

Abstract
Transitional cell carcinoma (TCC) is the one of the most commonly observed types of cancer globally. The identification of novel disease-associated genes in TCC has had a significant effect on the diagnosis and treatment of bladder cancer; however, there may be a large number of novel genes that have not been identified. In the present study, the exomes of two individuals who were diagnosed with muscle-invasive TCC (MI-TCC) were sequenced to investigate potential variants. Subsequently, following algorithm and filter analysis, Sanger sequencing was used to validate the results of deep sequencing. Immunohistochemistry (IHC) was employed to observe the differences in HECT, C2 and WW domain-containing E3 ubiquitin protein ligase 1 (HECW1) protein expression between tumor tissues and para-carcinoma tissues. A total of 6 nonsynonymous mutation genes were identified in MI-TCC, identified as copine VII, RNA binding motif protein, X-linked-like 3, acyl-CoA synthetase medium-chain family member 2A, HECW1, zinc finger protein 273 and trichohyalin. Furthermore, 5 cases were identified to possess a HECW1 gene mutation in 61 MI-TCC specimens, and all of these were point mutations located at exon 11 on chromosome 7. The mutation categories of HECW1 had 4 missense mutations and 1 nonsense mutation. IHC revealed that HECW1 protein was expressed at significantly increased levels in MI-TCC compared with normal bladder urothelium (P<0.001). The present study provided a novel approach for investigating genetic changes in the MI-TCC exome, and identified the novel mutant gene HECW1, which may possess a significant role in the pathogenesis of TCC.
Keywords: whole-exome sequencing, Sanger sequencing, mutation, HECT, C2 and WW domain-containing E3 ubiquitin protein ligase 1, transitional cell carcinoma
Introduction
Bladder cancer (BC) is the ninth most commonly observed cancer globally, with an estimated 386,300 novel cases and 150,200 mortalities in 2011 (1). Transitional cell carcinoma (TCC) is the most predominant type, accounting for 90% of all cases of diagnosed BC (1). Clinical studies have divided TCC patients into two distinct subtypes: Non-muscle-invasive TCCs (NMI-TCCs) and muscle-invasive TCCs (MI-TCCs). In total, 70% of patients present with NMI-TCC, while the remaining 30% present with MI-TCC. MI-TCCs are less common than NMI-TCCs, but are associated with an increased mortality rate (2). Therefore, the present study primarily focused on MI-TCC due to its poor prognosis and survival rates.
The occurrence and development of cancer frequently involves the accumulation of genomic alterations (3), therefore it is important to identify these alterations using various techniques. A number of researchers have employed next-generation sequencing technologies, including whole-genome, whole-exome and whole-transcriptome approaches. This enables the investigation of the main types of alterations to the somatic cancer genome, including point mutations, copy number alterations, microbial infections, small insertions and deletions, and chromosomal rearrangements), providing insights for improving our understanding of cancer biology, diagnosis and therapy in acute myeloid leukemia (4,5), breast cancer (6–8), melanoma (9), lung cancer (10,11), kidney cancer (12) and ovarian clear cell carcinoma (13), among others.
Previous research based on candidate gene approaches has suggested that BC may represent a heterogeneous disease (2). The two subgroups of TCCs may possess differential genetic backgrounds: NMI-TCCs frequently carry mutations in the fibroblast growth factor receptor 3 (FGFR3) and RAS genes, while MI-TCCs often carry mutations in the tumor protein p53 (TP53) and retinoblastoma 1 (RB1) genes (2). These findings provided significant insights into potential diagnoses and therapeutic applications, nevertheless, to the best of our knowledge, a comprehensive analysis of TCC has not been performed. Gui et al (14) identified genetic aberrations of unknown chromatin remodeling genes [Ubiquitously transcribed tetratricopeptide repeat, X chromosome, mixed-lineage leukemia protein 3, CREBBP-EP300, nuclear receptor corepressor 1, AT rich interactive domain 1A (SWI-like) and chromodomain helicase DNA binding protein 6] in TCC by whole-exome sequencing and proposed that aberrant chromatin regulation may be a hallmark of BC. Li et al (15) employed single-cell sequencing analysis to investigate the genetic properties of bladder tumor alterations at the single-cell level and to assess the evolution of bladder cancer at a cell-population level. Guo et al (16) observed frequent alterations in stromal antigen 2 and extra spindle pole bodies homolog 1, and recurrent fusion involving FGFR3 and transforming, acidic coiled-coil containing protein 3. These genes were identified to be involved in the sister chromatid cohesion and segregation (SCCS) process by whole-genome and whole-exome sequencing, and evidence was provided that genetic alterations affecting the SCCS process may be involved in bladder tumorigenesis, meaning that a novel therapeutic possibility for bladder cancer was identified.
