

ABSTRACT
Influenza A virus (IAV) affects the upper and lower respiratory tracts and rapidly induces the expression of mucins, which are common O-glycosylated proteins, on the epithelial surfaces of the respiratory tract. Although mucin production is associated with the inhibition of virus transmission as well as characteristic clinical symptoms, little is known regarding how mucins are produced on the surfaces of respiratory epithelial cells and how they affect IAV replication. In this study, we found that two microRNAs (miRNAs), miR-17-3p and miR-221, which target GalNAc transferase 3 (GALNT3) mRNA, are rapidly downregulated in human alveolar basal epithelial cells during the early stage of IAV infection. We demonstrated that the expression of GALNT3 mRNA is upregulated in an IAV replication-dependent fashion and leads to mucin production in bronchial epithelial cells. A lectin microarray analysis revealed that the stable expression of GALNT3 by human alveolar basal epithelial cells induces mucin-type O-glycosylation modifications similar to those present in IAV-infected cells, suggesting that GALNT3 promotes mucin-type O-linked glycosylation in IAV-infected cells. Notably, analyses using short interfering RNAs and miRNA mimics showed that GALNT3 knockdown significantly reduces IAV replication. Furthermore, IAV replication was markedly decreased in embryonic fibroblast cells obtained from galnt3-knockout mice. Interestingly, IAV-infected galnt3-knockout mice exhibited high mortality and severe pathological alterations in the lungs compared to those of wild-type mice. Our results demonstrate not only the molecular mechanism underlying rapid mucin production during IAV infection but also the contribution of O-linked glycosylation to the replication and propagation of IAV in lung cells.
IMPORTANCE Viral infections that affect the upper or lower respiratory tracts, such as IAV, rapidly induce mucin production on the epithelial surfaces of respiratory cells. However, the details of how mucin-type O-linked glycosylation is initiated by IAV infection and how mucin production affects viral replication have not yet been elucidated. In this study, we show that levels of two miRNAs that target the UDP-GalNAc transferase GALNT3 are markedly decreased during the early stage of IAV infection, resulting in the upregulation of GALNT3 mRNA. We also demonstrate that the expression of GALNT3 initiates mucin production and affects IAV replication in infected cells. This is the first report demonstrating the mechanism underlying the miRNA-mediated initiation of mucin-type O-glycosylation in IAV-infected cells and its role in viral replication. Our results have broad implications for understanding IAV replication and suggest a strategy for the development of novel anti-influenza approaches.
INTRODUCTION
Mucin-type O-linked glycosylation is a fundamental form of glycosylation that occurs in the Golgi apparatus in eukaryotes. This type of glycosylation is initiated by the UDP-GalNAc polypeptide:N-acetylgalactosaminyltransferase (ppGalNAcT) family and formed by GalNAc attachment to the serine/threonine (Ser/Thr) side chains of protein substrates (1–3). The generation of complex O-linked glycans is involved in a variety of biological molecular processes, such as cell motility, cell growth and death, cell-to-cell communication, the immune response, and host-pathogen interactions (4–6). On the other hand, abnormalities in O-linked glycan modifications have been shown to be linked to a variety of diseases, including cancers (7).
O-linked glycosylation also has been shown to be involved in various steps of viral infection. For some viruses, glycosylation of the cell surface plays a critical role in determining infectivity and transmissibility (8, 9). Furthermore, it has been reported that mutations and/or selection in the glycosylation sites of viral envelope glycoproteins during infection is a critical step to escape from the host immune system (10). These observations suggest that the patterns of the O-linked glycosylation of both the viral and host proteins are a key factor influencing viral virulence, in turn contributing to viral pathogenesis.
The most abundant and well-characterized O-glycosylation occurs on mucins, which are large cell-surface-bound and -secreted glycoproteins. In viral infections affecting the upper or lower respiratory tracts, such as influenza viruses and respiratory syncytial virus (RSV), the increased production of mucus, which is largely composed of mucins, is commonly induced in the epithelial surfaces of the respiratory tract (11, 12). Recent studies have revealed that mucins are upregulated in epithelial cells soon after infection by respiratory viruses, such as influenza A virus (IAV), both in vivo and in vitro. Moreover, mucin production in the respiratory tract during the early stage of viral infections is associated with characteristic clinical symptoms as well as viral elimination (12, 13). It also has been shown that the expression of MUC5B, which is the most abundant mucin in the airway epithelium in humans, is essential for mucociliary clearance and airway defense against microbial infections in mice (14). However, the details of how mucin expression is regulated during infection and how mucin production affects viral replication have not yet been fully elucidated.
In this study, we attempted to understand the early host cell response to IAV infection and its impact on IAV replication. Here, we show by microRNA (miRNA) microarray analysis that the expression of two miRNAs, miR-17-3p and miR-221, which target an UDP-GalNAc transferase, ppGalNAcT-3 (GALNT3), significantly decreases as early as 4.5 h postinfection with IAV, resulting in the upregulation of GALNT3 mRNA. The expression of GALNT3 was shown to be correlated with mucin production and O-linked glycosylation modification in human alveolar basal epithelial cells. Furthermore, we found that GALNT3 knockdown moderately but significantly reduced IAV replication. Notably, IAV replication was shown to be markedly decreased in mouse embryo fibroblast (MEF) cells obtained from galnt3-knockout (KO) mice. On the other hand, galnt3-KO mice exhibited severe pathological alterations in the lungs by IAV infection. Based on these results, we suggest that IAV infection induces the expression of a UDP-GalNAc transferase and mucin-type O-linked glycosylation modification via the miRNA machinery and also that GALNT3 plays a dual function in IAV infection, namely, enhancement of IAV replication and control of viral propagation in vivo.
MATERIALS AND METHODS
Cells and viruses.
A549, BEAS-2B (CRL-9609; American Type Culture Collection), MDCK, and HEK293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS). BEAS-2B cells stably expressing GALNT3 were generated as previously described (15). Briefly, an expression plasmid containing human GALNT3, pEF-hGALNT3, was transfected into BEAS-2B cells, and cell clones stably expressing GALNT3 were selected using 200 μg/ml zeocin (Invitrogen) and cultured in DMEM containing 10% FCS. HEK293T cells were transfected with the constructed plasmids using Lipofectamine 2000 (Invitrogen). A549, BEAS-2B, and HEK293T cells were infected with several IAV strains, including A/Puerto Rico/8/1934 (PR8 [H1N1]), A/WSN/1933 (WSN [H1N1]), A/Beijing/262/95 (Beijing [H1N1]), A/Suita/82/2011 (Suita [H3N2]), and A/Panama/2007/99 (Panama [H3N2]). UV-PR8 was generated by UV irradiation of PR8 for 15 min.
Plasmids.
The human GALNT3 expression plasmid pEF4-GALNT3 was used as reported previously (15). The Renilla luciferase reporter plasmid pRL-tk-GALNT3-3′ UTR, which contains the 3′ untranslated region (3′ UTR) of GALNT3 mRNA, was generated by inserting the 3′ UTR of GALNT3 using the In-fusion cloning system. The mutants of the miR-221 and miR-17-3p binding regions, pRL-tk-GALNT3-3′ UTR 221 mut and 17-3p mut, were generated from the wild-type plasmid using PCR-based mutagenesis. We determined the binding sites for miR-221 and miR-17-3p by using microRNA.org and GENETYX ver.10 software. The pPolI vector and pPolI-CAT-WSN, pCAGGS-PA, pCAGGS-PB1, pCAGGS-PB2, and pCAGGS-NP plasmids were used as described previously.
miRNA microarray analysis.
miRNA microarray analysis was carried out using an Agilent human miRNA microarray (V3). It contained 20 to 40 features targeting each of 866 human miRNAs and 89 viral miRNAs cataloged in the Sanger database (version 12.0; design ID 021827). Total RNA was extracted from infected cells at 0.5, 1.5, or 4.5 h postinfection using miRNeasy (Qiagen) and subjected to microarray analysis in duplicate. As a control, we used the total RNA extracted from uninfected A549 cells at one time point of 0.5 h. One hundred-nanogram aliquots of total RNA were used to make the miRNA probes as previously described (16). To identify significantly up- and downregulated miRNAs in the infected cells at 0.5, 1.5, or 4.5 h postinfection, one-way analysis of variance (ANOVA) (GeneSpring GX) with Tukey's honest-significant-difference (HSD) post hoc test was conducted to compare the differentially expressed miRNAs between IAV-infected and control cells (P < 0.05).
Titration of infectious units.
To determine viral titers, monolayers of MDCK cells in 96-well plates were infected with the trypsin-pretreated supernatants of IAV-infected cells for 1 h at 37°C, washed 3 times with phosphate-buffered saline (PBS), changed to DMEM-F12 containing 0.2% bovine serum albumin (BSA), and then incubated for 12 h at 37°C. At 12 h postinfection, the cells were washed 3 times with PBS and fixed with 100% ethanol for 3 min. Virus samples were pretreated with 1.0 μg/ml of acetylated trypsin for 1 h at 37°C. The viral titers were obtained using a focus-forming assay as previously described.
Immunofluorescence assay.
PR8-infected MDCK cells in 96-well plates were fixed with 100% ethanol, incubated with anti-NP antibody (C43; 1/1,000 dilution) for 1 h at 37°C, washed 4 times with PBS, and reacted with Alexa Fluor 488 anti-mouse antibody (1/1,000 dilution) for 45 min at 37°C. After being washed 4 times in PBS, the plates were coated with PBS containing 50% glycerol. To analyze the cell tropism of the WSN strain, differentiated human bronchial epithelial cells (HBECs), which are described in the three-dimensional cell culture subsection below, were infected with WSN for 9 h (multiplicity of infection [MOI] of 3.0), fixed with 4% paraformaldehyde for 15 min, and reacted with 0.4% Triton X-100 for 5 min. The fixed HBECs were transferred to glass slides and incubated with anti-MUC5AC (ab78660; 1/200 dilution) and anti-NP (C43; 1/500 dilution) for 1 h at 37°C. After 3 washes in PBS, the cells were incubated with Alexa Fluor 488 anti-rabbit and Alexa Fluor 555 anti-mouse antibodies (1/1,000 dilution) for 45 min at 37°C.
