ORF38 associates with membranes derived from these compartments. This result is also reminiscent of the proposed model in which herpesviruses employ the trans-Golgi network (TGN) membrane for virus maturation and egress (45). Therefore, we reasoned that ORF38 or the ORF38/ORF33 complex is involved in the formation of virion-containing vesicles. To confirm this prediction, the lysates of cells infected by the wild-type or mutant viruses were analyzed by membrane floatation assay. The tegument and capsid proteins, but not the nonvirion protein ORF59/PF8, consistently floated to the top fraction of lysates of wild-type virus-infected cells (Fig. 9A). However, we observed a dramatic reduction in the levels of virion proteins detected in the top vesicle-rich fractions following infection with mutant virus, with the exception of ORF38 (Fig. 9A). In concordance with this result, the percentage of viral DNA present in the vesicle-rich fractions was dramatically reduced by the loss of ORF33 and/or ORF38 (Fig. 9B). Collectively, these results suggest that ORF33 and ORF38 are involved in the packaging of virions into cytoplasmic vesicles, a critical process for viral maturation and egress.
FIG 8.

Membrane localization analysis of ORF38. HEK293T cells were seeded on coverslips in a 12-well plate. The next day, cells were cotransfected with plasmids expressing ORF38 and RFP-tagged endomembrane marker genes, including the endoplasmic reticulum gene (Sec61), Golgi gene (eNOS 1-33), early endosome gene (Rab5), late endosome gene (Rab7), and lysosome gene (Lamp1). At 24 h posttransfection, the cells were fixed, stained with anti-ORF38 antibody, and imaged with a Nikon A1R confocal microscope. Relative colocalization was quantified using Pearson's correlation coefficient (n > 6) and is shown on the right.
FIG 9.

ORF33 and ORF38 are required for vesicle transport of virions. (A and B) iSLK.BAC16 cells were induced with doxycycline and sodium butyrate. Cells were harvested at 60 h postinduction, osmotically disrupted, and then subjected to a membrane flotation assay. Six continuous fractions were collected. The viral components in each fraction were analyzed by Western blotting using the indicated antibodies (A), and the viral DNA of each fraction was determined by qPCR (B). The numbers in the top left of each graph (Fr1%) indicate the percentages of viral DNA detected in the first fraction relative to the total viral DNA.
DISCUSSION
Our recent discovery of ORF33 binding to the conserved C terminus of ORF45 prompted us to characterize ORF33 and its binding partner, ORF38, both of which are among 40 core herpesviral proteins that are conserved in all three subfamilies of herpesviruses (30, 31). We found that both proteins are expressed late during KSHV lytic replication and packaged in mature virions as tegument proteins (Fig. 1C and D). We confirmed that ORF33 and ORF38 of KSHV interact with each other (Fig. 2). Null mutation of either or both in BAC16 caused ∼10-fold reductions of progeny extracellular viruses (Fig. 5A and B). The mutant viruses also exhibit ∼16-fold lower infectivity than the revertants and wild-type viruses (Fig. 5C), suggesting that both ORF33 and ORF38 are required for optimal production of infectious viruses. In addition, the mutant viruses have altered virion protein components (Fig. 6), suggesting that both ORF33 and ORF38 play critical roles in the packaging of its partner and a subset of tegument proteins, including ORF45, into virions. ORF38 colocalizes with Golgi or early endosome markers (Fig. 8), suggesting ORF38 associates with the membranes of these organelles. Furthermore, we noticed that both ORF33 and ORF38 are involved in transporting newly assembled viral particles through cytoplasmic vesicles (Fig. 9), a critical process for virus maturation and egress.
Roles of ORF33 in KSHV lytic replication.