The identification of these novel disease-associated genes in TCC has exhibited a significant effect on the diagnosis and treatment of BC; however, there may be a large number of novel genes that have not yet been identified. Thus, the present study employed whole-exome sequencing methods to detect somatic mutations in two MI-TCC patients. The aim of the present study was to screen the MI-TCC patients systematically to identify any previously unidentified MI-TCC-associated genes. A total of 565 somatic mutation candidates were detected in the sequenced exomes of the two MI-TCC patients, and 8 nonsynonymous mutation genes were validated, including copine VII (CPNE7), serine/arginine repetitive matrix 5, HECT, C2 and WW domain-containing E3 ubiquitin protein ligase 1 (HECW1), zinc finger protein (ZNF)792, ZNF273, trichohyalin (TCHH), RNA binding motif protein, X-linked-like 3 (RBMXL3) and acyl-CoA synthetase medium-chain family member 2A (ACSM2A). These novel mutation genes may be associated with the mechanism of bladder tumorigenesis and development. Identification of these genes may have therapeutic implications and may assist with the development of future treatments for bladder cancer.
Materials and methods
Sample description and preparation
Samples were taken from two patients who had been newly diagnosed with primary MI-TCC of the bladder at the First Affiliated Hospital of Soochow University (Suzhou, China), according to the 2004 World Health Organization/International Society of Urological Pathology grading system (17). Each subject was provided with appropriate information prior to recruitment for the present study, according to the regulations of the Institutional Ethics Review Boards. Cancerous tissue samples and normal controls (morphologically adjacent healthy bladder tissue) were rapidly frozen in liquid nitrogen following collection and were stored at −80°C until subsequent study. The pathological type of bladder cancer was observed to be high-grade muscle invasive urothelial carcinoma (T2), and was microscopically validated by two independent pathologists. In the present study, only TCCs with malignant cell purities >80% were selected for DNA extraction and subsequent sequencing.
Genomic DNA extraction and whole-exome sequencing
Genomic DNA from tumor and matched para-carcinoma normal tissue samples for the two patients with MI-TCC was isolated using a DNA extraction kit (Wizard Genomic DNA Purification Kit®; Promega Corp., Madison, WI, USA), and DNA fragment libraries were sheared and constructed according to the manufacturer's protocols, provided using the 5500 SOLiD™ Fragment Library Core kit (Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA). Subsequently, the exome capture procedure was performed according to the manufacturer's protocols with the A14060 TargetSeq™ Exome Enrichment kit (Applied Biosystems; Thermo Fisher Scientific, Inc.).
Enriched DNA fragment libraries were subsequently sequenced on the 3730 DNA Analyzer (Applied Biosystems; Thermo Fisher Scientific, Inc.), according to the manufacturer's protocols, and 200-bp paired-end reads were generated.
Whole-exome sequencing read mapping and detection of somatic mutations
High-quality paired-end reads were gap aligned to the National Center for Biotechnology Information human reference genome (hg19) using Burrows-Wheeler Aligner (BWA; http://bio-bwa.sourceforge.net/) following elimination of whole-exome sequencing reads, including low-quality reads and polymerase chain reaction (PCR) duplicates with >5 unknown bases (18). Local realignment of the BWA-aligned reads was subsequently corrected with the Genome Analysis Toolkit (GATK; https://www.broadinstitute.org/gatk/index.php) (19). The raw lists of potential somatic substitutions were obtained by VarScan (v2.2; http://varscan.sourceforge.net/) based on the GATK-alignments (20,21). In order to remove germline variants, somatic mutations were referenced using the dbSNP database (version 135; http://www.ncbi.nlm.nih.gov/SNP/). The somatic mutation candidates were subsequently submitted and annotated using ANNOVAR tool (http://annovar.openbioinformatics.org/en/latest/) (22). For these software packages, a number of rules must be complied with: i) All samples must be covered sufficiently (5–100×) at the genomic position; ii) the average base reads for a given genomic position in tumor samples should be ≥2; iii) the variants should be supported by at least 10% of the total reads in the tumors, and no high-quality variant-supporting reads are allowed in normal controls (14); and iv) the variant P-values in the tumors should be ≤0.05. The shared single nucleotide polymorphisms (SNPs) and insertion/deletions (INDELs) were screened and selected from the sequence outcomes of the two samples, following filtering using the aforementioned criteria.
