

Abstract
Tolerance of glucose deprivation is an important factor for cancer proliferation, survival, migration and progression. To systematically understand adaptive responses under glucose starvation in cancers, we analyzed reverse phase protein array (RPPA) data of 115 protein antibodies across a panel of approximately 170 heterogeneous cancer cell lines, cultured under normal and low glucose conditions. In general, glucose starvation broadly altered levels of many of the proteins and phosphoproteins assessed across the cell lines. Many mTOR pathway components were selectively sensitive to glucose stress, although the change in their levels still varied greatly across the cell line set. Furthermore, lineage- and genotype-based classification of cancer cell lines revealed mutation-specific variation of protein expression and phosphorylation in response to glucose starvation. Decreased AKT phosphorylation (S473) was significantly associated with PTEN mutation under glucose starvation conditions in lung cancer cell lines. The present study (see TCPAportal.org for data resource) provides insight into adaptive responses to glucose deprivation under diverse cellular contexts.
Keywords: reverse phase protein array, glucose starvation, mutation, cancer cell line panel
Introduction
The reverse phase protein array (RPPA), as a high-throughput proteomic technique, provides quantitative measurement for protein expression and phosphorylation. The proteomic data-sets generated from RPPA represent abundance of proteins under various conditions and have been used to systematically evaluate protein alterations in signaling networks (1,2). The application of those proteomic datasets for expression and phosphorylation (activation status) of core signaling proteins have provided opportunities to expand understanding of the molecular characteristics of cancer cell lines at the systems level in resting and perturbed conditions (3,4). In order to efficiently integrate targeted therapeutics into clinical practice, it is critical to understand how signaling pathways function and how they are controlled by the intracellular and extracellular factors present in human tumors.
Glucose provides the basic fuel for cell survival, proliferation and function in both normal and cancer cells. An ability to tolerate glucose deprivation, which commonly occurs in the tumor microenvironment, contributes to cancer cell proliferation, migration, and progression (5). Thus, over the course of the past 20 years, multiple studies have yielded useful information on the role of energy homeostasis in cancer growth and survival (6,7). However, a systematic analysis of proteomic changes under conditions of glucose deprivation has not been performed across a large set of cancer cell lines representing a broad mutational and lineage background. Although adaptive responses to glucose deprivation are key to the survival of cancer cells, they have not yielded key therapeutic opportunities, partly due to diversity and flexibility of the adaptive mechanisms used by different cancer lineages and driven by different mutations in tumor cells.
Here, a large RPPA proteomic dataset was generated to facilitate evaluation of effects of glucose deprivation on cancer signaling across ~170 human cancer cell lines, derived from 15 lineage types. Both pan cell line analysis and combined categories of cancer lineage and mutational genotypes were used to identify associations with glucose-dependent regulation of protein expression and phosphorylation. This proteomic dataset and its analysis will provide an important tool to assist the implementation of approaches to target adaptive responses to glucose deprivation.
Materials and methods
Data acquisition
RPPA datasets for ~170 cancer cell lines in normal glucose and low glucose condition were generated in the Functional Proteomics Core of the M.D. Anderson Cancer Center, University of Texas. Cells were grown in RPMI-1640 medium with 10% fetal bovine serum (FBS) and penicillin/streptavidin (all from Gibco, Grand Island, NY, USA), and maintained at 37°C in a humidified atmosphere at 5% CO2. Before protein harvest, cell lines were starved for the indicated time in medium with 5% FBS, 0.63 g/l glucose plus 2 mM glutamine without Na pyruvate (low glucose) and cultured in medium with 10% FBS, 2 g/l glucose plus 4 mM glutamine and 1 mM Na pyruvate (normal glucose). RPPA assay was done as previously described (8). The two RPPA datasets were independently normalized and mean-centered.
Cell line culture and siRNA transfection
NCI-60 lung cancer cell lines (NCI-H460, A549 and EKVX) were obtained from National Cancer Institute (NCI DTP), USA. For siRNA transfection, 2×105 cells/well were plated in a 6-well plate. After adhering for 24 h, target siRNA (Thermo Fisher Scientific, Inc., Logan, UT, USA) were added in transfection medium (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) for 6 h at 37°C in a CO2 incubator. After transfection, cells were supplemented with RPMI-1640 containing FBS and cultured at 37°C/5% CO2 for another 24 h. Then cells were starved for 12 h under glucose deprived and replete conditions as described above. Protein supernatants were isolated using cell lysis buffer (#9803; Cell Signaling Technology, Inc., Beverly, MA, USA) with added PMSF.