ORF33 is a tegument protein that is conserved among all herpesviruses (13, 21, 22), but its actual function remains elusive. The ORF33 homologue in HSV-1 and pseudorabies virus (PrV), UL16, is not absolutely required for the production of infectious progeny (24, 46). In contrast, the homologue UL94 of HCMV and MCMV is essential, because no infectious viruses can be produced in its absence (15, 47). In all cases, the lack of UL16 or UL94 resulted in the inefficient envelopment of capsids in the cytoplasm (15, 48). In gammaherpesviruses, ORF33 of MHV-68 also has been shown to be essential (13). For KSHV, although ORF33 has been identified as a tegument protein by us and others (21, 49), its function in lytic replication has not been carefully examined. An earlier study showed that deletion of the entire ORF33 coding sequence from BAC36 yielded no detectable infectious viruses (50), providing the first clue that ORF33 plays an indispensable role in KSHV lytic replication. Presumably because of the relatively low efficiency of the system employed in that study, detailed analyses of the mutant viruses were not feasible. In the present study, we took advantage of a recently developed and greatly improved system, consisting of the highly infectious BAC16 (37), the seamless BAC genome editing tool (38, 39), and iSLK cells, which support robust lytic replication of KSHV (40). We introduced premature stop codons in ORF33 without affecting the coding sequence of the overlapping ORF32 (Fig. 3) and found that the loss of ORF33 reduced the yield of progeny viruses by ∼10-fold (Fig. 5A and B). The progeny viruses also appeared to be less infectious. On the basis of scoring GFP positive by FACS, we found that ∼16-fold more stop33 viral particles than wild-type viral particles were needed to achieve the same rate of infection (Fig. 5C). When the effects on viral yield and infectivity are both considered, the loss of ORF33 reduced the titer of infectious progeny by more than two orders of magnitude. Thus, while ORF33 is not absolutely essential for KSHV to replicate in iSLK cells, it is required for the optimal production of infectious virions.
Roles of ORF38 in KSHV lytic replication.
Like ORF33, ORF38 is a herpesvirus core protein (30, 31). At 61 aa, KSHV ORF38 is considerably smaller than its homologues in HSV-1 (UL11; 96 aa) and HCMV (UL99/pp28; 190 aa). UL11 and UL99 have been shown to be myristylated, membrane-associated, and tegument proteins (51, 52). Although the deletion of UL11 in HSV and PRV caused only a moderate reduction in viral titers (53, 54), UL99 of HCMV is absolutely essential for infectious virion production (55). In the absence of UL99 or UL11, nonenveloped nucleocapsids accumulated in the cytoplasm, suggesting an impairment in secondary envelopment (53, 55). Perhaps due to its small size and nonuniform distribution of lysine residues, ORF38 was not among the virion proteins of KSHV or MHV68 to be identified by mass spectrometry (21, 56, 57), although its homologues in RRV and EBV were detected in virions (16, 58).
In order to characterize the functions of ORF38 protein in KSHV replication, we first generated hybridomas against it. Recent studies revealed generally strong antibody responses to ORF38 among KSHV-infected patients (59, 60). In apparent agreement with this high immunogenicity in humans, most hybridoma clones recognized a strong and specific signal of ∼10 kDa in KSHV-infected cells (Fig. 1B), as well as in purified virions, confirming that ORF38 is an abundant virion component. Analyses of the virion-associated ORF38 in response to trypsin digestion and detergent treatment suggested that this protein resides interior to the viral envelope. More specifically, its sensitivity to detergents indicates that it is an outer tegument protein and likely associates with the viral envelope (Fig. 1C to E). Our data also provided the first experimental evidence that KSHV ORF38 is an abundant tegument protein. To determine the roles of ORF38 in KSHV lytic replication, we generated a BAC16 mutant in which the initial ATG of ORF38 was changed to GTG without changing the coding sequence of the overlapping ORF37 (Fig. 3). We found that the loss of ORF38 reduced viral yield by ∼10-fold and that the progeny are less infectious than the wild type or revertant (Fig. 5). We next generated a double mutant in which both ORF33 and ORF38 have been disrupted. Our results suggested that the double mutant has a phenotype similar to that of the single deletion of either ORF33 or ORF38. This is reminiscent of the virtually identical phenotypes of UL94-null and UL99-null HCMV (15, 55). Although ultrastructural data are lacking, our membrane flotation assay suggested that immature virions were assembled into cytoplasmic vesicles for final maturation and egress (Fig. 7). These virion-containing vesicles apparently were sensitive to detergent, because the number of virions was dramatically reduced in the vesicle-enriched fraction upon treatment (Fig. 7). It is worth noting that we observed only a small fraction of virion components or DNA in the top fraction (Fig. 7), suggesting a relatively low efficiency of viral maturation and egress, even in this greatly improved iSLK.BAC16 system. Since ORF38 colocalizes with the Golgi and early endosome organelles (Fig. 8), we postulate that ORF38 associates with the membranes of these organelles. This result, in combination with its critical role in transporting virions into cytoplasmic vesicles, is reminiscent of the process of HCMV final envelopment. It has been reported that HCMV final envelopment on membranes contains both TGN and endosome markers, suggesting that the immature virus buds into the transport vesicles which shuttle between endosomes and the TGN or bud into a novel hybrid compartment with both endosome and TGN components (61). Interestingly, MHV-68 ORF38 has been shown to be present only in virions that have been packaged into vesicles (29). Whether or not KSHV does the same is still unknown. However, it is possible for ORF38 to recruit the Golgi membrane or early endosome membrane for virion-containing vesicle formation to facilitate subsequent virion maturation and egress. The nature of these cytoplasmic vesicles and their roles in virion assembly await further investigation.
