

Abstract
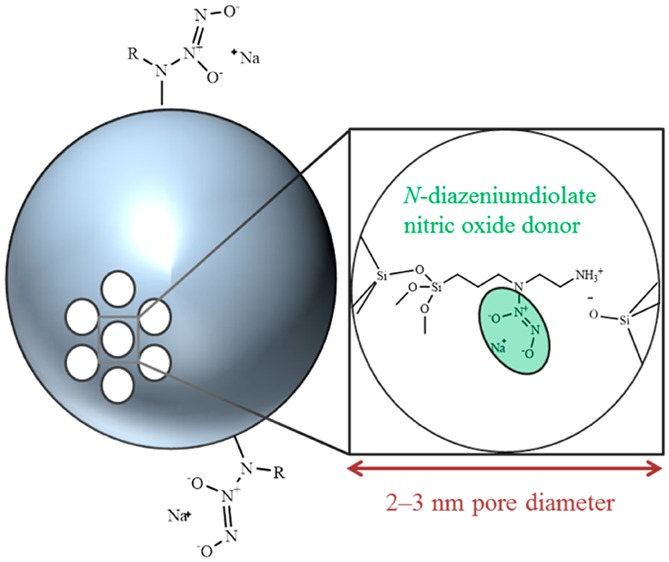
Nitric oxide-releasing mesoporous silica nanoparticles (MSNs) were prepared using an aminosilane-template surfactant ion exchange reaction. Initially, bare silica particles were synthesized under basic conditions in the presence of cetyltrimethylammonium bromide (CTAB). These particles were functionalized with nitric oxide (NO) donor precursors (i.e., secondary amines) via the addition of aminosilane directly to the particle sol and a commensurate ion exchange reaction between the cationic aminosilanes and CTAB. N-Diazeniumdiolate NO donors were formed at the secondary amines to yield NO-releasing MSNs. Tuning of the ion exchange-based MSN modification approach allowed for the preparation of monodisperse particles ranging from 30 to 1100 nm. Regardless of size, the MSNs stored appreciable levels of NO (0.4–1.5 μmol mg–1) with tunable NO release durations (1–33 h) dependent on the aminosilane modification. Independent control of NO release properties and particle size was achieved, demonstrating the flexibility of this novel MSN synthesis over conventional co-condensation and surface grafting strategies.
Keywords: mesoporous silica, nitric oxide, N-diazeniumdiolate, silica nanoparticle, drug release, macromolecular nitric oxide donor
Introduction
Nitric oxide (NO), an endogenous diatomic free radical, mediates multiple physiological processes, including angiogenesis,1 blood pressure regulation,2 wound healing,3,4 and the immune response.5,6 In vivo, nitric oxide synthase (NOS) enzymes generate NO at concentrations (nM−μM) and kinetics dependent on the enzyme location and purpose.3 For example, low concentrations of NO generated via calcium-dependent endothelial and neuronal NOS regulate neovascularization1 and serve roles in neurotransmission.7 Activation of the inducible NOS isoform by immunological stimuli (e.g., lipopolysaccharide, interferon-γ) causes sustained NO release at high concentrations to eradicate foreign pathogens as part of the innate immune response.5,6 The multifaceted roles of endogenous NO are attributable to precise spatiotemporal NO release by cells expressing the NOS enzymes. In addition, NOs short biological lifetime (seconds) restricts its action to <0.5 mm from the point of generation.8
Because of NO’s overwhelming presence in physiology, the administration of exogenous NO gas represents a potential therapy for many diseases.9 A significant body of research has focused on the development of donors that store and release NO under specific chemical conditions to address the concentration-dependent behavior of NO and avoid challenges associated with the administration of NO directly, such as the need for a pressurized gas cylinder and NO’s rapid reaction in biological media.9 In particular, N-diazeniumdiolate NO donors, formed by the reaction of gaseous NO with secondary amines, spontaneously release NO in physiological buffer upon reaction with hydronium ions. This class of molecules has accordingly received attention for biological applications because the breakdown of the NO donor and concomitant NO release occurs at rates dependent on pH, temperature, and the chemical structure of the precursor molecule used for N-diazeniumdiolate formation.
The potential utility of the N-diazeniumdiolate functional group originally inspired research on low molecular weight NO donors.10,11 Unfortunately, limited NO capacity and duration generally preclude the use of these small molecule NO donors for therapeutic applications. For enhancement of NO storage and for exerting additional control over NO release, much work has focused on the synthesis of N-diazeniumdiolate-modified macromolecular NO delivery scaffolds, including chitosan oligosaccharides,12 dendrimers,13−15 gold clusters,16,17 and silica nanoparticles.18−25 With respect to silica, surface grafting,21,26 co-condensation,20,27 and water-in-oil microemulsion19 methods have been used to prepare N-diazeniumdiolate-functionalized particles. Silica is attractive as an NO release scaffold as it is well-tolerated (i.e., nontoxic) and readily implemented as a drug delivery vehicle.11,19,26 For example, NO donor-modified silica particles have served as reinforcing fillers for NO-releasing polymeric coatings (i.e., for in vivo sensors) to promote angiogenesis and wound healing.28,29 Such materials have also proven effective as antimicrobial abrasives that may be integrated with oral hygiene technologies.18,27
Despite their value as potential therapeutics, current strategies for synthesizing NO-releasing silica nanoparticles remain limited by challenges associated with altering the physical properties of the particles and the NO release independent of one another. The use of mesoporous silica represents an attractive macromolecular scaffold for enhancing NO storage and release because of the inherently greater and modifiable surface area (500–1,200 m2 g–1) relative to previous nonporous silica systems.19−21,23,25−27 Control over pore formation and the silica mesophase is achieved via the synthesis of the nanoparticles around an ordered surfactant aggregate, generally an alkyltrimethylammonium salt, that serves as the structure-directing agent (SDA).30,31 Covalent attachment of secondary-amine containing silanes (i.e., NO donor precursors) to mesoporous silica is carried out by direct incorporation of the aminosilane into the particle backbone via co-condensation32−34 or postsynthetically through surface grafting.35−37 In the co-condensation approach, Coulombic repulsion between the cationic surfactant molecules and the protonated backbone amines destabilizes the template, resulting in materials with irregular morphology, even at low aminosilane concentrations.32,38 Postsynthetic surface grafting (after extracting the SDA) is generally the preferred method for functionalizing mesoporous silica, albeit at the cost of a multistep workflow and loss of control over the amount of aminosilane incorporated. Moreover, the grafting process requires a nonpolar aprotic solvent to avoid irreversible water-induced particle agglomeration,25 often resulting in heterogeneous amine distribution and batch-to-batch irreproducibility.37
Ion exchange between cationic organosilanes and common alkyltrimethylammonium SDAs represents a new MSN functionalization approach. To date, this strategy has been limited to postsynthetic modification in organic solvent, raising concerns regarding synthesis irreproducibility that is a similar detriment to the surface grafting approach. Herein, we report aminosilane ion exchange with cetyltrimethylammonium bromide (CTAB) in the aqueous particle solution. Initially, we prepare a diverse selection of monodisperse NO-releasing amine-functionalized mesoporous silica nanoparticles (MSNs). The surface- and pore-bound secondary amines are then converted to N-diazeniumdiolate moieties to yield the NO-releasing MSNs. Using the aqueous ion exchange approach, we demonstrate autonomous control over particle size and NO release capabilities (i.e., NO release rates and total NO storage), representing a significant advantage over conventional co-condensation and grafting methods. The relationship between NO release kinetics and particle mesophase ordering is also elucidated via detailed physicochemical analysis of the MSNs.