Western blot analysis
The total protein content (40 μg) from cell lysates was separated using SDS-PAGE (10%) and transferred to a 0.45-l M nitrocellulose membrane (Millipore) for 2 h. The membranes were washed with TBST containing 5% (w/v) BSA. The membranes were incubated overnight with specific PTEN and AKT_pS473 antibodies (Cell Signaling Technology, Inc.) and were exposed to secondary antibodies coupled to horseradish peroxidase for 2 h at room temperature. The membranes were then washed three times with TBST at room temperature. Antibody binding was detected using an enhanced chemiluminescent substrate from Thermo Fisher Scientific, Inc. (Logan, UT, USA) and analyzed with an LAS 3000 Luminescent Image Analyzer from Fujifilm (Tokyo, Japan). Equal protein loading was assessed by the level of α-actin protein (Cell Signaling Technology, Inc.).
Statistical analysis
Network construction was done using Cytoscape 2.6.3 (9) (www.cytoscape.org). Hierarchical cluster analysis was done using QCanvas (10) (http://compbio.sookmyung.ac.kr/~qcanvas/). The correlation for each pair of proteins was calculated by Pearson's correlation coefficients (PCCs) and its statistical significance (P-value). To compare protein levels of different genotypes and/or lineages, fold-change and Student's t-test P-value were calculated. The log2 fold-change of a protein is given by the difference between average of cell lines for each category and median value of total cell lines. To determine statistical significance, P-value from t-statistic was calculated. The different datasets were generated for cell lines by directly subtracting the logarithmic value in low condition from the logarithmic value of normal condition.
Results and Discussion
The RPPA dataset consisted of 77 antibodies against total protein and 38 antibodies against specific phosphorylation site. These proteins (89 unique protein symbols) were mainly included in 15 key pathways associated with cancer cell function in the KEGG database (Table I). These pathways were grouped into 4 functional categories, cancer related pathways (pathway in cancer and mTOR signaling pathway), glucose metabolism pathways (insulin signaling pathway and type II diabetes mellitus), growth and survival regulating pathways (focal adhesion, cell cycle, apoptosis, adherens junction and gap junction) and cell signaling events (ErbB, mTOR, VEGF, p53, Wnt and JAK-STAT signaling pathways). This pathway-oriented classification of RPPA proteins enabled us to explore the critical signaling networks associated with glucose starvation in cancers.
Table I.
Functional categories of proteins screened in the present RPPPA experiment. For a total of 89 proteins, 77 total protein antibodies and 38 phospho-antibodies were used in the screening. Top 15 KEGG pathways are displayed based on the number of included proteins. Thirteen proteins screened in the RPPA analysis were not found in these 15 categories.
| Pathway (total) | Count | % | Protein symbol |
|---|---|---|---|
| Pathways in cancer (328) | 34 | 10.4 | AKT, AR, β.Catenin, BCl2, c.JUN, c.KIT, c.Myc, Caspase.3, COX2, cRAF, Cyclin.D1, Cyclin.E1, E.Cadherin, EGFR, ERK2, FAK, Fibronectin, GSK3A_B,HER2,JNK2,MAPK, MEK1, mTOR, p21, p53, PI3K, PKCa, PTCH, PTEN, Rb, SMAD3, STAT3, STAT5, XIAP |
| ErbB signaling pathway (87) | 20 | 23.0 | 4EBP1, AKT, c.JUN, c.Myc, cRAF, EGFR, ERK2, FAK, GSK3A_B, HER2, JNK2, MAPK, MEK1, mTOR, p21, p70S6K, PI3K, PKCa, SRC, STAT5 |
| Focal adhesion (201) | 23 | 11.4 | AKT, β.Catenin, BCl2, c.JUN, Collagen.VI, cRAF, Cyclin.D1, EGFR, ERK2, FAK, Fibronectin, GSK3A_B, HER2, JNK2, MAPK, MEK1, PI3K, PKCa, PTEN, SRC, VASP,VEGFR2, XIAP |
