

Abstract
Hepatic ischemia and reperfusion (I/R) injury plays an active role in hepatic resection and transplantation. While the effects of protein kinase C (PKC)-βII activation and the role of PKC-β inhibitors are well understood in myocardial I/R in diabetes, they remain unclear in liver I/R. The aim of this study was to explore the effect of PKC-β inhibition and the potential mechanism by which PKC-β inhibitor treatment protects against hepatic I/R injury in diabetic rats. Diabetic rats were established and randomized into two groups. These were an untreated group (n=10), which did not receive any treatment, and a treatment group (n=10), orally treated with ruboxistaurin at a dose of 5 mg/kg/day for 2 weeks. The rats from the two groups were subjected to hepatic I/R. Aspartate transaminase (AST) and lactate dehydrogenase (LDH) levels were measured by enzymatic methods at 1, 3 and 5 h after I/R. Tumor necrosis factor-α (TNF-α) and intercellular adhesion molecule 1 (ICAM-1) were examined by enzyme-linked immunosorbent assay at the same time-points. Nuclear factor-κB (NF-κB) p65 expression was analyzed by immunofluorescence and western blotting. Apoptosis of hepatic cells was examined by the western blot analysis of caspase 3 expression and by DNA ladder analysis. Pathological changes were examined using light and electron microscopy. Serum AST and LDH levels in the PKC-β inhibitor treatment group were diminished compared with those in the untreated group (P<0.01). Serum TNF-α and ICAM-1 (P<0.01) levels were also decreased at different time-points in the PKC-β inhibitor treatment group. The relative expression of NF-κB p65 and caspase 3 in the hepatic tissue was weakened in the PKC-β inhibitor treatment group compared with that in the untreated group (P<0.01). Pathological changes in hepatic tissue were attenuated by the PKC-β inhibitor. In conclusion, PKC-β inhibitor treatment protected against liver I/R injury in diabetic rats. The mechanisms probably involved the attenuation of microvascular injury, reduced transport of injury-associated factors and diminishment of the activation of NF-κB p65.
Keywords: diabetes, hepatic ischemia/reperfusion injury, protein kinase C-β inhibitor
Introduction
Growing evidence from experimental studies in animals and clinical observations indicates that hepatic failure remains a most important cause of mortality following hepatic resection and transplantation (1–3). Hepatic ischemia and reperfusion (I/R) injury plays an active role in this process (4). Disordered glycometabolism is a common phenomenon in patients with liver disease (5). In severe cases, this is accompanied by diabetes. The reduction of I/R injury in patients with liver disease and diabetes is necessary. Routine strategies, such as controlling hepatic blood flow, ischemic preconditioning and the application of protecting agents are available (6–8). However, the identification of a novel approach that is able to reduce I/R injury in these patients is imperative.
Diabetic microvascular complications characterized by barrier dysfunction and increased permeability have been linked to the activation of protein kinase Cs (PKCs) (9). PKCs are serine/threonine kinases that participate in cellular signal transduction in diabetes-mediated vascular damage. Once activated, PKC is phosphorylated and translocated to the membrane, resulting in an elevated expression of PKC at the membrane. Ruboxistaurin (Rx) is a PKC-β inhibitor that is highly selective for the PKC-βII isoform. Rx has been shown to normalize endothelial function and prevent microvascular complications (10,11). While the effects of PKC-βII activation and the role of Rx are well understood in myocardial I/R (12), they remain unclear in liver I/R.
Therefore, in the present study the aims were to: i) evaluate the effectiveness of Rx in attenuating the liver damage and hepatic apoptotic injury induced by I/R in an rat model of diabetes; ii) elucidate whether Rx is able to attenuate liver microvascular dysfunction and attenuate inflammatory factor release; and iii) investigate the mechanism(s) by which Rx may modulate the liver damage and hepatic apoptotic injury induced by I/R in a rat model of diabetes with a focus on PKC-βII-dependent signaling.