Interaction between KSHV ORF33 and ORF38.
We confirmed that KSHV ORF33 interacts with ORF38 in vitro. The interaction is specific, because KSHV ORF38 interacts with KSHV ORF33 but not its homologues in other gammaherpesviruses, such as RRV and EBV (Fig. 2). This is in contrast to the relatively promiscuous binding between homologues of UL11 and UL16 in alphaherpesviruses, including HSV and PrV (62). KSHV ORF33 contains 14 cysteines, 9 of which are conserved among all herpesviruses. In congruence with previous analyses of HSV UL16, we found that NEM treatment abolished the retention of ORF33 to GST-ORF38 (Fig. 2D), suggesting roles of ORF33's cysteine residues in regulating their interaction. Moreover, the interaction of ORF33 with ORF38 appears to be more efficient in the oxidized than the reduced environment (Fig. 2E). Our membrane flotation experiment suggested that ORF33 resides within cytoplasmic vesicles in the presence of ORF38 (Fig. 2C, 7A, and 9A). These vesicles may provide an ideal oxidative folding environment for ORF33. However, the extent to which the interaction between KSHV ORF33 and ORF38 is regulated remains unclear. Additional studies are needed to further characterize their interaction and specific functions during KSHV lytic replication.
It has been speculated that HSV UL16 provides a bridging function between the capsid and the membrane during budding events through its interactions with both capsid and the membrane-bound tegument protein UL11 (63). Recent evidence from HCMV has shown that the interaction between UL99 and UL94 is required for the proper localization of each protein to the assembly complex (AC) and for virus replication. HCMV UL94 and UL99 were found to exhibit aberrant localization and to not accumulate at the AC in the absence of their interaction, resulting in defective virus maturation and egress (28). Similar results were obtained with mouse cytomegalovirus (MCMV) (47) and MHV-68 (13, 29). Recent reports also showed that MHV-68 ORF33 interacts with ORF38 during virus replication (13, 29). Together, these studies suggest that the conserved interaction between ORF33 and ORF38 homologues plays critical roles in the production of progeny virions.
Although ORF33 is conserved among the three subfamilies of herpesviruses, noticeable differences among its homologues have been observed. For example, UL16 protein is detected in extracellular virions of HSV-1 but not in that of HSV-2 (19, 22). Avian infectious laryngotracheitis virus, an alphaherpesvirus, lacks UL16 altogether (64). Furthermore, UL94 of HCMV and ORF33 of KSHV associate tightly with the capsid, unlike HSV-1 UL16, which is easily detachable (18, 65). Additionally, although ORF33 and ORF38 are conserved among all herpesviruses, their interaction is highly specific (Fig. 2B), which suggests their specific function during virus lytic replication.
Interactions between ORF33, ORF38, and ORF45: roles in viral assembly.
ORF33 homologues also differ in their interactions with other virion proteins. In addition to UL11, UL16 has been shown to interact with several other viral proteins, including another tegument protein, UL21, and glycoprotein E (gE) (66). These three tegument proteins (UL11, UL16, and UL21) and gE come together to form a highly efficient complex in vivo through coordinated interactions (66). Recent evidence also suggested its interaction with VP22 (48). In the absence of UL16, the packaging of UL11, VP22, and gE is defective (48). It has been hypothesized that the bridging interaction between capsid-bound UL16 and membrane-bound UL11 is required for envelopment during HSV assembly and maturation (14). Recent studies suggested that MHV-68 ORF33 directly associates with intranuclear capsids through interactions with capsid proteins ORF25 and ORF26 (67) and that ORF38 is associated with the membrane (29). Similarly, KSHV ORF33 appears to associate with the capsid, because it remained bound to capsids even after virions were treated with harsh detergent and proteinase, while ORF38 apparently is membrane associated, because it became soluble upon treatment with a low concentration of detergent. The association of ORF38 with the membrane was further confirmed by membrane flotation assays. Therefore, the basic properties of ORF33 and ORF38 homologues appear to be conserved among herpesviruses, and their interactions with each other and with other virion proteins are expected to play critical roles in the assembly of progeny virions.
