

Abstract
Two potential platform technologies for the oral delivery of protein therapeutics were synthesized and tested. pH-Responsive poly(itaconic acid-co-N-vinyl-2-pyrrolidone) (P(IA-co-NVP)) hydrogel microparticles were tested in vitro with model proteins salmon calcitonin, urokinase, and rituximab to determine the effects of particle size, protein size, and crosslinking density on oral delivery capability. Particle size showed no significant effect on overall delivery potential but did improve percent release of encapsulated protein over the micro-scale particle size range studied. Protein size was shown to have a significant impact on the delivery capability of the P(IA-co-NVP) hydrogel. We show that when using P(IA-co-NVP) hydrogel microparticles with 3 mol% tetra(ethylene glycol) dimethacrylate crosslinker, a small polypeptide (salmon calcitonin) loads and releases up to 45 μg/mg hydrogel while the mid-sized protein urokinase and large monoclonal antibody rituximab load and release only 19 and 24 μg/mg hydrogel, respectively. We further demonstrate that crosslinking density offers a simple method for tuning hydrogel properties to variously sized proteins. Using 5 mol% TEGDMA crosslinker offers optimal performance for the small peptide, salmon calcitonin, whereas lower crosslinking density of 1 mol% offers optimal performance for the much larger protein rituximab. Finally, an enzymatically-degradable hydrogels of P(MAA-co-NVP) crosslinked with the peptide sequence MMRRRKK were synthesized and tested in simulated gastric and intestinal conditions. These hydrogels offer ideal loading and release behavior, showing no degradative release of encapsulated salmon calcitonin in gastric conditions while yielding rapid and complete release of encapsulated protein within 1 h in intestinal conditions.
Keywords: Oral drug delivery, pH-responsive hydrogels, Protein therapeutics, Itaconic acid, Isoelectric point, Enzyme-responsive, Crosslinking density, Salmon calcitonin, Rituxan, Urokinase
Graphical abstract
1. Introduction
Protein therapy has emerged over the past few decades as one of the most impactful areas of medicine. After recombinant insulin was approved for use in 1982, over 150 different protein-based drugs have received FDA approval to treat an impressive array of diseases.[1] Protein therapeutics represents a large market, with over $108 billion in sales in 2010 with projected sales of $165 billion by 2018.[2–4] Furthermore, as seen in Table 1, 9 of the 20 best-selling drug products in 2013 were protein therapeutics.[5] Nevertheless, the technology remains young and growing: proteins are the most rapidly expanding class of new therapeutics, accounting for 23% of newly FDA-approved drugs in 2012,[6] and advances in protein manufacturing and discovery capabilities are driving this progress forward at ever increasing rates.[7–11] Proteins are experiencing this rapid increase in therapeutic use because the complexity of macromolecules enables complex functions with a high degree of specificity unmatched by traditional small molecule drugs, resulting in more effective medicines with fewer off-target side effects.
Table 1.
Protein Therapeutics Appearing in the Top 20 Best-Selling Drug Products List.
| Rank | Drug Name | Protein | Manufacturer | Indications | Sales (Q4 2013, $MM) |
|---|---|---|---|---|---|
| 3 | Humira | Adalimumab | AbbVie | Crohn’s disease; Rheumatoid arthritis; Plaque psoriasis; Ulcerative colitis; Ankylosing spondylitis | 1,461.9 |
| 6 | Enbrel | Etanercept | Amgen | Rheumatoid arthritis | 1,189.8 |
| 8 | Remicade | Infliximab | Centocor Ortho Biotech, Inc. | Crohn’s disease; Rheumatoid arthritis | 994.0 |
| 9 | Neulasta | Filgrastim (G-CSF) | Amgen | Neutropenia | 854.5 |
| 11 | Lantus Solostar | Insulin glargine | Sanofi | Diabetes mellitus | 839.7 |
| 12 | Rituxan | Rituximab | Genentech | Non-Hodgkin’s lymphoma; rheumatoid arthritis | 746.8 |
| 16 | Lantus | Insulin glargine | Sanofi | Diabetes mellitus | 675.5 |
| 17 | Avastin | Bevacizumab | Genentech | Cancers | 650.2 |
| 20 | Epogen | Epoetin alfa | Amgen | Anemia | 503.5 |
Nearly all protein therapeutics are currently administered by intravenous, subcutaneous, or intramuscular injection. Injection provides a straightforward and effective administration route; however, injections can be painful to the user and embarrassing to administer in public, leading to low patient compliance. In a recent study, over half of insulin users reported intentionally skipping injections at some point, and 20% reported regularly skipping injections, leading to a dangerous lapse in treatment.[12] A more amenable administration route would therefore improve quality of life for patients and increase treatment compliance and efficacy.
Delivery via the oral route is a preferable method of drug administration. It offers a much more convenient pathway that is very familiar to all users and eliminates the fear, pain, and embarrassment that is associated with injections. Unfortunately, there are significant obstacles that have prevented oral delivery of protein therapeutics thus far. Proteins are subject to enzymatic cleavage or acid-catalyzed denaturation in the harsh environment of the stomach, are further digested by enzymes in the small intestine, and are generally poorly absorbed across the intestinal epithelium into the bloodstream.[13–18] As such, unmodified and unprotected proteins typically display negligible bioavailability via the oral route.
Use of pH-responsive hydrogels as delivery systems for enabling the oral delivery of protein therapeutics has shown great promise.[19–32] The large and rapid pH change between the stomach (pH 1.7 in the fasted state) and the small intestine (pH 6.1)[33] offers a convenient triggering mechanism for initiating drug release to the site of the small intestine for transport into the bloodstream. Systems of poly(methacrylic acid-grafted-poly(ethylene glycol)) (P(MAA-g-EG)) take advantage of this trigger by forming hydrogen bonding complexes at low pH that result in small mesh size and swelling significantly at neutral pH with the loss of these complexes. As such, proteins may be encapsulated in hydrogel particles and protected through the stomach before being released in the small intestine by diffusion through the swollen hydrogel mesh.
Due to the anionic charges of the deprotonated carboxylic acids present in the hydrogel at neutral pH, cationically charged proteins (i.e., those exhibiting a high isoelectric point, pI > 7.4) have demonstrated limited bioavailability within these systems. We have previously demonstrated improved delivery capability of a high pI protein, salmon calcitonin, by using itaconic acid-based hydrogels copolymerized with N-vinylpyrrolidone (P(IA-co-NVP)) and low ionic strength conditions during drug loading.[25] In this work, we expand on our previous work with further testing of the P(IA-co-NVP) hydrogel system using additional high pI proteins of various size, varying hydrogel crosslinking density, and different hydrogel particle size, demonstrating tunable material properties for enabling the oral delivery of a wide range of therapeutic proteins. Furthermore, we report successful synthesis and in vitro use of an enzymatically-responsive P(MAA-co-NVP) hydrogel with peptide crosslinker that enables rapid and complete release of protein within small intestinal conditions.