| mTOR signaling pathway (52) | 13 | 25.0 | 4EBP1, AKT, AMPK, elF4E, ERK2, LKB1, MAPK, mTOR, p70S6K, p90RSK, PI3K, S6, TSC2 |
| Insulin signaling pathway (135) | 17 | 12.6 | 4EBP1, ACC, AKT, AMPK, cRAF, elF4E, ERK2, GSK3A_B, IRS.1, JNK2, MAPK, MEK1, mTOR, p70S6K, PI3K, S6, TSC2 |
| VEGF signaling pathway (75) | 13 | 17.3 | AKT, COX2, cRAF, ERK2, FAK, HSP27, MAPK, MEK1, p38, PI3K, PKCa, SRC, VEGFR2 |
| Cell cycle (125) | 12 | 9.6 | 14-3-3-Beta, 14-3-3-Zeta, c.Myc, Cyclin.B1, Cyclin.D1, Cyclin.E1, GSK3A_B, p21, p53, PCNA, Rb, SMAD3 |
| MAPK signaling pathway (267) | 17 | 6.4 | AKT, c.JUN, c.Myc, Caspase.3, cRAF, EGFR, ERK2, HSP27, JNK2, MAPK, MEK1, p38, p53, p90RSK, PKCa, Stathamin, TAU |
| p53 signaling pathway (68) | 9 | 13.2 | Caspase.3, Cyclin.B1, Cyclin.D1, Cyclin.E1, p21, PTEN, PAI1, TSC2, p53 |
| Apoptosis (87) | 8 | 9.2 | BCl2, XIAP, Caspase.3, Caspase.7, PI3K, p85_PI3K, p53, AKT |
| Type II diabetes mellitus (47) | 6 | 12.8 | IRS.1, mTOR, MAPK, JNK2, PI3K, ERK2 |
| Adherens junction (77) | 7 | 9.1 | β.Catenin, E.Cadherin, EGFR, HER2, MAPK, SMAD3, SRC |
| Wnt signaling pathway (151) | 9 | 6.0 | β.Catenin, c.JUN, c.Myc, Cyclin.D1, GSK3A_B, JNK2, p53, PKCa, SMAD3 |
| JAK-STAT signaling pathway (155) | 8 | 5.2 | AKT, c.Myc, Cyclin.D1, p85_PI3K, PI3K, STAT3, STAT5, STAT6 |
| Gap junction (89) | 7 | 7.9 | EGFR, MAPK, MEK1, PKCa, cRAF, SRC, ERK2 |
| etc. | 13 | BIM, GATA3, MGMT, YAP, N.Cadherin, ER, IGFBP1, P27, AIB, PAX2, PARP1, TAZ, Telomerase |
From the unsupervised hierarchical clustering of the differential total and phosphoprotein levels between low and normal glucose condition, we observed that proteins generally exhibited varied response to glucose starvation across all the cancer cell lines (Fig. 1A). Although proteins in a pathway tended to vary in parallel across the lines, in general cell lines in a single lineage demonstrate different patterns of protein response to glucose starvation and did not cluster together (Fig. 1A). To provide further insight into the effects of glucose deprivations across the cell lines, PCCs and corresponding P-value were calculated for each protein pair across all the cell lines. The network structure was generated using protein pairs with PCC>0.5 and P<0.01 for normal glucose condition (Fig. 1B). However, many of pair-wise correlations in the mTOR pathway were no longer apparent after glucose starvation, implying that mTOR signaling was differentially regulated across cell lines by glucose deprivation (Fig. 1B). In contrast, relationships in the insulin and ErbB pathways, with the exception of the relationship between AKT and PTEN were conserved under glucose deprivation.
Figure 1.

Change of protein expression and phosphorylation between low and normal glucose conditions in 170 diverse cancer cell lines. (A) Clustering of differential level of 77 total protein and 38 phosphoprotein levels between low and normal glucose condition. The lineage of the cancer cell lines are indicated above the heatmap. (B) Network presentation of correlation for total protein and phosphoprotein levels over 170 cancer cell lines. PCC was calculated for each pair of proteins using their expression (or phosphorylation) data on all cell lines. Black nodes represent correlations consistently found in both normal and low glucose conditions. Red presents correlations that disappeared under low glucose condition. The correlation cut off values for a node are 0.5 and −0.5 for positive and negative correlations, respectively. Total protein antibodies and phospho-antibodies are represented by open circle and filled circle, respectively.