Experimental Section
Materials
Tetraethylorthosilicate (TEOS), 3-aminopropyltriethoxysilane (APTES), 3-mercaptopropyltrimethoxysilane (MPTMS), 3-(trimethoxysilylpropyl)diethylenetriamine (DET3), N-methylaminopropyltrimethoxysilane (MAP3), N-(6-aminohexyl)aminopropyltrimethoxysilane (AHAP3), N-(2-aminoethyl)-3-aminopropyltrimethoxysilane (AEAP3), and isobutyltrimethoxysilane (BTMS) were purchased from Gelest (Morrisville, PA) and stored under a nitrogen atmosphere. Sodium methoxide (NaOMe; 5.4 M in methanol), anhydrous N,N-dimethylformamide (DMF), anhydrous methanol (MeOH), ethanol (EtOH), aqueous ammonium hydroxide (30 wt %; NH4OH), concentrated hydrochloric acid (HCl), and all salts were purchased from Fisher Scientific (Fair Lawn, NJ). Cetyltrimethylammonium bromide was purchased from Sigma (St. Louis, MO). Nitrogen (N2), argon (Ar), and nitric oxide (NO) calibration (25.87 ppm in nitrogen) gases were purchased from Airgas National Welders (Raleigh, NC). Pure NO gas was purchased from Praxair (Danbury, CT). Water was purified using a Millipore Milli-Q UV Gradient A10 system (Bedford, MA) to a resistivity of 18.2 MΩ cm and a total organic content of ≤10 ppb. Unless specified, all chemicals were used as received without further purification.
Nanoparticle Synthesis
Particle synthesis was achieved by addition of tetraethylorthosilicate as a bolus to a stirred solution of water, EtOH, NH4OH, and CTAB, allowing the reaction to proceed for 2 h. For synthesis of the 30, 150, and 450 nm diameter particles, 2.500 mL of TEOS in EtOH (0.88, 1.06, and 1.33 M, respectively) was added to the reaction mixture, whereas 1.395 mL of concentrated TEOS was used for the synthesis of the larger 1100 nm particles. Synthesis conditions for the MSNs are provided in Table 1. In all cases, reaction solutions appeared turbid within 15 min of silane introduction. Following particle formation, additional organosilane (AEAP3, AHAP3, APTES, BTMS, MAP3, MPTMS, or DET3) was introduced directly to the colloidal solution dropwise for 5 min using a Kent Scientific Genie Plus syringe pump (Torrington, CT). The reaction was then aged overnight (∼18 h) with stirring. Unless specified, an optimized TEOS:organosilane molar ratio of 1.56:1.00 was used. Following functionalization, particles were collected by centrifugation (6540g, 4 °C, 15 min), washed three times with EtOH, and dried under vacuum. For both the 30 and 150 nm particles, EtOH (one volume per two volumes of the reaction mixture) was added to the solution to induce particle flocculation during the collection procedure and enhance the overall yield. Bare MSNs were synthesized and collected similarly but without the organosilane functionalization step.
Table 1. Synthesis Conditions and Nitrogen Physisorption Data for MSNs of Varying Sizea.
| particle size | [H2O] (M) | [NH3] (M) | [CTAB] (mM) | reaction volume (mL) | temperature (oC) | specific surface area (m2 g–1)b | average pore width (Å)c | nitrogen wt %d |
|---|---|---|---|---|---|---|---|---|
| 30 nm | 54.5 | 0.267 | 5.30 | 150 | 68 ± 1 | 1290 ± 90 | 23.6 ± 2.3 | ≤0.01 |
| 150 nm | 39.4 | 0.267 | 5.30 | 150 | 38 ± 1 | 1170 ± 80 | 21.9 ± 0.6 | 1.11 ± 0.12 |
| 450 nm | 35.0 | 0.267 | 5.30 | 150 | 23 ± 1 | 1280 ± 120 | 20.4 ± 0.2 | 0.13 ± 0.06 |
| 1100 nm | 25.5 | 0.521 | 2.20 | 350 | 23 ± 1 | 1170 ± 70 | 19.5 ± 0.3 | ≤0.01 |
Error bars represent standard deviation for n ≥ 3 separate syntheses.
Determined by BET analysis of the nitrogen sorption isotherms (0.05 ≤ p/p0 ≤ 0.20).
Calculated via BJH analysis of the nitrogen adsorption isotherm (p/p0 ≤ 0.60).
Nitrogen wt % measured by elemental analysis.
Following MSN synthesis, residual CTAB was removed by ion exchange with hydrochloric acid (HCl). Particles (∼200 mg) were suspended in 30 mL of 10% v/v HCl in EtOH, agitated in an ultrasonicator bath for 30 min, and collected by centrifugation (6540g, 4 °C, 15 min). This process was repeated three times to ensure complete CTAB removal followed by two additional EtOH washes. The particles were dried under vacuum to yield the surfactant-free nanoparticles. Typical yields for the amine-modified 30, 150, 450, and 1100 nm MSNs were 150, 175, 275, and 650 mg, respectively.