Materials and methods
Animal preparation
All animal procedures were performed in accordance with the National Institutes of Health Guidelines on the Use of Laboratory Animals, and the study was approved by the Ethics Committee on Animal Care of Tianjin Third Central Hospital (Tianjin, China). Diabetes was induced in 40 male Sprague-Dawley rats (weight 250–280 g) by a single intraperitoneal injection of streptozotocin (STZ; 50 mg/kg in 0.9% saline; Sigma-Aldrich, St. Louis, MO, USA). Blood glucose levels were measured after 24 h and then daily for a total of 5 days. Animals with glucose levels ≥16.5 mmol/l were classified as diabetic. The diabetic rats were randomized into two groups. These were an untreated group, which did not receive any treatment (n=10) and a treatment group (n=10), orally treated with Rx (Eli Lilly, Indianapolis, IN, USA) at a dose of 5 mg/kg/day for 2 weeks.
Hepatic I/R
Hepatic I/R procedures were performed in the rats of the two groups. In brief, rats were anesthetized with 3% pentobarbital (30 mg/kg intraperitoneally) after fasting for 12 h. Laparotomy via a middle incision exposed the liver lobes. Following surgical exposure of the portal vein, the rats were injected with heparin (100 U/kg) via the tail vein to prevent the formation of blood clots during the ischemia period. The portal vein and hepatic artery were occluded for 60 min with a metallic clamp to induce hepatic ischemia. Blood samples were collected from the vena cava before and 1, 3 and 5 h after I/R, prior to rapid excision of the liver. Portions of liver tissue were fixed in 10% neutralized formalin for histological evaluation or snap frozen in liquid nitrogen and maintained at −80°C.
Hepatic enzyme assays
Serum levels of aspartate transaminase (AST) and lactate dehydrogenase (LDH) were analyzed using a commercially available diagnostic kit (BD Biosciences, Franklin Lakes, NJ, USA) and automatic biochemical equipment (Abbott C800; Abbott Diagnostics, Santa Clara, CA, USA). The serum levels of tumor necrosis factor-α (TNF-α) and intercellular adhesion molecule 1 (ICAM-1) were measured using commercial enzyme-linked immunosorbent assay (ELISA) kits (BD Biosciences) according to the manufacturer's instructions.
Histological studies
Fixed liver specimens were embedded in paraffin, sectioned at 4-µm thickness, and stained with hematoxylin and eosin (H&E) for the evaluation of liver injury. Photomicrographs were taken with a digital camera under a microscope (CKX41; Olympus Corporation, Tokyo, Japan).
Examination of liver tissue with a scanning electron microscope (SEM)
After fixing, tissue samples were examined via scanning electron microscopy to assess the microvascular integrity of the liver. After liver tissue was collected, regular pretreatments in accordance with standard protocols were employed, including dehydration, desiccation and gilding (13). Prepared samples were subsequently examined with an SEM (S-3400N; Hitachi, Ltd., Tokyo, Japan).
Western blot analysis
Proteins from ischemic liver tissue were extracted using Protein Extraction Reagent kit (Pierce Biotechnology, Inc., Rockford, IL, USA) for western blot analysis. Protein homogenates were separated on 10% SDS-PAGE gels, transferred to nitrocellulose membranes, and western blotting with mouse anti-human monoclonal antibodies against nuclear factor-κB (NF-κB p65; 1:1,000; cat. no. 610869, BD Biosciences) and caspase 3 (1:1,000; cat. no. 9664, Cell Signaling Technology, Inc., Danvers, MA, USA) was performed at 4°C overnight. Nitrocellulose membranes were then incubated with Texas Red AffiniPure goat anti-mouse immunoglobulin G (ZSGB-BIO, Beijing, China) secondary antibodies for 2 h at room temperature, and the blots were developed with a SuperSignal chemiluminescence detection kit (Pierce Biotechnology, Inc.). The resulting immunoblotting was visualized with an Image Station 400 (Kodak, Tokyo, Japan).
Detection of NF-κB p65 expression
To reveal the location of NF-κB p65 expression in ischemic liver tissue, immunohistochemical staining was performed. Paraformaldehyde-fixed liver tissues were cut into 4–5-µm semi-thin sections and underwent antigen retrieval. Briefly, the sections were incubated in a water bath at 80°C for 30 min in 50 mM sodium citrate (pH 8.5–9.5). Subsequently, the sections were stained with a mouse anti-human monoclonal antibody against NF-κB p65 (1:250; BD Biosciences) at 4°C for 24 h. An IX71 confocal microscope (Olympus Corporation, Tokyo, Japan) was used to examine the images.