TaqMan microRNA assay.
The TaqMan microRNA reverse transcription (RT) kit (Life Technologies) was used for the reverse transcriptase reaction in a 15-μl mixture containing 10 ng RNA, 0.15 μl deoxynucleoside triphosphates (dNTPs) (100 mmol/liter), 1 μl MultiScribe RTase, 1.5 μl 10× RT buffer, 0.19 μl RNase inhibitor, 4.16 μl RNase-free water, and 3 μl RT primers. The reaction conditions were 16°C for 30 min, 42°C for 30 min, and 85°C for 5 min. Real-time quantitative PCR (qPCR) was carried out in a 20-μl mixture containing 10 μl TaqMan 2× Universal master mix (ABI), 1 μl 20× TaqMan microRNA mix, 7.67 μl distilled water, and 1.33 μl RT reaction product. The reaction conditions were 95°C for 10 min followed by 40 cycles of amplification (95°C for 15 s and 60°C for 60 s).
Transfection of miRNA mimics and siRNA.
After the harvested HEK293T cells were incubated in 12-well plates for 24 h at 37°C, 5 nmol/liter of miR-221 mimic, miR-17-3p mimic, or scrambled small RNA (si control) was transfected using HiPerfect reagent (Qiagen) at a concentration of 6 μl/ml. At 48 h posttransfection, total RNA was isolated using TRIzol reagent (Invitrogen). BEAS-2B cells were seeded in 12-well plates and incubated for 48 h at 37°C, and then 5-nmol/liter aliquots of siRNA against GALNT3 and GALNT1 (siGALNT3 and siGALNT1), miR-221 mimic, miR-17-3p mimic, and si control were transfected into the cells using HiPerfect reagent.
Dual-luciferase reporter assay.
The harvested HEK293T cells were transfected with pRL-tk-GALNT3-3′ UTR, pRL-tk-GALNT3-3′ UTR-221 mut, and 17-3p mut plasmids using Lipofectamine 2000 in 24-well plates. At 24 h posttransfection, the cell lysates were collected using a dual-luciferase reporter assay kit (Promega). We measured the luciferase activity using a dual-luciferase reporter assay. The relative luciferase activities were expressed as the ratio of the pRL-tk reporter activity to the pGL3 control plasmid activity.
Quantitative RT-PCR.
A Verso cDNA synthesis kit (Thermo) was used for reverse transcription in a 20-μl reaction mixture containing 1 μg RNA, 4 μl 5× buffer, 2 μl dNTP mix, 1 μl RT enhancer, 1 μl enzyme mix, and either 1 μl oligo(dT) primer or 1 μl Uni12 (an IAV genome-specific primer). The reverse transcriptase reaction was carried out at 70°C for 5 min, 50°C for 60 min (Uni12, 42°C for 60 min), and then 95°C for 2 min. qPCR was performed in a 20-μl mixture containing 5 μl cDNA, 10 μl Thunderbird SYBR qPCR mix (Toyobo), 1 μl forward primer, 1 μl reverse primer, and 3 μl distilled water. The reaction conditions were 95°C for 5 s and 60°C for 30 s for 40 cycles of amplification.
Isolation of murine bronchial and alveolar epithelial cells.
Female C57BL/6 mice (weight, 17 to 20 g) were intranasally inoculated with 103 PFU of PR8 virus. At 0 to 7 days postinfection, primary bronchial and alveolar epithelial cells were isolated as described previously (17–19). B6 mice were sacrificed by an overdose of mixed anesthesia (0.75 mg/kg of body weight of medetomidine, 4 mg/kg midazolam, and 5 mg/kg butorphanol tartrate). They were exsanguinated by cutting the inferior vena cava. The lungs were perfused with 10 ml 0.9% NaCl using a 10-ml syringe fitted with a 21-gauge needle through the right ventricle until the lungs were visually free of blood. A 20-gauge intravenous catheter was inserted into the trachea and secured tightly with a suture. The lungs were filled with 1.5 ml dispase (Corning) through the tracheal catheter. Low-melting-point agarose (1%, 0.2 ml, stored in a 45°C water bath) was infused through the catheter. The lungs were immediately covered with crushed ice, incubated for 2 min, removed from the ice, and incubated for 45 min at room temperature. They were transferred to 4 ml DMEM containing 1% penicillin-streptomycin, 25 mM HEPES, 10% FCS, and 100 μl of 1 mg/ml DNase (Sigma-Aldrich). The digested tissue was homogenized with gentleMACS (Miltenyi Biotec) and successively filtered through 70-μm and 40-μm Falcon cell strainers. The filtered suspension was centrifuged at 1,300 rpm for 8 min at 8°C.
The cells were incubated with 1 μg biotinylated anti-CD45, 0.5 μg biotinylated anti-CD31, 1 μg biotinylated of anti-CD16/32, and 0.5 μg biotinylated anti-CD90.2 for 20 min at 4°C. The cells were washed with magnetically activated cell sorting (MACS) buffer (Miltenyi Biotec) and centrifuged at 1,300 rpm for 5 min at 8°C. The pellet was resuspended with MACS buffer and incubated with 20 μl anti-biotin microbeads (Miltenyi Biotec) for 20 min at 4°C. The cells were washed again and centrifuged at 1,300 rpm for 5 min at 8°C. The cell suspensions were added to LS columns (Miltenyi Biotec) attached to a MidiMACS separator (Miltenyi Biotec). The columns were washed three times with 3 ml MACS buffer, and the eluates were collected in sterile tubes.
Western blot analysis.
The cells were mixed with SDS sample buffer and heated for 5 min at 98°C. The lysates were separated by SDS-PAGE on an acrylamide gel and blotted onto a polyvinylidene difluoride membrane (PVDF; Millipore). Immunoblot analyses were performed according to a standard protocol using Tris-buffered saline (TBS) containing 0.1% Tween 20 and 5% skim milk as a blocking solution and for antibody dilution. The following antibody dilutions were used: anti-GALNT3 antibody (Sigma-Aldrich), 1/1,000 dilution; anti-NP antibody, 1/1,000 dilution; anti-tubulin antibody, 1/1,000 dilution; and horseradish peroxidase (HRP)-conjugated secondary antibodies, 1/10,000 dilutions. The antibodies were visualized with ECL prime Western blotting detection reagents (GE Healthcare) and exposed in an ImageQuant LAS-4000 imager (Fujifilm). The protein levels on the Western blots were measured by densitometric scanning of the corresponding bands using Image Reader LAS-4000 software. The densitometric values for tubulin were used to standardize for equal protein loading among the samples.
Three-dimensional cell culture.
Primary HBECs were obtained from a commercial source (CC-2540S; Lonza) and cultured in growth medium (Lonza) supplemented with a B-ALI medium SingleQuot kit in T25 flasks. After two passages, the HBECs were placed in the upper chamber of transwell plates in differentiation medium (Lonza) and maintained for 30 days. During culture, the cells differentiated into ciliated cells and goblet cells, and increasing amounts of mucus were secreted on the surfaces of the cells.
Lectin microarray analysis.
Lectin microarray analysis (LecChip) was performed as previously described (20, 21) using membrane fractions extracted from IAV-infected A549 and BEAS-2B and recombinant GALNT3-expressing BEAS-2B cells. Briefly, a small aliquot of the protein fraction (200 ng) was labeled with Cy3-succinimidyl ester (Cy3-labeled glycoprotein). The LecChip lectin chip with 45 lectins (GP BioSciences) was incubated with the Cy3-labeled glycoprotein solution (100 μl) at concentrations of 0.25 and 0.5 μg/ml in probing buffer at 4°C until the binding reached equilibrium. The net intensity value for each spot on the microarray was calculated by subtracting a background value from the signal intensity, and then the net signal intensity values of the three spots were averaged. The lectin microarray data for each cell type were processed by the microarray system using a maximum normalization procedure after a gain-merging process.
Minireplicon assay.
HEK293T cells were seeded in 12-well plates and transfected with plasmids containing 0.125 μg pCAGGS-PA, 0.125 μg pCAGGS-PB1, 0.125 μg pCAGGS-PB2, 0.125 μg pCAGGS-NP, and 0.5 μg pPolI-CAT-WSN minigenome template using Lipofectamine 2000. At 24 h posttransfection, the cells were lysed and the lysates were prepared for chloramphenicol acetyltransferase (CAT) assay using a CAT enzyme-linked immunosorbent assay (ELISA) kit (Roche).
Primary mouse embryo fibroblast cells.
Primary MEFs were isolated from E17 wild-type (WT) and galnt3-KO mouse embryos by trypsin digestion at 37°C for 10 min. The cell suspension was cultured in DMEM with 10% FCS. After genotyping the embryos, the MEF cells were seeded in 12-well culture dishes and inoculated with PR8 (H1N1) and Suita (H3N2) viruses at multiplicities of infection (MOI) of 3.0 and 1.0, respectively.
Infection of mice.
WT and KO (Galnt3−/−) C57BL/6 mice (6- to 7-week-old females) were challenged with 103 PFU of PR8 virus. For each mouse, 30 μl of virus solution diluted in PBS was inoculated intranasally. The same volume of PBS was used for mock infection. For each experimental group, the body weights of 5 mice were monitored daily. Lung homogenates were collected at 0 to 7 days postinoculation (dpi) to measure the viral titers activated in vivo by focus formation assay. Three mice were used for each experimental group, and samples of their right or left lungs were separately collected from individual mice. For histopathological examinations, WT and galnt3-KO mice were infected with 103 PFU of the PR8 strain and autopsied at 6 dpi. Three mice were used for each experimental group.