Mesoporous Silica Nanoparticle Characterization
Particle morphology was characterized using a JEOL 2010F transmission electron microscope (Peabody, MA). Particles were suspended in MeOH at 1 mg mL–1 via brief agitation with an ultrasonicator. Subsequently, 5 μL of the resulting dispersion was cast onto a Formvar-coated copper grid (Ted Pella, Inc.; Redding, CA). The geometric size distribution of the particles was estimated from the electron micrographs using ImageJ software (Bethesda, MD). The solution-phase behavior of the nanoparticles in water was investigated using dynamic light scattering (Malvern Zetasizer Nano-ZS; Westborough, MA) to determine MSN hydrodynamic diameter (Z-average size) and polydispersity index. Aqueous colloidal nanoparticle suspensions were prepared by dispersing particles at a concentration of 0.5 mg mL–1 via probe sonication at 7 W for 45 s using a Misonix S-4000 ultrasonicator (Farmingdale, NY). Nitrogen sorption isotherms were collected on a Micromeritics Tristar II 3020 surface area and porosity analyzer (Norcross, GA). Samples were dried under a stream of N2 gas at 110 °C overnight and then degassed for 2 h prior to analysis. Brunauer–Emmett–Teller (BET) analysis of physisorption data was used to calculate MSN specific surface area for p/p0 values of 0.05–0.20. Pore size analysis using the adsorption branch of the sorption isotherm (0.05 < p/p0 < 0.60) was accomplished using the Barrett–Joyner–Halenda (BJH) method. Data obtained at relative pressures >0.60 p/p0 were not considered for pore size determination as nitrogen capillary condensation occurred in the interparticle volumes for the 30 and 150 nm particles, inflating the calculated pore width. Pore structure/ordering information was obtained by small-angle X-ray scattering analysis of the dry MSN powder. The Cu Kα line (1.54 Å) was used as the source radiation, and scattering profiles were collected on a SAXSLab Ganesha point collimated pinhole system equipped with a moveable Dectris Pilatus 300 K 2-dimensional single-photon-counting detector (Northampton, MA). Scattering vector (q) calibration was accomplished using the first-order ring for silver behenate, and data was collected for q-values of 0.005–0.724 Å–1. Covalent incorporation of aminosilanes into the MSN backbone was confirmed via solid-state cross-polarization/magic angle spinning (CP/MAS) 29Si nuclear magnetic resonance spectroscopy using a Bruker DMX 360 wide-bore spectrometer at a resonance frequency of 71.548 Hz. Samples were carefully ground in a mortar and pestle, packed into a 4 mm ZrO2 rotor, and spun at 10 kHz. All chemical shifts were determined relative to an external tetramethylsilane standard. Elemental analysis was used to quantify the nitrogen weight percent of particles before and after functionalization with secondary amine-containing silanes using a PerkinElmer 2400 CHNS/O analyzer (Waltham, MA) operated in CHN mode.
N-Diazeniumdiolate Modification and Nitric Oxide Release Measurements
The aminosilane-modified MSNs (∼15 mg) were suspended in 9:1 DMF:MeOH at 5 mg mL–1 in a glass vial and dispersed by ultrasonication for 20 min. After forming a homogeneous particle dispersion, NaOMe (5.4 M in MeOH; 9.0 μmol per mg MSN) was added to the solution and mixed. The MSN-containing vials were equipped with stir bars, placed in a stainless steel reaction bottle (Parr Instrument Co.; Moline, IL), and connected to an in-house NO reactor. The Parr bottle was flushed six times (three rapid, three 10 min) with 8 bar Ar gas to remove atmospheric oxygen and minimize the formation of NO byproducts. The vessel was subsequently pressurized with 10 bar NO gas, and the reaction proceeded for 72 h. Of note, the NO gas used for N-diazeniumdiolate formation was purified over solid potassium hydroxide for at least 4 h prior to reaction. After 72 h, the Parr bottle was vented, and the vessel was flushed six more times (three short, three 10 min) to remove unreacted NO. The particles were again collected by centrifugation (6540g, 4 °C, 15 min), washed three times with EtOH, and dried under vacuum for 1–2 h. The resulting N-diazeniumdiolate-modified particles were stored in a vacuum-sealed bag at −20 °C until further use.
Nitric oxide release measurements were carried out using a Sievers 280i NO analyzer (Boulder, CO). Generation of NO from the proton-labile N-diazeniumdiolate NO donors was detected indirectly via chemiluminescence from excited state nitrogen dioxide formed upon the reaction of NO with ozone. The NOA was calibrated using a two-point linear calibration; air passing through a Sievers NO zero filter served as the blank value, and 25.87 ppm of NO in N2 was used as the second calibration point. Particles (∼1 mg) were added to the NOA sample flask containing 30 mL of deoxygenated phosphate buffered saline (PBS, 0.010 M, pH 7.41) at 37 °C. A stream of N2 gas (80 mL min–1) was continuously bubbled through solution to carry liberated NO to the analyzer. Supplemental nitrogen flow was provided to the flask to match the instrument collection rate of 200 mL min–1. Instantaneous NO concentrations were measured at a sampling frequency of 1 Hz, providing near real-time information regarding MSN NO release kinetics. The NO measurements were terminated when NO release from the particles was below 10 ppb mg–1.
Statistical Analysis
One-way analysis of variance was used for multiple comparisons of MSN physicochemical properties (e.g., surface area, pore size, NO-release total amounts, NO-release kinetics) with provided p-values. Individual comparisons were carried out using a two-tailed Student’s t test with α = 0.05 considered as the threshold for statistical significance. All results presented represent data from three or more separate synthesis experiments.
Results and Discussion
The synthesis of NO-releasing nanoparticles has been previously reported18−27 but without autonomous control over particle size, NO release kinetics, and NO storage. Generally, total NO storage for silica-based materials is limited to <0.40 μmol mg–1 due to low aminosilane incorporation. Limited NO storage often is further compounded by a lack of morphological control and poor synthesis yields. Mesoporous silica was thus selected as a new scaffold in an attempt to exert greater control over particle NO release properties. Mesoporous silica nanoparticles were prepared via a supramolecular liquid-crystal templating approach. Cationic, amphiphilic CTAB aggregates were used as the structure-directing agent for particle synthesis.31 The synthesis of four different-sized MSNs was achieved using TEOS as the backbone silane by altering the reaction temperature and reactant concentrations (Table 1). Surfactant was removed by ion exchange in ethanolic HCl to yield the bare mesoporous scaffolds. Although other methods (e.g., calcination) have been used for CTAB removal, irreversible particle agglomeration often results.39,40 Surfactant removal from the MSNs after agitation in HCl was evaluated using elemental analysis. The measured nitrogen wt % for the bare particles was <0.2% in all cases (indicating complete CTAB removal) with the exception of the 150 nm system (∼1.11%). The significant nitrogen content was attributed to trapped ammonia because the low carbon content (5.48 ± 1.00%) did not reflect the presence of CTAB (∼80.3% carbon by mass). Indeed, the 150 nm particles had a propensity to aggregate and did not disperse into solution during the washing and CTAB removal processes, whereas all three remaining particle systems (e.g., 30, 450, and 1100 nm) readily suspended with sonication. The heterogeneity of these wash steps is likely at fault for residual nitrogen content in the 150 nm MSNs.
The surface areas and pore sizes of the unmodified MSNs were calculated from the affiliated nitrogen sorption isotherms (Figure 1). Each of the physisorption isotherms exhibited steep inflections at ∼0.2–0.4 p/p0 and >0.8 p/p0 corresponding to capillary condensation of nitrogen in the particle mesopores and interparticle volumes, respectively.41 All isotherms were classified as Type IV isotherms without hysteresis according to the conventions adopted by the International Union of Pure and Applied Chemistry (IUPAC).42 Nitrogen gas adsorption/desorption on CTAB-templated mesoporous silica has consistently yielded similar results.43 Importantly, MSN surface areas calculated using the Brunauer–Emmett–Teller (BET) method exceeded 1000 m2 g–1 in all cases (Table 1) regardless of particle size. Average pore sizes were evaluated using Barrett–Joyner–Halenda (BJH) analysis of a portion of the nitrogen adsorption branch (Figure 1E) and yielded calculated pore widths in the range of 19.5–23.6 Å, which are comparable to those reported in the literature.32,44,45
Figure 1.