Assessment of apoptosis in the liver tissue
The liver tissue was collected following I/R injury. Apoptosis was detected by a DNA ladder assay and the analysis of cleaved caspase 3 expression. DNA was extracted from the ischemic liver tissue by repeated cell disruption in 0.15 M NaCl and SDS-proteinase K (Beyotime Biotechnology, Shanghai, China), followed by purification with hydroxybenzen-chloroform-isoamyl alcohol (14). The DNA was incubated with protein enzyme K (Guangzhou Dongsheng Biotech Co., Ltd., Guangzhou, China) for 10 h in a 37°C environment and then precipitated using ethanol. DNA ladders were subjected to gel electrophoresis on a 2% agarose gel using a voltage of 50 V. Western blotting with a monoclonal antibody against cleaved caspase 3 (1:250; Cell Signaling Technology, Inc.) was performed.
Statistical analysis
Data expressed as mean values ± standard error of the mean were evaluated by analysis of variance (ANOVA) and the Newman-Keuls tests for multiple comparisons among groups. P<0.05 was considered to indicate a statistically significant result.
Results
Effect of treatment with PKC-β inhibitor on serum AST and LDH levels
As shown in Tables I and II, as the time after hepatic I/R was prolonged, marked increases in serum AST and LDH levels occurred. Treatment with the PKC-β inhibitor markedly decreased the serum AST and LDH levels at each time-point. It appeared that treatment with the PKC-β inhibitor provided protection against I/R injury. These findings are consistent with the histological observations shown in Fig. 1.
Table I.
Effect of treatment with Rx on serum AST levels in diabetic rats at different time-points following hepatic I/R injury (U/l).
| Groups | 1 h | 3 h | 5 h |
|---|---|---|---|
| DM + I/R | 83.2±27.1 | 92.8±26.5a | 145.8±25.3b |
| DM + Rx + I/R | 64.3±24.6c | 83.2±20.9a,d | 122.1±23.8b,c |
Rx, ruboxistaurin; AST, aspartate transaminase; I/R, ischemia and reperfusion; DM, diabetes mellitus.
P<0.01 vs. the 1 h time-point
P<0.01 vs. the 3 h time-point
P<0.01 vs. the DM + I/R group and
P<0.05 vs. the DM+I/R group. Data are presented as the mean ± standard error of the mean (n=10).
Table II.
Effect of treatment with Rx on serum LDH levels in diabetic rats at different time-points following hepatic I/R injury (U/l).
| Groups | 1 h | 3 h | 5 h |
|---|---|---|---|
| DM + I/R | 123.2±36.1 | 284.5±41.5a | 360.8±32.1b |
| DM + Rx + I/R | 114.8±27.9c | 135.6±38.6a,d | 213.2±34.5b,d |
Rx, ruboxistaurin; LDH, lactate dehydrogenase; I/R, ischemia and reperfusion; DM, diabetes mellitus.
P<0.01 vs. the 1 h time-point
P<0.01 vs. the 3 h time-point
P<0.05 vs. the DM + I/R group and
P<0.01 vs. the DM + I/R group. Data are presented as the mean ± standard error of the mean (n=10).
Figure 1.

Pathological changes in liver tissue. Hepatic cell and microvascular injury were observed under a light microscope and scanning electron microscope (SEM). With hematoxylin and eosin staining, disordered hepatic lobules, swelling cells and vacuoles in liver specimens were exhibited in each group, which implied hepatic I/R injury. As compared with (A) the untreated group, I/R injury was ameliorated in the (B) Rx treatment group. Scale bar, 30 µm. Under the SEM, red blood cells were observed to be blocked, clustered and conglutinated in capillary vessels, and normal hepatic plates were not evident in (C) the untreated group. (D) The situation was improved in the treated group. Scale bar, 10 µm. Arrows indicate: (A) impaired cells; (B) mildly impaired cells; (C) conglutinated red blood cells in capillary vessels; (D) lack of conglutinated red blood cells in capillary vessels. DM, diabetes mellitus; I/R, ischemia/reperfusion; Rx, ruboxistaurin.