By analogy to HSV UL16, ORF33 homologues of gammaherpesviruses are expected to interact with additional tegument and/or envelope proteins, either directly or through interaction with ORF38. However, such interacting partners have not been identified to date. Some of UL16's binding partners, such as gE, are alphaherpesvirus specific, and their functional counterparts in gammaherpesviruses are unknown. We recently found that ORF33 binds to the conserved C terminus of ORF45. We demonstrated that deletion of this region diminished ORF33 accumulation and production of progeny virions (12). Because the accumulation of ORF33 depends on its interaction with ORF45, it was unclear whether the defect we observed could be attributed to the loss of ORF45-ORF33 interaction or simply the loss of ORF33 protein accumulation. Here, we found that the entire deletion of ORF33 caused only an ∼10-fold reduction of progeny virus yield (Fig. 5A), which is less severe than the ∼50- to 100-fold defect caused by ORF45 C-terminal deletion and ORF45-null mutants. These results indicate that ORF45 has additional functions other than stabilizing ORF33. Indeed, our recent phosphoproteomic studies revealed multiple roles of ORF45-activated ribosomal S6 kinases in KSHV lytic replication (32, 68). Notwithstanding this, the interactions of ORF45-ORF33 and ORF38-ORF33 apparently are critical for the packaging of some tegument proteins into virions. Because ORF33-ORF38 interaction is conserved among all three subfamilies of herpesviruses, it is not unexpected that deletion of either one affected the assembly of mature virions. In agreement with this, deletion of ORF33 resulted in the failure of packaging of ORF45 and ORF38 into extracellular virions. Interestingly, while ORF45 also was not detected in ORF38-null virions, a small amount of ORF33 remained, suggesting that the assembly of ORF33 precedes the assembly of ORF38. This is consistent with recent work showing that MHV-68 ORF33 is associated with intranuclear capsids (67). Substantial evidence supports that ORF38 and its homologues are involved in viral secondary envelopment and maturation, a process during which additional tegument proteins are added to immature virions (45, 53, 55, 69). It is at this stage that we expect ORF33-ORF38 interaction to be critical. It is currently unknown whether ORF45 is assembled with ORF33 in the nucleus or is added during secondary envelopment. Further studies are required to uncover the regulatory mechanisms governing the viral assembly process. Importantly, ORF33/ORF38 mutant virions apparently were less infectious, presumably due to their lack of other virion protein components, such as ORF45, which is required for optimal de novo infection (70). The failure of packaging of ORF45 and potentially other tegument proteins into virions in the absence of ORF33 or ORF38 highlights the critical importance of viral-viral protein interactions, including those between ORF33-ORF45 and ORF33-ORF38, for the proper assembly of progeny virions. Overall, our comprehensive characterization of the functional interaction between ORF33 and ORF38 sheds light on their essential roles in virion assembly at the late stage of KSHV lytic replication.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants R01DE016680 (to F.Z.) and F31 CA183250 (to D.A.) and in part by the National Cancer Institute, NIH (contract number HHSN261200800001E).
We thank Brian Washburn, Rani Dhanarajan, and Kristina Poduch for making hybridomas. We thank David Meckes for providing us endomembrane gene markers. We thank Ruth Didier at the Florida State University Flow Cytometry Facility for assistance with flow cytometry. We thank Timothy Megraw for assistance with the Nikon A1R confocal microscope. We are grateful to Klaus Osterrieder for providing pEPKan-S plasmid, Gregory Smith for providing E. coli strain GS1783, Rolf Renne for providing E. coli strain GS1783 carrying BAC16, and Jinjong Myoung and Don Ganem for providing the iSLK cells.