Nitrogen adsorption and desorption isotherms for bare (A) 1100, (B) 450, (C) 150, and (D) 30 nm MSNs. The estimated pore width distributions calculated via BJH analysis of the adsorption branch are presented in (E) for the (i) 1100, (ii) 450, (iii) 150, and (iv) 30 nm particles.
The particles were modified with secondary amines by direct organosilane addition to the reaction solution following completion of the particle synthesis reaction (≤2 h as determined by dynamic light scattering). Residual surfactant SDA was removed in a subsequent step similar to unmodified particles. The aminosilane N-(2-aminoethyl)-3-aminopropyltrimethoxysilane (AEAP3) was selected to optimize this process, initially using the largest (1100 nm) particles. As expected, lower specific surface areas were observed due to increased particle aminosilane content (Table 2). Pore size analysis of the nitrogen adsorption isotherms indicated a clear decrease in mesopore volume with increasing AEAP3 concentration, whereas the pore width remained invariable (p > 0.50). At AEAP3 concentrations at or exceeding 5.7 mM, the gas sorption isotherm abruptly transitioned from a type IV to a type I isotherm (Figure S1) consistent with bound organic groups on the silica network.46
Table 2. Characterization of AEAP3-Modified 1100 nm Mesoporous Silica Particles as a Function of Reaction Aminosilane Concentrationa.
| [AEAP3] (mM) | specific surface area (m2 g–1)b | cumulative pore volume (cm3 g–1)c | average pore width (Å)c | nitrogen wt %d |
|---|---|---|---|---|
| 0 | 1200 ± 70 | 0.47 ± 0.09 | 19.5 ± 0.3 | ≤0.01 |
| 1.4 | 790 ± 60 | 0.13 ± 0.02 | 19.4 ± 0.7 | 2.41 ± 0.25 |
| 2.9 | 520 ± 130 | 0.05 ± 0.01 | 20.0 ± 0.7 | 3.38 ± 0.41 |
| 5.7 | 5 ± 1 | 0.01 ± 0.00 | 20.7 ± 2.0 | 4.38 ± 0.33 |
| 11.5 | 3 ± 1 | 0.00 ± 0.00 | N.D.e | 4.87 ± 0.04 |
Error bars represent standard deviation for n ≥ 3 separate syntheses.
Determined by BET analysis of the nitrogen sorption isotherms (0.05 ≤ p/p0 ≤ 0.20).
Calculated via BJH analysis of the nitrogen adsorption isotherm (p/p0 ≤ 0.60).
Nitrogen wt % measured by elemental analysis.
Pore width could not be calculated from the adsorption isotherm.
Solid-state cross-polarization (1H/29Si)/magic angle spinning (CP/MAS) nuclear magnetic resonance spectroscopy (NMR) confirmed covalent incorporation of AEAP3 into the inorganic TEOS backbone (Figure 2).47 The Q-band peaks at −94, −103, and −112 ppm were assigned to backbone Si atoms present as geminal silanol (Q2), lone silanol (Q3), and siloxane (Q4) species, respectively. The T-band, indicative of the bound organosilane (AEAP3), consisted of peaks at −60 and −69 ppm that were assigned to T2 and cross-linked T3 species, respectively. The intensity of the T-band increased with aminosilane concentration, corresponding to progressively greater amine incorporation in the final product. However, quantitative CP/MAS NMR analysis was not pursued due to signal intensity dependence on the location of 1H atoms relative to 29Si. Interestingly, we noted a considerable amount of cross-linked (T3) surface-bound aminosilanes that was attributed to the large water concentration (>20 M) in the reaction mixture, driving condensation between aminosilanes.47 For comparison, MCM-41 materials produced through postsynthetic surface grafting in anhydrous solvents are primarily bidentate T2 species and exhibit limited cross-linking (T3).48
Figure 2.

Solid-state CP/MAS 29Si NMR spectra of 1100 nm MSNs at AEAP3 reaction concentrations of (i) 0.0, (ii) 1.4, (iii) 2.9, (iv) 5.7, and (v) 11.5 mM.
Although CP/MAS 29Si NMR confirmed covalent aminosilane incorporation, elemental analysis was used as an adjunct experiment to quantitatively assess amine incorporation. As expected, nitrogen content increased with the overall reaction aminosilane concentration. At the highest AEAP3 concentration tested (11.5 mM; Table 2), a maximum nitrogen content of 4.87 wt % was measured. Taken together, the nitrogen sorption, NMR, and elemental analysis experiments indicated covalent incorporation of AEAP3 and suggested mesopore infiltration at aminosilane concentrations of ≤11.5 mM.
Powder small-angle X-ray scattering (SAXS) was used to gain insight into potential alterations to MSN pore structure as a function of reaction AEAP3 concentration (Figure 3).49 The SAXS profile for the bare 1100 nm MSNs (Figure 3) exhibited an intense scattering peak at 0.170 Å–1 (2θ = 2.41 o; hkl 100), and two weaker, larger-angle peaks in the scattering profile were assigned to the 110 (0.292 Å–1) and 200 (0.339 Å–1) reflections indexed on a hexagonal lattice (lattice constant a = 43.1 ± 1.5 Å). Although the absence of higher-order peaks indicated only modest mesoscopic ordering, the scattering profile consisted of the prominent structural lines for MCM-41-type (hexagonal) silica.31,50−52 Broadening of the 100 reflection (Figure 3A) and the gradual disappearance of the 110/200 scattering peaks (Figure 3B) was observed for the amine-modified MSNs (relative to the bare particles), representing a loss of long-range ordering with increasing AEAP3 concentration. These results further verify aminosilane localization within the pores.
Figure 3.

Small-angle X-ray scattering profiles for 1100 nm MSNs at AEAP3 reaction concentrations of (i) 0, (ii) 1.4, (iii) 2.9, (iv) 5.7, and (v) 11.5 mM. The scattering profile is presented in (A), and an enlarged view of the 110/200 scattering lines is displayed in (B).