Serum TNF-α and ICAM-1 levels in diabetic rats with hepatic I/R injury
Serum TNF-α levels in untreated diabetic rats increased as the time following the hepatic I/R procedure was prolonged before reaching a peak at 3 h, and then decreasing at 5 h. The TNF-α levels in the rats treated with the PKC-β inhibitor were significantly lower than those in the untreated rats at all time-points (P<0.01; Table III). Serum ICAM-1 levels in the untreated diabetic rats gradually increased as the time following the hepatic I/R procedure was prolonged. The ICAM-1 levels in the rats treated with the PKC-β inhibitor were significantly lower than those in the untreated rats at all time-points (P<0.01; Table IV).
Table III.
Effect of treatment with Rx on serum TNF-α levels in diabetic rats at different time-points following hepatic I/R injury (ng/l).
| Groups | 1 h | 3 h | 5 h |
|---|---|---|---|
| DM + I/R | 1260±128 | 1862±145a | 1567±132b |
| DM + Rx + I/R | 1026±114c | 1326±152a,c | 1307±148b,c |
Rx, ruboxistaurin; TNF-α, tumor necrosis factor-α; I/R, ischemia and reperfusion; DM, diabetes mellitus.
P<0.01 vs. the 1 h time-point
P<0.01 vs. the 3 h time-point and
P<0.01 vs. the DM + I/R group. Data are presented as the mean ± standard error of the mean (n=10).
Table IV.
Effect of treatment with Rx on serum ICAM-1 levels in diabetic rats at different time-points following hepatic I/R injury (ng/l).
| Groups | 1 h | 3 h | 5 h |
|---|---|---|---|
| DM + I/R | 263.3±38.2 | 465.7±31.4a | 689.3±37.8b |
| DM + Rx + I/R | 189.1±31.8c | 326.4±26.7a,c | 495.2±29.4b,c |
Rx, ruboxistaurin; ICAM-1, intercellular adhesion molecule 1; I/R, ischemia and reperfusion; DM, diabetes mellitus.
P<0.01 vs. the 1 h time-point
P<0.01 vs. the 3 h time-point and
P<0.01 vs. the DM + I/R group. Data are presented as the mean ± standard error of the mean (n=10).
Expression of NF-κB in the liver tissue of diabetic rats with hepatic I/R injury
The expression of NF-κB in the liver tissue was examined by immunohistochemical staining of the tissue with an antibody against NF-κB p65. Proteins from the ischemic liver tissue of the two groups were collected in order to perform western blot analysis with a monoclonal antibody against NF-κB p65. The immunohistochemical staining results revealed that the activation and expression of NF-κB, mainly located in the hepatic sinusoids and portal area, was present in the two groups of rats at 5 h after I/R. The staining was intense in the untreated group, and that in the treatment group was markedly lower (arrows in Fig. 2). Similarly, the activation and expression of NF-κB p65 in the nuclear extracts from the liver tissue were identified to be significantly lower in the Rx treatment group compared with those in the untreated group by western blot analysis (0.46±0.10 vs. 0.82±0.09, respectively; P<0.01).
Figure 2.