REFERENCES
- 1.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 2.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 332:1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- 3.Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d'Agay MF, Clauvel JP, Raphael M, Degos L, Sigaux F. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86:1276–1280. [PubMed] [Google Scholar]
- 4.Greene W, Kuhne K, Ye F, Chen J, Zhou F, Lei X, Gao SJ. 2007. Molecular biology of KSHV in relation to AIDS-associated oncogenesis. Cancer Treat Res 133:69–127. doi: 10.1007/978-0-387-46816-7_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Speck SH, Ganem D. 2010. Viral latency and its regulation: lessons from the gamma-herpesviruses. Cell Host Microbe 8:100–115. doi: 10.1016/j.chom.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trus BL, Heymann JB, Nealon K, Cheng N, Newcomb WW, Brown JC, Kedes DH, Steven AC. 2001. Capsid structure of Kaposi's sarcoma-associated herpesvirus, a gammaherpesvirus, compared to those of an alphaherpesvirus, herpes simplex virus type 1, and a betaherpesvirus, cytomegalovirus. J Virol 75:2879–2890. doi: 10.1128/JVI.75.6.2879-2890.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo H, Shen S, Wang L, Deng H. 2010. Role of tegument proteins in herpesvirus assembly and egress. Protein Cell 1:987–998. doi: 10.1007/s13238-010-0120-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelly BJ, Fraefel C, Cunningham AL, Diefenbach RJ. 2009. Functional roles of the tegument proteins of herpes simplex virus type 1. Virus Res 145:173–186. doi: 10.1016/j.virusres.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 9.Kalejta RF. 2008. Tegument proteins of human cytomegalovirus. Microbiol Mol Biol Rev 72:249–265. doi: 10.1128/MMBR.00040-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sathish N, Wang X, Yuan Y. 2012. Tegument proteins of Kaposi's sarcoma-associated herpesvirus and related gamma-herpesviruses. Front Microbiol 3:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jia Q, Chernishof V, Bortz E, McHardy I, Wu TT, Liao HI, Sun R. 2005. Murine gammaherpesvirus 68 open reading frame 45 plays an essential role during the immediate-early phase of viral replication. J Virol 79:5129–5141. doi: 10.1128/JVI.79.8.5129-5141.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gillen J, Li W, Liang Q, Avey D, Wu J, Wu F, Myoung J, Zhu F. 2015. A survey of the interactome of Kaposi's sarcoma-associated herpesvirus ORF45 revealed its binding to viral ORF33 and cellular USP7, resulting in stabilization of ORF33 that is required for production of progeny viruses. J Virol 89:4918–4931. doi: 10.1128/JVI.02925-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo H, Wang L, Peng L, Zhou ZH, Deng H. 2009. Open reading frame 33 of a gammaherpesvirus encodes a tegument protein essential for virion morphogenesis and egress. J Virol 83:10582–10595. doi: 10.1128/JVI.00497-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chadha P, Han J, Starkey JL, Wills JW. 2012. Regulated interaction of tegument proteins UL16 and UL11 from herpes simplex virus. J Virol 86:11886–11898. doi: 10.1128/JVI.01879-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phillips SL, Bresnahan WA. 2011. The human cytomegalovirus (HCMV) tegument protein UL94 is essential for secondary envelopment of HCMV virions. J Virol 86:2523–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johannsen E, Luftig M, Chase MR, Weicksel S, Cahir-McFarland E, Illanes D, Sarracino D, Kieff E. 2004. Proteins of purified Epstein-Barr virus. Proc Natl Acad Sci U S A 101:16286–16291. doi: 10.1073/pnas.0407320101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nalwanga D, Rempel S, Roizman B, Baines JD. 1996. The UL 16 gene product of herpes simplex virus 1 is a virion protein that colocalizes with intranuclear capsid proteins. Virology 226:236–242. doi: 10.1006/viro.1996.0651. [DOI] [PubMed] [Google Scholar]
