

Abstract
Purpose
Congenital cataract is a leading cause of childhood blindness. Mutations in the EPHA2 gene are one of the causes of inherited congenital cataract. The EPHA2 gene encodes a membrane-bound tyrosine kinase receptor and is highly expressed in epithelial cells, including in the ocular lens. Signaling through the EPHA2 receptor plays a pivotal role in epithelial cell homeostasis. The aim of this study was to determine the effect of congenital cataract causing mutations in the EPHA2 gene on the encoded protein in epithelial cells.
Methods
The effect of five disease-causing mutations, p.P584L (c.1751C>T), p.T940I (c.2819C>T), p.D942fsXC71 (c.2826–9G>A), p.A959T (c.2875G>A), and p.V972GfsX39 (c.2915_2916delTG), on localization of the protein was examined in two in vitro epithelial cell culture systems: Madin-Darby Canine Kidney (MDCK) and human colorectal adenocarcinoma (Caco-2) epithelial cells. Myc-tagged mutant constructs were generated by polymerase chain reaction (PCR)-based mutagenesis. The Myc-tagged wild-type construct was used as a control. The Myc-tagged wild-type and mutant proteins were ectopically expressed and detected by immunofluorescence labeling.
Results
Two of the mutations, p.T940I and p.D942fsXC71, located within the cytoplasmic sterile-α-motif (SAM) domain of EPHA2, led to mis-localization of the protein to the perinuclear space and co-localization with the cis-golgi apparatus, indicating sub-organellar/cellular retention of the mutant proteins. The mutant proteins carrying the remaining three mutations, similar to the wild-type EPHA2, localized to the cell membrane.
Conclusions
Mis-localization of two of the mutant proteins in epithelial cells suggests that some disease-causing mutations in EPHA2 likely affect lens epithelial cell homeostasis and contribute to cataract. This study suggests that mutations in EPHA2 contribute to congenital cataract through diverse mechanisms.
Introduction
Cataract is an opacification of the ocular lens; it may develop at birth or within the first two decades of life, where it is termed congenital cataract [1]. Congenital cataract is one of the leading causes of childhood blindness in the world. It occurs at a frequency of 1–15/10,000 live births and is a phenotypically and genotypically heterogeneous disease [2-4]. At least a quarter of congenital cataracts are inherited, with more than 27 causative genes known so far [1]. EPHA2 is one of the recently identified causative genes for congenital cataract [5-9]. Mutations in EPHA2 can lead to both autosomal dominant and recessive forms of cataract [6,7]. We reported that mutations in this gene account for ~5% of inherited cataracts in the South-Eastern Australian population [10], indicating that mutations in EPHA2 are a major contributor to congenital cataract. Furthermore, EPHA2 deficiency leads to adult-onset cataract in mice [11]. Hence, this gene is important in mammalian lens development and lens maintenance. The EPHA2 gene encodes a transmembrane tyrosine kinase receptor of the EPH receptor family. The protein comprises a ligand binding, a cysteine-rich and two fibronectin type III repeats in the extracellular region, a transmembrane segment, and a juxtamembrane region, a tyrosine kinase, a sterile-α-motif (SAM) and a PSD-95, DLG, ZO-1 (PDZ) domain in the cytoplasmic region [12]. Most of the causative mutations identified so far reside in the SAM domain of the protein, and a mutation each in the fibronectin type III repeats, tyrosine kinase domain, between the tyrosine kinase and SAM domain and the PDZ domain.
EPHA2 signaling is involved in several biological processes, such as cell-cell adhesion and repulsion, cell migration, cell spreading, and epithelial-to-mesenchymal transformation [13]. These cellular processes are important in lens development, maintenance, and function [14]. Consistently, EPHA2 is highly expressed during development [15-18], including lens development [19]. In the developing lens, the strongest expression has been reported in fiber cells in the bow region and in the lens epithelium [20]. It is also expressed in a variety of other epithelial cells and is important for maintenance of epithelia [13,21].
Epithelial cells are connected with the neighboring cells through three types of junctions in the lateral cell membrane: tight junctions in the apical region, adherence junctions (AJs) in the lateral region, and desmosomes in the basal region [22]. Interaction of EPHA2 with the junctional proteins provides evidence for its role in regulating cellular junctions [23-27]. The integrity of cellular junctions plays a critical role in maintaining cell-cell communication and homeostasis in the lens [28]. EPHA2 plays an important role at cell-cell junctions in the lens, as EPHA2−/− mice exhibit altered localization of the AJ protein, E-cadherin, and the AJ-associated protein beta(β)-catenin in lens epithelial cells [29]. N-cadherin, an AJ protein homologous to E-cadherin, shows diffused localization in lens fiber cells in EPHA2−/− mice [11]. Therefore, congenital cataract causing mutations in EPHA2 may affect cell-cell contacts in the lens and in turn lead to cataract.
In the present study, we investigated the effect of congenital cataract-causing mutations in EPHA2 on subcellular localization of the protein in epithelial cells that form well-established intercellular contacts in culture. We previously showed that EPHA2 protein localizes in the cytoplasm in human SRA01/04 and mouse αTN4 lens epithelial cells, and that intercellular contacts between these cells are less defined than in other epithelial cell lines [30]. We also showed that EPHA2 localizes to the cell periphery in polarized epithelial cells, such as Madin Darby Canine Kidney (MDCK) cells [30], similar to that seen in the lens in vivo [11,19]. Likewise, EPHA2 localizes to the cell membrane in human adenocarcinoma (Caco-2) epithelial cells, which also polarize in culture [15,31]. Therefore, the effect of causative mutations on protein localization was examined in MDCK and Caco-2 epithelial cells. We found that two of the disease-causing mutations led to altered localization of the protein that may impair intercellular contacts in the lens of the affected individuals carrying these mutations. The study provides an insight into the likely mechanisms of congenital cataract due to mutations in the EPHA2 gene.
Methods
Generation of EPHA2 mutant expression constructs
EPHA2 was amplified from SRA01/04 human lens epithelial cell cDNA and cloned into pcDNA3.1 Myc/HisA (Clontech Laboratories Inc., CA) at XhoI and HindIII sites. EPHA2-Myc was then amplified using the forward primer (5’-3′) CAC ACA CAC ACA TTA ATT AAA CCG AGA GCG AGA AGC GCG GCA TGG AG and reverse primer (5′-3′) CAC ACA CAC ACA TTA ATT AAA CTC AGA TCC TCT TCT GAG ATG AG, which introduced PacI sites on either end of the amplicon and was cloned into pTOPO® TA (Invitrogen, Life Technologies Australia Pty Ltd., VIC, Australia). From the resulting clone, the EPHA2-Myc cDNA flanked by PacI sites was cloned into pQCXIP (Clontech Laboratories Inc.) at the PacI site. Expression of the C-terminal Myc-tagged wild-type EPHA2 was driven by the human cytomegalovirus (CMV) promoter. The construct also encoded enhanced green fluorescent protein (EGFP) gene under the phosphoglycerate kinase (PGK) promoter. The EGFP and EPHA2-Myc open reading frames (ORFs) were transcribed in the opposite directions.