Histology.
For light microscopy analysis, female mice were anesthetized by intraperitoneal injection of mixed anesthesia and fixed with Bouin solution at 4°C overnight. Dissected right or left lungs were embedded in paraffin. The sections were stained with hematoxylin and eosin (H&E) according to a standard protocol. Histological evaluations were carried out by the New Histo Science Laboratory Company. The inflammation symptoms of the lungs were scored based on the method of a previous study (22).
Primers and probes.
The primers and probes used in this study are available upon request.
Ethics statement.
Experimental infection of the galnt3-KO mice was conducted by following the guidelines for the Care and Use of Laboratory Animals of the Ministry of Education, Culture, Sports, Science and Technology, Japan. Animal experiments were carried out at the Institute for Virus Research in Kyoto University (IVRKU) (protocol numbers D15-11, D14-11, and D13-07) after approval by the Committee on the Ethics of Animal Experiments of IVRKU in accordance with the guidelines for animal experiments at IVRKU.
Microarray data accession number.
The microarray data were deposited in the NCBI GEO database under accession number GSE57508.
RESULTS
Downregulation of miRNAs targeting GALNT3 mRNA during the early stage of influenza A virus infection.
An increasing number of studies have suggested that cellular miRNAs play an important role in various host cell responses to viral infection (23–25). To understand the host cell responses during the early stage of IAV infection, we performed an miRNA microarray analysis using a human alveolar adenocarcinoma cell line, A549, infected with influenza A/Puerto Rico/8/34 H1N1 (PR8) virus. We isolated the cellular RNAs at 0.5, 1.5, and 4.5 h postinfection and detected significant changes in the global profile of miRNA expression after infection with IAV. Specifically, we detected 23 miRNAs that were differentially expressed at a statistically significant level (P < 0.05) between IAV-infected cells and uninfected control cells (Fig. 1A). Among the differentially expressed miRNAs, 20 and 3 miRNAs were significantly downregulated or upregulated, respectively, at 4.5 h postinfection. Heat map analysis showed the changes in the miRNA expression during the observation periods in IAV-infected cells (Fig. 1A). By using Ingenuity pathway analysis (IPA; Ingenuity Systems) to ensure the quality of the data, we predicted the pathways controlled by the miRNAs and identified two Homo sapiens (hsa) miRNAs, miR-17-3p and miR-221, that showed a high degree of homology to the sequences of a UDP-GalNAc transferase (GALNT3) mRNA.
FIG 1.
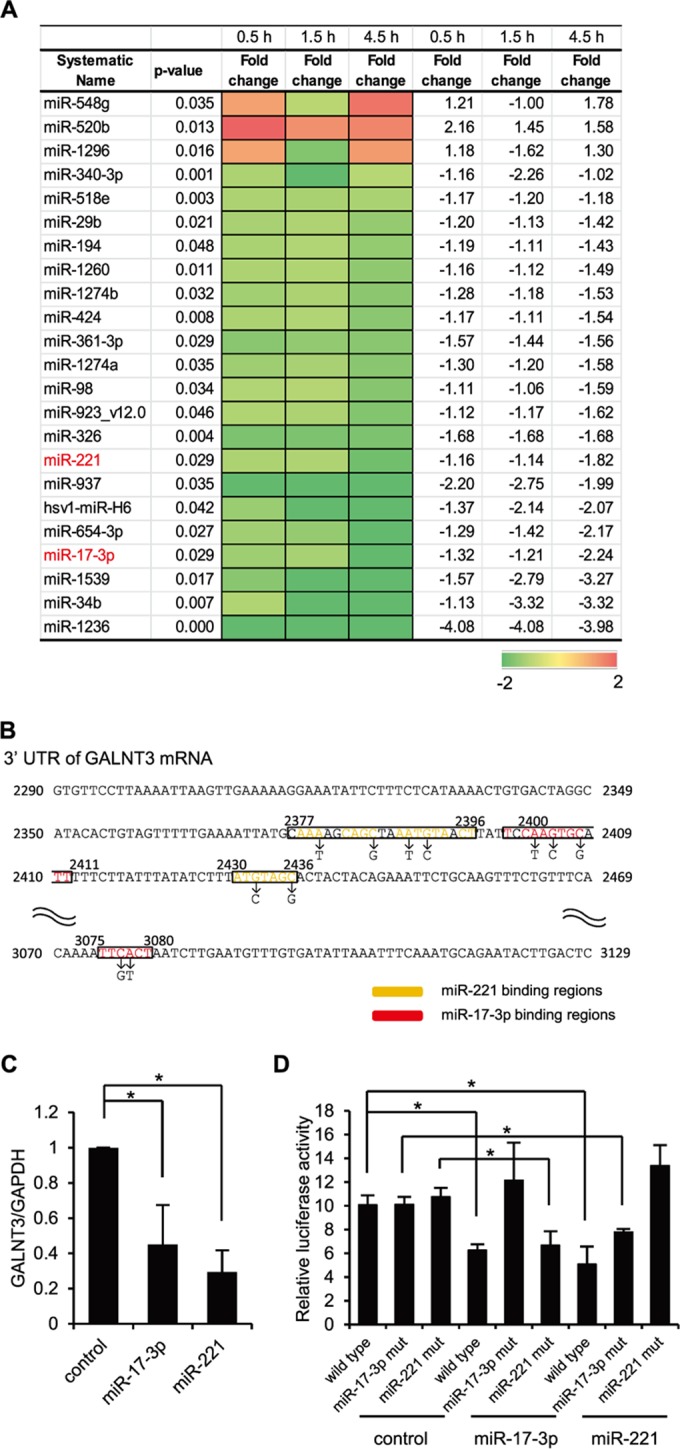
Downregulation of miR17-3p and miR-221 during the early stage of IAV infection. (A) A heat map comparing the fold changes of microRNAs with significantly higher (red) or lower (green) expression in A549 cells infected with PR8 virus at an MOI of 1.0. The P values for the fold changes are indicated. (B) Sequences of predicted miR-17-3p and miR-221 binding sites in the 3′ UTR of GALNT3 mRNA. The black letters below the predicted binding sites (boxed) indicate the nucleic acid substitutions in the mutant plasmids used for the luciferase assay. (C) The reduction of GALNT3 mRNA expression by miR-17-3p and miR-221 mimic oligonucleotides. HEK293T cells were transfected with oligonucleotides mimicking miR-17-3p and miR-221, and the expression of GALNT3 mRNA was detected by real-time PCR. The relative expression levels of GALNT3 and GAPDH are shown (control, 1.0). Control indicates scrambled control miRNA. (D) miR-17-3p and miR-221 target the 3′ UTR of GALNT3 mRNA. Plasmids containing point mutations in the putative miR-17-3p and miR-221 binding sites or miR-17-3p mut and miR-221 mut were transfected into HEK293T cells after the introduction of the mimic oligonucleotide. The luciferase activity in the transfected cells was monitored 24 h after transfection. The y axis indicates the luciferase activity of the pRL-tk reporter relative to that of the pGL3 control plasmid. All values are expressed as the means (n > 3) ± standard errors of the means (SEM). Statistical significance was analyzed by Student's t test. *, P < 0.05.
GALNT3 belongs to the largest family of UDP-GalNAc transferases catalyzing the O-glycosylation of mucin or mucin-like proteins. Although the involvement of GALNT3 in viral infections has not been demonstrated yet, recent evidence showed that the UDP-GalNAc transferase families may play important roles in viral replication (26, 27). Therefore, we next investigated the regulation of GALNT3 in IAV infection. At first, we analyzed the expression of GALNT3 mRNA by real-time PCR after the transfection of the oligonucleotide mimicking the identified miRNA into HEK293T cells to investigate whether miR-17-3p and miR-221 actually regulate the expression of GALNT3 mRNA. As shown in Fig. 1C, the mimic sequences significantly decreased the expression of GALNT3 mRNA. We next searched for the putative miRNA-binding sites in the GALNT3 gene and found that the 3′ UTR of GANLT3 mRNA contains the putative sites for both miR-17-3p and miR-221 (Fig. 1B). To confirm the miRNA binding and their function in regulating the GALNT3 gene, we designed a reporter plasmid, pRL-tk-GALNT3-3′UTR, in which the 3′ UTR of GALNT3 mRNA was subcloned downstream of the luciferase open reading frame. The mutant plasmids, pRL-tk-GALNT3-3′UTR 17-3p mut and 221 mut, which contain point mutations in the putative binding sites for miR-221 and miR-17-3p, also were generated (Fig. 1B). HEK293T cells were transfected with mimic oligonucleotides of miR-17-3p or miR-221, and then the pRL-tk-GALNT3 vectors, either the wild type or the mutants, were introduced. As shown in Fig. 1D, the expression of miRNA mimic sequences significantly reduced the luciferase activity in cells transfected with the wild-type pRL-tk-GALNT3-3′UTR plasmid. In contrast, the luciferase activity of the 3′ UTR mutant plasmids was not decreased in the cells transfected with the mimic sequences corresponding to the mutants (Fig. 1D). These results demonstrated that the binding sites in the 3′ UTR of GALNT3 mRNA are critical for the miR-17-3p- and miR-221-mediated repression of luciferase activity from the pRL-tk vectors and also that both miR-17-3p and miR-221 could be the regulatory miRNAs for GALNT3 mRNA in IAV-infected cells.
Replication of influenza A virus rapidly induces the expression of GALNT3 mRNA.