A global analysis of average alteration and corresponding variation of total and phosphoprotein levels after glucose deprivation identified diverse regulation patterns across cell lines (Fig. 2A). A total of 11 protein probes (i.e., antibodies) including 9 downregulated (AKT_pS473, S6_pS240, S6_pS235, S6, IRS1_pS307, 4EBP1_pT37, β.Catenin, VEGFR2 and Cyclin D1) and two upregulated (MAPK_pT202 and YAP), presented relatively large changes and variation in total or phosphoprotein levels across all cell lines after glucose deprivation. Among them, 5 unique protein symbols for 7 antibodies were mainly enriched in the mTOR signaling pathway (Fig. 2B). The mTOR pathway has already been implicated in maintaining glucose homeostasis and carbohydrate, lipid and protein metabolism (11). Our analysis demonstrates a selective role for components of the mTOR pathway in the adaptive response of a cell line to glucose deprivation.
Figure 2.
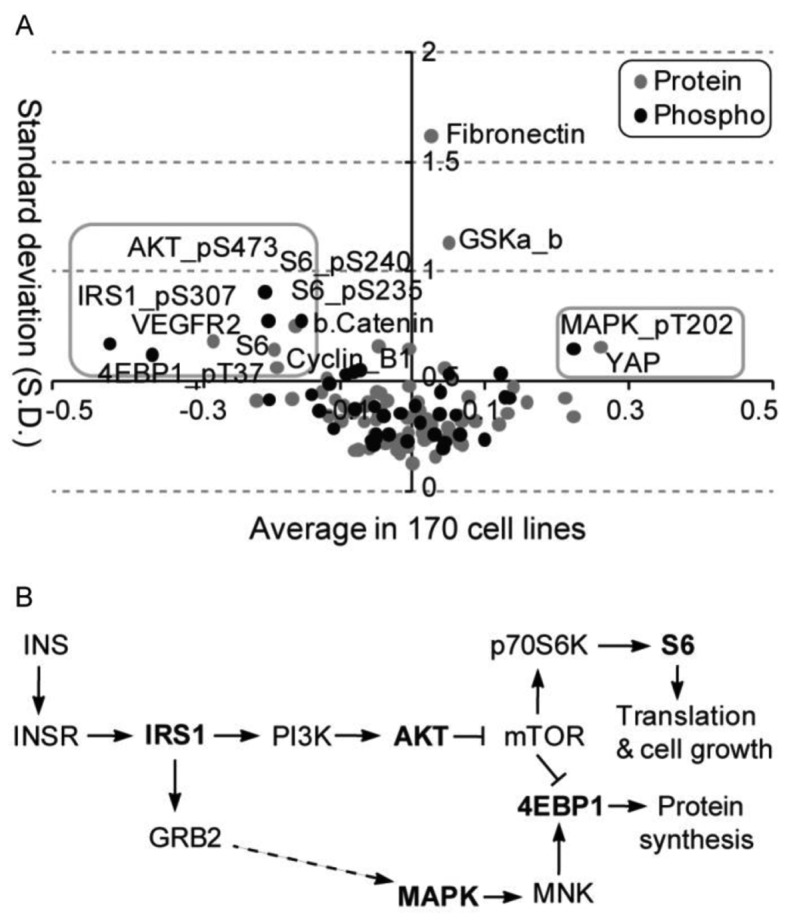
Major variation of protein expression and phosphorylation under glucose starvation condition in the cell line panel. (A) Average alteration and the standard deviation (SD) of protein expression and phosphorylation after glucose starvation across 170 cancer cell lines. Grey boxes included 11 proteins with most variation after glucose starvation. (B) mTOR signaling pathway is enriched for 11 significantly altered proteins. Five protein symbols, labeled in black, of the total 9 unique symbols are found in mTOR signaling pathway.