Five mutations, c.1751C>T, c.2819C>T, c.2826–9G>A, c.2875G>A, and c.2915_2916delTG, were individually introduced in the wild-type EPHA2-Myc cDNA by PCR-based mutagenesis, as previously described [32]. The primer sequences are listed in Appendix 1, and annealing temperatures used for PCR are given in Appendix 2. The GC-RICH PCR System (Roche Diagnostics Australia Pty Ltd, NSW, Australia) was used to perform all rounds of PCR. Wild-type EPHA2-Myc cDNA was used as a template for PCR 1 and 2 to introduce c.1751C>T, c.2819C>T, and c.2875G>A mutations. The mutations c.2826–9G>A and c.2915_2916delTG lead to a frameshift due to the addition of 71 and 39 aberrant amino acids, respectively, in the mutant protein. For introducing these mutations, total cDNA from SRA01/04 cells was used as template for PCR.
For PCR 1 and 2, the enzyme mix was activated at 95 °C for 3 min. Denaturation was performed at 95 °C for 30 s, annealing for 30 s at the temperatures indicated in Appendix 2, and extension at 68 °C for 30 s for 25 cycles. Equimolar amounts of PCR 1 and PCR 2 products were used as template for PCR3, using the same cycling conditions as described above, except PCR was performed for 20 cycles.
For cloning the mutant cDNA fragments, with the exception of the c.2875G>A mutation, the corresponding wild-type EPHA2 cDNA fragment was replaced with the mutated fragment in the pQCXIP-EPHA2-Myc construct. Each mutant fragment, except that carrying the c.1751C>T mutation, was digested using restriction enzymes BstZ17I and BglII (New England Biolabs Inc., MA) and cloned in the wild-type construct at BstZ17I/BamHI sites. The fragment carrying the c.2875G>A mutation was cloned in the mutant construct carrying the c.2819C>T mutation, using the same cloning strategy. The mutant fragment carrying the c.1751C>T mutation was cloned in the wild-type construct at the BstXI site. All the clones were confirmed by sequencing before use in experiments.
Cell culture and transfection
Human embryonic kidney (HEK293A; Appendix 3) fibroblast cells were used to analyze expression from mutant constructs and were available in the laboratory. MDCK epithelial cells were a kind gift from Dr. Stephen A. Wood (Griffith University, Queensland, Australia), and Caco-2 epithelial cells from Ms. Monica Dreimanis (Department of Haematology, Flinders University). All the cell lines were cultured in Dulbecco’s Modified Eagle’s medium (DMEM; GIBCO, Life Technologies Australia Pty Ltd., VIC, Australia) supplemented with 10% fetal bovine serum and penicillin/streptomycin (10,000 U/ml penicillin and 10,000 µg/ml streptomycin; Life Technologies Australia Pty Ltd). Cell cultures were maintained in a humidified atmosphere at 37 °C with 5% CO2. We previously reported the expression and peripheral localization of the tight junction proteins ZO-1 and occludin in confluent cultures of MDCK cells from this source, demonstrating their epithelial characteristic and polarization in culture [33]. Peripheral localization of the NHS-A protein that interacts with ZO-1 at tight junctions [33] and similar intense localization at sites of tricellular junctions of tricellulin, a tricellular tight junction protein, in confluent monolayers of Caco-2 cells from this source (Appendix 4) demonstrate their polarized epithelial characteristic in culture. Low passages of each cell line were used for experiments.
For protein expression studies, 4×105 HEK293A cells were seeded in six-well tissue culture plates. For protein localization studies, 4×105 MDCK or Caco-2 cells were seeded onto coverslips in six-well tissue culture plates. Seventy to 80% confluent HEK293A cells and 90–95% confluent MDCK and Caco-2 cells were transfected with 4 µg of wild-type or mutant EPHA2-Myc construct using Lipofectamine® 2000 (Invitrogen, Life Technologies Australia Pty Ltd.) as per the manufacturer’s protocol.
Western blotting
HEK293A cells were harvested 24 h and 48 h after transfection for protein extraction. Total cellular proteins were extracted in a radio-immunoprecipitation assay (RIPA) buffer, size-separated by SDS–PAGE [30] and then transferred onto Hybond-C Extra (GE Healthcare Australia Pty Ltd., NSW, Australia) or Polyvinylidine fluoride-low fluorescence (PVDF-LF) membranes (BioRad Laboratories Pty Ltd, NSW, Australia). The blots were hybridized with the mouse anti-Myc (1:500, Cell Signaling Technology, Inc., MA) primary antibody, followed by donkey anti-mouse IgG conjugated with horseradish peroxidase (HRP; 1:500, Jackson ImmunoResearch, Laboratories, Inc., PA) secondary antibody, and developed using SuperSignal West Pico Chemiluminescent substrate (Thermo Fisher Scientific, VIC, Australia) or Amersham™ ECL™ Prime western blotting reagent (GE Healthcare Australia Pty Ltd.).
Immunofluorescence labeling
MDCK or Caco-2 cells were immunolabeled 24 h after transfection that is, 3–4 days after seeding. For immunolabeling, the cells were fixed in 4% paraformaldehyde/phosphate buffered saline (PBS) and permeabilized with 0.4% Triton X-100/PBS for 5 min. The cells were blocked with 5% goat or donkey serum/PBS and hybridized with the mouse anti-Myc (1:2000 for MDCK and 1:1500 for Caco-2 cells; Cell Signaling Technology) primary antibody, followed by hybridization with a goat anti-mouse IgG conjugated with Alexa Flour 594 (1:500; Invitrogen) or donkey anti-mouse IgG conjugated with Cy5 (1:50; Jackson ImmunoResearch, Laboratories) secondary antibody.