In order to filter out identical mutations, the SNP data sets were referenced against the Standard Nucleotide Basic Local Alignment Search Tool (BLAST; blast.cbi.nlm.nih.gov/Blast.cgi) and SNP BLAST (blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&PAGE_TYPE=BlastSearch &LINK_LOC=blasthom). The remaining mutations were subjected to subsequent analyses. Bam files were aligned to the Integrative Genomics Viewer 2.0 tool (www.broadinstitute.org/igv/) (23,24).
Validation of somatic substitutions by Sanger sequencing
Sanger sequencing based on PCR amplification was used to validate the non-silent somatic variants. PCR primers for putative somatic variants were designed by Primer Premier 5.0 software (PREMIER Biosoft International, Palo Alto, CA, USA). If variants were successfully confirmed in tumors by reverse transcription (RT)-PCR, identical primer pairs were used to validate the putative mutation by Sanger sequencing in 61 MI-TCC formalin-fixed, paraffin-embedded (FFPE) samples. Total RNA was isolated from tissue samples using Invitrogen TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer's protocol. RT-PCR was carried out using the OneStep RT-PCR kit (catalog no. 210212; Qiagen, Hilden, Germany), 500 ng total RNA template. The total volume of the reaction was 50 µl and the following PCR cycling parameters were used: 50°C for 30 min to reverse transcribe the RNA; 95°C for 15 min; 94°C for 45 sec, 56°C for 1 min and 72 °C for 1 min, for 28 cycles; and then 72°C for 10 min. Equal volumes of PCR products were resolved on agarose gel, visualized with ethidium bromide, and photographed. Images were analyzed with AlphaImager 2200 (Alpha Innotech Co., San Leandro, CA, USA).
Immunohistochemistry (IHC) of the putative mutated gene
A total of 10 matched tissue samples, including cancer and adjacent normal tissues, were obtained from pathologically validated MI-TCC surgical specimens and were immediately fixed in 4% 3-heptanone in Gibco phosphate-buffered saline (PBS; Thermo Fisher Scientific) for 24 h, and then embedded in paraffin. Subsequently, HECW1 staining was accomplished with EnVision™ IHC (Dako, Glostrup, Denmark) protocols. The 5-µm thick paraffin-embedded tissue sections were prepared and subsequently deparaffinized using xylene (Beyotime Institute of Biotechnology, Haimen, China) and rehydrated with graded ethanol (Beyotime Institute of Biotechnology). Antigen retrieval was performed at 95°C in an oven for 45 min. The slides were dewaxed using xylene and rehydrated with an ethanol series. The slides were then treated with 3% hydrogen peroxide in methanol for 10 min at room temperature to inactivate endogenous peroxidase activity, following by washing with PBS 3 times for 3 min each. The samples were incubated with rabbit anti-human polyclonal HECW1 antibody (dilution, 1:200; catalog no. ab-121264; Abcam, Cambridge, UK) overnight at 4°C, followed by washing with PBS 3 times for 3 min each. The sections were incubated with goat anti-rabbit IgG secondary antibody (dilution, 1:200; catalog no. ab-97051; Abcam) for 30 min at room temperature, washed with PBS 3 times for 3 min each and stained with color reagent 3,3′-diaminobenzidine, followed by rinsing in water and counterstaining with hematoxylin (Beyotime Institute of Biotechnology). The slides were mounted using permanent mounting medium. Each set of experiments was performed in triplicate and completed under identical experimental conditions.