To confirm the downregulation of miR-17-3p and miR-221 by IAV infection, A549 cells were infected with PR8 virus, and the expression levels of the miRNAs were monitored for up to 4.5 h postinfection. Consistent with the microarray analysis, the expression of both miR-17-3p and miR-221 was significantly downregulated by the infection (Fig. 2A). We next investigated whether IAV infection actually affects the expression of GALNT3. We utilized real-time PCR to detect the mRNA levels of GALNT3 in A549 and BEAS-2B cells infected with the PR8 virus. At an MOI of 3.0, the expression of GALNT3 mRNA was significantly upregulated in the cells by 4.5 h postinfection (Fig. 2B). Notably, BEAS-2B cells appeared to vastly induce the expression of GALNT3 mRNA by IAV infection because of the low-level expression in the uninfected status. The infection of A549 cells with H1N1 (A/Beijing/262/95) and H3N2 (A/Panama/2007/99 and A/Suita/82/2011) IAV (MOI of 3.0) also was shown to induce GALNT3 mRNA by 4.5 h postinfection (Fig. 2C, D, and E). We confirmed the increase of GALNT3 protein in the bronchial and alveolar epithelial cells isolated from C57BL/6 mice that were inoculated with the PR8 virus at 103 PFU. As shown in Fig. 2F, the expression of GALNT3 protein gradually increased with the replication of IAV in lung cells during the observation period of 7 days postinoculation. We also observed that infection with the PR8 virus did not induce the upregulation of GALNT1, GALNT2, GALNT6, or GALNT7 (Fig. 2G). These observations suggest that both GALNT3 mRNA and protein are specifically upregulated by IAV infection.
FIG 2.

Upregulation of GALNT3 mRNA by IAV replication. (A) Downregulation of miR-221 and miR-17-3p in A549 cells infected with PR8 virus. A549 cells were infected with PR8 virus at an MOI of 3.0, and the expression levels of miR-221 and miR-17-3p were monitored at the indicated time points by real-time RT-PCR. (B to E) IAV infection upregulates GALNT3 mRNA. (B) A549 and BEAS-2B cells were infected with H1N1 PR8 virus at an MOI of 3.0. A549 cells were infected with H1N1 Beijing (C), H3N2 Panama (D), and H3N2 Suita (E) viruses at an MOI of 3.0. The expression of GALNT3 was monitored by real-time PCR at the indicated times postinfection. Samples were normalized to GAPDH mRNA, and the relative expression levels of GALNT3 are shown (0 h = 1.0). (F) Upregulation of GALNT3 protein in bronchial and alveolar epithelial cells isolated from IAV-infected mice. After the intranasal infection of C57BL/6 mice with H1N1 PR8 virus at 103 PFU, the expression of the GALNT3 and IAV NP proteins in lung cells was detected by Western blotting. The line graphs indicate the relative band intensities of each protein compared to that of tubulin. The intensities of the bands were determined using a LAS-4000 imager (Fujifilm). (Left) GALNT3 protein; (right) IAV NP protein. (G) IAV infection does not change the mRNA expression levels of GALNT1, GALNT2, GALNT6, or GALNT7. A549 cells were infected with PR8 virus at an MOI of 3.0, and the mRNA expression levels of GALNT1, GALNT2, GALNT6, and GALNT7 were monitored at the indicated times by real-time RT-PCR. Samples were normalized to GAPDH mRNA, and the relative expression levels of GALNTs are shown (mock, 1.0). (H and I) Viral replication is necessary for the upregulation of GALNT3 mRNA. A549 cells were inoculated with UV-irradiated PR8 virus (UV-PR8) at an MOI of 3.0, and the replication of the inoculated virus was detected by immunofluorescence analysis using an anti-NP antibody at 12 h postinfection. Scale bar, 30 μm. The expression of GALNT3 mRNA was determined via real-time RT-PCR. All values are expressed as the means (n > 3) ± SEM. Statistical significance was analyzed by Student's t test. *, P < 0.05; **, P < 0.01.
We next sought to determine whether IAV replication in the cells is essential for the upregulation of GALNT3 mRNA. To this end, the UV-irradiated PR8 virus was inoculated into A549 cells, and the expression of GALNT3 mRNA was determined at 4.5 h postinfection. An immunofluorescence analysis with anti-NP antibody revealed that the UV-irradiated PR8 virus could not replicate in the cells (Fig. 2H). Notably, under this condition, the increased expression of GALNT3 mRNA was not observed in the cells (Fig. 2I). We showed that the adsorption of UV-irradiated virus to the cells was not different from that of wild-type virus, suggesting that the replication of IAV within the cells is necessary for the upregulation of GALNT3.
GALNT3 expression is associated with mucin production and the modification of O-linked glycosylation in IAV-infected cells.
The aforementioned results indicate that IAV infection rapidly induces GALNT3 expression in infected cells. Because GALNT3 initiates the O-glycosylation of mucin or mucin-like proteins, it is conceivable that the upregulation of GALNT3 by IAV replication leads to mucin production, as well as to O-glycosylation modification, in infected cells. To understand the roles of the induction of GALNT3 in IAV-infected cells, we next monitored mucin production in differentiated HBECs infected with IAV using an air-liquid interface culture system. Previous studies have revealed that the HBECs gradually differentiate into ciliated epithelial and goblet cells. It has been shown that goblet cells are a main source of mucins in the airways and that IAV primarily infects the goblet cells that express an abundance of MUC5AC (28). In fact, we detected IAV infection only in the MUC5AC-positive goblet cells in the HBEC culture (Fig. 3A). To investigate mucin production following IAV infection, the HBECs infected with the WSN strain were analyzed by real-time PCR. This analysis revealed that the infection significantly induced the expression of the mRNAs encoding GALNT3 and MUC1 (Fig. 3B and C). There was no difference between the numbers of goblet cells in the infected and uninfected differentiated HBECs, suggesting that the production of GALNT3 following IAV infection most likely is associated with mucin production in infected goblet cells.
FIG 3.

GALNT3 affects mucin production and O-linked glycosylation in IAV-infected cells. (A) IAV infects goblet cells expressing MUC5AC. Differentiated HBECs were infected with WSN virus at an MOI of 3.0. Nine hours after inoculation, virus-infected cells were detected by immunofluorescence analysis using anti-MUC5AC and anti-NP antibodies. Scale bar, 30 μm. A high-magnification image of an infected cell (square) is also shown. (B and C) The relative expression levels of GALNT3 (B) and MUC1 (C) mRNAs in HBECs were detected at 9 h postinfection. Statistical significance was analyzed by Student's t test. *, P < 0.05. (D and E) Lectin microarray analysis of PR8 virus-infected A549 (upper) and BEAS-2B (lower) cells (D) and recombinant GALNT3-expressing BEAS-2B cells (E). The cells were infected with PR8 virus at an MOI of 3.0, and samples were isolated at 12 h postinfection. Each graph was generated according to lectin group. The lectin groups are colored according to their recognition of the sugar chains indicated to the right of the graphs. Fuc, l-fucose; Man, d-mannose; SA, sialic acid; GlcNAc, N-acetyl-d-glucosamine; Gal/GalNAc, d-galactose/N-acetyl-d-galactosamine. The membrane fractions extracted from infected and uninfected cells were labeled and incubated with a LecChip representing 45 lectins. The net intensity value for each spot of the microarray was calculated by subtracting a background value from the signal intensity. The net signal intensity values of three spots were averaged. All values are expressed as the means (n > 3) ± SEM. *, P < 0.05.
To understand whether GALNT3 expression modifies O-linked glycosylation, we next performed lectin microarray analysis (LecChip) with the membrane proteins from the IAV-infected and uninfected A549 and BEAS-2B cells. In addition to the alteration of glycosylation by the IAV infection, we also monitored the modification in BEAS-2B cells stably expressing GALNT3 (15). The binding of Cy3-labeled glycoprotein was performed in triplicate for the 45 different lectins. The data then were analyzed with the Array-Pro analyzer (Media Cybernetics). The results were shown with the gain set to register a maximum net intensity of <50,000 for the most intense spots. In the IAV-infected cells, we observed that the lectins that recognize sialic acids, such as MAL (Maackia amurensis lectin), MAH (Maackia amurensis hemagglutinin), and TJA-I (Trichosanthes japonica agglutinin I), were significantly decreased, indicating that the membrane-bound sialic acids presumably are cleaved by the viral neuraminidase (NA) in the infected cells (Fig. 3D; also see Table S1 in the supplemental material). In association with the cleavage of the membrane-bound sialic acids, microarray analysis revealed that lectins specifically recognizing O-linked glycosylation, including RCA120 (Ricinus communis agglutinin 120), BPL (Bauhinia purpurea lectin), TJA-II (Trichosanthes japonica agglutinin), PNA (peanut agglutinin), and WFA (Wisteria floribunda lectin), were markedly increased in the cells infected with the PR8 virus (Fig. 3D; also see Table S1). Notably, the membrane glycoproteins from the cells stably expressing GALNT3 showed significant increases mainly in the lectins recognizing O-linked glycosylation, the upregulation of which also was observed in the IAV-infected cells (Fig. 3E; also see Table S1). These observations suggest that the upregulation of GALNT3 is involved in the modification of O-linked glycosylation, as well as in mucin production, in IAV-infected cells.
Expression of GALNT3 positively regulates influenza virus replication.
A previous study suggested that during IAV infection, mucin acts as an immobilization substrate for the virus on the cell surface (29). Furthermore, a recent study revealed that increased MUC5AC secretion appears to play a protective role against influenza virus infection (12). However, it remains unknown whether the modification of mucin-type O-linked glycosylation in infected cells affects IAV replication as well as viral production. Therefore, we next sought to determine the effect of GALNT3 expression on IAV replication. First, we monitored the time course of the expression levels of GALNT3 mRNA and viral genomic RNA in A549 and BEAS-2B cells after infection with the PR8 virus. As shown in Fig. 4A and B, the induction of the GALNT3 mRNA was highly correlated with the release of viral genomic RNA in the culture supernatants, suggesting that the expression of GALNT3 is associated with IAV replication in these cells.
FIG 4.