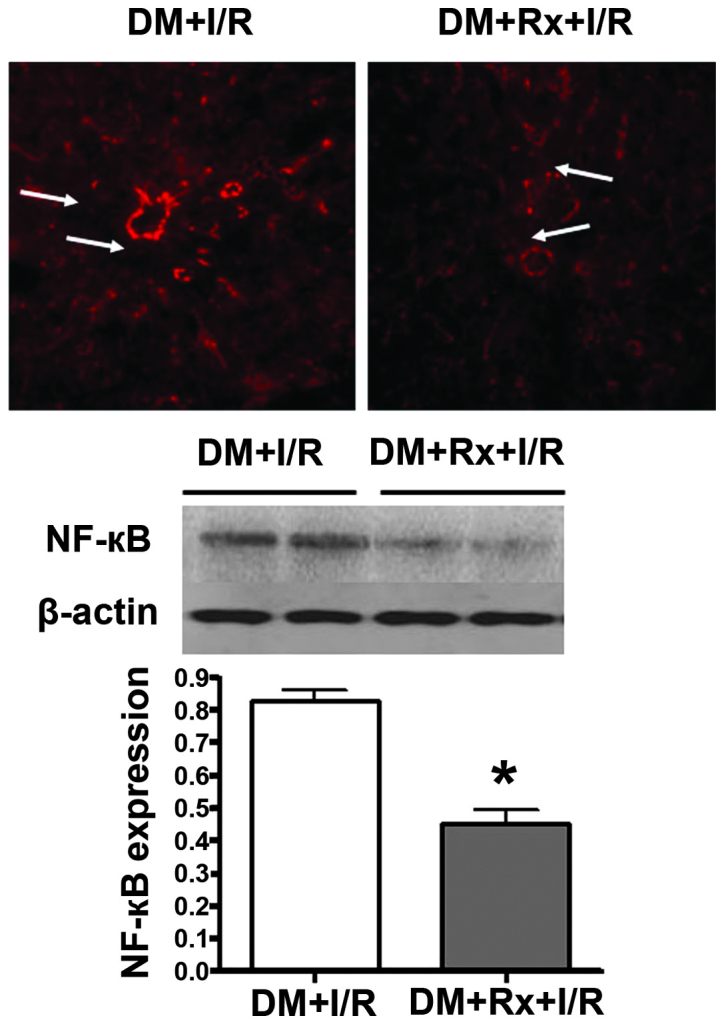
Expression of nuclear factor (NF)-кB in the liver tissue of diabetic rats with hepatic I/R injury. NF-кB in liver tissue was immunohistochemically stained with an antibody against NF-кB p65. Expression of NF-кB, mainly located in the hepatic sinusoids and portal area, was detected in the two groups of rats at 5 h after I/R. It was intense in the untreated group and clearly lower in the Rx treatment group (see arrows). Proteins from the ischemic liver tissue of the two groups were subjected to western blot analysis with monoclonal antibody against NF-кB p65. Activation and expression of NF-кB p65 in the nuclear extracts was significantly lower in the Rx treatment group (n=8) compared with that in the untreated group (n=8) by western blot analysis (0.46±0.10 vs. 0.82±0.09, respectively; P<0.01). *P<0.01 vs. the DM + I/R group. DM, diabetes mellitus; I/R, ischemia/reperfusion; Rx, ruboxistaurin.
Cell apoptosis in the liver tissue
Apoptosis was evaluated by DNA ladder assay and the analysis of cleaved caspase 3 expression. DNA agarose electrophoresis revealed the presence of DNA degradation in the liver tissue in the two groups of rats following I/R. DNA segments, at regular lengths of 180–200 bp, appearing as a DNA ladder, were more evident in the untreated group compared with the treatment group. (Fig. 3A) The expression of caspase 3 in the liver tissue was revealed to be significantly lower in the treatment group than in the untreated group by western blot analysis (0.52±0.09 vs. 1.36±0.12, respectively; P<0.01; Fig. 3B).
Figure 3.

Cell apoptosis in liver tissue. Liver tissue was collected 5 h after I/R. Apoptosis was detected by DNA ladder assay and the analysis of cleaved caspase 3 expression. (A) DNA collected from ischemic liver tissue was incubated with protein enzyme K for 10 h at 37°C and then precipitated with ethanol. DNA ladders were run on a 2% agarose gel using a voltage of 50V. DNA fragments, appearing as a DNA ladder, were evident in the untreated group (lane 1) compared with the untreated group (lane 2). (B) Western blotting with monoclonal antibody against cleaved caspase 3 was performed. Expression of caspase 3 in liver tissue was significantly lower in the Rx treatment group compared with the untreated group (n=10 per group; 0.52±0.09 vs. 1.36±0.12, respectively; P<0.01). *P<0.01 vs. the DM + I/R group. DM, diabetes mellitus; I/R, ischemia/reperfusion; Rx, ruboxistaurin.
Pathological changes in liver tissue
Hepatic cell and hepatic microvascular injury were observed by light microscopy and scanning electron microscopy. With H&E staining, disordered hepatic lobules, swelling cells and vacuoles in the liver specimens were visible in each group, which implied that hepatic I/R injury had occurred. The I/R injury was ameliorated in the treatment group. Under the SEM, it was observed that red blood cells were blocked, clustered and conglutinated in capillary vessels, and normal hepatic plates were not evident. The situation was observed to be improved in the treatment group (arrows in Fig. 1).