For evaluation of HECW1 expression in MI-TCC and normal tissues, the positive staining cell counting method was employed. A total of 10 visual fields were randomly selected for each sample and examined by a light microscope (IX53; Olympus, Tokyo, Japan) under high magnification (×400). Bladder cancer cells that were immunoreactive to anti-HECW1 demonstrated brown staining in the nucleus and cytoplasm. Samples were scored by counting five high-power fields per slice and represented the average of three independent experiments. A score of ≥10% brown stained cells in the total number of five high-power fields was considered to indicate the expression of HECW1, whereas <10% brown stained cells indicated no or negative expression.
Statistical analysis
Data were compared using Fisher's exact test in SPSS version 13.0 (SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
An extensive repertoire of somatic mutations is identifiable using whole-genome sequencing
Whole-exome sequencing of genomic DNA from tissue samples of two individuals exhibiting MI-TCC (stage, ≥T2) and their matched para-carcinoma normal tissue samples was performed. Using the SOLiD™ 5500 platform, 75-bp short-read sequences were generated. An average coverage depth of 63.29× was gained for all the samples sequenced, with 88.64% of the targeted bases being sufficiently covered.
Following the use of bioinformatic algorithms and stringent criteria to validate somatically acquired genetic variation from the raw sequencing data, 121 predicated candidate somatic mutations were identified, and 34 somatic substitutions and 3 INDELS were confirmed. C:G>T:A transitions were the predominant mutation spectrum in the two MI-TCC samples. A total of 6 nonsynonymous mutation genes were identified (CPNE7, RBMXL3, ACSM2A, HECW1, ZNF273 and TCHH) by comparing the standard nucleotide BLAST and SNP BLAST. The 6 nonsynonymous mutation genes were subjected to rigorous validation by RT-PCR and Sanger sequencing in 61 FFPE MI-TCC cases. Mutations obtained from whole-exome sequencing outcomes were compared with those obtained from Sanger sequencing outcomes in the 6 nonsynonymous mutation genes, in order to validate the recurrently mutated genes that may be involved in TCC tumorigenesis. It was identified that there were no mutations at the same point or near to 5 of the nonsynonymous mutation genes (CPNE7, RBMXL3, ACSM2A, ZNF273, TCHH). However, 4 missense and 1 nonsense point mutations were identified in the HECW1 gene, which was altered in 6.6% of TCC cases (Fig. 1). In addition, it was identified that the HECW1 mutant gene was located at exon 11 on chromosome 7, and the mutation point aggregate was in PS5011 (cysteine-rich) and PS50324 (serine-rich) of exon 11 (asia.ensembl.org/Homo_sapiens/Transcript/ProteinSummary?db=core; g=ENSG00000002746; r=7:43112599–43566001; t=ENST00000395891) (Fig. 2).
Figure 1.

HECW1 mutation revealed by (A) whole-exome sequencing and (B-F) Sanger sequencing. (A) Sequencing analysis of HECW1 was aligned by Integrative Genomics Viewer 2.0 tool, which indicated a base substitution (G to T). (B) A case of missense mutation (GCC to ACC) in HECW1 was validated at the same mutation point by Sanger sequencing. (C-E) A total of three cases of missense mutation (CCC to ACC) in HECW1 were detected in nearby areas by Sanger sequencing. (F) A case of nonsense mutation (TCG to TAG) was validated in a nearby area by Sanger sequencing. The mutation points are presented by an arrow and were confirmed with the reverse strand (not shown). HECW1, HECT, C2 and WW domain-containing E3 ubiquitin protein ligase 1.
Figure 2.

Mutation point of the HECT, C2 and WW domain-containing E3 ubiquitin protein ligase 1 gene aggregating in PS50311 (cysteine-rich) and PS50324 (serine-rich) of exon 11 (http://www.ensembl.org/index/html/). SMART; simple modular architecture research tool.
HECW1 expression is detectable in bladder tissue specimens
Immunostaining analysis revealed that HECW1 protein was localized to the cytoplasm and nucleus, and was highly expressed in MI-TCC bladder tissue specimens, with a total positive rate of 100% (10/10 cases). However, HECW1 protein exhibited almost no expression in all normal bladder urothelium samples (Fig. 3). Fisher's exact test revealed that HECW1 expression was statistically significant in MI-TCC specimens compared with control tissues (P<0.001).