GALNT3 expression upregulates IAV replication. (A and B) Correlation between the expression of GALNT3 mRNA and viral replication. The induction of GALNT3 mRNA and viral RNA expression in PR8 virus-infected A549 (A) and BEAS-2B (B) cells was monitored by real-time RT-PCR at the indicated time points. The viral NP segment was used for quantification. The virus was infected at an MOI of 3.0. The viral titers in the culture supernatants were determined by focus-forming unit assays. (C to F) Knockdown of GALNT3 mRNA reduces IAV replication. BEAS-2B cells treated with siRNAs against GALNT3 (siGALNT3) and GALNT1 (siGALNT1) and with mimic oligonucleotides (miR-17-3p and miR-221). At 48 h posttransfection, the levels of GALNT3 (C) and GALNT1 (D) were monitored by qRT-PCR. The cells treated with siRNA or mimic oligonucleotides were infected with PR8 virus at an MOI of 3.0, and the levels of the viral genome and viral titer in the culture supernatants were determined at 4.5 h (E) and 24 h (F) after infection by qRT-PCR and FFU assays, respectively. For viral RNAs, samples were normalized to GAPDH mRNA. The results are expressed in arbitrary units. The scramble siRNA (si control) was used as a control. (G) Overexpression of GALNT3. BEAS-2B cells were transfected with a human GALNT3 expression plasmid, and at 48 h after transfection, the level of GALNT3 mRNA was monitored by quantitative RT-PCR. (H) GALNT3 overexpression does not affect IAV replication. Cells were infected with PR8 virus at an MOI of 3.0, and IAV replication was monitored by the level of the viral genome and the viral titer at 4.5 h postinoculation. The values are presented as means ± SEM from at least three independent experiments. *, P < 0.05; **, P < 0.01.
To elucidate the role of GALNT3 in IAV replication, the effects of small interfering RNAs (siRNAs) against GALNT3 and of the oligonucleotides mimicking miR-17-3p and miR-221 on PR8 viral replication were examined in BEAS-2B cells. We examined the level of viral genomic RNA and the viral titers in the infected cells at 4.5 and 24 h after inoculation. The effects of GALNT3 and GALNT1 knockdown by the siRNAs and mimic miRNAs were verified (Fig. 4C and D). At 4.5 h postinoculation, the knockdown of GALNT3, but not GALNT1, markedly reduced the viral genomic RNA and viral titer by approximately 50% and 75%, respectively, compared to those of the control cells (Fig. 4E). Consistent with this result, treatment with the sequences mimicking miR-17-3p and miR-221 prior to viral inoculation also reduced the level of viral RNA and the viral titer compared to those of untreated cells (Fig. 4E). However, we could not detect significant differences in the viral genomic RNA and viral titer at 24 h postinfection (Fig. 4F). We found that the overexpression of GALNT3 did not appear to significantly influence IAV replication at 4.5 h postinoculation (Fig. 4G and H), indicating that the expression level of endogenous GALNT3 is sufficient to stimulate viral replication in the cells. These results suggested that the upregulation of GALNT3 significantly affects the efficiency of the early infection of IAV.
We next investigated whether GALNT3 is directly involved in IAV polymerase activity. To this end, we used an IAV minireplicon assay system, which is the reconstitution of the three viral ribonucleoprotein complex polymerase subunits, NP, and synthetic viral RNA encoding the CAT gene in transfected cells. Prior to transfection with the minireplicon plasmids, we introduced either GALNT3 or GALNT1 siRNA into HEK293T cells and measured the CAT activity in the cells at 24 h after transfection. Notably, treatment with GALNT3 siRNA, but not GALNT1 siRNA, significantly reduced the CAT activity compared to that of the control scrambled siRNA (Fig. 5). Together, these observations suggest that the upregulation of GALNT3 affects IAV polymerase activity in the cells, thereby facilitating viral replication.
FIG 5.
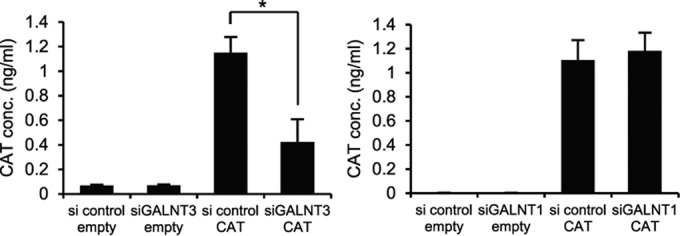
Knockdown of GALNT3 mRNA affects IAV minigenome replication. The IAV minireplicon assay was performed in HEK293T cells transfected with siGALNT3 and siGALNT1. Twenty-four hours after minigenome transfection, CAT activity was determined using a CAT ELISA system. conc., concentration. As a control, an empty vector (empty) was transfected instead of the minigenome plasmid. All values are presented as the means (n > 3) ± SEM. Statistical significance was analyzed by the Student's t test. *, P < 0.05.
Galnt3−/− mice reveal the dual function of GALNT3 during influenza virus infection.
To confirm the effect of GALNT3 expression on IAV replication, we obtained Galnt3−/− mice (30) and analyzed IAV replication in MEF cells. We used PR8 and H3N2-Suita viruses to infect MEF cells derived from WT and Galnt3−/− mice and monitored the levels of viral genomic RNA and titers for up to 4.5 h postinfection. As shown in Fig. 6A and B, both the viral genomic RNA and the viral titer were significantly decreased in the Galnt3−/− MEF cells from the early time points after inoculation, suggesting that GALNT3 positively regulates IAV replication during the early stage of infection.
FIG 6.
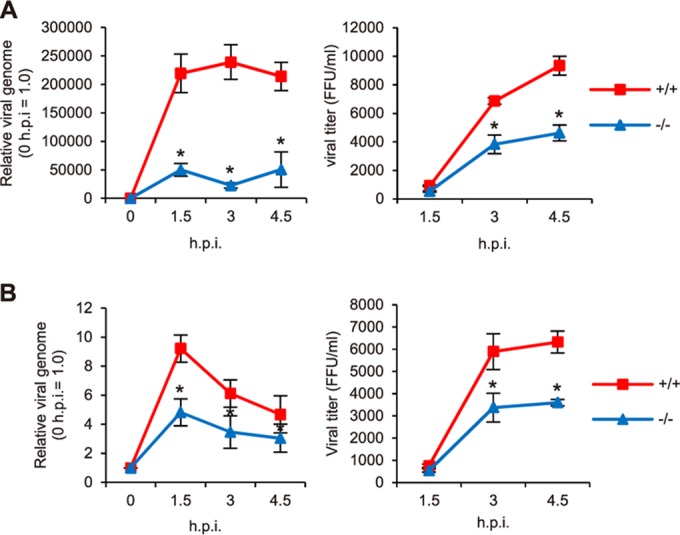
IAV replication in Galnt3−/− MEF cells. (A and B) The replication of PR8 (A) and Suita (B) viruses in MEF cells derived from WT and Galnt3−/− mice was monitored by the level of viral genomic RNA and the titer at the indicated times. Samples were normalized to GAPDH mRNA. +/+, WT; −/−, Galnt3−/− mice. All values are presented as the means (n > 3) ± SEM. *, P < 0.05.
We experimentally infected Galnt3−/− mice with the PR8 virus. The virus was intranasally inoculated into 6- to 7-week-old WT and Galnt3−/− mice at 103 PFU. We confirmed the lack of GALNT3 expression in the lungs of Galnt3−/− mice by Western blotting (Fig. 7A) and observed no histological difference between WT and Galnt3−/− mouse lungs before inoculation (Fig. 7B). Following inoculation with the PR8 virus, interestingly, a significant body weight loss (<40%) occurred in all KO mice by 7 dpi, whereas WT mice showed only a <15% reduction by 14 dpi (Fig. 7D). Furthermore, all KO mice inoculated with 103 PFU of PR8 virus died by 11 dpi. In contrast, no fatality was observed in WT mice during the observation period of 14 dpi (Fig. 7E).
FIG 7.

Role of GALNT3 in IAV pathogenicity in vivo. (A) The expression of GALNT3 protein in WT and Galnt3−/− mice. The lungs of 6-week-old WT and KO mice were removed and subjected to Western blot analysis using anti-Galnt3 antibody. GALNT3 protein size, 73 kDa. (B) Histological analysis of the lungs of uninfected, 6-week-old WT and Galnt3−/− mice. The data were obtained by H&E staining (magnification, ×10). (C) Histopathological findings in the lungs of WT and Galnt3−/− mice infected with PR8 IAV. The data obtained by H&E staining (magnification, ×4) are shown. The inflammation scores of individual mice (n = 3) are shown at the bottom of each panel: 0, no apparent changes; 1, minimal changes or bronchiolitis; 2, bronchiolitis and/or slight alveolitis; 3, mild alveolitis with neutrophils, monocytes/macrophages, or lymphocytes; 4, moderate alveolitis. (D and E) WT and Galnt3−/− mice were intranasally inoculated with H1N1 PR8 virus at 103 PFU (n = 3). (D) Body weights were measured daily. Error bars represent standard deviations. (E) Survival curves of IAV-infected mice. Each experimental group consisted of 3 mice. d.p.i, days postinoculation. (F and G) Viral titers in the lungs of IAV-infected mice. Lung homogenates were obtained every day and were used for virus titration. For each day, 3 mice were used. Error bars represent standard deviations. Panels F and G are independent experiments. (F) Viral titers during the early inoculation periods up to 3 dpi.
Figure 7C shows the histopathology of lungs of mice infected with 103 PFU of the PR8 virus. The Galnt3−/− mice showed alveolus and peribronchial inflammation with the infiltration of inflammatory cells, especially macrophages, and also developed alveolitis by 6 dpi. In contrast, the lungs of WT mice infected with the same virus exhibited significantly reduced levels of inflammation (Fig. 7C). To verify the viral propagation in the lungs, lung homogenates were harvested every day and used to determine the viral titers. Although the lungs of Galnt3−/− mice exhibited delayed increases in the viral titers until 3 dpi (Fig. 7F), the viral titer in the lungs of KO mice rapidly increased and were much higher than those in the lungs of WT mice after 4 dpi (Fig. 7G). These data suggested the efficient propagation of the virus in the lungs of Galnt3−/− mice.