2. Materials and Methods
2.1 pH-Responsive Hydrogel Synthesis and Purification
The hydrogel synthesis procedure was carried out as described previously by Koetting and Peppas.[25] Itaconic acid (IA) (Acros Organics, Fair Lawn, NJ) and N-vinylpyrrolidone (NVP) (Sigma-Aldrich, St. Louis, MO) were dissolved at a 1:2 molar ratio of IA:NVP in a 50:50 w/w mixture of aqueous sodium hydroxide (NaOH) (Fisher Scientific, Fair Lawn, NJ) and ethanol (Fisher Scientific). The NaOH was of appropriate concentration to yield a 1:2 molar ratio of NaOH to IA. The mixture was prepared at a 30/70 w/w ratio of monomers to co-solvent. Tetra(ethylene glycol) dimethacrylate (TEGDMA) (Sigma-Aldrich) was added as a crosslinker at 3 mol%, and Irgacure 2959 (Ciba Specialty Chemicals Corp., Tarrytown, NY) was added as a photoinitiator at 1 mol%, relative to the total moles of monomers and crosslinker. The solution was mixed, purged with nitrogen, and polymerized in a 0.7 mm thick film using 35 mW/cm2 UV light for 75 min. The film was washed pure of reactants using 18.2 MΩ-cm deionized water changed daily for 10 days, dried at 30 °C under vacuum, crushed into microparticles using a mortar and pestle, and sieved to size ranges of 45–75 μm, 75–90 μm, and 90–150 μm. These purified microparticles were used in all subsequent studies.
2.2 pH-Responsive In Vitro Loading and Release Experiments
2.2.1 Effect of Hydrogel Particle Size on Oral Delivery
A solution of salmon calcitonin (sCT) (Selleck Chemicals, Houston, TX) was prepared at a concentration of 0.40 mg/mL in 0.00150 M PBS buffer (pH 7.4), and 1.5 mL of this solution was added to each of 15 × 2.0 mL, low-adhesion microcentrifuge tubes. Microparticles of the 1:2 P(IA-co-NVP) hydrogel were added to this solution at 10 mg of dry hydrogel per tube, with particles sieved into size ranges of 45–75 μm, 75–90 μm, and 90–150 μm (n = 5 per size range). The mixture was agitated for 24 h, allowing the microparticles to swell to equilibrium and sCT to diffuse into the interior of the particles. The particles were collapsed using 25 μL of 1 N HCl and isolated by centrifugation and decanting. The supernatant was collected for analysis. The particles were resuspended in two subsequent washes consisting of 1.0 mL of 0.01 N HCl to remove surface-bound protein. After each wash, the particles were isolated by centrifugation and decanting, and the rinse was collected for analysis. The isolated microparticles with encapsulated sCT were lyophilized until dry. Samples of the stock solution, the supernatant following particle collapse, and the two acid rinses were analyzed by a Micro BCA protein concentration assay (Thermo Fisher Scientific, Rockford, IL).
Once the microparticles were dry from lyophilization, 1.5 mL of 0.150 M PBS buffer at pH 3 (from adding HCl) was added to each microcentrifuge tube and agitated for 1 h at 37 °C. A 150 μL sample was removed for analysis and replaced with 150 μL fresh, pH 3.0 PBS before raising the pH back to 7.4 by addition of 1 N NaOH. The neutralized mixture was agitated for 4 h at 37 °C. A 250 μL sample was collected after 4 h to test end point release. All samples were analyzed for sCT concentration using a Micro BCA protein concentration assay (Thermo Scientific Pierce, Rockford, IL).
2.2.2 Effect of Protein Size on Oral Delivery
Salmon calcitonin (Selleck Chemicals), urokinase (ProSpec, East Brunswick, NJ), and rituximab (Genentech, San Francisco, CA) were used as high pI proteins to test the effect of protein size on delivery capability. Solutions of the three proteins were prepared at 200 μg protein/mL concentration in 1.5 mM PBS (0.01x). In 2.0 mL low adhesion tubes, 5 mg of 1:2 P(IA-co-NVP) hydrogel microparticles (90–150 μm in size) were added to each tube and incubated with 1.5 mL of protein solution (n = 5 per protein) for 24 h at pH 7.4. The solutions were collapsed using 1 N HCl to reduce the pH to 2, isolated by centrifugation and decanting, washed two times with 0.5 mL of 0.01 N HCl, and lyophilized. Following drying, 1.0 mL of 150 mM PBS (1x) at pH 3 was added to each tube and incubated at 37 °C for 1 h. A 150 μL sample was taken and replaced with pH 3 PBS, and the solutions were then raised to pH 7.4 using 1 N NaOH. The solutions were stirred at 37 °C for 24 h, with 150 μL samples acquired and replaced with fresh PBS at time points of 1, 2, 4, and 24 h. All loading and release samples were analyzed for protein concentration using a MicroBCA assay (Thermo Scientific Pierce).
2.2.3 Effect of Hydrogel Crosslinking Density on Oral Delivery
2.2.3.1 Loading and Release of Salmon Calcitonin
A solution of salmon calcitonin (sCT) (Selleck Chemicals, Houston, TX) was prepared at a concentration of 0.20 mg/mL in 0.00150 M PBS buffer (pH 7.4), and 1.0 mL of this solution was added to each of 12 × 2.0 mL, low-adhesion microcentrifuge tubes. Microparticles of three hydrogel formulations were added to this solution at 5 mg of dry hydrogel per tube. The hydrogels used were 1:2 IA:NVP monomer molar ratio P(IA-co-NVP) hydrogels, prepared as described in Section 2.1 and sieved to 90–150 μm in size, but with varying crosslinker mole percent in the monomer feed, with 1%, 5%, and 10% TEGDMA crosslinking (n = 4 per formulation). The mixture was agitated for 24 h, allowing the microparticles to swell to equilibrium and sCT to diffuse into the interior of the particles. The particles were collapsed using 1 N HCl to reduce pH to 2.0 and isolated by centrifugation and decanting. The supernatant was collected for protein quantification. The particles were resuspended in two subsequent washes consisting of 1.0 mL of 0.01 N HCl to remove surface-bound protein. After each wash, the particles were isolated by centrifugation and decanting, and the rinse was collected for analysis. The isolated microparticles with encapsulated sCT were dried overnight in air. Samples of the stock solution, the supernatant following particle collapse, and the two acid rinses were analyzed by a Micro BCA protein concentration assay.