Importantly, the particles retained excellent sphericity and monodispersity (Figure 4A) upon amine modification as indicated by transmission electron microscopy (TEM) even at the highest AEAP3 concentration presented in Table 2 (11.5 mM). Undesirable particle agglomeration was routinely noted at greater AEAP3 concentrations (≥14.3 mM). Interparticle bridging was occurring at these higher concentrations, revealing a practical maximum in the attainable aminosilane incorporation (Figure S2). On the basis of the results for the 1100 nm particles, the optimal TEOS:aminosilane molar ratio of 1.56:1.00 (corresponding to 11.5 mM AEAP3 in Table 2) was used to synthesize smaller AEAP3 particles. Regardless of the intended size, this approach resulted in well-defined nanomaterials (Figure 4B–D). Dynamic light scattering (DLS) analysis of aqueous MSN dispersions (Table 3) supported TEM observations. The low observed polydispersity indices (PDIs; 0.12, 0.02, and 0.04 for the 30, 150, and 450 nm particles, respectively) affirmed narrow particle size distributions. The DLS/TEM data also verified covalent bonding of aminosilanes to the particle surface rather than the formation of discrete entities and likely large agglomerates. Elemental analysis (Table 3) and CP/MAS NMR (Figure S3) confirmed aminosilane incorporation with significant measured nitrogen content (>4.50% N) for each particle system.
Figure 4.

Transmission electron micrographs of (A) 1100, (B) 450, (C) 150, and (D) 30 nm AEAP3-modified mesoporous silica nanoparticles.
Table 3. Physicochemical Characterization of AEAP3-Functionalized MSNs of Varying Sizea.
| geometric diameter (nm)b | Z-average size (nm)c | PDIc | nitrogen wt %d | specific surface area (m2 g–1)e | pore width (Å)f |
|---|---|---|---|---|---|
| 36 ± 8 | 74 ± 6 | 0.12 ± 0.06 | 4.65 ± 0.19 | 210 ± 40 | 25.1 ± 1.1 |
| 149 ± 13 | 223 ± 17 | 0.02 ± 0.01 | 5.91 ± 0.13 | 69 ± 13 | 24.8 ± 0.6 |
| 450 ± 50 | 564 ± 66 | 0.04 ± 0.02 | 5.07 ± 0.10 | 68 ± 20 | 21.5 ± 0.8 |
| 1110 ± 210 | n/ag | n/ag | 4.87 ± 0.04 | 3 ± 1 | n/ah |
Error bars represent standard deviation for n ≥ 3 separate syntheses.
Estimated using electron micrographs.
Measured via dynamic light scattering.
Nitrogen wt % measured by elemental analysis.
Determined by BET analysis of the nitrogen sorption isotherms (0.05 ≤ p/p0 ≤ 0.20).
Calculated via BJH analysis of the nitrogen adsorption isotherm (p/p0 ≤ 0.60).
Particle sedimentation interfered with DLS measurement.
Pore width could not be calculated from the adsorption isotherm.
Nitric Oxide Release
Different-sized AEAP3-modified particles were functionalized with N-diazeniumdiolate moieties by reaction with NO gas at high pressure in the presence of sodium methoxide. Nitric oxide release was evaluated in real-time via chemiluminescent analysis of the NO-releasing particles in physiological buffer (PBS, pH 7.4) at 37 °C (Table 4). Upon immersion into aqueous solution, the AEAP3/NO MSNs were characterized by a large instantaneous NO flux corresponding to reaction of the proton-labile N-diazeniumdiolate with water to generate NO.53,54 Despite large total NO storage (>0.8 μmol mg–1) for all four particle systems, total NO storage (p < 0.01), NO release half-lives (p < 0.01), and release durations (p = 0.02) were unexpectedly diverse. The 1100 nm particles exhibited large NO storage (1.41 μmol mg–1) and rapid release (t1/2 = 25.6 min). Similarly, the 30 nm AEAP3/NO particles released their total NO payload rapidly (t1/2 = 27.4 min) but stored only a fraction of the NO measured for the 1100 nm particles ([NO]t = 0.88 μmol mg–1). Although the 450 nm MSNs were characterized with low NO storage (0.82 μmol mg–1), they were associated with the longest NO release half-life (88.2 min). Relative to the 1100 nm AEAP3/NO particles, the 150 nm MSNs exhibited comparable NO storage (1.30 μmol mg–1) and intermediate NO release rates (t1/2 = 41.9 min).
Table 4. Chemiluminescent NO Release Measurements in Physiological Buffer (PBS, pH 7.4, 37 °C) from AEAP3/NO MSNs of Varying Sizea.
| particle size (nm) | [NO]max (ppm mg–1)b | t1/2 (min)c | td (h)d | [NO]t (μmol mg–1)e | N-diazeniumdiolate formation efficiency (%)f |
|---|---|---|---|---|---|
| 30 | 18.7 ± 2.2 | 27.4 ± 8.9 | 12.2 ± 3.0 | 0.88 ± 0.05 | 26.6 ± 1.8 |
| 150 | 22.6 ± 4.4 | 40.7 ± 11.0 | 16.7 ± 1.4 | 1.30 ± 0.11 | 30.9 ± 2.7 |
| 450 | 6.6 ± 1.8 | 88.2 ± 10.5 | 14.0 ± 0.3 | 0.82 ± 0.08 | 22.8 ± 2.3 |
| 1100 | 32.8 ± 9.8 | 25.6 ± 5.0 | 11.1 ± 0.7 | 1.41 ± 0.19 | 40.7 ± 5.2 |
Error bars represent standard deviation for n ≥ 3 separate syntheses.
Maximum instantaneous NO concentration.
Half-life of NO release.
NO release duration; time required for NO concentrations to reach ≤10 ppb mg–1.
Total NO release.
Calculated using total NO release and nitrogen wt % determined by elemental analysis (Table 3) according to equation provided in the Supporting Information.
The difference in NO release kinetics between particle systems was not anticipated as all particles were functionalized with the same N-diazeniumdiolate precursor (AEAP3). For shedding further light on these effects, total NO release from the AEAP3/NO particles were compared to the degree of nitrogen incorporation measured by elemental analysis (Table 3) to determine N-diazeniumdiolate formation efficiencies. As expected based on the NO release data, the 1100 nm MSNs exhibited the greatest NO donor formation efficiency (40.7%), far greater than that reported by Carpenter et al. (<27%).19 The NO donor formation efficiencies for the other three particle sizes were calculated at 23–31%.
The wide range of NO release kinetics (half-lives of 27–88 min) and NO donor formation efficiencies (23–41%) suggested additional factors were controlling NO release. One possible influence to NO release kinetics is particle size and the position of the N-diazeniumdiolates within the pore network, which may affect NO donor accessibility by water and thus impact NO release kinetics. However, aminosilane (N-diazeniumdiolate) location alone does not account for the differences in NO release kinetics. For example, the largest particles, which could have NO donors buried as far as 0.5 μm from their external surface, also had the shortest NO release half-life (t1/2 = 25.6 min) and duration (td = 11.1 h). We hypothesized that the structure and ordering of the particle pore network may also affect NO release kinetics and partially account for these variations, particularly because a link between mesoscopic ordering and diffusion-based drug release has been demonstrated previously.55 For example, decreased organization may impede sodium methoxide access to pore-bound secondary amines, hindering N-diazeniumdiolate formation. As an extension of the same logic, altered water diffusion into the pores would give rise to differences in NO release kinetics between AEAP3/NO MSNs of different sizes.