- 18.Wing BA, Lee GC, Huang ES. 1996. The human cytomegalovirus UL94 open reading frame encodes a conserved herpesvirus capsid/tegument-associated virion protein that is expressed with true late kinetics. J Virol 70:3339–3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loret S, Guay G, Lippe R. 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J Virol 82:8605–8618. doi: 10.1128/JVI.00904-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Varnum SM, Streblow DN, Monroe ME, Smith P, Auberry KJ, Pasa-Tolic L, Wang D, Camp DG II, Rodland K, Wiley S, Britt W, Shenk T, Smith RD, Nelson JA. 2004. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J Virol 78:10960–10966. doi: 10.1128/JVI.78.20.10960-10966.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu FX, Chong JM, Wu L, Yuan Y. 2005. Virion proteins of Kaposi's sarcoma-associated herpesvirus. J Virol 79:800–811. doi: 10.1128/JVI.79.2.800-811.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oshima S, Daikoku T, Shibata S, Yamada H, Goshima F, Nishiyama Y. 1998. Characterization of the UL16 gene product of herpes simplex virus type 2. Arch Virol 143:863–880. doi: 10.1007/s007050050338. [DOI] [PubMed] [Google Scholar]
- 23.Yeh PC, Han J, Chadha P, Meckes DG Jr, Ward MD, Semmes OJ, Wills JW. 2011. Direct and specific binding of the UL16 tegument protein of herpes simplex virus to the cytoplasmic tail of glycoprotein E. J Virol 85:9425–9436. doi: 10.1128/JVI.05178-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baines JD, Roizman B. 1991. The open reading frames UL3, UL4, UL10, and UL16 are dispensable for the replication of herpes simplex virus 1 in cell culture. J Virol 65:938–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Phillips SL, Bresnahan WA. 2011. Identification of binary interactions between human cytomegalovirus virion proteins. J Virol 85:440–447. doi: 10.1128/JVI.01551-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song MJ, Hwang S, Wong WH, Wu TT, Lee S, Liao HI, Sun R. 2005. Identification of viral genes essential for replication of murine gamma-herpesvirus 68 using signature-tagged mutagenesis. Proc Natl Acad Sci U S A 102:3805–3810. doi: 10.1073/pnas.0404521102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yeh PC, Meckes DG Jr, Wills JW. 2008. Analysis of the interaction between the UL11 and UL16 tegument proteins of herpes simplex virus. J Virol 82:10693–10700. doi: 10.1128/JVI.01230-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Phillips SL, Cygnar D, Thomas A, Bresnahan WA. 2012. Interaction between the human cytomegalovirus tegument proteins UL94 and UL99 is essential for virus replication. J Virol 86:9995–10005. doi: 10.1128/JVI.01078-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shen S, Guo H, Deng H. 2014. Murine gammaherpesvirus-68 ORF38 encodes a tegument protein and is packaged into virions during secondary envelopment. Protein Cell 5:141–150. doi: 10.1007/s13238-013-0005-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mocarski E., Jr 2007. Betaherpes viral genes and their functions, p 204–230. In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom. [PubMed] [Google Scholar]
- 31.Roizman B. 2007. Family herpesviridae: a brief introduction, p 2479–2499. In Knipe DM, Howley PM, Griffin DE, Lam AR, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 32.Fu B, Kuang E, Li W, Avey D, Li X, Turpin Z, Valdes A, Brulois K, Myoung J, Zhu F. 2015. Activation of p90 ribosomal S6 kinases by ORF45 of Kaposi's sarcoma-associated herpesvirus is critical for optimal production of infectious viruses. J Virol 89:195–207. doi: 10.1128/JVI.01937-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu JJ, Li W, Shao Y, Avey D, Fu B, Gillen J, Hand T, Ma S, Liu X, Miley W, Konrad A, Neipel F, Sturzl M, Whitby D, Li H, Zhu F. 2015. Inhibition of cGAS DNA sensing by a herpesvirus virion protein. Cell Host Microbe 18:333–344. doi: 10.1016/j.chom.2015.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harlow E, Lane D. 2006. Staining immunoblots for total protein using Ponceau S. CSH Protoc 2006:pdb.prot4629. [DOI] [PubMed] [Google Scholar]
- 35.Leelawong M, Lee JI, Smith GA. 2012. Nuclear egress of pseudorabies virus capsids is enhanced by a subspecies of the large tegument protein that is lost upon cytoplasmic maturation. J Virol 86:6303–6314. doi: 10.1128/JVI.07051-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leelawong M, Guo D, Smith GA. 2011. A physical link between the pseudorabies virus capsid and the nuclear egress complex. J Virol 85:11675–11684. doi: 10.1128/JVI.05614-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brulois KF, Chang H, Lee AS, Ensser A, Wong LY, Toth Z, Lee SH, Lee HR, Myoung J, Ganem D, Oh TK, Kim JF, Gao SJ, Jung JU. 2012. Construction and manipulation of a new Kaposi's sarcoma-associated herpesvirus bacterial artificial chromosome clone. J Virol 86:9708–9720. doi: 10.1128/JVI.01019-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. doi: 10.2144/000112096. [DOI] [PubMed] [Google Scholar]