Once the microparticles were dry, 1.0 mL of 0.150 M PBS buffer at pH 3 was added to each microcentrifuge tube and agitated for 1 h at 37 °C using an Eppendorf Thermomixer. A 150 μL sample was removed for analysis and replaced with 150 μL fresh, pH 3 PBS before raising the pH back to 7.4 by addition of 1 N NaOH. The neutralized mixture was agitated for 24 h at 37 °C, with 150 μL samples collected at time points of 1, 2, 4, and 24 h, each time being replaced by 150 μL of fresh PBS (pH 7.4). All samples were analyzed for sCT concentration using a Micro BCA protein concentration assay.
2.2.3.2 Loading and Release of Rituxan
A protocol identical to that presented in Section 2.2.3.1 was used, with a 0.40 mg/mL solution of Rituxan (rituximab) (Genentech) substituted for the 0.20 mg/mL sCT. Rituxan was generously provided for our research by Genentech. The same 1%, 5%, and 10% TEGDMA crosslinked, 1:2 P(IA-co-NVP) microparticles were used in identical loading and release conditions. Protein samples were quantified using a MicroBCA assay.
2.3 Enzymatically-Degradable Hydrogels
2.3.1 Synthesis and Purification
Enzymatically degradable hydrogels using peptide crosslinks were prepared as described by Knipe et al.[34] Uncrosslinked P(MAA-co-NVP) polymer was prepared by UV-initiated free radical polymerization. Methacrylic acid (Sigma-Aldrich) and N-vinylpyrrolidone (Sigma-Aldrich) were added to a 50:50 w/w mixture of water and ethanol at a molar ratio of 1:1 MAA:NVP and 30 wt% monomer to total solution weight. Irgacure 2959 (Ciba Specialty Chemicals Corp.) was added as a photoinitiator at 1 wt% relative to total monomer weight. The solution was purged with nitrogen for 5 min and polymerized for 30 min in 35 mW/cm2 UV light using an IntelliRay 600 UV flood source (Uvitron International). After polymerization, the polymer was purified using 1 N hydrochloric acid (Fisher Scientific, Fair Lawn, NJ) and acetone to cause precipitation of the polymer, followed by centrifugation and resuspension in deionized water 3 times. The purified polymer was dried by lyophilization.
The degradable P(MAA-co-NVP) hydrogel was formed by crosslinking using a synthetic peptide. Uncrosslinked P(MAA-co-NVP) polymer was dissolved in a 50:50 w/w mixture of water and ethanol at a concentration of 50 mg/mL. 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) (Sigma-Aldrich) was dissolved in ethanol at 50 mg/mL concentration. N-hydroxysuccinimide (NHS) (Thermo Scientific Pierce, Waltham, MA) was dissolved separately in ethanol at 16 mg/mL concentration. The EDC and NHS solutions were added to the polymer solution at a 6:3:1 ratio of polymer:EDC:NHS by weight, vortexed briefly, and reacted for 3 min. The solution pH was raised to pH 8 using 1 N sodium hydroxide (Fisher Scientific). A synthetic peptide with the sequence MMRRRKK (CHI Scientific, Maynard, MA) was dissolved in ethanol at a concentration of 100 mg/mL and added to the polymer solution at a 2:1 weight ratio of polymer:peptide. The mixture reacted for 12 h, and the product was isolated by centrifugation. The polymer was resuspended in deionized water and then isolated by centrifugation 3 times to remove remaining reactants. The hydrogel was then dried by lyophilization, crushed using a mortar and pestle, and sieved to microparticles 90–150 μm in size for use in studies.
2.3.2 Loading and Release of Salmon Calcitonin from Enzymatically-Responsive Hydrogels
In a release study using salmon calcitonin, 10 mg of purified, degradable P(MAA-co-NVP) microparticles crosslinked with MMRRRKK and sieved to 90–150 μm in size were added to 1.5 mL of a 400 μg/mL solution of salmon calcitonin (Selleck Chemicals) in 1.50 mM PBS buffer at pH 7.4 (n = 3). The mixture was agitated for 24 h using an Eppendorf Thermomixer, allowing the microparticles to swell to equilibrium and sCT to diffuse into the interior of the particles. The particles were collapsed using 25 μL of 1 N HCl and isolated by centrifugation and decanting. The supernatant was collected for analysis. The particles were resuspended in two subsequent washes consisting of 1.0 mL of 0.01 N HCl to remove surface-bound protein. After each wash, the particles were isolated by centrifugation and decanting, and the rinse was collected for analysis. The isolated microparticles with encapsulated sCT were lyophilized until dry. Samples of the stock solution, the supernatant following particle collapse, and the two acid rinses were analyzed for sCT concentration using high performance liquid chromatography.
Once the microparticles were dry from lyophilization, 1.5 mL of USP-standard simulated gastric fluid (containing 3.2 mg/mL pepsin) at pH 1.2 was added to each microcentrifuge tube and agitated for 1 h at 37 °C. The particles were isolated by centrifugation, and the simulated gastric fluid supernatant was removed. The microparticles were resuspended in 1.5 mL of USP-standard simulated intestinal fluid (containing 10 mg/mL pancreatin) at pH 6.8 and agitated for 4 h at 37 °C. 250 μL samples were collected at times of 1 h, 4 h, and 48 h after adding simulated intestinal fluid and subsequently replaced with fresh simulated intestinal fluid. All samples were analyzed for sCT concentration using high performance liquid chromatography.
3. Results and Discussion
3.1 Effect of Hydrogel Particle Size on Oral Delivery
The mechanism for release of the therapeutic proteins from the hydrogel microparticles is based on diffusion through the hydrogel network, so it is reasonable to assume that length scales will be an important factor in drug delivery capability. During release, the protein must overcome charge interactions by diffusion or osmotic pressure-induced convection. The further the drug must travel by diffusion through the hydrogel mesh to the exterior of the particle, the more likely it is to encounter strong ionic interactions that overwhelm the diffusional “force” and prevent its release. Likewise, the average time scale required for ordinary diffusion at a given distance scales as
where l is the diffusional distance [m] and D is the diffusional coefficient [m2/s]. As a result, even in the case where coulombic interactions are not present and therefore do not impede release, a longer diffusional distance will require significantly more time, scaling by the square of the distance.