Discussion
Diabetic microvascular complications characterized by barrier dysfunction have been linked to the activation of PKC-β (15), with disordered endothelial function and the subsequently increased transport of injury-associated factors as characteristics. Increased permeability of proteins and macromolecules has been observed in tissues of the retina, kidney and arterial aorta in diabetic animals (16). Activation of PKC-β is commonly considered as an important initial cause of microvascular damage. Correspondingly, microvascular complications induced by the activation of PKC-β in target organs, such as increased glomerular filtration rate and retinal edema, could be diminished by treatment with the PKC-β inhibitor Rx (17).
The activation and expression of NF-κB plays a specific role in the development of I/R injury in the heart, brain, liver, kidney and other organs, which is described as cyclic activation (18,19). NF-κB regulates the genetic expression of ICAM-1 of the adhesion molecule family, the proinflammatory factor TNF-α and P-selectin. These inflammatory factors promote the activation of NF-κB (20,21).
Apoptosis of hepatocytes is the detrimental consequence of the elevated release of inflammatory factors and overexpression of adhesion molecules (22,23), and significantly contributes to hepatic I/R injury. Reperfusion of the ischemic liver has been reported to activate Kupffer cells, increase the release of TNF-α, trigger apoptotic genes, activate caspase and cause degradation, leading to the emergence of DNA fragments and apoptotic bodies (24). Previous investigators have documented that liver endotheliocyte damage, microvascular dysfunction and the activation of Kupffer cells are involved in caspase-mediated apoptosis (25–27). These observations provide an explanation for the results of the present study.
The results of the present study documented that serum AST, LDH, TNF-α and ICAM-1 levels exhibited marked increases in the untreated I/R group and clear reductions in the group treated with the PKC-β inhibitor Rx. Pathological damage observed under a light microscope and TEM, and NF-κB expression detected by immunohistochemical staining, were attenuated in the treatment group. These results indicate that the activation of PKC-β in diabetic rats may result in damage to the hepatic microvascular barrier, thereby increasing the transport of injury-associated factors in liver tissues, leading to the activation of NF-κB. Conversely, NF-κB stimulates the genetic transcription of TNF-α and ICAM-1. Elevated expression of TNF-α and ICAM-1 has been documented to induce apoptosis mediated by caspase, accelerate hepatocellular apoptosis and aggravate pathological damage during hepatic I/R injury (28). The present results are consistent with previous studies (29,30). Xu et al (31) provided definitive evidence of the activation of NF-κB and expression of TNF-α and ICAM-1 mRNA in whole liver extracts during hepatic I/R injury; the activity of NF-κB was elevated in a time-dependent manner. This is a vicious circle, which may be inhibited with PKC-β inhibitor treatment.
The results obtained in the present study support the hypothesis that treatment with a PKC-β inhibitor may play a key role in controlling the microvascular injury induced by the activation of PKC-β, by attenuating damage to the barrier function and endothelial cells as well as by reducing the transport of TNF-α and the adhesion of ICAM-1 during hepatic I/R injury. This should break the vicious circle caused by the activation of NF-κB and the overexpression of TNF-α. Accordingly, the success rate of hepatic surgery in diabetic patients is likely to be improved as a result of decreased hepatic apoptosis and pathological injury during hepatic I/R injury. Confirmation of the hypothesis that PKC-β inhibitor treatment prior to hepatic surgery will improve clinical outcomes in diabetic patients with liver disease requires evaluation in further studies.
Intriguing points remain for research in our ongoing studies. The details of the correlation of PKC-β activation with diabetic hepatic microvascular injury may be determined with co-immunoprecipitation. Hepatic I/R injury is precisely regulated by a variety of proteins and phosphorylated proteins; the specific proteins involved in this pathology may be further explored.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (NSFC; no. 81200158), Science Foundation of Tianjin Health Bureau (no. 2013KZ011) and Key Research Projects of Tianjin Health Bureau (no. 13KG115).