For co-localization experiments, after fixing and permeabilizing the cells, EPHA2-Myc was detected using the mouse anti-Myc primary antibody (1:2000 for MDCK and 1:1500 for Caco-2 cells) and donkey anti-mouse IgG conjugated with Cy5 (1:50) secondary antibody; the cis-golgi apparatus was labeled using the rabbit anti-GM130 antibody (1:1000; Abcam®, MA) and donkey anti-rabbit IgG conjugated with Alexa Flour 555 (1:1000; Invitrogen) secondary antibody. Cells labeled with mismatched secondary antibodies and by omitting the primary antibodies were used as controls for demonstrating the specificity of signals.
Labeled cells were mounted in Prolong Gold Antifade reagent with DAPI (Invitrogen) and imaged on a Leica SP5 confocal microscope (Leica Microsystems Pty Ltd, NSW, Australia) using a 63× objective. The images were captured using Leica Application Suite (LAS) microscope software (Leica Microsystems Pty Ltd). A 405 nm diode was used to excite DAPI, a DPSS 561 laser for Alexa Flour 594 and Alexa Flour 555, and an HeNe 633 laser to excite Cy5. The emission spectra for DAPI, Alexa Flour 594, Alexa Flour 555, and Cy5 were set at 408–488 nm, 585–700 nm, 565–620 nm, and 645–730 nm, respectively.
Results
We analyzed the effects of five disease-causing mutations in EPHA2 on localization of the resulting mutant proteins in two epithelial cell lines. The mutations included p.P584L (c.1751C>T) in the juxtamembrane domain identified in an Australian family, three mutations in the SAM domain, p.T940I (c.2819C>T) identified in a Chinese family, p.D942fsXC71 (c.2826–9G>A) reported in three Australian families and p.A959T (c.2875G>A) found in one Australian family, and a p.V972GfsX39 (c.2915_2916delTG) mutation identified in the PDZ domain in a British family [6,10]. The mutations were individually introduced in the Myc-tagged wild-type EPHA2 cDNA, and the mutant constructs were used for the study.
First, the expression of mutant proteins from the mutant constructs was determined in transiently transfected HEK293A cells by western blotting. The transiently expressed protein was detected using an anti-Myc antibody. Three clones each of the constructs carrying a p.P584L, p.T940I, p.D942fsXC71, or p.V972GfsX39 mutation and a clone of the construct carrying the p.A959T mutation were analyzed. All the analyzed clones of each mutant construct were found to express the protein of the expected size of ∼130 kDa (Figure 1). The cells transfected with the wild-type EPHA2-Myc construct, used as a positive control, also showed the presence of the same-sized protein that was absent in mock transfected cells. Hence, all the mutant constructs expressed the mutant EPHA2-Myc protein.
Figure 1.
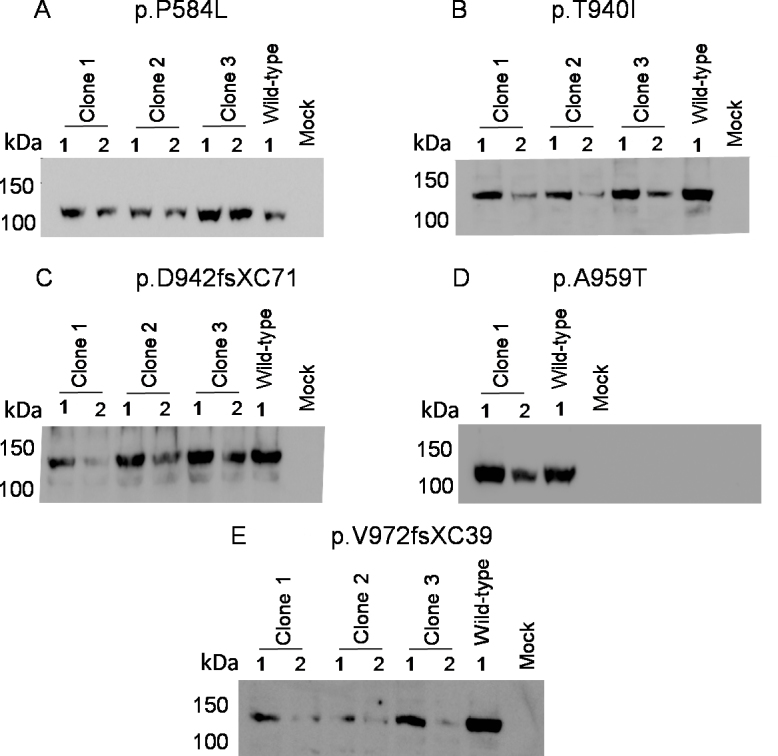
Expression of the mutant EPHA2-Myc proteins from the mutant constructs. Cell lysates of HEK293A cells transiently transfected with the mutant EPHA2–Myc constructs were analyzed at 24 h (1) and 48 h (2) post-transfection. The proteins were size-separated by SDS–PAGE. The transiently expressed protein was detected by western blotting with the anti-Myc primary antibody and HRP-conjugated secondary antibody. Three clones each of the recombinant EPHA2-Myc carrying A: p.P584L, B: p.T940I, C: p.D942fsXC71, and E: p.V972GfsX39 mutation and a clone carrying D: p.A959T mutation show protein of the expected size (∼130 kDa). Cells transfected with the wild-type EPHA2-Myc construct were used as a positive control. Protein extracts of mock transfected cells were used as a negative control. Masses of protein standards are indicated in kilo Daltons (kDa).
For the localization study, a single clone of each mutant construct was ectopically expressed in MDCK and Caco-2 epithelial cells. The wild-type construct was used as a positive control. Expression of the ectopically expressed wild-type and mutant proteins in confluent cultures of these cells was analyzed by immunofluorescence labeling using the anti-Myc antibody. The wild-type EPHA2-Myc protein was found to discretely localize at the cell periphery in MDCK cells (Figure 2A); the protein was also observed in the cytoplasm in some cells. As EPHA2 is an integral membrane protein [34], the peripheral localization is consistent with integration of the protein in the cell membrane and localization of the endogenous EPHA2 in these cells [30]. EPHA2 is packaged in secretory vesicles for its transport to the cell membrane and may be endocytosed and degraded in endocytic vesicles [35]. Localization of the wild-type EPHA2-Myc in the cytoplasm can be explained by its presence in the secretory or endocytic vesicles. The p.P584L, p.A959T, and p.V972GfsX39 mutant EPHA2-Myc proteins were similarly detected in the cell periphery and the cytoplasm (Figure 2B,E,F). In addition to peripheral localization, a few cells also exhibited perinuclear localization of these proteins. In contrast, p.T940I and p.D942fsXC71 mutant EPHA2-Myc proteins exhibited predominantly perinuclear localization with some cytoplasmic and minimal peripheral localization (Figure 2C,D). Similar localization patterns of the wild-type and mutant EPHA2-Myc proteins to those in MDCK cells were observed in confluent Caco-2 cells (Figure 3).