For example, if encapsulated proteins are distributed uniformly through a particle, those proteins near the center of a 100 μm particle would be expected to take 4 times as long to escape as those near the center of a 50 μm particle. Because there is a limited time frame available for release in the small intestine (approximately 4 h total through the duodenum, jejunum, and ileum [35]), the diffusional time is limited, and particles too large in size will waste encapsulated protein due to incomplete release of protein within the biologically-imposed time constraints. Therefore, it is logical to hypothesize that using smaller particles, thus increasing the surface area to volume ratio and decreasing diffusional distance, could enhance protein delivery capabilities.
To study the effect of decreased particle size on oral protein delivery, P(IA-co-NVP) microparticles were crushed and sieved into size ranges of 45–75 μm, 75–90 μm, and 90–150 μm and used in a loading and release experiment using salmon calcitonin as the model high pI drug. The results of this experiment are shown in Figure 1.
Figure 1.

Loading and Release of Salmon Calcitonin from P(IA-co-NVP) Microparticles of Varying Sizes. All microparticles are comprised of 1:2 P(IA-co-NVP). Acidic release data reported after 1 h in PBS buffer at pH 3, and neutral release data reported after 4 h in same PBS buffer neutralized to pH 7.4.
The results do not show any statistically significant difference in the final delivery potential of the differently-sized hydrogel samples (p > 0.23), either in acidic conditions or neutral conditions. In terms of loading levels, the 90–150 μm size range loaded the most amount of protein (29.0 μg sCT/mg hydrogel), significantly more than the 45–75 μm size range (p = 0.03) but not significantly more than the 75–90 μm size range, which is to be expected given the juxtaposed size boundaries. Thus, although there was no statistically significant difference in overall delivery levels on a per mass basis, the 45–75 μm size range exhibited a statistically significant increase in percent release (29.3%) of encapsulated protein compared to the 90–150 μm size range (22.3%).
The improved percent release does support the previous hypothesis that smaller particles could improve drug delivery, but the degree of improvement at this length scale is small. Nevertheless, the improved percent release is beneficial in that less protein is lost due to not releasing in the small intestine. Protein not loaded can be recovered and recycled for loading into other particles, but protein that is not released is forever lost, so percent release is important from the perspective of reducing cost of such a delivery system. Therefore, from a cost perspective, these results indicate moderate benefit to using smaller size microparticles.
That the overall release levels per mass of hydrogel are so similar is more difficult to explain. Presumably, the percent release is an accurate verification of the hypothesis that smaller particles improve delivery due to smaller diffusional distance and increased surface area to volume. However, the larger size range seems to make up for the lower percent release by having a higher loading level. This result is most likely due to the washing steps employed during loading to minimize the burst release of surface-bound protein. A uniform weight of hydrogel particles is used across the different size ranges, meaning equal volume of hydrogel, since all particles are from the same formulation and therefore have uniform density. As a result, the particles will load approximately the same amount of protein on a per mass basis if protein loads uniformly through the particles. However, the smaller particles will have a larger cumulative surface area than the larger particles, meaning more of the volume is made up of surface or near surface level binding sites. When the wash steps occur, therefore, a larger proportion of the protein is washed away, making the overall loading appear to be lower than with larger particles.
Therefore, the results of this experiment suggest that there is some benefit associated with use of smaller particles, although it may be limited to higher percent release rather than higher delivery per mass of hydrogel. This further shows that the protein release is limited by the diffusional time expected in the small intestine, so experiments that seek to increase the residence time of the particles in the small intestine, such as those by Wood et al.[36,37] or Schoener et al.,[38–40] can potentially improve the delivery system at the microparticle scale. However, even though the improvement in percent release is statistically significant, it is still not a large increase in efficiency. Large improvement may not be observed from reducing the size of the particles within the micro-scale; improvement may be observed by using nano-scale particles, but these particles will require alternative synthesis procedures.
3.2 Effect of Protein Size on Oral Delivery
The loading and release of salmon calcitonin, urokinase, and Rituxan were compared using P(IA-co-NVP) microparticles synthesized with a monomer ratio of 1:2 IA to NVP. This experiment utilized the intermediately-sized protein urokinase (pI = 8.66, MW = 54.0 kDa) and the large monoclonal antibody rituximab (pI = 8.86, MW = 144 kDa), allowing us to see the effects of protein size on oral delivery across a wide range of sizes. Urokinase is used clinically as a thrombolytic agent for myocardial infarction, pulmonary embolism, and deep vein thrombosis. As such, it is not a strong candidate for therapeutic use with our systems, due to the need for very fast application to the bloodstream. However, for the purposes of testing drug delivery capability, it works well as a commercially available, medium-sized, high pI-exhibiting model drug. Rituximab (Rituxan) is a monoclonal antibody that is used for treatment of rheumatoid arthritis, non-Hodgkin’s lymphoma, chronic lymphocytic leukemia, granulomatosis with polyangiitis, and microscopic polyangiitis. Rituximab has a much larger molecular weight than salmon calcitonin or urokinase, and therefore works well as a model for large molecular weight, high pI proteins, specifically monoclonal antibodies.
The results of the experiment are shown in Figure 2 and Table 2. The loading level of the proteins vary considerably: salmon calcitonin had high loading levels with 44.8 μg sCT/mg hydrogel, while urokinase (19.0 μg/mg) and rituximab (24.1 μg/mg) exhibited similar, but significantly lower loading levels. This is likely due to mesh size limitations of the hydrogel that prevent easy access into the particle for larger proteins. Accordingly, both the amount of protein released and the percent release of urokinase and rituximab were lower than those of salmon calcitonin. This result demonstrates that protein size is a strong determining factor in the delivery potential of these hydrogels, with larger proteins requiring larger mesh sizes in order to facilitate their diffusive transport through the tortuous paths into and out of the hydrogel mesh.
Figure 2.

Release Profile of Calcitonin, Urokinase, and Rituxan from 1:2 P(IA-co-NVP) Hydrogel Microparticles. For t = 0–1 h, release conditions were acidic at pH 3. The release solutions were then neutralized, to pH 7.4 for t > 1 h.
Table 2.