To analyze the association between common cancer mutations and adaptive responses to glucose deprivation, differential RPPA data (fold-change) between normal and low glucose conditions were collapsed into lineage and mutation classes (Fig. 3A). A total of 10 protein probes detected significant alterations (P<0.01) specific to glucose starvation among 23 combined categories of lineage and mutation. AKT phosphorylation (S473 and T308) were selectively downregulated in PTEN mutant cell lines by glucose deprivation. The phosphorylation of GSK3A&B (S21) and expression of IGFBP2 were also down regulated in a similar pattern with AKT. AMPK-α phosphorylation (T173) showed a distinct downregulation in lung cancer cell lines particularly with those with CTNNB1 mutations. 4E-BP1 phosphorylation (T37) and VEGFR2 expression showed a similar pattern in lung cancer cell lines. ER expression was uniquely upregulated in the PTEN mutated lung cancer cells. Increased expression of COX2 was observed in CDKN2A-mutated melanoma cell lines. PTEN expression was increased in PTEN mutant but decreased in P53-mutant cell lines by glucose starvation. In terms of combinations of mutations and lineage, the most striking observation was that AKT phosphorylation (S473) was significantly decreased in lung cancer cell lines with PTEN mutation (Fig. 3B). The importance of AKT in glucose metabolism has been indicated in previous studies, however, the effects of glucose deprivation have been disparate across cell lines. Glucose deprivation inhibited AKT phosphorylation of S473 site in an ovarian cancer cell line (12) and leukemic T cells (13), while transient glucose deprivation activated AKT at both T308 and S473 in HeLa cells (14). To confirm that glucose deprivation downregulates AKT phosphorylation (S473) in lung cancer cells lacking functional PTEN, the effect of knockdown of PTEN on AKT phosphorylation levels was assessed in three PTEN wild-type lung cancer cell lines (NCI-H460, EKVX, and A549) (Fig. 3C and D). Levels of AKT (S473) phosphorylation were increased after glucose starvation, when PTEN was present. Furthermore, as expected AKT phosphorylation was increased by a functional loss of PTEN under normal glucose conditions. We confirmed that AKT phosphorylation (S473) was significantly downregulated by PTEN knockdown under glucose deprivation conditions in the three lung cancer cell lines, albeit to a lesser degree in A549 cells. This unexpected observation suggests that the effects of PTEN on AKT phosphorylation are context-dependent with PTEN limiting AKT activation under glucose replete conditions, but increasing AKT activation by an unknown mechanism under glucose deprived conditions.
Figure 3.

Mutation-oriented analysis of protein regulation under glucose starvation. (A) Clustering of fold-change for 10 proteins using lineage and mutation categories. (B) AKT (Ser473) phosphorylation was significantly decreased after glucose starvation in lung cancer cell lines with PTEN mutation. (C) Western blot analysis of AKT_pS473 phosphorylation level in the PTEN knockdown NCI-H460, EKVX and A549 cancer cell lines. (D) Quantitative bar graph of western blot analysis (C). α-actin was measured as internal control protein. The data represent mean ± SEM (n=3). *P<0.05 and **P<0.01 between the compared data.
Cancer cells typically exhibit high demands for glucose to provide the energy necessary for cell growth (15,16). Multiple signaling pathways contributed to the regulation of glucose metabolism and balance of cellular energy under diverse contexts. However, in most studies AKT proto-oncogene has been implicated in increased glucose transportation and aerobic glycolysis in cancer cells (17,18). In this study, we analyzed adaptive responses to glucose deprivation across tumor lineage and mutational genotypes. Combined categorization of a large cell line panel by lineage and mutation criteria has been successfully used to find new molecular signatures from different omics datasets (19,20).
Protein expression and phosphorylation are dependent on the cell origin, genomic changes, and environment factors. Measurement of total and phosphoprotein levels in this large cell line set provided the opportunity to elucidate comprehensive interactions among functional signaling pathways. Importantly, the data set generated in this study, which will be made available in The Cancer Proteome Atlas (TCPAportal. org), will provide a useful community resource to explore adaptive responses to glucose stress under many different cancer relevant contexts.
Acknowledgements
The present study was supported by the Sookmyung Women's University Research grant no. 1-1203-0226.