Figure 2.
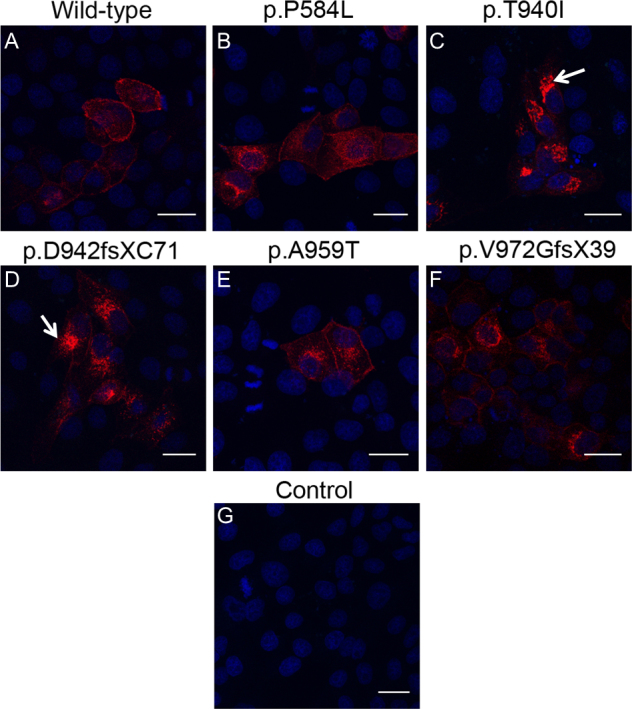
Localization of the mutant EPHA2-Myc proteins in MDCK cells. The ectopically expressed EPHA2-Myc protein (red) in MDCK cells was detected using anti-Myc primary antibody and Alexa Flour 594-conjugated secondary antibody. Nuclei (blue) were labeled with DAPI. Cells expressing the wild-type EPHA2-Myc (A) and p.P584L (B), p.A959T (E), or pV972fsX39 (F) mutant EPHA2-Myc proteins show peripheral and cytoplasmic localization of the protein. In cells expressing the p.T940I (C) and p.D942fsXC71 (D) mutant EPHA2-Myc proteins, the protein can be seen predominantly in the perinuclear region (arrow) and some in the cytoplasm. Mock transfected cells were used as a negative control (G). Representative images from two independent experiments are shown. Scale-bar=20 µm.
Figure 3.
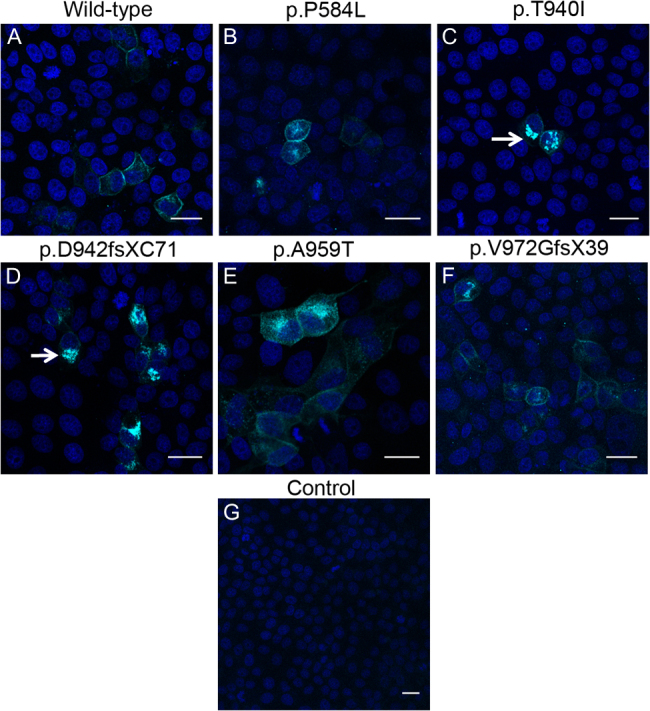
Localization of the mutant EPHA2-Myc proteins in Caco-2 cells. The ectopically expressed EPHA2-Myc protein (cyan) was detected in Caco-2 cells using an anti-Myc primary antibody and Cy5-conjugated secondary antibody. Nuclei (blue) were labeled with DAPI. Cells expressing the wild-type EPHA2-Myc (A) and p.P584L (B), p.A959T (E), or pV972fsX39 (F) mutant EPHA2-Myc show peripheral and some cytoplasmic localization of the protein. In the cells expressing p.T940I (C) or p.D942fsXC71 (D) mutant EPHA2-Myc protein, the protein is mostly seen in the perinuclear region (arrow). Mock transfected cells were used as a negative control (G). Representative images from two independent experiments are shown. Scale-bar=20 µm.
The perinuclear localization of some of the mutant proteins resembled that of the golgi apparatus. Thus, to further examine the fate of the mutant proteins, we performed co-localization analysis with a cis-golgi marker, GM130. Confluent cultures of MDCK cells ectopically expressing the mutant EPHA2-Myc proteins were immunolabeled with the anti-Myc and anti-GM130 antibodies. The double-immunolabeling experiments did not reveal any co-localization of the wild-type protein or the p.P584L, p.A959T, and p.V972GfsX39 mutant proteins with the cis-golgi marker (Figure 4A). These mutant proteins and the wild-type protein localized to the cell periphery and cytoplasm, as was observed in the single-labeling experiments. However, upon dual labeling of cells transfected with p.T940I or p.D942fsXC71 encoding EPHA2-Myc mutant construct, we observed co-localization of the mutant proteins with the cis-golgi marker in the majority of cells (Figure 4B, arrows). Additionally, approximately a quarter of cells showed peripheral localization of these two mutant proteins (Figure 4B, rows 2 and 4). These experiments indicated that the p.T940I and p.D942fsXC71 mutant proteins were retained in the cis-golgi apparatus, and that their recruitment to the cell membrane in MDCK cells was impaired. A similar pattern of co-localization of the wild-type and mutant EPHA2-Myc proteins with the cis-golgi marker was observed in confluent Caco-2 cells (Figures 5A,B). However, as observed in the single-labeling experiments, p.A959T and p.V972GfsX39 mutant proteins exhibited some perinuclear localization and co-localized with the cis-golgi apparatus in a few Caco-2 cells (Figure 5A). As these cells also exhibited peripheral localization of these mutant proteins, the observed co-localization may be due to overexpression of the ectopically expressed proteins. No labeling in cells hybridized with the two primary antibodies and mismatched secondary antibodies demonstrated specificity of each signal. Minimal background labeling in cells labeled by omitting the primary antibodies further demonstrated signal specificity (Appendix 5 and Appendix 6). Collectively, these data show that, of the five mutations analyzed in the present study, p.T940I and p.D942fsXC71 mutations have a more severe effect on localization of the EPHA2 protein in confluent, hence polarized, epithelial cells.