Loading and Release of Salmon Calcitonin, Urokinase, and Rituxan from 1:2 P(IA-co-NVP) Microparticles.
| Protein | Loading Level (μg/mg hydrogel) | Protein Released, t = 24 h (μg/mg hydrogel) | Percent Release, t = 24 h (%) |
|---|---|---|---|
| Salmon Calcitonin | 44.8 ± 4.7 | 15.5 ± 1.8 | 35.2 ± 7.2 |
| Urokinase | 19.0 ± 1.1 | 2.7 ± 0.5 | 14.2 ± 3.2 |
| Rituxan | 24.1 ± 1.5 | 4.8 ± 1.1 | 20.0 ± 3.6 |
3.3 Effect of Crosslinking Density on Oral Delivery
Another factor that could have a significant impact on the protein delivery capability of the hydrogel systems is the crosslinking density. Crosslinking density directly affects the value of , the average molecular weight between crosslinks, which as described by the Brannon-Peppas swelling model shown graphically in Figure 3,[41] directly affects the swelling of the hydrogel. Lower crosslinking density will lead to larger , which in turn leads to larger swelling ratios and larger mesh sizes. This effect can be highly beneficial in enabling the delivery of macromolecules and should grant the ability to tailor mesh sizes for delivery of differently sized molecules. For example, a small protein like salmon calcitonin (3.4 kDa) may benefit from high crosslinking density to prevent diffusion out of the mesh at low pH, whereas much larger monoclonal antibodies like rituximab (144 kDa) may require low crosslinking density to achieve sufficiently large mesh size for diffusion in and out of the hydrogel.
Figure 3.
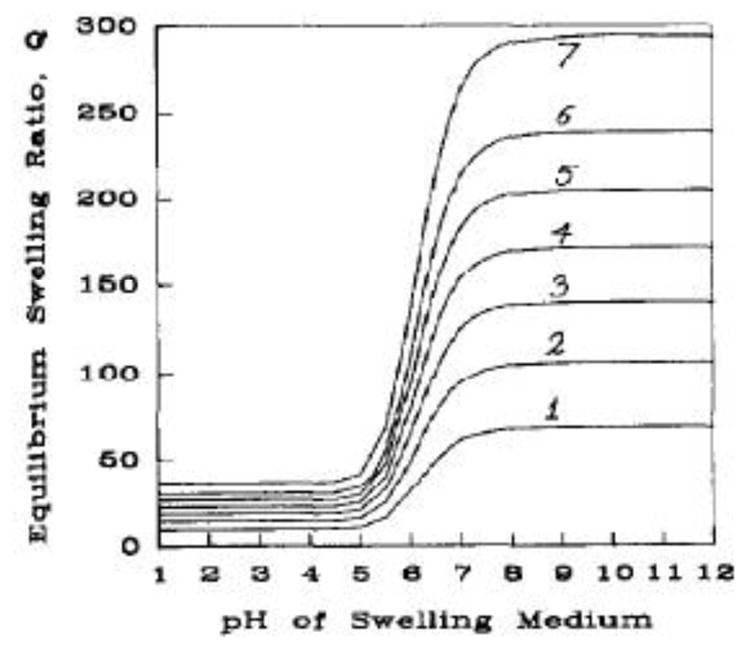
Theoretical Swelling of Anionic Hydrogel with Varying Crosslinking Density. Swelling profiles obtained by solution of Brannon-Peppas equation for varying values of : (1) ; (2) ; (3) ; (4) ; (5) ; (6) ; and (7) . Reprinted from Brannon-Peppas and Peppas[43] with permission from Elsevier.
To test the effect of crosslinking density on protein delivery capability, a standard loading and release experiment was conducted using salmon calcitonin and Rituxan as test proteins loaded into 90–150 μm microparticles of 1:2 P(IA-co-NVP) made using 1%, 5%, or 10% TEGDMA crosslinker. As expected, the hydrogels with higher crosslinking density exhibited qualitatively greater mechanical toughness; the 10% crosslinked gel was easily formed as a film and remained as a film throughout all wash steps, while the 5% broke after multiple water changes, and the 1% formulation rapidly broke into pieces sufficiently small to necessitate the use of a sieve during washes after hydration, even with minimal shear stress. This implies successful incorporation of higher crosslinking densities in the gels as expected.
The results of the salmon calcitonin loading and release are shown in Table 3 and Figure 4. As shown by the release profiles, the 5% crosslinked formulation achieves the highest total release of sCT within 4 h at neutral conditions, while releasing no more than the 10% formulation in the 1 h at acidic conditions. The 1% formulation, although achieving similar loading level as the 10% formulation, did not release as much sCT at either acidic conditions or in neutral conditions. This may seem counterintuitive that a hydrogel with less crosslinker would release a lower percentage of its payload than more crosslinked hydrogels. However, lower crosslinking density also equates to higher concentrations of anions per unit of polymer backbone, yielding more binding sites for coulombic interactions. Thus, this implies that for a small, high pI protein like sCT, the release is determined by an interplay of mesh size and coulombic interactions, with the optimal crosslinking density appearing to be around 5% (somewhere between 1 and 10%).
Table 3.
Loading and Release of Salmon Calcitonin from 1:2 P(IA-co-NVP) Microparticles with Varying Crosslinking Density.
| Crosslinking Density | Loading Level (μg sCT/mg hydrogel) | Protein Released, t=4 h (μg sCT/mg hydrogel) | Percent Release, t=4 h (%) |
|---|---|---|---|
| 1% TEGDMA | 33.7 ± 0.4 | 4.53 ± 1.13 | 13.4 ± 3.2 |
| 5% TEGDMA | 36.2 ± 0.4 | 7.08 ± 0.63 | 19.5 ± 1.6 |
| 10% TEGDMA | 33.7 ± 0.3 | 6.17 ± 0.30 | 18.3 ± 0.9 |
Figure 4.

Effect of Crosslinking Density on Delivery of Salmon Calcitonin, Release Profiles. For t = 0–1 h, release conditions were acidic at pH 3. The release solutions were then neutralized, to pH 7.4 for t > 1 h.
The results of the Rituxan loading and release experiment with different crosslinking densities are shown in Figure 5 and Table 4. Interestingly, despite the much larger size of Rituxan (144 kDa), the release levels and percent release seen in this experiment are higher than were observed with salmon calcitonin. This result is due to higher loading levels (56–64 μg/mg hydrogel) that result primarily from the higher protein loading concentration used (400 μg/mL rather than 200 μg/mL), although other possible contributors could be greater coulombic interactions with the carriers, reduced penetration of the protein into the carriers (due to the size), or minor shifts in the pH during the loading step. Further experimentation with confocal microscopy or simply altering the loading pH could potentially elucidate the mechanisms for this result.
Figure 5.

Release of Rituxan (rituximab) from 1:2 P(IA-co-NVP) Microparticles of Varying Crosslinking Density. For t = 0–1 h, release conditions were acidic at pH 3. The release solutions were then neutralized, to pH 7.4 for t > 1 h.