References
- 1.Rahbari NN, Reissfelder C, Koch M, et al. The predictive value of postoperative clinical risk scores for outcome after hepatic resection: a validation analysis in 807 patients. Ann Surg Oncol. 2011;18:3640–3649. doi: 10.1245/s10434-011-1829-6. [DOI] [PubMed] [Google Scholar]
- 2.Karatzas T, Neri AA, Baibaki ME, Dontas IA. Rodent models of hepatic ischemia-reperfusion injury, time and percentage-related pathophysiological mechanisms. J Surg Res. 2014;191:399–412. doi: 10.1016/j.jss.2014.06.024. [DOI] [PubMed] [Google Scholar]
- 3.Rahbari NN, Garden OJ, Padbury R, et al. Posthepatectomy liver failure: a definition and grading by the International Study Group of Liver Surgery (ISGLS) Surgery. 2011;149:713–724. doi: 10.1016/j.surg.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 4.Schmidt R. Hepatic organ protection: from basic science to clinical practice. World J Gastroenterol. 2010;16:6044–6045. doi: 10.3748/wjg.v16.i48.6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Su AP, Cao SS, Le Tian B, et al. Effect of transjugular intrahepatic portosystemic shunt on glycometabolism in cirrhosis patients. Clin Res Hepatol Gastroenterol. 2012;36:53–59. doi: 10.1016/j.clinre.2011.09.011. [DOI] [PubMed] [Google Scholar]
- 6.Çekın AH, Gür G, Türkoğlu S, et al. The protective effect of L-carnitine on hepatic ischemia-reperfusion injury in rats. Turk J Gastroenterol. 2013;24:51–56. [PubMed] [Google Scholar]
- 7.Yang J, Sun H, Takacs P, et al. The effect of octreotide on hepatic ischemia-reperfusion injury in a rabbit model. Transplant Proc. 2013;45:2433–2438. doi: 10.1016/j.transproceed.2013.02.112. [DOI] [PubMed] [Google Scholar]
- 8.Jin LM, Liu YX, Zhou L, et al. Ischemic preconditioning attenuates morphological and biochemical changes in hepatic ischemia/reperfusion in rats. Pathobiology. 2010;77:136–146. doi: 10.1159/000292647. [DOI] [PubMed] [Google Scholar]
- 9.Clarke M, Dodson PM. PKC inhibition and diabetic microvascular complications. Best Pract Res Clin Endocrinol Metab. 2007;21:573–586. doi: 10.1016/j.beem.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 10.Wei L, Yin Z, Yuan Y, et al. A PKC-beta inhibitor treatment reverses cardiac microvascular barrier dysfunction in diabetic rats. Microvasc Res. 2010;80:158–165. doi: 10.1016/j.mvr.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 11.Budhiraja S, Singh J. Protein kinase C beta inhibitors: a new therapeutic target for diabetic nephropathy and vascular complications. Fundam Clinical Pharmacol. 2008;22:231–240. doi: 10.1111/j.1472-8206.2008.00583.x. [DOI] [PubMed] [Google Scholar]
- 12.Wei L, Sun D, Yin Z, et al. A PKC-beta inhibitor protects against cardiac microvascular ischemia reperfusion injury in diabetic rats. Apoptosis. 2010;15:488–498. doi: 10.1007/s10495-009-0439-2. [DOI] [PubMed] [Google Scholar]
- 13.Yin Z, Fan L, Wei L, et al. FTY720 protects cardiac microvessels of diabetes: a critical role of S1P1/3 in diabetic heart disease. PLoS One. 2012;7:e42900. doi: 10.1371/journal.pone.0042900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldenberger D, Perschil I, Ritzler M, Altwegg M. A simple “universal” DNA extraction procedure using SDS and proteinase K is compatible with direct PCR amplification. PCR Methods Appl. 1995;4:368–370. doi: 10.1101/gr.4.6.368. [DOI] [PubMed] [Google Scholar]
- 15.Gutterman DD. Vascular dysfunction in hyperglycemia: is protein kinase. C the culprit? Circ Res. 2002;90:5–7. [PubMed] [Google Scholar]
- 16.Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res. 2010;106:1319–1331. doi: 10.1161/CIRCRESAHA.110.217117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joy SV, Scates AC, Bearelly S, et al. Ruboxistaurin, a protein kinase C beta inhibitor, as an emerging treatment for diabetes microvascular complications. Ann Pharmacother. 2005;39:1693–1699. doi: 10.1345/aph.1E572. [DOI] [PubMed] [Google Scholar]