Figure 4.

Co-localization of the mutant EPHA2-Myc proteins with the cis-golgi apparatus in MDCK cells. Ectopically expressed EPHA2-Myc protein (cyan) in MDCK cells was detected using an anti-Myc primary antibody and Cy5-conjugated secondary antibody; the cis-golgi apparatus (red) was detected using an anti-GM130 primary antibody against the cis-golgi marker, GM130, and Alexa Flour 555-conjugated secondary antibody. Nuclei (blue) were labeled with DAPI. A: Cells expressing EPHA2-Myc protein with p.P584L (second row), p.A959T (third row), or p.V972GfsX39 (fourth row) mutation show peripheral and cytoplasmic localization of the protein similar to that of the wild-type protein (first row). No co-localization with the cis-golgi apparatus was observed in cells expressing these mutant proteins (Merge). B: Cells expressing EPHA2-Myc protein carrying a p.T940I or p.D942fsXC71 mutation show mis-localization of the protein in the perinuclear space (first and third row) and co-localization (white) with the cis-golgi apparatus (Merge, first and third row; arrow). A few cells expressing these mutant proteins show peripheral and cytoplasmic localization of the protein (second and fourth row), with some co-localization with the cis-golgi apparatus (Merge, fourth row, arrow). Representative images from two independent experiments are shown. Scale-bar=20 µm.
Figure 5.

Co-localization of the mutant EPHA2-Myc proteins with the cis-golgi apparatus in Caco-2 cells. Transiently transfected Caco-2 cells were labeled with an anti-Myc primary antibody and Cy5-conjugated secondary antibody to detect EPHA2-Myc protein (cyan), and with an anti-GM130 primary antibody and Alexa Flour 555-conjugated secondary antibody to detect the cis-golgi apparatus (red). Nuclei (blue) were labeled with DAPI. A: Cells expressing the EPHA2-Myc protein carrying p.P584L (second row), p.A959T (third row), or p.V972GfsX39 (fourth row) mutation showed peripheral and cytoplasmic localization of the protein comparable to that of the wild-type EPHA2-Myc protein (first row). The cells expressing mutant EPHA2-Myc protein carrying p.A959T and p.V972GfsX39 mutation also display some perinuclear localization of the protein (third and fourth row). These cells show some co-localization with the cis-golgi apparatus (white in the merged images), possibly due to the overexpression of the mutant proteins. B: Cells expressing mutant EPHA2-Myc proteins with p.T940I and p.D942fsXC71 mutations predominantly show mis-localization of the protein in the perinuclear region and co-localization with the cis-golgi apparatus (white in the merged image in the first and third rows; arrows). A few cells expressing these mutant proteins also exhibited peripheral and cytoplasmic localization (second and fourth rows). Representative images from two independent experiments are shown. Scale-bar=20 µm.
Discussion
In this study, we investigated the effect of congenital cataract causing mutations in EPHA2 on subcellular localization of the resulting mutant proteins in polarized epithelial cells. Functional effects of four causative mutations—p.G948W, p.T940I, p.D942fsXC71, and p.V972GfsX39—located in the SAM domain of EPHA2 protein have been previously reported [36]. These mutations reportedly lead to destabilization of the transiently expressed mutant proteins and impair cell migration in human fibroblast and mouse lens epithelial cells. The mutant proteins were reported to have a reduced half-life and undergo degradation through the proteasomal pathway. These mutations did not have any effect on receptor activation, but led to reduced phosphorylation of Akt, a downstream effector molecule in EPHA2 signaling. Additionally, the ectopically expressed wild-type protein upon stimulation by the EPHA2 ligand Ephrin-A5-Fc evenly distributed in small protein aggregates throughout the cell, whereas the mutant proteins were found to form large aggregates in EPHA2−/− mouse embryonic fibroblast (MEF) cells [36]. The EPHA2 protein localizes to the cell membrane in both lens fiber cells and lens epithelial cells in vivo [11,19,29]. Hence, in the present study, we investigated the effects of the causative mutations on localization of the protein in epithelial cells that represent EPHA2 localization in lens epithelial cells in vivo.
We found that ectopically expressed p.T940I and p.D942fsXC71 mutant EPHA2 proteins mis-localized to the perinuclear space and co-localized with the cis-golgi apparatus in both MDCK and Caco-2 epithelial cells. Membrane proteins such as EPHA2 are translated in the cytoplasm and transported to the endoplasmic reticulum (ER) for folding and glycosylation, and then to the golgi apparatus for further glycosylation [35]. The golgi apparatus comprises cisternae that are organized in cis, medial, and trans fashions, providing a distinct polarity to the organelle [35]. The cis-cisternae proximal to the ER receive the folded glycosylated protein from the ER, whereas the mis-folded proteins are retained in the ER. The proteins are modified by a series of enzymes as they pass through the medial and trans-golgi network, and are finally transported to their destination, the cell membrane, through packaging in the secretary vesicles. Therefore, co-localization of the two EPHA2 mutant proteins with the cis-golgi apparatus likely indicates their altered folding or glycosylation.
The secondary structure of the SAM domain of the EPHA2 protein is composed of five helical loops (H1–H5; Figure 6) [37]. The p.T940I and p.D942fsXC71 mutations alter the residues in the SAM domain that are located within or in proximity to the H4 loop. The p.T940I mutation alters a neutral polar amino acid residue, threonine, preceding the loop H4, into a highly hydrophobic amino acid residue, isoleucine, which may impact the EPHA2 structure. The frameshift due to the p.D942fsXC71 mutation alters residues not only in loop H4 but also in loop H5, including residues predicted to be critical for intramolecular interactions [37]. Thus, these mutations likely affect protein folding and post-translational modification, leading to retention of the mutant proteins in the perinuclear region in the ER/Golgi network. This may explain the previously reported reduced solubility of both these mutant proteins [36]. As these mutant proteins showed co-localization with the cis-golgi apparatus in some areas in the perinuclear region, this suggests that they may also be retained in the ER or in medial or trans-golgi networks during transport to the cell membrane. Co-localization studies with ER and medial or trans-golgi-specific markers may shed further light on impaired transport of these two mutant proteins. Whether the aggregated localization leads to degradation of the misfolded proteins via the proteasomal pathway in these cells as reported previously needs further investigation [36]. Nevertheless, mis-localization of these mutant EPHA2 proteins likely abolishes or delays their recruitment to the cell membrane and negatively impacts intercellular contacts in the lens epithelium that leads to cataract.