Figure 3.
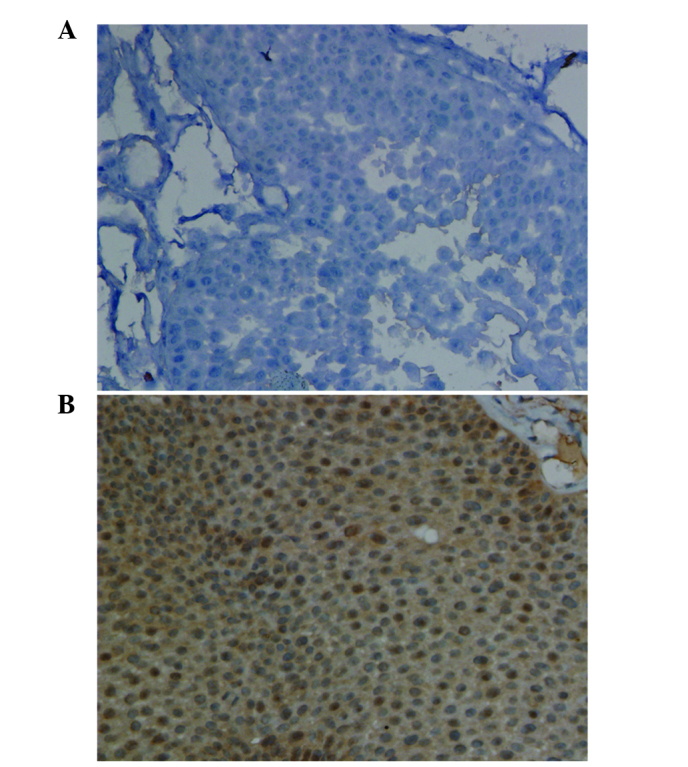
Representative immunohistochemical staining for HECW1 in (A) normal bladder urothelial tissue and (B) MI-TCC tissue under high magnification (×400). HECW1 protein levels in MI-TCC tissues were significantly upregulated compared with normal bladder urothelial tissues. HECW1, HECT, C2 and WW domain-containing E3 ubiquitin protein ligase 1; MI-TCC, muscle-invasive transitional cell carcinoma.
Discussion
In the present study, the sequencing and analysis of two MI-TCC cancer genomes was performed using whole-exome sequencing technology. Unlike whole-genome and whole-transcriptome genetic analysis, the present study did not detect copy number alterations, chromosomal translocations or epigenetic changes. However, a number of novel mutated genes were identified, which may be relevant for TCC. C:G>T:A transitions were the most common substitution spectrum in the two MI-TCC samples, as has previously been reported in TCC cases (14) and a number of other human cancers (10,25,26). The six somatic mutations that were discovered in the present study were single base changes, and none of them had previously been identified in the TCC genome. The novel nonsynonymous mutation genes that were detected did not include the well-known bladder cancer genes (FGFR3, RAS, TP53 and RB1) and contrasted with the findings of previous studies (14–16). This discrepancy may be due to differing experimental conditions, and algorithm and filter use. Subsequently, the mutation results were validated by Sanger sequencing and a recurrent mutant HECW1 gene was identified. HECW1 mutations were detected at exon 11 on chromosome 7 in more than one MI-TCC genome, suggesting that these mutations did not occur randomly and potentially possessed a significant role in the pathogenesis of TCC. In the present study, the remaining five nonsynonymous mutation genes were not validated by Sanger sequencing. Small quantities of tissue samples may have contributed to these results. Therefore, additional studies with increased sample sizes are required to achieve conclusive results.