DISCUSSION
In this study, we demonstrated that IAV upregulates the expression of GALNT3 via the miRNA machinery in human lung epithelial cell lines and HBECs. This report is the first to show that IAV infection affects the expression of GalNAc transferase, which is responsible for the initial step of mucin-type O-glycosylation. We also found that the upregulation of GalNAc transferase is involved in both O-linked glycosylation and IAV replication in infected cells. Although it has been known that the alteration of mucin production and/or the O-glycosylation of cellular receptors is critical for the adsorption of virions onto the cell surface, elucidating the role of mucin-type glycosylation in viral replication may provide new insights into the life cycle of IAV.
Understanding the initial step of IAV infection is important for the development of a strategy to suppress the multiplication of IAV in vivo. We used an miRNA microarray analysis to investigate the cellular responses during the early steps of IAV infection and found that several miRNA pathways were markedly altered in human lung epithelial carcinoma cells (Fig. 1). Recent studies have demonstrated the changes in miRNA expression that are directed by IAV infection (23, 25). Buggele et al. revealed the induction of primary miRNA expression in human respiratory cell lines, including IAV-infected A549 and BEAS-2B cells, but they detected miRNAs beginning only at 8 h postinfection (23). Furthermore, many studies have focused mainly on the regulation of antiviral responses by the miRNA machinery in the cells infected with viruses, including IAV (31). In this study, we focused on the miRNAs that mediate posttranslational modifications, such as glycosylation, of cellular proteins during the early stage of IAV infection and investigated two specific miRNAs, miR-17-3p and miR-221, both of which were significantly downregulated by IAV infection. We found that both miRNAs target the GalNAc transferase GALNT3 and are downregulated as early as 4.5 h postinfection. Note that it is unclear how viral infection suppresses these miRNAs in infected cells. Because these miRNAs are downregulated at an early time point following infection, it is conceivable that host responses to virus infection, such as innate immune responses, trigger the suppression of these miRNAs in infected cells.
We demonstrated that the 3′ UTR of GALNT3 mRNA is the target of the miRNAs and that the expression level of GALNT3 mRNA is actually reduced by the expression of the miRNA mimics (Fig. 1 and 2), providing evidence for the miRNA-mediated regulation of GALNT3 expression. In previous studies, it has been revealed that both miRNAs regulate cellular molecules other than GALNT3. Interestingly, miR-17-3p is also thought to target the mRNA of another UDP-GalNAc transferase, GALNT7, and may induce the development of hepatocellular carcinoma (32). This observation suggests that miR-17-3p is a key regulator of GalNAc transferase expression in other cell or tissue types, although we did not observe changes in GALNT7 mRNA expression in A549 cells infected with IAV (Fig. 2). In addition, miR-17-3p has been reported to have several potential targets, including GFRA2 (GDNF family receptor alpha 2) (33), Par4 (Prostate apoptosis response-4) (34), TIMP3 (TIMP metallopeptidase inhibitor 3) (35), MDM2 (murine double minute 2) (36), and Olig2 (Oligodendrocyte transcription factor) (37), all of which are known to be involved in cell proliferation or viability. In this study, however, we did not assess the involvement of these genes in IAV replication. On the other hand, it is noteworthy that the expression of miR-221 also is downregulated in HEBCs infected with RSV, resulting in the upregulation of nerve growth factor (NGF) and the promotion of viral replication in infected cells (24). This finding suggests that the miR-221 pathway commonly is regulated during infection with respiratory viruses, such as IAV and RSV. A recent study showed that miR-221 accentuates the effect of interferon (IFN) on the hepatitis C virus (HCV) by targeting SOCS1 and SOCS3 (38). Furthermore, miR-221 is known to be an oncogenic miRNA that targets many genes associated with cellular proliferations, including PTEN (39), p27 (Kip1) (40), and TIMP2 (41). Therefore, it would be interesting to investigate whether the downregulation of miR-221 also affects the expression of such molecules in IAV-infected cells, particularly respiratory epithelial cells.
Our observation suggested a functional role for O-linked glycosylation modification in the early steps of IAV infection. In this study, we demonstrated that the altered expression of GALNT3 protein in mouse lung cells infected with IAV, but not in infected cell lines, such as A549 and BEAS-2B cells, likely was due to its basal expression level in the cell lines. Nevertheless, mucin production and O-linked glycosylation were detected in cultured cells infected with IAVs (Fig. 3). The lectin microarray analysis demonstrated similar alterations to the O-linked glycosylation between the cells infected with IAV and those stably expressing recombinant GALNT3, strongly suggesting that IAV infection affects the posttranslational modification and formation of mucin-type glycoproteins by regulating the GALNT3 machinery. Our results suggest a mechanism that explains how O-linked glycosylation modification or mucin production is rapidly initiated in respiratory epithelial cells infected with IAV, which could be the first example of the molecular regulation of mucin production during respiratory virus infections.
Recent evidence has demonstrated that O-linked glycosylation modifications in virus-infected cells are critically involved in the various stages of infection. In many studies, glycosylation has been shown to affect the viral attachment or penetration steps via the modification of cellular receptors or viral envelope glycoproteins. For instance, the O-linked glycoproteins of α2,3- or α2,6-linked sialic acids were shown to play important roles during infection by IAV and other viruses, including the coxsackievirus A24 variant and enterovirus 70 (42–45). Furthermore, the envelope proteins of many viruses, such as human immunodeficiency virus type 1, HCV, Ebola virus, and herpes simplex virus 1, have been shown to be modified by O-linked glycosylation, which influences the attachment of viral virions to the cell surface (42, 46–48). These observations suggest that O-linked glycosylation is an important modification in virus-infected cells that directly or indirectly controls various steps of viral replication. Therefore, it would be interesting to investigate changes in GalNAc transferase expression during other viral infections to understand the regulation of O-linked glycosylation in infected cells.
In this study, we showed that UV-treated IAV virions could not induce GALNT3 expression in A549 cells (Fig. 2). Furthermore, the upregulation of GALNT3 appeared to be correlated with the multiplication of IAV (Fig. 4). These findings suggested that viral replication, but not the cellular association of viral particles, is necessary for the induction of GALNT3 and also that the upregulation of GALNT3 affects the IAV replication. In fact, using siRNA and mimic miRNA we demonstrated that the expression of GALNT3 markedly increases the efficiency of virus replication, although the effect of GALNT3 knockdown was modest. In addition, we showed that the replication of IAV is significantly impaired in MEF cells derived from the galnt3-KO mice (Fig. 6). From all these observations, it could be reliably stated that the expression of GALNT3 facilitates viral replication at early time points postinfection, suggesting an important role for GALNT3 in IAV infection. The upregulation of GALNT3 in IAV-infected cells (Fig. 4), in addition to the IAV minigenome assay (Fig. 5), revealed that O-linked glycosylation via GALNT3 expression might be required at multiple steps of the viral life cycle. It is possible that the O-linked glycosylation of certain host factors is involved in late steps of viral replication, including nuclear transport, assembly, and viral RNP budding.
GALNT3 conceivably is involved in IAV replication via the O-linked glycosylation of specific cellular proteins. The O-linked glycosylation of specific cellular substrates is known to affect various cellular processes, such as protein folding, cell adhesion, signal transduction, protein trafficking, cell proliferation, apoptosis, and cancer (2, 49, 50). At present, only a few specific substrates for GALNT3 have been identified. Fibroblast growth factor 23 (FGF23), which is a bone marrow-derived hormone that regulates and is regulated by the blood levels of phosphate and active vitamin D (51), is a well-known cellular substrate of GALNT3. The posttranslational glycosylation of FGF23 by GALNT3 results in the prevention of FGF23 proteinase processing (52), suggesting that GALNT3 is important for controlling the pathophysiological level of FGF23 (53, 54). Although galnt3-KO mice exhibit testicular calcifications and male infertility (30), it is unlikely that FGF23 is directly involved in the replication of IAV due to the fact that the expression of FGF23 has not been demonstrated in respiratory epithelial cells. However, a recent study revealed that GALNT3 suppression results in the up- or downregulation of numerous genes associated with the pathways involved in transcriptional regulation, signal transduction, cell proliferation, immune and inflammatory responses, and mucin O-glycosylation modification in epithelial ovarian cancer cells, demonstrating that GALNT3 contributes to various cellular processes through the O-glycosylation of many unknown substrates (55). In this respect, it is conceivable that GALNT3 affects IAV replication by modifying the O-linked glycosylation of unknown cellular factor(s) that may be involved in the activity of viral polymerase. In fact, alteration of the O-linked glycosylation is known to be correlated with the antiviral response to certain virus infections. It has been reported recently that variants of a GalNAc transferase, GALNT8, may contribute to the response to IFN therapy against chronic HCV infection and that the overexpression of GALNT8 attenuated IFN-α-induced gene transcription via the IFN-stimulated response element (26). We are currently working to identify specific substrates of GALNT3 in the respiratory tracts to better understand the mechanism underlying the effect of GALNT3 on IAV replication during early stages of infection.
In this study, we demonstrated that compared to WT mice, galnt3-KO mice exhibit high mortality with severe pathological alterations in the lungs following infection with PR8 (Fig. 7). Interestingly, the viral titers in the lungs of galnt3-KO mice were significantly higher than those of WT mice after 4 dpi. Considering the fact that mucin plays essential roles in airway defense against microbial infection and propagation in vivo, it is conceivable that GALNT3 exerts a dual function during IAV infection; GALNT3 may enhance the intracellular viral replication and may control viral propagation in vivo for IAV infection. We are conducting in vivo experiments to understand the dual function of GALNT3 and mucin in IAV infection.