- 39.Tischer BK, Smith GA, Osterrieder N. 2010. En passant mutagenesis: a two step markerless red recombination system. Methods Mol Biol 634:421–430. doi: 10.1007/978-1-60761-652-8_30. [DOI] [PubMed] [Google Scholar]
- 40.Myoung J, Ganem D. 2011. Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: maintenance of tight latency with efficient reactivation upon induction. J Virol Methods 174:12–21. doi: 10.1016/j.jviromet.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mettenleiter TC. 2002. Herpesvirus assembly and egress. J Virol 76:1537–1547. doi: 10.1128/JVI.76.4.1537-1547.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mori Y, Koike M, Moriishi E, Kawabata A, Tang H, Oyaizu H, Uchiyama Y, Yamanishi K. 2008. Human herpesvirus-6 induces MVB formation, and virus egress occurs by an exosomal release pathway. Traffic 9:1728–1742. doi: 10.1111/j.1600-0854.2008.00796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Homman-Loudiyi M, Hultenby K, Britt W, Soderberg-Naucler C. 2003. Envelopment of human cytomegalovirus occurs by budding into Golgi-derived vacuole compartments positive for gB, Rab 3, trans-Golgi network 46, and mannosidase II. J Virol 77:3191–3203. doi: 10.1128/JVI.77.5.3191-3203.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Girard M, Allaire PD, Blondeau F, McPherson PS. 2005. Isolation of clathrin-coated vesicles by differential and density gradient centrifugation. Curr Protoc Cell Biol Chapter 3:Unit 3.13. [DOI] [PubMed] [Google Scholar]
- 45.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol 9:382–394. doi: 10.1038/nrmicro2559. [DOI] [PubMed] [Google Scholar]
- 46.Klupp BG, Bottcher S, Granzow H, Kopp M, Mettenleiter TC. 2005. Complex formation between the UL16 and UL21 tegument proteins of pseudorabies virus. J Virol 79:1510–1522. doi: 10.1128/JVI.79.3.1510-1522.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maninger S, Bosse JB, Lemnitzer F, Pogoda M, Mohr CA, von Einem J, Walther P, Koszinowski UH, Ruzsics Z. 2011. M94 is essential for the secondary envelopment of murine cytomegalovirus. J Virol 85:9254–9267. doi: 10.1128/JVI.00443-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Starkey JL, Han J, Chadha P, Marsh JA, Wills JW. 2014. Elucidation of the block to herpes simplex virus egress in the absence of tegument protein UL16 reveals a novel interaction with VP22. J Virol 88:110–119. doi: 10.1128/JVI.02555-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bechtel JT, Winant RC, Ganem D. 2005. Host and viral proteins in the virion of Kaposi's sarcoma-associated herpesvirus. J Virol 79:4952–4964. doi: 10.1128/JVI.79.8.4952-4964.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Budt M, Hristozova T, Hille G, Berger K, Brune W. 2011. Construction of a lytically replicating Kaposi's sarcoma-associated herpesvirus. J Virol 85:10415–10420. doi: 10.1128/JVI.05071-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.MacLean CA, Clark B, McGeoch DJ. 1989. Gene UL11 of herpes simplex virus type 1 encodes a virion protein which is myristylated. J Gen Virol 70(Part 12):3147–3157. doi: 10.1099/0022-1317-70-12-3147. [DOI] [PubMed] [Google Scholar]
- 52.Jones TR, Lee SW. 2004. An acidic cluster of human cytomegalovirus UL99 tegument protein is required for trafficking and function. J Virol 78:1488–1502. doi: 10.1128/JVI.78.3.1488-1502.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baines JD, Roizman B. 1992. The UL11 gene of herpes simplex virus 1 encodes a function that facilitates nucleocapsid envelopment and egress from cells. J Virol 66:5168–5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kopp M, Granzow H, Fuchs W, Klupp BG, Mundt E, Karger A, Mettenleiter TC. 2003. The pseudorabies virus UL11 protein is a virion component involved in secondary envelopment in the cytoplasm. J Virol 77:5339–5351. doi: 10.1128/JVI.77.9.5339-5351.