Small-angle X-ray scattering (SAXS) was used to assess pore ordering of the bare (Figure 5) and amine-modified (Figure S4) MSNs. As determined previously, the SAXS profile for the 1100 nm MSNs corresponded to a hexagonal lattice with modest ordering. In contrast to the observed MCM-41 structure for the largest particles, the scattering profile for the 30 nm MSNs alluded to an alternative mixed pore structure. Analysis of the smallest particles revealed three scattering peaks at 0.155, 0.301, and 0.552 Å–1, which represented an intermediate to typical hexagonal and lamellar (layered) pore ordering.51,52 The appearance of the high angle reflection (0.552 Å–1) was evidence for a significant degree of pore ordering; this peak is seldom observed for intermediate products. The electron micrographs for the 30 nm MSNs (Figure 4D) were in good agreement with the scattering data and provided further confirmation of a mixed pore structure. X-ray scattering patterns obtained for the 150 and 450 nm particles were representative of a greater degree of pore disorder. Only a broad peak centered at ∼0.32 Å–1 was observed in both scattering profiles (in addition to the 100 line at ∼0.17–0.19 Å–1). The absence of an additional reflection confirmed mesophase irregularity for these particles. The scattering profiles for the 150 and 450 nm particles were characteristic of mesopore arrangements between hexagonal and lamellar structures.31 Although pore disorder was not as extensive for the 150 nm particles, the skewed peak at ∼0.18 Å–1 for 450 nm MSNs suggested a more heterogeneous pore structure. In fact, the irregular peak shape was likely the superimposition of two separate low order reflections. Of note, the X-ray scattering data for the amine-functionalized particles 1100 nm particles pointed to a slight loss in long-range ordering (Figure 5A) due to pore filling by AEAP3.56,57 Only broad reflections were evident in the scattering profiles for the 30, 150, and 450 nm AEAP3-modified MSNs, indicating that the aminosilane modification impacted the pore structures of these particle systems. Potential interference to pore structure determination by scattering contributions from the particles themselves (i.e., as monodisperse spheres) was unlikely because the raw scattering profile did not display typical interference fringes in the 0.01–0.1 Å–1 region (Figure S4).
Figure 5.

Small-angle X-ray scattering profiles for (A) 1100, (B) 450, (C) 150, and (D) 30 nm (i) bare and (ii) AEAP3-modified MSNs.
Particle X-ray scattering data provided insight into the relationship between MSN pore structure and NO release kinetics. The more ordered pore system (i.e., amine-modified 1100 nm particles) enables unrestricted (pore) access of sodium methoxide and water, resulting in large NO storage and rapid NO release, respectively. The 150 and 450 nm particles were capable of more sustained NO release, likely due to mesophase disruption with aminosilane addition. In contrast, the smallest 30 nm MSNs exhibited rapid NO release and lower NO storage (0.88 μmol mg–1) despite collapse of the pore system. In this case, location of the N-diazeniumdiolates within the pore network and water access to the NO donors may more significantly affect NO release than particle mesoporosity.
Organosilane Modification
Although aminosilanes are highly reactive with the silanol groups that populate the surface of silica nanoparticles, they also readily undergo hydrolysis and autocondensation in aqueous conditions to form new, discrete entities. For this reason, addition of organosilane directly to the colloidal solution (i.e., particle reaction mixture) generally yields amorphous materials with heterogeneous functional group distribution. Postsynthetic grafting approaches thus require active steps for water removal from the reaction mixture to avoid undesirable particle agglomeration.38
In addition to anhydrous conditions, efficient particle modification is contingent upon successful removal of the pore-resident surfactant prior to reaction with aminosilanes, as the positively charged template molecule stabilizes the anionic surface silanols and may impede diffusion of external species into the pores.58,59 De Juan and co-workers previously exploited the stability of the surfactant CTAB template for selective derivatization of the outer and inner mesoporous silica surfaces using a step-by-step functionalization approach.60 In our study, the large degree of particle functionalization suggested that the aminosilanes likely displaced CTAB before undergoing autocondensation. We rationalized that this phenomenon might be due to an ion-exchange process between the surfactant and protonated aminosilanes (Figure 6). Both Dai et al.61 and Bourlinos et al.62 have described ion exchange between cationic species (metal ions and aminosilanes, respectively) and the CTAB template as a method for particle modification. In both cases, the uncalcined (i.e., CTAB-containing) silica was modified in a separate reaction rather than a one-step procedure.
Figure 6.

Proposed mechanism for MSN functionalization with aminosilanes. Positively charged aminosilanes undergo ion exchange with the template surfactant to stabilize anionic silanol species anchored to the mesopore walls.
We sought to verify that only MSN modification with cationic species would preserve particle morphology. Using the 150 nm particle system, the MSNs were functionalized with either isobutyl(trimethoxy)silane (BTMS) or (3-mercaptopropyl)trimethoxysilane (MPTMS) at concentrations equal to those employed for the 150 nm AEAP3 particles. As the colloidal solution is formed under basic conditions, the BTMS alkyl groups remain neutral, whereas a significant fraction of the MPTMS side chains would exist as the anionic thiolate species (pKa ∼ 10)63,64 in both cases preventing ion exchange. 3-Aminopropyltriethoxysilane (APTES) was used as a positive control, as APTES is similar in size to BTMS and MPTMS but should undergo efficient ion exchange with CTAB due to the presence of a basic primary amine.
The morphology of the 150 nm APTES, BTMS, and MPTMS particles was examined using transmission electron microscopy (Figure 7). As expected, 150 nm particles functionalized with APTES exhibited uniform morphology with excellent sphericity consistent with that of the 150 nm AEAP3 MSNs. Evaluation of aqueous APTES particle suspensions by DLS indicated that the monodispersity of the particles (PDI = 0.03 ± 0.02) was preserved upon aminosilane modification. In contrast, undesirable silane bridging and particle agglomeration were evident in electron micrographs of the MPTMS- and BTMS-modified particles. A significant increase in the carbon wt % (measured by elemental analysis; Table S2) for all particle systems indicated that the silanes were incorporated into the final product. The morphological differences between particles observed using TEM were due to reaction with organosilanes. Although this data does not exclude the possibility of alternative reaction mechanisms, the particle analyses presented provide clear support of ion exchange reactions between cationic organosilanes and CTAB.
Figure 7.

Transmission electron micrographs of 150 nm MSNs modified with (A) APTES, (B) BTMS, and (C) MPTMS. Particles in (A) exhibit smooth morphology, whereas agglomeration is observed in (B) and (C).
Aminosilane Modification and Nitric Oxide-Release Kinetics
As the structure of the precursor amine for N-diazeniumdiolate formation influences NO release kinetics from both small molecules11 and nonporous silica particles,27 we sought to alter the NO release kinetics from the MSNs using different organosilanes. The 30 nm particle system was systematically modified with several aminosilanes, including AHAP3, DET3, and MAP3.