In conclusion, the results of our study may improve our knowledge of the regulation of IAV replication during early stages of viral infection, as well as our understanding of the role of mucin production in IAV infection. Understanding the regulation of IAV replication via GALNT3 expression, especially regarding specific substrates of GALNT3, would be useful for establishing strategies to control IAV multiplication in vivo and for developing novel anti-influenza virus approaches.
Supplementary Material
ACKNOWLEDGMENTS
We thank Atsuyasu Sato for technical assistance with the isolation of murine bronchial and alveolar epithelial cells.
This study was supported in part by KAKENHI grant numbers 26253027 and 26670225 (K.T.), Core-to-Core Program A, Advanced Research Networks (K.T.), a Grant-in-Aid for JSPS Fellows (13J05979) (S.N.) from the Japan Society for the Promotion of Science (JSPS), the Science and Platform Technology Program for Innovative Biological Medicine (K.T.) from the Ministry of Education, Culture, Sports, Science & Technology (MEXT), and grants from the Sumitomo Foundation (K.T.) and the Daiichi Sankyo Foundation of Life Science (K.T.).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02246-15.
REFERENCES
- 1.Kong Y, Joshi HJ, Schjoldager KT, Madsen TD, Gerken TA, Vester-Christensen MB, Wandall HH, Bennett EP, Levery SB, Vakhrushev SY, Clausen H. 2015. Probing polypeptide GalNAc-transferase isoform substrate specificities by in vitro analysis. Glycobiology 25:55–65. doi: 10.1093/glycob/cwu089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yoshida CA, Kawane T, Moriishi T, Purushothaman A, Miyazaki T, Komori H, Mori M, Qin X, Hashimoto A, Sugahara K, Yamana K, Takada K, Komori T. 2014. Overexpression of Galnt3 in chondrocytes resulted in dwarfism due to the increase of mucin-type O-glycans and reduction of glycosaminoglycans. J Biol Chem 289:26584–26596. doi: 10.1074/jbc.M114.555987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Song L, Bachert C, Schjoldager KT, Clausen H, Linstedt AD. 2014. Development of isoform-specific sensors of polypeptide GalNAc-transferase activity. J Biol Chem 289:30556–30566. doi: 10.1074/jbc.M114.599563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ioannidis I, McNally B, Willette M, Peeples ME, Chaussabel D, Durbin JE, Ramilo O, Mejias A, Flano E. 2012. Plasticity and virus specificity of the airway epithelial cell immune response during respiratory virus infection. J Virol 86:5422–5436. doi: 10.1128/JVI.06757-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rabinovich GA, Toscano MA, Jackson SS, Vasta GR. 2007. Functions of cell surface galectin-glycoprotein lattices. Curr Opin Struct Biol 17:513–520. doi: 10.1016/j.sbi.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsuboi S, Fukuda M. 2001. Roles of O-linked oligosaccharides in immune responses. Bioessays 23:46–53. doi: 10.1002/1521-1878(200101)23:1<46::AID-BIES1006>3.3.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 7.Stuchlova Horynova M, Raska M, Clausen H, Novak J. 2013. Aberrant O-glycosylation and anti-glycan antibodies in an autoimmune disease IgA nephropathy and breast adenocarcinoma. Cell Mol Life Sci 70:829–839. doi: 10.1007/s00018-012-1082-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Go EP, Liao HX, Alam SM, Hua D, Haynes BF, Desaire H. 2013. Characterization of host-cell line specific glycosylation profiles of early transmitted/founder HIV-1 gp120 envelope proteins. J Proteome Res 12:1223–1234. doi: 10.1021/pr300870t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mazurov D, Ilinskaya A, Heidecker G, Filatov A. 2012. Role of O-glycosylation and expression of CD43 and CD45 on the surfaces of effector T cells in human T cell leukemia virus type 1 cell-to-cell infection. J Virol 86:2447–2458. doi: 10.1128/JVI.06993-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Das SR, Hensley SE, David A, Schmidt L, Gibbs JS, Puigbo P, Ince WL, Bennink JR, Yewdell JW. 2011. Fitness costs limit influenza A virus hemagglutinin glycosylation as an immune evasion strategy. Proc Natl Acad Sci U S A 108:E1417–E1422. doi: 10.1073/pnas.1108754108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hotard AL, Lee S, Currier MG, Crowe JE Jr, Sakamoto K, Newcomb DC, Peebles RS Jr, Plemper RK, Moore ML. 2014. Identification of residues in the human respiratory syncytial virus fusion protein that modulate fusion activity and pathogenesis. J Virol 89:512–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ehre C, Worthington EN, Liesman RM, Grubb BR, Barbier D, O'Neal WK, Sallenave JM, Pickles RJ, Boucher RC. 2012. Overexpression mouse model demonstrates the protective role of Muc5ac in the lungs. Proc Natl Acad Sci U S A 111:5753–5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barbier D, Garcia-Verdugo I, Pothlichet J, Khazen R, Descamps D, Rousseau K, Thornton D, Si-Tahar M, Touqui L, Chignard M, Sallenave JM. 2012. Influenza A induces the major secreted airway mucin MUC5AC in a protease-EGFR-extracellular regulated kinase-Sp1-dependent pathway. Am J Respir Cell Mol Biol 47:149–157. doi: 10.1165/rcmb.2011-0405OC. [DOI] [PubMed] [Google Scholar]
- 14.Roy MG, Livraghi-Butrico A, Fletcher AA, McElwee MM, Evans SE, Boerner RM, Alexander SN, Bellinghausen LK, Song AS, Petrova YM, Tuvim MJ, Adachi R, Romo I, Bordt AS, Bowden MG, Sisson JH, Woodruff PG, Thornton DJ, Rousseau K, De la Garza MM, Moghaddam SJ, Karmouty-Quintana H, Blackburn MR, Drouin SM, Davis CW, Terrell KA, Grubb BR, O'Neal WK, Flores SC, Cota-Gomez A, Lozupone CA, Donnelly JM, Watson AM, Hennessy CE, Keith RC, Yang IV, Barthel L, Henson PM, Janssen WJ, Schwartz DA, Boucher RC, Dickey BF, Evans CM. 2014. Muc5b is required for airway defence. Nature 505:412–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakamura S, Horie M, Fujino K, Matsumoto Y, Honda T, Tomonaga K. 2012. Generation of human bronchial epithelial cell lines expressing inactive mutants of GALNT3. J Vet Med Sci 74:1493. doi: 10.1292/jvms.12-0199. [DOI] [PubMed] [Google Scholar]
- 16.Oneyama C, Ikeda J, Okuzaki D, Suzuki K, Kanou T, Shintani Y, Morii E, Okumura M, Aozasa K, Okada M. 2011. microRNA-mediated downregulation of mTOR/FGFR3 controls tumor growth induced by Src-related oncogenic pathways. Oncogene 30:3489–3501. doi: 10.1038/onc.2011.63. [DOI] [PubMed] [Google Scholar]
- 17.Corti M, Brody AR, Harrison JH. 1996. Isolation and primary culture of murine alveolar type II cells. Am J Respir Cell Mol Biol 14:309–315. doi: 10.1165/ajrcmb.14.4.8600933. [DOI] [PubMed] [Google Scholar]
- 18.Herold S, von Wulffen W, Steinmueller M, Pleschka S, Kuziel WA, Mack M, Srivastava M, Seeger W, Maus UA, Lohmeyer J. 2006. Alveolar epithelial cells direct monocyte transepithelial migration upon influenza virus infection: impact of chemokines and adhesion molecules. J Immunol 177:1817–1824. doi: 10.4049/jimmunol.177.3.1817. [DOI] [PubMed] [Google Scholar]
- 19.Marsh LM, Cakarova L, Kwapiszewska G, von Wulffen W, Herold S, Seeger W, Lohmeyer J. 2009. Surface expression of CD74 by type II alveolar epithelial cells: a potential mechanism for macrophage migration inhibitory factor-induced epithelial repair. Am J Physiol Lung Cell Mol Physiol 296:L442–L452. doi: 10.1152/ajplung.00525.2007. [DOI] [PubMed] [Google Scholar]
- 20.Kuno A, Uchiyama N, Koseki-Kuno S, Ebe Y, Takashima S, Yamada M, Hirabayashi J. 2005. Evanescent-field fluorescence-assisted lectin microarray: a new strategy for glycan profiling. Nat Methods 2:851–856. doi: 10.1038/nmeth803. [DOI] [PubMed] [Google Scholar]
- 21.Kuno A, Solenkova NV, Solodushko V, Dost T, Liu Y, Yang XM, Cohen MV, Downey JM. 2008. Infarct limitation by a protein kinase G activator at reperfusion in rabbit hearts is dependent on sensitizing the heart to A2b agonists by protein kinase C. Am J Physiol Heart Circ Physiol 295:H1288–H1295. doi: 10.1152/ajpheart.00209.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sakai K, Ami Y, Tahara M, Kubota T, Anraku M, Abe M, Nakajima N, Sekizuka T, Shirato K, Suzaki Y, Ainai A, Nakatsu Y, Kanou K, Nakamura K, Suzuki T, Komase K, Nobusawa E, Maenaka K, Kuroda M, Hasegawa H, Kawaoka Y, Tashiro M, Takeda M. 2014. The host protease TMPRSS2 plays a major role in in vivo replication of emerging H7N9 and seasonal influenza viruses. J Virol 88:5608–5616. doi: 10.1128/JVI.03677-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buggele WA, Johnson KE, Horvath CM. 2012. Influenza A virus infection of human respiratory cells induces primary microRNA expression. J Biol Chem 287:31027–31040. doi: 10.1074/jbc.M112.387670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Othumpangat S, Walton C, Piedimonte G. 2012. MicroRNA-221 modulates RSV replication in human bronchial epithelium by targeting NGF expression. PLoS One 7:e30030. doi: 10.1371/journal.pone.0030030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song L, Liu H, Gao S, Jiang W, Huang W. 2010. Cellular microRNAs inhibit replication of the H1N1 influenza A virus in infected cells. J Virol 84:8849–8860. doi: 10.1128/JVI.00456-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakano R, Maekawa T, Abe H, Hayashida Y, Ochi H, Tsunoda T, Kumada H, Kamatani N, Nakamura Y, Chayama K. 2013. Single-nucleotide polymorphisms in GALNT8 are associated with the response to interferon therapy for chronic hepatitis C. J Gen Virol 94:81–89. doi: 10.1099/vir.0.044396-0. [DOI] [PubMed] [Google Scholar]