- 18.Galloway E, Shin T, Huber N, et al. Activation of hepatocytes by extracellular heat shock protein 72. Am J Physiol Cell Physiol. 2008;295:C514–C520. doi: 10.1152/ajpcell.00032.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang H, Li ZY, Wu HS, et al. Endogenous danger signals trigger hepatic ischemia/reperfusion injury through toll-like receptor 4/nuclear factor-kappa B pathway. Chin Med J (Engl) 2007;120:509–514. [PubMed] [Google Scholar]
- 20.Zhang W, An J, Jawadi H, et al. Sphingosine-1-phosphate receptor-2 mediated NFκB activation contributes to tumor necrosis factor-α induced VCAM-1 and ICAM-1 expression in endothelial cells. Prostaglandins Other Lipid Mediat. 2013;106:62–71. doi: 10.1016/j.prostaglandins.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu YP, Shen T, Lin YJ, et al. Astragalus polysaccharides suppress ICAM-1 and VCAM-1 expression in TNF-α-treated human vascular endothelial cells by blocking NF-κB activation. Acta Pharmacol Sin. 2013;34:1036–1042. doi: 10.1038/aps.2013.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen X, Ding WX, Ni HM, et al. Bid-independent mitochondrial activation in tumor necrosis factor alpha-induced apoptosis and liver injury. Mol Cell Biol. 2007;27:541–553. doi: 10.1128/MCB.01166-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hatano E. Tumor necrosis factor signaling in hepatocyte apoptosis. J Gastroenterol Hepatol. 2007;22((Suppl 1)):S43–S44. doi: 10.1111/j.1440-1746.2006.04645.x. [DOI] [PubMed] [Google Scholar]
- 24.Martin J, Romanque P, Maurhofer O, et al. Ablation of the tumor suppressor histidine triad nucleotide binding protein 1 is protective against hepatic ischemia/reperfusion injury. Hepatology. 2011;53:243–252. doi: 10.1002/hep.23978. [DOI] [PubMed] [Google Scholar]
- 25.Giakoustidis DE, Giakoustidis AE, Iliadis S, et al. Attenuation of liver ischemia/reperfusion induced apoptosis by epigallocatechin-3-gallate via down-regulation of NF-kappaB and c-Jun expression. J Surg Res. 2010;159:720–728. doi: 10.1016/j.jss.2008.08.038. [DOI] [PubMed] [Google Scholar]
- 26.Huet PM, Nagaoka MR, Desbiens G, et al. Sinusoidal endothelial cell and hepatocyte death following cold ischemia-warm reperfusion of the rat liver. Hepatology. 2004;39:1110–1119. doi: 10.1002/hep.20157. [DOI] [PubMed] [Google Scholar]
- 27.Teoh NC, Farrell GC. Hepatic ischemia reperfusion injury: pathogenic mechanisms and basis for hepatoprotection. J Gastroenterol Hepatol. 2003;18:891–902. doi: 10.1046/j.1440-1746.2003.03056.x. [DOI] [PubMed] [Google Scholar]
- 28.Guo JY, Yang T, Sun XG, et al. Ischemic postconditioning attenuates liver warm ischemia-reperfusion injury through Akt-eNOS-NO-HIF pathway. J Biomed Sci. 2011;18:79. doi: 10.1186/1423-0127-18-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taki-Eldin A, Zhou L, Xie HY, et al. Triiodothyronine attenuates hepatic ischemia/reperfusion injury in a partial hepatectomy model through inhibition of proinflammatory cytokines, transcription factors, and adhesion molecules. J Surg Res. 2012;178:646–656. doi: 10.1016/j.jss.2012.05.069. [DOI] [PubMed] [Google Scholar]
- 30.Coito AJ. Leukocyte transmigration across endothelial and extracellular matrix protein barriers in liver ischemia/reperfusion injury. Curr Opin Organ Transplant. 2011;16:34–40. doi: 10.1097/MOT.0b013e328342542e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu J, Xie J, Bao M, et al. NF-kappaB/I-kappaB pathway during ischemia reperfusion injury of rat liver. Chin Med J (Engl) 2003;116:1146–1149. [PubMed] [Google Scholar]