The characterization of the precursor- and NO donor-modified MSNs is provided in Table 5. Both the geometric size (∼35–43 nm) and PDI (<0.20) of the particles remained approximately constant (p > 0.5), indicating that the small particle size and monodispersity were conserved during the chemical modification procedure regardless of aminosilane type. The measured hydrodynamic diameter (Z-average size) of each particle system (75–130 nm) was dependent on the composition of the aminosilane but agreed well with the corresponding geometric sizes. Nitrogen content for each MSN system varied expectedly based on the elemental composition of the aminosilane reactant. Particles functionalized with the monoamine MAP3 incorporated the least amount of nitrogen (3.26%), whereas the nitrogen wt % was greatest for the triamine DET3 modification (5.60%). Intermediate nitrogen content was measured for MSNs with attached AHAP3 (4.18%) and AEAP3 (4.65%), which are diaminosilanes of differing carbon content.
Table 5. Physicochemical Characterization of 30 nm NO-Releasing MSNs as a Function of Aminosilane Modificationa.
| particle
characterization |
NO release |
||||||
|---|---|---|---|---|---|---|---|
| aminosilane modification | geometric size (nm)b | Z-average size (nm)c | PDIc | nitrogen wt %d | t1/2 (min)e | td (h)f | [NO]t (μmol mg–1)g |
| MAP3 | 37.1 ± 8.3 | 91.2 ± 8.8 | 0.16 ± 0.05 | 3.26 ± 0.15 | 2.2 ± 0.2 | 1.8 ± 0.4 | 1.39 ± 0.10 |
| AHAP3 | 42.3 ± 8.1 | 131.8 ± 9.4 | 0.17 ± 0.04 | 4.18 ± 0.05 | 4.7 ± 2.3 | 5.9 ± 0.2 | 1.20 ± 0.10 |
| AEAP3 | 35.7 ± 8.1 | 74.1 ± 6.2 | 0.12 ± 0.06 | 4.65 ± 0.19 | 27.4 ± 8.9 | 12.2 ± 3.0 | 0.88 ± 0.05 |
| DET3 | 34.5 ± 7.6 | 83.0 ± 7.6 | 0.17 ± 0.04 | 5.60 ± 0.31 | 47.0 ± 11.9 | 33.2 ± 4.7 | 1.37 ± 0.19 |
Error bars represent standard deviation for n ≥ 3 separate syntheses.
Estimated using electron micrographs.
Measured via dynamic light scattering.
Nitrogen wt % measured via elemental analysis.
Half-life of NO release.
NO release duration; time required for NO concentrations to reach ≤10 ppb mg–1.
Total NO release.
The large degree of aminosilane incorporation translated to excellent particle NO storage, exceeding 1.00 μmol mg–1 for all particle systems tested except AEAP3/NO. Lower total NO storage for AEAP3/NO was expected based on previous results, as intramolecular hydrogen bonding between the side chain amines hinders N-diazeniumdiolate formation.11,65 Although these interactions are also possible for DET3, the presence of two secondary amines resulted in greater NO storage. As anticipated, the MSN NO release kinetics were markedly different between the four particle systems (p < 0.01). The MAP3/NO and AHAP3/NO particles were characterized with rapid initial NO release (t1/2 of 2.2 and 4.7 min, respectively), whereas the NO release for the AEAP3/NO and DET3/NO particles was more sustained (t1/2 of 27.4 and 47.0 min, respectively) as a result of N-diazeniumdiolate charge stabilization by neighboring protonated amines (Table 5; Figure S5).11,65 The NO release durations covered ∼2–33 h, rendering these particles especially useful as NO delivery vehicles where tuning NO release kinetics is critical to efficacy.9
It should be noted that others have reported macromolecular NO donor scaffolds with total NO release values exceeding ∼1.5 μmol mg–1. For example, several porous metal organic frameworks (MOFs) have been developed with NO storage approaching 1–7 μmol mg–1 through direct adsorption of NO gas.66 However, NO release from MOFs is generally rapid, restricting their utility to applications in which the NO donor scaffold is in contact with humidified gas. Both dendrimers67 and silica particles24 modified with S-nitrosothiol (RSNO) NO donors also exhibit large NO payloads (2 and 4 μmol mg–1, respectively) with NO release durations exceeding 2 days in deoxygenated PBS buffer. Unfortunately, RSNOs are unstable NO donors, readily decomposing to yield NO under multiple triggers (e.g., light, heat, reaction with Cu+ ions or ascorbate). S-nitrosothiol stability is further compromised in the presence of oxygen, where reaction with NO produces nitrogen trioxide, an RSNO-reactive species that initiates excessively rapid autocatalytic decomposition.68 In contrast, N-diazeniumdiolate NO donors alleviate the issue of uncontrolled decomposition, liberating NO at rates dependent on both the structure of the aminosilane and the solution pH. Although poor NO storage and difficult synthetic procedures have traditionally excluded N-diazeniumdiolate-modified macromolecular NO donors from therapeutic evaluation, the preparation of NO-releasing mesoporous silica particles was achieved in high yields via ion exchange reactions. Excellent NO storage and diverse NO release kinetics from the MSNs were obtained by simply changing the aminosilane without further synthetic optimization, representing a significant improvement to N-diazeniumdiolate-based NO delivery vehicles.
Conclusions
Nitric oxide-releasing mesoporous silica nanoparticles with a range of sizes (30, 150, 450, and 1100 nm) were successfully prepared using a straightforward aminosilane-CTAB ion exchange approach. The resulting MSNs were well-defined and exhibited a large degree of surface modification, which translated to competitive NO storage with other macromolecular NO donors (e.g., MOFs, RSNOs). Particle NO storage and release kinetics were dependent on both the structure of the pores and the identity of the precursor aminosilane. This report is the first to detail the dependence of NO release kinetics on the architectural properties of mesoporous silica. Further understanding of the intricate relationships between pore ordering and NO release kinetics remains an exciting area of research, as controlled mesophase structure would also provide an additional degree of control in macromolecular NO donor design. Moreover, the ability to easily modify the MSNs with different aminosilanes enabled tuning of NO release kinetics without sacrificing control over either total NO storage or particle size.
Acknowledgments
The authors express their gratitude to Dr. Amar Kumbhar at the UNC Chapel Hill Analytical and Nanofabrication Laboratory (CHANL) for expertise and assistance with the TEM. We acknowledge Dr. Marc ter Horst for assistance with solid-state 29Si NMR. We also thank Dr. Mark Walters and Jimmy Thostenson at Duke University’s Shared Materials Instrumentation Facility (SMIF) for their technical help with the Ganesha SAXS system.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsami.5b10942.