Table 4.
Loading and Release of Rituxan (rituximab) from 1:2 P(IA-co-NVP) Microparticles with Varying Crosslinking Density.
| Crosslinking Density | Loading Level (μg Rituxan/mg hydrogel) | Protein Released, t=4 h (μg Rituxan/mg hydrogel) | Percent Release, t=4 h (%) |
|---|---|---|---|
| 1% TEGDMA | 56.3 ± 0.7 | 64.1 ± 3.7 | 113.8 ± 5.2 |
| 5% TEGDMA | 60.4 ± 3.4 | 19.1 ± 2.5 | 31.5 ± 2.6 |
| 10% TEGDMA | 64.1 ± 3.9 | 16.8 ± 3.4 | 26.4 ± 6.2 |
What this experiment clearly demonstrates, however, is the greatly enhanced delivery resulting from use of the 1% crosslinked hydrogels as compared to the 5% or 10% formulations. Although all profiles are nearly ideal in terms of limited release at the 1 h, acidic time point and enhanced release over a physiological time at neutral conditions, the release level observed by the 1% crosslinked formulation significantly exceeds that of the 5% and 10% formulations (p = 0.0004). Meanwhile, the 5% and 10% crosslinked formulations are not significantly different at any time point (p > 0.35). Unlike with the much smaller sCT, the release seems to be very strongly affected by the crosslinking density with the much larger rituximab protein. This further demonstrates that the release is determined by a competing combination of mesh size and coulombic interactions. While the coulombic interactions seemed to dominate with the much smaller protein, likely due to the large size of all the tested hydrogels’ meshes compared to the protein, the mesh size appears to dominate with the much larger protein.
In conclusion, these tests demonstrate two important points. First, these results indicate that the delivery potential for any given protein is largely specific to that protein due to wide differences in charge and size between proteins. While the 5% and 10% crosslinked formulations proved best for the small peptide salmon calcitonin, the 1% crosslinked formulation was vastly better for the large antibody rituximab. Second, these results also demonstrate that these P(IA-co-NVP) hydrogel systems may be easily tuned using varying crosslinking density to accommodate a wide range of proteins, ranging from the 3.4 kDa salmon calcitonin to the 144 kDa rituximab. Crosslinking density is a facile tuning knob controlling the molecular-scale morphology of the hydrogels that enables delivery of many proteins of varying sizes without changing the fundamental properties of the hydrogel, thus making these systems a potential platform technology for oral delivery of protein therapeutics rather than a single, protein-specific system.
3.4 Loading and Release of Salmon Calcitonin from Enzymatically-Degradable Hydrogels
The results of the enzymatically-sensitive hydrogel loading and release study are shown in Figure 6 and Table 5. The degradable hydrogels displayed reduced loading levels of sCT compared to what has been observed with the P(IA-co-NVP) systems, achieving only 12.4 μg sCT/mg hydrogel of encapsulated protein despite the relatively high concentration of sCT in the loading solution (400 μg/mL). The percent release of encapsulated protein, however, was approximately 100% at all time points in the simulated intestinal conditions. Although the calculated averages are slightly above 100%, the values are not significantly different from 100% (p > 0.064), indicating complete release of encapsulated sCT.
Figure 6.

Salmon Calcitonin Release Profile from Enzymatically Degradable P(MAA-co-NVP) Hydrogel Microparticles Crosslinked with MMRRRKK Peptide. For t = 0–1 h, protein release occurred in USP-standard simulated gastric fluid (pH 1.2, 3.2 mg/mL pepsin). For t > 1 h, protein release occurred in USP-standard simulated intestinal fluid (pH 6.8, 10 mg/mL pancreatin). Salmon calcitonin release reported as average ± standard deviation (n = 3).
Table 5.
Loading and Release of Salmon Calcitonin from Enzymatically Degradable P(MAA-co-NVP) Microparticles Crosslinked with MMRRRKK Peptide. Microparticles were incubated in USP simulated gastric fluid (pH 1.2, 3.2 mg/mL pepsin) for 1 h, then incubated in USP simulated intestinal fluid (pH 6.8, 10 mg/mL pancreatin) for 48 h. All values reported as average ± standard deviation (n = 3).
| Release Conditions | Time in Release Conditions (h) | Loading Level (μg sCT/mg hydrogel) | Protein Released (μg sCT/mg hydrogel) | Percent Release (%) |
|---|---|---|---|---|
| Gastric | 1 | 12.4 ± 0.7 | 0 | 0 |
|
| ||||
| 1 | 12.9 ± 0.7 | 103.8 ± 2.6 | ||
| Intestinal | 4 | 12.4 ± 0.7 | 12.8 ± 1.5 | 102.8 ± 9.5 |
| 48 | 13.4 ± 1.2 | 107.9 ± 7.6 | ||
The observed overall delivery level was only 13 μg sCT/mg hydrogel, necessitating the use of more hydrogel to accommodate a therapeutic dose of protein compared to the P(IA-co-NVP) system. However, because the entirety of the encapsulated protein is released by degradation of the hydrogel, the bioavailability of the protein by this method would be higher, as no protein drug would be wasted due to incomplete release in the small intestine. Furthermore, no release was observed in the simulated gastric fluid as measured by HPLC. Therefore, this system performed very well in terms of delivering protein to the small intestine, losing none of the encapsulated protein to degradation in the stomach conditions and achieving complete release in the small intestine conditions. As such, this hydrogel is very promising for achieving the highest possible bioavailability of therapeutic proteins via the oral route.
The primary limitation of the system, however, is that is difficult and costly to synthesize large quantities of the peptide crosslinker. Peptides are not easily manufactured at large scales and therefore generally are not. One notable exception is the HIV fusion inhibitor peptide, Fuzeon, which was manufactured at near-ton annual quantities because of the large daily dose needed.[42] However, the excessive cost of the manufacturing process prevented the drug from reaching blockbuster status. As a result of the high cost of peptide synthesis, this hydrogel is likely too costly to make a financially viable product at the current time. The system could find utility with protein drugs that are sufficiently expensive to manufacture that complete release in the small intestine makes up for the increased cost of the hydrogel. However, until technological improvements in peptide synthesis are made, making inexpensive, large-scale synthesis of peptides a reality, this system may be limited to academic interest, despite its ideal performance in this in vitro experiment.