Figure 6.

Structural domains in the EPHA2 protein and location of the mutations analyzed in the study. Schematic diagram of the EPHA2 protein showing the ligand binding (brown), fibronectin III repeats (purple), transmembrane segment (light blue), kinase (dark blue), SAM (red), and PDZ (gray) domains. The helical loops (H1–H5) formed in the secondary structure of the SAM domain are also shown. The positions of the mutations analyzed in this study are marked. The mutations that mis-localized in MDCK and Caco-2 cells are highlighted in orange, and those that showed normal localization are highlighted in green. The p.P584L mutation affects a residue in the juxtamembrane region of the protein, but does not affect protein localization. The mutant EPHA2 proteins that mis-localize to the perinuclear region carry p.T940I and p.D942fsXC71 mutations that affect helix 4 (H4) in the SAM domain. The p.A959T mutation alters a residue in helix 5 (H5) in the SAM domain, and the p.V972GfsXC39 mutation affects residues in the SAM and PDZ domains of EPHA2 but do not affect protein localization.
The p.P584L, p.A959T, and p.V972GfsX39 mutant EPHA2 proteins localized to the cell periphery or cytoplasm, similar to the wild-type protein. In addition, the wild-type (data not shown) and these mutant EPHA2 proteins (Figure 2 and Figure 3) were detected in the perinuclear space in some transfected cells. The perinuclear localization of these mutant proteins is different than that of the p.T940I and p.D942fsXC71 mutant EPHA2 proteins, as the latter showed minimal localization to the periphery. Thus, we propose that the perinuclear localization of the former mutant proteins indicates their trafficking to the cell membrane. The p.V972GfsX39 mutant protein reportedly aggregates in MEF cells [36]. The difference in its localization between the present and the reported study may be due to the difference in cell types used in the two studies. Although the p.P584L, p.A959T, and p.V972GfsX39 mutant proteins did not affect protein localization, they may alter EPHA2 signaling, resulting in congenital cataract. The p.P584L mutant protein may alter phosphorylation states at serine 579 and tyrosine 588, the residues proximal to the mutated residue that may affect protein activation and signaling. The p.A959T mutation alters a residue downstream of p.I958, predicted to be important for intramolecular interactions [37]. Therefore this mutation could potentially disrupt such interactions. The p.V972GfsX39 mutation, in addition to disrupting the C-terminal residues in the SAM domain, disrupts the PDZ domain of EPHA2. PDZ domains of proteins are key regulators of their interactions with other molecules [38]. Hock et al. reported that the addition of 15 amino acids to the C-terminal of EPHB3 results in disruption of the PDZ domain and leads to a loss of interaction with a ras-binding protein Afadin (AF6), known to play a role in cellular junctions [39]. Thus, the p.V972GfsX39 mutation may similarly disrupt interaction of EPHA2 with downstream signaling molecules.
The two mutations that exhibited the most severe effects on protein localization in this study, p.T940I and p.D942fsXC71, lead to severe posterior polar and total/nuclear congenital cataract, respectively [6]. The presence of p.P584L, p.A959T, and p.V972GfsXC39 mutations, with mild or no effect on EPHA2 localization, leads to nuclear, mild sub-capsular and cortical/posterior sub-capsular, and severe posterior polar cataract, respectively [6,10]. Thus, there is no obvious correlation between localization of the mutant EPHA2 proteins in epithelial cells and congenital cataract phenotype or severity in affected individuals carrying these mutations. This observation is consistent with phenotypic heterogeneity of congenital cataract and the involvement of additional modifiers in the disease. Furthermore, it suggests that mutations in EPHA2 likely contribute to congenital cataract through various mechanisms. Finally, this study highlights the effect of the cell type used for investigation on the behavior of the mutant proteins. Similar studies in lens-derived cell lines, such as human HLE-B3 or mouse 17EM15 and 21EM15 cells, if wild-type EPHA2 localizes in those cell lines as in the lens in vivo, may provide further insights into the effects of the mutations in a more relevant environment to the lens. In summary, this study suggests that some of the causative mutations in the EPHA2 gene contribute to congenital cataract by affecting intercellular contacts between epithelial cells during lens development.
Acknowledgments
This work was funded by the National Health and Medical Research Council (NHMRC; Australia) project grant number GNT1009955. JEC and KPB are recipients of the NHMRC practitioner and research fellowships, respectively. The authors do not have any conflict of interest related to this work. This work was presented at the Asia-ARVO 2013 and International Congress for Eye Research 2014 meetings.
Appendix 1. Sequences of primers used for PCR-based mutagenesis. Underlined nucleotides indicate incorporated restriction enzyme sites.
To access the data, click or select the words “Appendix 1.”
Appendix 2. Primer combinations used for generating mutant EPHA2 cDNA fragments and the respective annealing temperatures employed for PCR.
To access the data, click or select the words “Appendix 2.”
Appendix 3. STR analysis.
To access the data, click or select the words “Appendix 3.”
Appendix 4. Localization of the NHS-A and tricellulin proteins in confluent Caco-2 cells demonstrating their polarized epithelial characteristic.
To access the data, click or select the words “Appendix 4.” Confluent cultures of Caco-2 cells were immunolabeled with the rabbit anti-NHS and anti-tircelllin antibody, as indicated. The epithelial cell specific isoform of NHS, NHS-A, localizes to the cell periphery in polarized epithelial cells where it interacts with the tight junction protein ZO-1 [33]. Peripheral immuno-positive labeling in Caco-2 cells with the anti-NHS antibody indicates their epithelial characteristic and polarization in culture (left panel). Tricellulin, a tricellular tight junction protein, localizes to tricellular tight junctions and bicellular junctions in epithelial cells [40]. Intense immune-positive labeling with anti-tricellulin antibody in Caco-2 cells at sites where three cells meet (tricellular tight junctions) and positive labeling at sites where two cells meet (bicellular tight junctions) also demonstrate their polarised epithelial characteristic (right panel).
Appendix 5. Negative controls included in the experiments for determining co-localization of the EPHA2-Myc mutant proteins with the cis-golgi apparatus in MDCK cells.