References
- 1.Tibes R, Qiu Y, Lu Y, Hennessy B, Andreeff M, Mills GB, Kornblau SM. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther. 2006;5:2512–2521. doi: 10.1158/1535-7163.MCT-06-0334. [DOI] [PubMed] [Google Scholar]
- 2.Gujral TS, Karp RL, Finski A, Chan M, Schwartz PE, MacBeath G, Sorger P. Profiling phospho-signaling networks in breast cancer using reverse-phase protein arrays. Oncogene. 2013;32:3470–3476. doi: 10.1038/onc.2012.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nishizuka S, Charboneau L, Young L, Major S, Reinhold WC, Waltham M, Kouros-Mehr H, Bussey KJ, Lee JK, Espina V, et al. Proteomic profiling of the NCI-60 cancer cell lines using new high-density reverse-phase lysate microarrays. Proc Natl Acad Sci USA. 2003;100:14229–14234. doi: 10.1073/pnas.2331323100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boyd ZS, Wu QJ, O'Brien C, Spoerke J, Savage H, Fielder PJ, Amler L, Yan Y, Lackner MR. Proteomic analysis of breast cancer molecular subtypes and biomarkers of response to targeted kinase inhibitors using reverse-phase protein microarrays. Mol Cancer Ther. 2008;7:3695–3706. doi: 10.1158/1535-7163.MCT-08-0810. [DOI] [PubMed] [Google Scholar]
- 5.He L, Li X, Luo HS, Rong H, Cai J. Possible mechanism for the regulation of glucose on proliferation, inhibition and apoptosis of colon cancer cells induced by sodium butyrate. World J Gastroenterol. 2007;13:4015–4018. doi: 10.3748/wjg.v13.i29.4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elf SE, Chen J. Targeting glucose metabolism in patients with cancer. Cancer. 2014;120:774–780. doi: 10.1002/cncr.28501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee AS. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer. 2014;14:263–276. doi: 10.1038/nrc3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park ES, Rabinovsky R, Carey M, Hennessy BT, Agarwal R, Liu W, Ju Z, Deng W, Lu Y, Woo HG, et al. Integrative analysis of proteomic signatures, mutations, and drug responsiveness in the NCI 60 cancer cell line set. Mol Cancer Ther. 2010;9:257–267. doi: 10.1158/1535-7163.MCT-09-0743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu H, Baroukh C, Dannenfelser R, Chen EY, Tan CM, Kou Y, Kim YE, Lemischka IR, Ma'ayan A. ESCAPE: database for integrating high-content published data collected from human and mouse embryonic stem cells. Database (Oxford) 20132013:bat045. doi: 10.1093/database/bat045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim N, Park H, He N, Lee HY, Yoon S. QCanvas: an advanced tool for data clustering and visualization of genomics data. Genomics Inform. 2012;10:263–265. doi: 10.5808/GI.2012.10.4.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 12.Priebe A, Tan L, Wahl H, Kueck A, He G, Kwok R, Opipari A, Liu JR. Glucose deprivation activates AMPK and induces cell death through modulation of Akt in ovarian cancer cells. Gynecol Oncol. 2011;122:389–395. doi: 10.1016/j.ygyno.2011.04.024. [DOI] [PubMed] [Google Scholar]
- 13.Coloff JL, Mason EF, Altman BJ, Gerriets VA, Liu T, Nichols AN, Zhao Y, Wofford JA, Jacobs SR, Ilkayeva O, et al. Akt requires glucose metabolism to suppress puma expression and prevent apoptosis of leukemic T cells. J Biol Chem. 2011;286:5921–5933. doi: 10.1074/jbc.M110.179101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao M, Liang J, Lu Y, Guo H, German P, Bai S, Jonasch E, Yang X, Mills GB, Ding Z. Site-specific activation of AKT protects cells from death induced by glucose deprivation. Oncogene. 2014;33:745–755. doi: 10.1038/onc.2013.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taubes G. Cancer research. Ravenous for glucose. Science. 2012;335:31. doi: 10.1126/science.335.6064.31. [DOI] [PubMed] [Google Scholar]
- 16.Adekola K, Rosen ST, Shanmugam M. Glucose transporters in cancer metabolism. Curr Opin Oncol. 2012;24:650–654. doi: 10.1097/CCO.0b013e328356da72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 18.Neary CL, Pastorino JG. Akt inhibition promotes hexokinase 2 redistribution and glucose uptake in cancer cells. J Cell Physiol. 2013;228:1943–1948. doi: 10.1002/jcp.24361. [DOI] [PubMed] [Google Scholar]
- 19.Kim N, He N, Kim C, Zhang F, Lu Y, Yu Q, Stemke-Hale K, Greshock J, Wooster R, Yoon S, et al. Systematic analysis of genotype-specific drug responses in cancer. Int J Cancer. 2012;131:2456–2464. doi: 10.1002/ijc.27529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He N, Kim N, Song M, Park C, Kim S, Park EY, Yim HY, Kim K, Park JH, Kim KI, et al. Integrated analysis of transcriptomes of cancer cell lines and patient samples reveals STK11/LKB1-driven regulation of cAMP phosphodiesterase-4D. Mol Cancer Ther. 2014;13:2463–2473. doi: 10.1158/1535-7163.MCT-14-0297. [DOI] [PubMed] [Google Scholar]