4. Conclusions
In this work, the effects of several material properties on the oral delivery capability of high isoelectric point therapeutic proteins using pH-responsive P(IA-co-NVP) microparticles or enzymatically-responsive P(MAA-co-NVP) particles with MMRRRKK peptide crosslinker were determined. The particle size of the pH-responsive system did not show a statistically significant effect on overall protein delivery at the micro-scale sizes used. However, protein size had a significant impact on protein delivery capability. Hydrogel crosslinking density was shown to enable facile tuning of the hydrogel properties to enable effective delivery of protein therapeutics across a wide range of sizes. The small protein salmon calcitonin (3.4 kDa) benefitted from higher crosslinking density (5 mol% TEGDMA) compared to the much larger protein Rituxan (144 kDa), which benefitted significantly from low crosslinking density (1 mol% TEGDMA). Effective use of the crosslinking density as a tuning variable enabled high delivery levels while limiting the release of protein in acidic conditions.
Furthermore, enzymatically-responsive hydrogels comprised of P(MAA-co-NVP) crosslinked with a trypsin and chymotrypsin degradable peptide crosslinker of the sequence MMRRRKK were successfully prepared and studied for in vitro protein loading and release. These hydrogels displayed rapid and complete release of salmon calcitonin within 1 h in simulated intestinal conditions while showing minimal degradation and undetectable protein release in simulated gastric conditions. As such, these hydrogels demonstrate ideal behavior for enabling high bioavailability of orally-delivered protein therapeutics to the small intestine.
Acknowledgments
This work is funded by the National Institutes of Health, grant R01-EB000246-21. Rituxan used in this research was generously donated by Genentech.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Michael Clinton Koetting, Email: mkoetting@utexas.edu.
Joseph Frank Guido, Email: josephguido@utexas.edu.
Malvika Gupta, Email: malvika@utexas.edu.
Annie Zhang, Email: anniezhang@utexas.edu.
References
- 1.Leader B, Baca QJ, Golan DE. Protein therapeutics: a summary and pharmacological classification. Nat Rev Drug Discov. 2008;7:21–39. doi: 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- 2.Dimitrov DS. In: Therapeutic Proteins. Voynov V, Caravella JA, editors. Humana Press; 2012. [accessed January 30, 2015]. http://link.springer.com/protocol/10.1007%2F978-1-61779-921-1_1. [Google Scholar]
- 3.RNCOS. Global Protein Therapeutics Market Outlook 2018. 2014 http://marketpublishers.com/report/healthcare/therapy/global-protein-therapeutics-market-outlook-2018.html?vsmaid=1318.
- 4.Global Protein Therapeutics Market Analysis. RNCOS; 2011. [accessed January 21, 2015]. http://www.rncos.com/Report/IM557.htm. [Google Scholar]
- 5.Drugs.com. Top 100 Drugs for Q4 2013 by Sales - US Pharmaceutical Statistics. n.d http://www.drugs.com/stats/top100/sales.
- 6.Mullard A. 2012 FDA drug approvals. Nat Rev Drug Discov. 2013;12:87–90. doi: 10.1038/nrd3946. [DOI] [PubMed] [Google Scholar]
- 7.Strohl WR, Strohl LM. Therapeutic Antibody Engineering: Current and Future Advances Driving the Strongest Growth Area in the Pharmaceutical Industry. Elsevier; 2012. [Google Scholar]
- 8.Fischer N. Sequencing antibody repertoires. mAbs. 2011;3:17–20. doi: 10.4161/mabs.3.1.14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Georgiou G, Ippolito GC, Beausang J, Busse CE, Wardemann H, Quake SR. The promise and challenge of high-throughput sequencing of the antibody repertoire. Nat Biotechnol. 2014;32:158–168. doi: 10.1038/nbt.2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lai T, Yang Y, Ng SK. Advances in Mammalian Cell Line Development Technologies for Recombinant Protein Production. Pharmaceuticals. 2013;6:579–603. doi: 10.3390/ph6050579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boutureira O, Bernardes GJL. Advances in Chemical Protein Modification. Chem Rev. 2015;115:2174–2195. doi: 10.1021/cr500399p. [DOI] [PubMed] [Google Scholar]
- 12.Peyrot M, Rubin RR, Kruger DF, Travis LB. Correlates of Insulin Injection Omission. Diabetes Care. 2010;33:240–245. doi: 10.2337/dc09-1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morishita M, Peppas NA. Is the oral route possible for peptide and protein drug delivery? Drug Discov Today. 2006;11:905–910. doi: 10.1016/j.drudis.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 14.Renukuntla J, Vadlapudi AD, Patel A, Boddu SHS, Mitra AK. Approaches for enhancing oral bioavailability of peptides and proteins. Int J Pharm. 2013;447:75–93. doi: 10.1016/j.ijpharm.2013.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gupta S, Jain A, Chakraborty M, Sahni JK, Ali J, Dang S. Oral delivery of therapeutic proteins and peptides: a review on recent developments. Drug Deliv. 2013;20:237–246. doi: 10.3109/10717544.2013.819611. [DOI] [PubMed] [Google Scholar]
- 16.Fink AL, Calciano LJ, Goto Y, Kurotsu T, Palleros DR. Classification of Acid Denaturation of Proteins: Intermediates and Unfolded States. Biochemistry (Mosc) 1994;33:12504–12511. doi: 10.1021/bi00207a018. [DOI] [PubMed] [Google Scholar]
- 17.Artursson P, Karlsson J. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem Biophys Res Commun. 1991;175:880–885. doi: 10.1016/0006-291X(91)91647-U. [DOI] [PubMed] [Google Scholar]
- 18.Yee S. In Vitro Permeability Across Caco-2 Cells (Colonic) Can Predict In Vivo (Small Intestinal) Absorption in Man—Fact or Myth. Pharm Res. 1997;14:763–766. doi: 10.1023/A:1012102522787. [DOI] [PubMed] [Google Scholar]
- 19.Kavimandan NJ, Losi E, Peppas NA. Novel delivery system based on complexation hydrogels as delivery vehicles for insulin–transferrin conjugates. Biomaterials. 2006;27:3846–3854. doi: 10.1016/j.biomaterials.2006.02.026. [DOI] [PubMed] [Google Scholar]