HECW1, also known as NEDD4-like ubiquitin protein ligase 1 (NEDL1), which encodes HECT-type E3 ubiquitin ligase, is primarily detected in human neuronal tissues (27,28) and may regulate the bone morphogenetic protein signaling pathway during embryonic development and bone remodeling (29). HECW1 may combine misfolded superoxide dismutase 1, translocon-associated protein-δ, and dishevelled-1 to form an ubiquitinated protein complex that mutually affect their functions and may be included in potentially cytotoxic protein aggregates, leading to motor neuron death in familial amyotrophic lateral sclerosis (ALS) (27). Notably, Zhang et al (30) identified that human NEDL1 transgenic mice may exhibit ALS-like symptoms. The HECW1 gene has been identified to be significantly upregulated in favorable neuroblastoma compared with unfavorable neuroblastoma (27). HECW1 cooperates with p53 to enhance its transcriptional pro-apoptotic activity, which may be capable of inducing apoptosis in cancerous cells possessing wild-type p53 independent of E3 ligase activity (31). Furthermore, small interfering RNA-mediated knockdown of endogenous HECW1 reduced the transcription of p53 target genes induced by Adriamycin (ADR) and allowed the resistance of U2OS cells to ADR (28). In addition, HECW1 has been observed to negatively regulate ErbB4 activity leading to suppression of its expression and functioning in breast cancer (32). The results of these previous studies indicate that HECW1 may act as a tumor suppressor gene in neuroblastoma, osteosarcoma and breast cancer. This may provide a novel insight for the investigation of HECW1 chemosensitivity in bladder cancer.
In the present study, it was identified that HECW1 protein was expressed at significantly increased levels in MI-TCC compared with normal bladder urothelium. This was in complete contrast to the expression of HECW1 in neuroblastoma, osteosarcoma and breast cancer. Therefore, the present study proposed that the HECW1 gene may have an alternative role in TCC, which may be opposite to its role in neuroblastoma and breast cancer. However, the role of the HECW1 gene mutation in bladder cancer, and whether this mutation affected the expression and functioning of the protein remain to be elucidated.
In conclusion, the present study successfully used a next-generation whole-exome sequencing approach to identify novel candidate genes that may be relevant for bladder cancer pathogenesis. The novel mutant HECW1 gene has been identified to have a significant role and may act as a tumor suppressor in various tumors other than bladder cancer. The results of the present study demonstrated the power of whole-exome sequencing in identifying mutational genes in cancer. It is predicted that this technology may lead to the identification of previously unknown mutant genes that may have potential as future therapeutic targets.
Acknowledgements
The present study was supported by the National Natural Science Foundation of China (grant nos. 81272839, 81300537 and 81472401), the Outstanding Medical Academic Leader Program of Jiangsu Province (grant no. LJ201138), the Key Discipline of Medicine of Jiangsu Province (grant nos. XK201151 and BL2014038) and the Key Laboratory Foundation of Suzhou (grant no. SZS201001).
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Wu XR. Urothelial tumorigenesis: A tale of divergent pathways. Nat Rev Cancer. 2005;5:713–725. doi: 10.1038/nrc1697. [DOI] [PubMed] [Google Scholar]
- 3.Haimovich AD. Methods, challenges, and promise of next-generation sequencing in cancer biology. Yale J Biol Med. 2011;84:439–446. [PMC free article] [PubMed] [Google Scholar]
- 4.Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, Dooling D, Dunford-Shore BH, McGrath S, Hickenbotham M, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stephens PJ, McBride DJ, Lin ML, Varela I, Pleasance ED, Simpson JT, Stebbings LA, Leroy C, Edkins S, Mudie LJ, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. 2009;462:1005–1010. doi: 10.1038/nature08645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ding L, Ellis MJ, Li S, Larson DE, Chen K, Wallis JW, Harris CC, McLellan MD, Fulton RS, Fulton LL, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464:999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shah SP, Morin RD, Khattra J, Prentice L, Pugh T, Burleigh A, Delaney A, Gelmon K, Guliany R, Senz J, et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009;461:809–813. doi: 10.1038/nature08489. [DOI] [PubMed] [Google Scholar]