- 27.Wu Q, Liu HO, Liu YD, Liu WS, Pan D, Zhang WJ, Yang L, Fu Q, Xu JJ, Gu JX. 2015. Decreased expression of hepatocyte nuclear factor 4alpha (Hnf4alpha)/microRNA-122 (miR-122) axis in hepatitis B virus-associated hepatocellular carcinoma enhances potential oncogenic GALNT10 protein activity. J Biol Chem 290:1170–1185. doi: 10.1074/jbc.M114.601203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matrosovich MN, Matrosovich TY, Gray T, Roberts NA, Klenk HD. 2004. Neuraminidase is important for the initiation of influenza virus infection in human airway epithelium. J Virol 78:12665–12667. doi: 10.1128/JVI.78.22.12665-12667.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lieleg O, Lieleg C, Bloom J, Buck CB, Ribbeck K. 2012. Mucin biopolymers as broad-spectrum antiviral agents. Biomacromolecules 13:1724–1732. doi: 10.1021/bm3001292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miyazaki T, Mori M, Yoshida CA, Ito C, Yamatoya K, Moriishi T, Kawai Y, Komori H, Kawane T, Izumi S, Toshimori K, Komori T. 2013. Galnt3 deficiency disrupts acrosome formation and leads to oligoasthenoteratozoospermia. Histochem Cell Biol 139:339–354. doi: 10.1007/s00418-012-1031-3. [DOI] [PubMed] [Google Scholar]
- 31.Zhang X, Dong C, Sun X, Li Z, Zhang M, Guan Z, Duan M. 2014. Induction of the cellular miR-29c by influenza virus inhibits the innate immune response through protection of A20 mRNA. Biochem Biophys Res Commun 450:755–761. doi: 10.1016/j.bbrc.2014.06.059. [DOI] [PubMed] [Google Scholar]
- 32.Shan SW, Fang L, Shatseva T, Rutnam ZJ, Yang X, Du W, Lu WY, Xuan JW, Deng Z, Yang BB. 2013. Mature miR-17-5p and passenger miR-17-3p induce hepatocellular carcinoma by targeting PTEN, GalNT7 and vimentin in different signal pathways. J Cell Sci 126:1517–1530. doi: 10.1242/jcs.122895. [DOI] [PubMed] [Google Scholar]
- 33.Song R, Liu Q, Liu T, Li J. 2015. Connecting rules from paired miRNA and mRNA expression data sets of HCV patients to detect both inverse and positive regulatory relationships. BMC Genomics 16(Suppl 2):S11. doi: 10.1186/1471-2164-16-S2-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Du WW, Li X, Li T, Li H, Khorshidi A, Liu F, Yang BB. 2015. The microRNA miR-17-3p inhibits mouse cardiac fibroblast senescence by targeting Par4. J Cell Sci 128:293–304. doi: 10.1242/jcs.158360. [DOI] [PubMed] [Google Scholar]
- 35.Yang X, Du WW, Li H, Liu F, Khorshidi A, Rutnam ZJ, Yang BB. 2013. Both mature miR-17-5p and passenger strand miR-17-3p target TIMP3 and induce prostate tumor growth and invasion. Nucleic Acids Res 41:9688–9704. doi: 10.1093/nar/gkt680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H, Yang BB. 2012. Stress response of glioblastoma cells mediated by miR-17-5p targeting PTEN and the passenger strand miR-17-3p targeting MDM2. Oncotarget 3:1653–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen JA, Huang YP, Mazzoni EO, Tan GC, Zavadil J, Wichterle H. 2011. mir-17-3p controls spinal neural progenitor patterning by regulating Olig2/Irx3 cross-repressive loop. Neuron 69:721–735. doi: 10.1016/j.neuron.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu G, Yang F, Ding CL, Wang J, Zhao P, Wang W, Ren H. 2014. miR-221 accentuates IFNs anti-HCV effect by downregulating SOCS1 and SOCS3. Virology 462–463:343–350. [DOI] [PubMed] [Google Scholar]
- 39.Zhao G, Cai C, Yang T, Qiu X, Liao B, Li W, Ji Z, Zhao J, Zhao H, Guo M, Ma Q, Xiao C, Fan Q, Ma B. 2013. microRNA-221 induces cell survival and cisplatin resistance through PI3K/Akt pathway in human osteosarcoma. PLoS One 8:e53906. doi: 10.1371/journal.pone.0053906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller TE, Ghoshal K, Ramaswamy B, Roy S, Datta J, Shapiro CL, Jacob S, Majumder S. 2008. microRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27Kip1. J Biol Chem 283:29897–29903. doi: 10.1074/jbc.M804612200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu Q, Li P, Chen X, Zong L, Jiang Z, Nan L, Lei J, Duan W, Zhang D, Li X, Sha H, Wu Z, Ma Q, Wang Z. 2015. miR-221/222 induces pancreatic cancer progression through the regulation of matrix metalloproteinases. Oncotarget 6:14153–14164. doi: 10.18632/oncotarget.3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lennemann NJ, Rhein BA, Ndungo E, Chandran K, Qiu X, Maury W. 2014. Comprehensive functional analysis of N-linked glycans on Ebola virus GP1 mBio 5:e00862-00813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nycholat CM, Peng W, McBride R, Antonopoulos A, de Vries RP, Polonskaya Z, Finn MG, Dell A, Haslam SM, Paulson JC. 2013. Synthesis of biologically active N- and O-linked glycans with multisialylated poly-N-acetyllactosamine extensions using P. damsela alpha2-6 sialyltransferase. J Am Chem Soc 135:18280–18283. doi: 10.1021/ja409781c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mistry N, Inoue H, Jamshidi F, Storm RJ, Oberste MS, Arnberg N. 2011. Coxsackievirus A24 variant uses sialic acid-containing O-linked glycoconjugates as cellular receptors on human ocular cells. J Virol 85:11283–11290. doi: 10.1128/JVI.05597-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nokhbeh MR, Hazra S, Alexander DA, Khan A, McAllister M, Suuronen EJ, Griffith M, Dimock K. 2005. Enterovirus 70 binds to different glycoconjugates containing alpha2,3-linked sialic acid on different cell lines. J Virol 79:7087–7094. doi: 10.1128/JVI.79.11.7087-7094.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang W, Shah P, Toghi Eshghi S, Yang S, Sun S, Ao M, Rubin A, Jackson JB, Zhang H. 2014. Glycoform analysis of recombinant and human immunodeficiency virus envelope protein gp120 via higher energy collisional dissociation and spectral-aligning strategy. Anal Chem 86:6959–6967. doi: 10.1021/ac500876p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brautigam J, Scheidig AJ, Egge-Jacobsen W. 2013. Mass spectrometric analysis of hepatitis C viral envelope protein E2 reveals extended microheterogeneity of mucin-type O-linked glycosylation. Glycobiology 23:453–474. doi: 10.1093/glycob/cws171. [DOI] [PubMed] [Google Scholar]
- 48.Wang J, Fan Q, Satoh T, Arii J, Lanier LL, Spear PG, Kawaguchi Y, Arase H. 2009. Binding of herpes simplex virus glycoprotein B (gB) to paired immunoglobulin-like type 2 receptor alpha depends on specific sialylated O-linked glycans on gB. J Virol 83:13042–13045. doi: 10.1128/JVI.00792-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vasudevan D, Haltiwanger RS. 2014. Novel roles for O-linked glycans in protein folding. Glycoconj J 31:417–426. doi: 10.1007/s10719-014-9556-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma Z, Vosseller K. 2014. Cancer metabolism and elevated O-GlcNAc in oncogenic signaling. J Biol Chem 89:34457–34465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tagliabracci VS, Engel JL, Wiley SE, Xiao J, Gonzalez DJ, Nidumanda Appaiah H, Koller A, Nizet V, White KE, Dixon JE. 2014. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc Natl Acad Sci U S A 111:5520–5525. doi: 10.1073/pnas.1402218111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kato K, Jeanneau C, Tarp MA, Benet-Pages A, Lorenz-Depiereux B, Bennett EP, Mandel U, Strom TM, Clausen H. 2006. Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O-glycosylation. J Biol Chem 281:18370–18377. [DOI] [PubMed] [Google Scholar]
- 53.Abbasi F, Ghafouri-Fard S, Javaheri M, Dideban A, Ebrahimi A, Ebrahim-Habibi A. 2014. A new missense mutation in FGF23 gene in a male with hyperostosis-hyperphosphatemia syndrome (HHS). Gene 542:269–271. doi: 10.1016/j.gene.2014.03.052. [DOI] [PubMed] [Google Scholar]
- 54.Ichikawa S, Gray AK, Padgett LR, Reilly AM, Unsicker TR. 2014. High dietary phosphate intake induces development of ectopic calcifications in a murine model of familial tumoral calcinosis. J Bone Miner Res 29:2017–2023. doi: 10.1002/jbmr.2242. [DOI] [PubMed] [Google Scholar]
- 55.Wang ZQ, Bachvarova M, Morin C, Plante M, Gregoire J, Renaud MC, Sebastianelli A, Bachvarov D. 2014. Role of the polypeptide N-acetylgalactosaminyltransferase 3 in ovarian cancer progression: possible implications in abnormal mucin O-glycosylation. Oncotarget 5:544–560. doi: 10.18632/oncotarget.1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.