- 20.Lowman AM, Morishita M, Kajita M, Nagai T, Peppas NA. Oral delivery of insulin using pH-responsive complexation gels. J Pharm Sci. 1999;88:933–937. doi: 10.1021/js980337n. [DOI] [PubMed] [Google Scholar]
- 21.Carr DA, Gómez-Burgaz M, Boudes MC, Peppas NA. Complexation Hydrogels for the Oral Delivery of Growth Hormone and Salmon Calcitonin. Ind Eng Chem Res. 2010;49:11991–11995. doi: 10.1021/ie1008025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carr DA, Peppas NA. Assessment of poly(methacrylic acid-co-N-vinyl pyrrolidone) as a carrier for the oral delivery of therapeutic proteins using Caco-2 and HT29-MTX cell lines. J Biomed Mater Res A. 2010;92A:504–512. doi: 10.1002/jbm.a.32395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Foss AC, Peppas NA. Investigation of the cytotoxicity and insulin transport of acrylic-based copolymer protein delivery systems in contact with caco-2 cultures. Eur J Pharm Biopharm. 2004;57:447–455. doi: 10.1016/j.ejpb.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 24.Kamei N, Morishita M, Chiba H, Kavimandan NJ, Peppas NA, Takayama K. Complexation hydrogels for intestinal delivery of interferon β and calcitonin. J Controlled Release. 2009;134:98–102. doi: 10.1016/j.jconrel.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koetting MC, Peppas NA. pH-Responsive poly(itaconic acid-co-N-vinylpyrrolidone) hydrogels with reduced ionic strength loading solutions offer improved oral delivery potential for high isoelectric point-exhibiting therapeutic proteins. Int J Pharm. 2014;471:83–91. doi: 10.1016/j.ijpharm.2014.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Torres-Lugo M, García M, Record R, Peppas NA. Physicochemical behavior and cytotoxic effects of p(methacrylic acid–g-ethylene glycol) nanospheres for oral delivery of proteins. J Controlled Release. 2002;80:197–205. doi: 10.1016/S0168-3659(02)00027-5. [DOI] [PubMed] [Google Scholar]
- 27.Torres-Lugo M, García M, Record R, Peppas NA. pH-Sensitive Hydrogels as Gastrointestinal Tract Absorption Enhancers: Transport Mechanisms of Salmon Calcitonin and Other Model Molecules Using the Caco-2 Cell Model. Biotechnol Prog. 2002;18:612–616. doi: 10.1021/bp0101379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Torres-Lugo M, Peppas NA. Molecular Design and in Vitro Studies of Novel pH-Sensitive Hydrogels for the Oral Delivery of Calcitonin. Macromolecules. 1999;32:6646–6651. doi: 10.1021/ma990541c. [DOI] [Google Scholar]
- 29.Shin HS, Kim SY, Lee YM. Indomethacin release behaviors from pH and thermoresponsive poly(vinyl alcohol) and poly(acrylic acid) IPN hydrogels for site-specific drug delivery. J Appl Polym Sci. 1997;65:685–693. doi: 10.1002/(SICI)1097-4628(19970725)65:4<685::AID-APP7>3.0.CO;2-G. [DOI] [Google Scholar]
- 30.Bell CL, Peppas NA. Water, solute and protein diffusion in physiologically responsive hydrogels of poly(methacrylic acid-g-ethylene glycol) Biomaterials. 1996;17:1203–1218. doi: 10.1016/0142-9612(96)84941-6. [DOI] [PubMed] [Google Scholar]
- 31.Madsen F, Peppas NA. Complexation graft copolymer networks: swelling properties, calcium binding and proteolytic enzyme inhibition. Biomaterials. 1999;20:1701–1708. doi: 10.1016/S0142-9612(99)00071-X. [DOI] [PubMed] [Google Scholar]
- 32.Lowman AM, Peppas Nicholas A, Morishita M, Nagai T. Tailored Polym Mater Control Deliv Syst. American Chemical Society; 1998. [accessed March 8, 2015]. Novel Bioadhesive Complexation Networks for Oral Protein Drug Delivery; pp. 156–164. http://dx.doi.org/10.1021/bk-1998-0709.ch012. [Google Scholar]
- 33.Dressman JB, Berardi RR, Dermentzoglou LC, Russell TL, Schmaltz SP, Barnett JL, et al. Upper Gastrointestinal (GI) pH in Young, Healthy Men and Women. Pharm Res. 1990;7:756–761. doi: 10.1023/A:1015827908309. [DOI] [PubMed] [Google Scholar]
- 34.Knipe JM, Chen F, Peppas NA. Enzymatic Biodegradation of Hydrogels for Protein Delivery Targeted to the Small Intestine. Biomacromolecules. 2015;16:962–972. doi: 10.1021/bm501871a. [DOI] [PubMed] [Google Scholar]
- 35.Dressman JB, Krämer J. Pharmaceutical dissolution testing. Taylor & Francis; Boca Raton, FL: 2005. [Google Scholar]
- 36.Wood KM, Stone, Gregory M, Peppas NA. Wheat Germ Agglutinin Functionalized Complexation Hydrogels for Oral Insulin Delivery. Biomacromolecules. 2008;9:1293–1298. doi: 10.1021/bm701274p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wood KM, Stone, Gregory M, Peppas NA. In vitro investigation of oral insulin delivery systems using lectin functionalized complexation hydrogels. Adv Med Eng Drug Deliv Syst Ther Syst. 2006:75–83. [Google Scholar]
- 38.Schoener CA, Hutson HN, Peppas NA. pH-responsive hydrogels with dispersed hydrophobic nanoparticles for the oral delivery of chemotherapeutics. J Biomed Mater Res A. 2013;101A:2229–2236. doi: 10.1002/jbm.a.34532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schoener CA, Hutson HN, Peppas NA. Amphiphilic Interpenetrating Polymer Networks for the Oral Delivery of Chemotherapeutics. AIChE J. 2013;59:1472–1478. doi: 10.1002/aic.14077. [DOI] [Google Scholar]
- 40.Schoener CA, Peppas NA. pH-Responsive hydrogels containing PMMA nanoparticles: an analysis of controlled release of a chemotherapeutic conjugate and transport properties. J Biomater Sci Polym Ed. 2013;24:1027–1040. doi: 10.1080/09205063.2012.731376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brannon-Peppas L, Peppas NA. Equilibrium swelling behavior of dilute ionic hydrogels in electrolytic solutions. J Controlled Release. 1991;16:319–329. doi: 10.1016/0168-3659(91)90009-3. [DOI] [Google Scholar]
- 42.Thayer AM. Making Peptides at Large Scale. Chem Eng News. 2011;89:21–25. [Google Scholar]
- 43.Brannon-Peppas L, Peppas NA. Equilibrium swelling behavior of pH-sensitive hydrogels. Chem Eng Sci. 1991;46:715–722. doi: 10.1016/0009-2509(91)80177-Z. [DOI] [Google Scholar]