To access the data, click or select the words “Appendix 5.” Top row, MDCK cells transfected with the EPHA2-Myc construct encoding the p.T940I mutation hybridized with the mouse anti-Myc primary antibody and anti-rabbit IgG conjugated with Alexa Flour 555 secondary antibody as a control for cross-reactivity of the anti-rabbit secondary antibody to the primary antibody generated in mouse. Middle row, cells hybridized with the rabbit anti-GM130 primary antibody (marker for cis-golgi apparatus) and Cy5 conjugated anti-mouse IgG secondary antibody as a control for cross-reactivity of the anti-mouse secondary antibody to the primary antibody generated in rabbit. Bottom row, mock transfected cells hybridized with both the secondary antibodies without a primary antibody used as a control for any background due to the secondary antibodies. Nuclei (blue) were stained with DAPI. Representative images of controls from two independent experiments are shown. Scale-bar 20 µm.
Appendix 6. Negative controls included in the experiments for determining co-localization of the EPHA2-Myc mutant proteins with the cis-golgi apparatus in Caco-2 cells.
To access the data, click or select the words “Appendix 6.” Top row, Caco-2 cells transfected with the EPHA2-Myc construct encoding the p.T940I mutation hybridized with the mouse anti-Myc primary antibody and anti-rabbit IgG conjugated with Alexa Flour 555 secondary antibody as a control for cross-reactivity of the anti-rabbit secondary antibody to the primary antibody generated in mouse. Middle row, cells hybridized with the rabbit anti-GM130 primary antibody (marker for cis-golgi apparatus) and anti-mouse IgG conjugated with Cy5 secondary antibody as a control for cross-reactivity of the anti-mouse secondary antibody to the primary antibody generated in rabbit. Bottom row, mock transfected cells hybridized with both the secondary antibodies without a primary antibody used as a negative control. The secondary antibodies demonstrated minimal cross-reactivity and background. Nuclei (blue) were stained with DAPI. Representative images of controls from two independent experiments are shown. Scale-bar 20 µm.
References
- 1.Shiels A, Bennett TM, Hejtmancik JF. Cat-Map: putting cataract on the map. Mol Vis. 2010;16:2007–15. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=21042563&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 2.Apple DJ, Ram J, Foster A, Peng Q. Elimination of cataract blindness: a global perspective entering the new millenium. Surv Ophthalmol. 2000;45(Suppl 1):S1–196. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=11291895&dopt=Abstract [PubMed] [Google Scholar]
- 3.Gilbert C, Foster A. Childhood blindness in the context of VISION 2020–the right to sight. Bull World Health Organ. 2001;79:227–32. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=11285667&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 4.Rahi JS, Sripathi S, Gilbert CE, Foster A. Childhood blindness in India: causes in 1318 blind school students in nine states. Eye (Lond) 1995;9:545–50. doi: 10.1038/eye.1995.137. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=8543070&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 5.Shiels A, Bennett TM, Knopf HL, Maraini G, Li A, Jiao X, Hejtmancik JF. The EPHA2 gene is associated with cataracts linked to chromosome 1p. Mol Vis. 2008;14:2042–55. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=19005574&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang T, Hua R, Xiao W, Burdon KP, Bhattacharya SS, Craig JE, Shang D, Zhao X, Mackey DA, Moore AT, Luo Y, Zhang J, Zhang X. Mutations of the EPHA2 receptor tyrosine kinase gene cause autosomal dominant congenital cataract. Hum Mutat. 2009;30:E603–11. doi: 10.1002/humu.20995. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=19306328&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 7.Kaul H, Riazuddin SA, Shahid M, Kousar S, Butt NH, Zafar AU, Khan SN, Husnain T, Akram J, Hejtmancik JF, Riazuddin S. Autosomal recessive congenital cataract linked to EPHA2 in a consanguineous Pakistani family. Mol Vis. 2010;16:511–7. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=20361013&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 8.Shentu XC, Zhao SJ, Zhang L, Miao Q. A novel p.R890C mutation in EPHA2 gene associated with progressive childhood posterior cataract in a Chinese family. Int J Ophthalmol. 2013;6:34–8. doi: 10.3980/j.issn.2222-3959.2013.01.07. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23447127&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aldahmesh MA, Khan AO, Mohamed JY, Hijazi H, Al-Owain M, Alswaid A, Alkuraya FS. Genomic analysis of pediatric cataract in Saudi Arabia reveals novel candidate disease genes. Genet Med. 2012;14:955–62. doi: 10.1038/gim.2012.86. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=22935719&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 10.Dave A, Laurie K, Staffieri SE, Taranath D, Mackey DA, Mitchell P, Wang JJ, Craig JE, Burdon KP, Sharma S. Mutations in the EPHA2 gene are a major contributor to inherited cataracts in South-Eastern Australia. PLoS One. 2013;8:e72518. doi: 10.1371/journal.pone.0072518. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=24014202&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jun G, Guo H, Klein BE, Klein R, Wang JJ, Mitchell P, Miao H, Lee KE, Joshi T, Buck M, Chugha P, Bardenstein D, Klein AP, Bailey-Wilson JE, Gong X, Spector TD, Andrew T, Hammond CJ, Elston RC, Iyengar SK, Wang B. EPHA2 is associated with age-related cortical cataract in mice and humans. PLoS Genet. 2009;5:e1000584. doi: 10.1371/journal.pgen.1000584. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=19649315&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pasquale EB. Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat Rev Cancer. 2010;10:165–80. doi: 10.1038/nrc2806. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=20179713&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pasquale EB. Eph-ephrin bidirectional signaling in physiology and disease. Cell. 2008;133:38–52. doi: 10.1016/j.cell.2008.03.011. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=18394988&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 14.McAvoy JW, Chamberlain CG, de Iongh RU, Hales AM, Lovicu FJ. Lens development. Eye (Lond) 1999;13(Pt 3b):425–37. doi: 10.1038/eye.1999.117. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=10627820&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 15.Rosenberg IM, Goke M, Kanai M, Reinecker HC, Podolsky DK. Epithelial cell kinase-B61: an autocrine loop modulating intestinal epithelial migration and barrier function. Am J Physiol. 1997;273:G824–32. doi: 10.1152/ajpgi.1997.273.4.G824. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=9357823&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 16.Tessier-Lavigne M. Eph receptor tyrosine kinases, axon repulsion, and the development of topographic maps. Cell. 1995;82:345–8. doi: 10.1016/0092-8674(95)90421-2. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=7634322&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 17.Mori T, Wanaka A, Taguchi A, Matsumoto K, Tohyama M. Differential expressions of the eph family of receptor tyrosine kinase genes (sek, elk, eck) in the developing nervous system of the mouse. Brain Res Mol Brain Res. 1995;29:325–35. doi: 10.1016/0169-328x(94)00263-e. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=7609620&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 18.Sefton M, Araujo M, Nieto MA. Novel expression gradients of Eph-like receptor tyrosine kinases in the developing chick retina. Dev Biol. 1997;188:363–8. doi: 10.1006/dbio.1997.8638. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=9268581&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 19.Shi Y, De Maria A, Bennett T, Shiels A, Bassnett S. A Role for Epha2 in Cell Migration and Refractive Organization of the Ocular Lens. Invest Ophthalmol Vis Sci. 2012;53:551–9. doi: 10.1167/iovs.11-8568. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=22167091&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Son AI, Cooper MA, Sheleg M, Sun Y, Kleiman NJ, Zhou R. Further analysis of the lens of ephrin-A5−/− mice: development of postnatal defects. Mol Vis. 2013;19:254–66. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23401654&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 21.Lindberg RA, Hunter T. cDNA cloning and characterization of eck, an epithelial cell receptor protein-tyrosine kinase in the eph/elk family of protein kinases. Mol Cell Biol. 1990;10:6316–24. doi: 10.1128/mcb.10.12.6316. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=2174105&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miyoshi J, Takai Y. Molecular perspective on tight-junction assembly and epithelial polarity. Adv Drug Deliv Rev. 2005;57:815–55. doi: 10.1016/j.addr.2005.01.008. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=15820555&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 23.Tanaka M, Kamata R, Sakai R. EphA2 phosphorylates the cytoplasmic tail of Claudin-4 and mediates paracellular permeability. J Biol Chem. 2005;280:42375–82. doi: 10.1074/jbc.M503786200. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=16236711&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 24.Zantek ND, Azimi M, Fedor-Chaiken M, Wang B, Brackenbury R, Kinch MS. E-cadherin regulates the function of the EphA2 receptor tyrosine kinase. Cell Growth Differ. 1999;10:629–38. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=10511313&dopt=Abstract [PubMed] [Google Scholar]
- 25.Orsulic S, Kemler R. Expression of Eph receptors and ephrins is differentially regulated by E-cadherin. J Cell Sci. 2000;113:1793–802. doi: 10.1242/jcs.113.10.1793. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=10769210&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 26.Fang WB, Ireton RC, Zhuang G, Takahashi T, Reynolds A, Chen J. Overexpression of EPHA2 receptor destabilizes adherens junctions via a RhoA-dependent mechanism. J Cell Sci. 2008;121:358–68. doi: 10.1242/jcs.017145. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=18198190&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 27.Wakayama Y, Miura K, Sabe H, Mochizuki N. EphrinA1-EphA2 signal induces compaction and polarization of Madin-Darby canine kidney cells by inactivating ezrin through negative regulation of RhoA. J Biol Chem. 2011;286:44243–53. doi: 10.1074/jbc.M111.267047. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=21979959&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bassnett S, Shi Y, Vrensen GF. Biological glass: structural determinants of eye lens transparency. Philos Trans R Soc Lond B Biol Sci. 2011;366:1250–64. doi: 10.1098/rstb.2010.0302. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=21402584&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng C, Ansari MM, Cooper JA, Gong X. EphA2 and Src regulate equatorial cell morphogenesis during lens development. Development. 2013;140:4237–45. doi: 10.1242/dev.100727. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=24026120&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dave A, Craig JE, Sharma S. The status of intercellular junctions in established lens epithelial cell lines. Mol Vis. 2012;18:2937–46. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23288986&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 31.Hidalgo IJ, Raub TJ, Borchardt RT. Characterization of the human colon carcinoma cell line (Caco-2) as a model system for intestinal epithelial permeability. Gastroenterology. 1989;96:736–49. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=2914637&dopt=Abstract [PubMed] [Google Scholar]
- 32.Sharma S, Burdon KP, Dave A, Jamieson RV, Yaron Y, Billson F, Van Maldergem L, Lorenz B, Gecz J, Craig JE. Novel causative mutations in patients with Nance-Horan syndrome and altered localization of the mutant NHS-A protein isoform. Mol Vis. 2008;14:1856–64. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=18949062&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 33.Sharma S, Koh KS, Collin C, Dave A, McMellon A, Sugiyama Y, McAvoy JW, Voss AK, Gecz J, Craig JE. NHS-A isoform of the NHS gene is a novel interactor of ZO-1. Exp Cell Res. 2009;315:2358–72. doi: 10.1016/j.yexcr.2009.05.008. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=19447104&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 34.Pasquale EB. Eph receptor signalling casts a wide net on cell behaviour. Nat Rev Mol Cell Biol. 2005;6:462–75. doi: 10.1038/nrm1662. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=15928710&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 35.Lewin B. Protein Trafficking. In: Carlson G, editor. Genes VIII: Pearson Prentice Hall; 2004. p. 787–791. [Google Scholar]
- 36.Park JE, Son AI, Hua R, Wang L, Zhang X, Zhou R. Human Cataract Mutations in EPHA2 SAM Domain Alter Receptor Stability and Function. PLoS One. 2012;7:e36564. doi: 10.1371/journal.pone.0036564. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=22570727&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smalla M, Schmieder P, Kelly M, Ter Laak A, Krause G, Ball L, Wahl M, Bork P, Oschkinat H. Solution structure of the receptor tyrosine kinase EphB2 SAM domain and identification of two distinct homotypic interaction sites. Protein Sci. 1999;8:1954–61. doi: 10.1110/ps.8.10.1954. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=10548040&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harris BZ, Lim WA. Mechanism and role of PDZ domains in signaling complex assembly. J Cell Sci. 2001;114:3219–31. doi: 10.1242/jcs.114.18.3219. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=11591811&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 39.Hock B, Bohme B, Karn T, Yamamoto T, Kaibuchi K, Holtrich U, Holland S, Pawson T, Rubsamen-Waigmann H, Strebhardt K. PDZ-domain-mediated interaction of the Eph-related receptor tyrosine kinase EphB3 and the ras-binding protein AF6 depends on the kinase activity of the receptor. Proc Natl Acad Sci USA. 1998;95:9779–84. doi: 10.1073/pnas.95.17.9779. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=9707552&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Furuse M, Izumi Y, Oda Y, Higashi T, Iwamoto N. Molecular organization of tricellular tight junctions. Tissue Barriers. 2014;2:e28960. doi: 10.4161/tisb.28960. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=25097825&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]