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Silva MC, Yu QC, Enquist L, Shenk T. 2003. Human cytomegalovirus UL99-encoded pp28 is required for the cytoplasmic envelopment of tegument-associated capsids. J Virol 77:10594–10605. doi: 10.1128/JVI.77.19.10594-10605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bechtel JT, Liang Y, Hvidding J, Ganem D. 2003. Host range of Kaposi's sarcoma-associated herpesvirus in cultured cells. J Virol 77:6474–6481. doi: 10.1128/JVI.77.11.6474-6481.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bortz E, Whitelegge JP, Jia Q, Zhou ZH, Stewart JP, Wu TT, Sun R. 2003. Identification of proteins associated with murine gammaherpesvirus 68 virions. J Virol 77:13425–13432. doi: 10.1128/JVI.77.24.13425-13432.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.O'Connor CM, Kedes DH. 2006. Mass spectrometric analyses of purified rhesus monkey rhadinovirus reveal 33 virion-associated proteins. J Virol 80:1574–1583. doi: 10.1128/JVI.80.3.1574-1583.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Labo N, Miley W, Marshall V, Gillette W, Esposito D, Bess M, Turano A, Uldrick T, Polizzotto MN, Wyvill KM, Bagni R, Yarchoan R, Whitby D. 2014. Heterogeneity and breadth of host antibody response to KSHV infection demonstrated by systematic analysis of the KSHV proteome. PLoS Pathog 10:e1004046. doi: 10.1371/journal.ppat.1004046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zheng D, Wan J, Cho YG, Wang L, Chiou CJ, Pai S, Woodard C, Zhu J, Liao G, Martinez-Maza O, Qian J, Zhu H, Hayward GS, Ambinder RF, Hayward SD. 2011. Comparison of humoral immune responses to Epstein-Barr virus and Kaposi's sarcoma-associated herpesvirus using a viral proteome microarray. J Infect Dis 204:1683–1691. doi: 10.1093/infdis/jir645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cepeda V, Esteban M, Fraile-Ramos A. 2010. Human cytomegalovirus final envelopment on membranes containing both trans-Golgi network and endosomal markers. Cell Microbiol 12:386–404. doi: 10.1111/j.1462-5822.2009.01405.x. [DOI] [PubMed] [Google Scholar]
- 62.Harper AL, Meckes DG Jr, Marsh JA, Ward MD, Yeh PC, Baird NL, Wilson CB, Semmes OJ, Wills JW. 2010. Interaction domains of the UL16 and UL21 tegument proteins of herpes simplex virus. J Virol 84:2963–2971. doi: 10.1128/JVI.02015-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Loomis JS, Courtney RJ, Wills JW. 2003. Binding partners for the UL11 tegument protein of herpes simplex virus type 1. J Virol 77:11417–11424. doi: 10.1128/JVI.77.21.11417-11424.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Thureen DR, Keeler CL Jr. 2006. Psittacid herpesvirus 1 and infectious laryngotracheitis virus: comparative genome sequence analysis of two avian alphaherpesviruses. J Virol 80:7863–7872. doi: 10.1128/JVI.00134-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Meckes DG Jr, Wills JW. 2007. Dynamic interactions of the UL16 tegument protein with the capsid of herpes simplex virus. J Virol 81:13028–13036. doi: 10.1128/JVI.01306-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Han J, Chadha P, Starkey JL, Wills JW. 2012. Function of glycoprotein E of herpes simplex virus requires coordinated assembly of three tegument proteins on its cytoplasmic tail. Proc Natl Acad Sci U S A 109:19798–19803. doi: 10.1073/pnas.1212900109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shen S, Jia X, Guo H, Deng H. 2015. Gammaherpesvirus tegument protein ORF33 is associated with intranuclear capsids at an early stage of the tegumentation process. J Virol 89:5288–5297. doi: 10.1128/JVI.00079-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Avey D, Tepper S, Li W, Turpin Z, Zhu F. 2015. Phosphoproteomic analysis of KSHV-infected cells reveals roles of ORF45-activated RSK during lytic replication. PLoS Pathog 11:e1004993. doi: 10.1371/journal.ppat.1004993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kopp M, Granzow H, Fuchs W, Klupp B, Mettenleiter TC. 2004. Simultaneous deletion of pseudorabies virus tegument protein UL11 and glycoprotein M severely impairs secondary envelopment. J Virol 78:3024–3034. doi: 10.1128/JVI.78.6.3024-3034.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhu FX, Li X, Zhou F, Gao SJ, Yuan Y. 2006. Functional characterization of Kaposi's sarcoma-associated herpesvirus ORF45 by bacterial artificial chromosome-based mutagenesis. J Virol 80:12187–12196. doi: 10.1128/JVI.01275-06. [DOI] [PMC free article] [PubMed] [Google Scholar]