- 9.Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, Varela I, Lin ML, Ordóñez GR, Bignell GR, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–196. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pleasance ED, Stephens PJ, O'Meara S, McBride DJ, Meynert A, Jones D, Lin ML, Beare D, Lau KW, Greenman C, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463:184–190. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Campbell PJ, Stephens PJ, Pleasance ED, O'Meara S, Li H, Santarius T, Stebbings LA, Leroy C, Edkins S, Hardy C, et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat Genet. 2008;40:722–729. doi: 10.1038/ng.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu X, Hou Y, Yin X, Bao L, Tang A, Song L, Li F, Tsang S, Wu K, Wu H, et al. Single-cell exome sequencing reveals single-nucleotide mutation characteristics of a kidney tumor. Cell. 2012;148:886–895. doi: 10.1016/j.cell.2012.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones S, Wang TL, Shih IEM, Mao TL, Nakayama K, Roden R, Glas R, Slamon D, Diaz LA, Jr, Vogelstein B, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228–231. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao S, Wu R, Chen C, Li X, Zhou L, et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet. 2011;43:875–878. doi: 10.1038/ng.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, Xu X, Song L, Hou Y, Li Z, Tsang S, Li F, Im KM, Wu K, Wu H, et al. Single-cell sequencing analysis characterizes common and cell-lineage-specific mutations in a muscle-invasive bladder cancer. Gigascience. 2012;1:12. doi: 10.1186/2047-217X-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo G, Sun X, Chen C, Wu S, Huang P, Li Z, Dean M, Huang Y, Jia W, Zhou Q, et al. Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nat Genet. 2013;45:1459–1463. doi: 10.1038/ng.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El Gendy H, Madkour B, Abdelaty S, Essawy F, Khattab D, Hammam O, El Kholy A, Nour HH. Galectin 3 for the diagnosis of bladder cancer. Arab J Urol. 2014;12:178–181. doi: 10.1016/j.aju.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koboldt DC, Chen K, Wylie T, Larson DE, McLellan MD, Mardis ER, Weinstock GM, Wilson RK, Ding L. VarScan: Variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009;25:2283–2285. doi: 10.1093/bioinformatics/btp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang K, Li M, Hakonarson H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pflueger D, Terry S, Sboner A, Habegger L, Esgueva R, Lin PC, Svensson MA, Kitabayashi N, Moss BJ, MacDonald TY, Cao X, et al. Discovery of non-ETS gene fusions in human prostate cancer using next-generation RNA sequencing. Genome Res. 2011;21:56–67. doi: 10.1101/gr.110684.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan MD, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miyazaki K, Fujita T, Ozaki T, Kato C, Kurose Y, Sakamoto M, Kato S, Goto T, Itoyama Y, Aoki M, Nakagawara A. NEDL1, a novel ubiquitin-protein isopeptide ligase for dishevelled-1, targets mutant superoxide dismutase-1. J Biol Chem. 2004;279:11327–11335. doi: 10.1074/jbc.M312389200. [DOI] [PubMed] [Google Scholar]
- 28.Donovan P, Poronnik P. Nedd4 and Nedd4-2: Ubiquitin ligases at work in the neuron. Int J Biochem Cell Biol. 2013;45:706–710. doi: 10.1016/j.biocel.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 29.Cui Y, He S, Xing C, Lu K, Wang J, Xing G, Meng A, Jia S, He F, Zhang L. SCFFBXL15 regulates BMP signalling by directing the degradation of HECT-type ubiquitin ligase Smurf1. EMBO J. 2011;30:2675–2689. doi: 10.1038/emboj.2011.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang L, Haraguchi S, Koda T, Hashimoto K, Nakagawara A. Muscle atrophy and motor neuron degeneration in human NEDL1 transgenic mice. J Biomed Biotechnol. 2011;2011:831092. doi: 10.1155/2011/831092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y, Ozaki T, Kikuchi H, Yamamoto H, Ohira M, Nakagawara A. A novel HECT-type E3 ubiquitin protein ligase NEDL1 enhances the p53-mediated apoptotic cell death in its catalytic activity-independent manner. Oncogene. 2008;27:3700–3709. doi: 10.1038/sj.onc.1211032. [DOI] [PubMed] [Google Scholar]
- 32.Li Y, Zhou Z, Alimandi M, Chen C. WW domain containing E3 ubiquitin protein ligase 1 targets the full-length ErbB4 for ubiquitin-mediated degradation in breast cancer. Oncogene. 2009;28:2948–2958. doi: 10.1038/onc.2009.162. [DOI] [PubMed] [Google Scholar]