Additional electron micrographs and NO release properties of the 1100 nm AEAP3-modified MSNs, nitrogen sorption isotherms for 1100 nm AEAP3 particles, 29Si NMR spectra for AEAP3-modified MSNs, SAXS profiles for AEAP3-modified MSNs, and elemental analysis of 150 nm APTES, BTMS, and MPTMS particles (PDF)
This research was supported by the National Science Foundation (DMR 1104892) and the National Institutes of Health (DK108318). R.J.S. thankfully acknowledges a John Motley Morehead graduate fellowship from the Royster Society of Fellows at the University of North Carolina-Chapel Hill.
The authors declare the following competing financial interest: Mark Schoenfisch is a cofounder, a member of the board of directors, and maintains a financial interest in Novan Therapeutics, Inc. Novan is commercializing macromolecular nitric oxide storage and release vehicles for dermatological indications.
Supplementary Material
References
- Cooke J. P. NO and Angiogenesis. Atheroscler. Suppl. 2003, 4, 53–60. 10.1016/S1567-5688(03)00034-5. [DOI] [PubMed] [Google Scholar]
- Ignarro L. J.; Buga G. M.; Wood K. S.; Byrns R. E.; Chaudhuri G. Endothelium-Derived Relaxing Factor Produced and Released from Artery and Vein Is Nitric Oxide. Proc. Natl. Acad. Sci. U. S. A. 1987, 84, 9265–9269. 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J.-d.; Chen A. F. Nitric Oxide: A Newly Discovered Function on Wound Healing. Acta Pharmacol. Sin. 2005, 26, 259–264. 10.1111/j.1745-7254.2005.00058.x. [DOI] [PubMed] [Google Scholar]
- Schwentker A.; Vodovotz Y.; Weller R.; Billiar T. R. Nitric Oxide and Wound Repair: Role of Cytokines?. Nitric Oxide 2002, 7, 1–10. 10.1016/S1089-8603(02)00002-2. [DOI] [PubMed] [Google Scholar]
- Bogdan C. Nitric Oxide and the Immune Response. Nat. Immunol. 2001, 2, 907–916. 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- MacMicking J.; Xie Q.-w.; Nathan C. Nitric Oxide and Macrophage Function. Annu. Rev. Immunol. 1997, 15, 323–350. 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- Kiss J. P. Role of Nitric Oxide in the Regulation of Monoaminergic Neurotransmission. Brain Res. Bull. 2000, 52, 459–466. 10.1016/S0361-9230(00)00282-3. [DOI] [PubMed] [Google Scholar]
- Hetrick E. M.; Schoenfisch M. H. Analytical Chemistry of Nitric Oxide. Annu. Annu. Rev. Anal. Chem. 2009, 2, 409–433. 10.1146/annurev-anchem-060908-155146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter A. W.; Schoenfisch M. H. Nitric Oxide Release Part Ii. Therapeutic Applications. Chem. Soc. Rev. 2012, 41, 3742–3752. 10.1039/c2cs15273h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetrick E. M.; Shin J. H.; Stasko N. A.; Johnson C. B.; Wespe D. A.; Holmuhamedov E.; Schoenfisch M. H. Bactericidal Efficacy of Nitric Oxide-Releasing Silica Nanoparticles. ACS Nano 2008, 2, 235–246. 10.1021/nn700191f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keefer L. K. Fifty Years of Diazeniumdiolate Research. From Laboratory Curiosity to Broad-Spectrum Biomedical Advances. ACS Chem. Biol. 2011, 6, 1147–1155. 10.1021/cb200274r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.; Shah A.; Hunter R. A.; Soto R. J.; Schoenfisch M. H. S-Nitrosothiol-Modified Nitric Oxide-Releasing Chitosan Oligosaccharides as Antibacterial Agents. Acta Biomater. 2015, 12, 62–69. 10.1016/j.actbio.2014.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.; Slomberg D. L.; Shah A.; Schoenfisch M. H. Nitric Oxide-Releasing Amphiphilic Poly(Amidoamine) (Pamam) Dendrimers as Antibacterial Agents. Biomacromolecules 2013, 14, 3589–98. 10.1021/bm400961r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.; Sun B.; Li C.; Schoenfisch M. H. Structurally Diverse Nitric Oxide-Releasing Poly(Propylene Imine) Dendrimers. Chem. Mater. 2011, 23, 4227–4233. 10.1021/cm201628z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasko N. A.; Schoenfisch M. H. Dendrimers as a Scaffold for Nitric Oxide Release. J. Am. Chem. Soc. 2006, 128, 8265–8271. 10.1021/ja060875z. [DOI] [PubMed] [Google Scholar]
- Polizzi M. A.; Stasko N. A.; Schoenfisch M. H. Water-Soluble Nitric Oxide-Releasing Gold Nanoparticles. Langmuir 2007, 23, 4938–4943. 10.1021/la0633841. [DOI] [PubMed] [Google Scholar]
- Rothrock A. R.; Donkers R. L.; Schoenfisch M. H. Synthesis of Nitric Oxide-Releasing Gold Nanoparticles. J. Am. Chem. Soc. 2005, 127, 9362–9363. 10.1021/ja052027u. [DOI] [PubMed] [Google Scholar]
- Backlund C. J.; Sergesketter A. R.; Offenbacher S.; Schoenfisch M. H. Antibacterial Efficacy of Exogenous Nitric Oxide on Periodontal Pathogens. J. Dent. Res. 2014, 93, 1089–1094. 10.1177/0022034514529974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter A. W.; Slomberg D. L.; Rao K. S.; Schoenfisch M. H. Influence of Scaffold Size on Bactericidal Activity of Nitric Oxide-Releasing Silica Nanoparticles. ACS Nano 2011, 5, 7235–7244. 10.1021/nn202054f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter A. W.; Worley B. V.; Slomberg D. L.; Schoenfisch M. H. Dual Action Antimicrobials: Nitric Oxide Release from Quaternary Ammonium-Functionalized Silica Nanoparticles. Biomacromolecules 2012, 13, 3334–3342. 10.1021/bm301108x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slomberg D. L.; Lu Y.; Broadnax A. D.; Hunter R. A.; Carpenter A. W.; Schoenfisch M. H. Role of Size and Shape on Biofilm Eradication for Nitric Oxide-Releasing Silica Nanoparticles. ACS Appl. Mater. Interfaces 2013, 5, 9322–9329. 10.1021/am402618w. [DOI] [PubMed] [Google Scholar]
- Frost M. C.; Meyerhoff M. E. Synthesis, Characterization, and Controlled Nitric Oxide Release from S-Nitrosothiol-Derivatized Fumed Silica Polymer Filler Particles. J. Biomed. Mater. Res., Part A 2005, 72A, 409–419. 10.1002/jbm.a.30275. [DOI] [PubMed] [Google Scholar]
- Kafshgari M. H.; Cavallaro A.; Delalat B.; Harding F. J.; McInnes S. J. P.; Mäkilä E.; Salonen J.; Vasilev K.; Voelcker N. H. Nitric Oxide-Releasing Porous Silicon Nanoparticles. Nanoscale Res. Lett. 2014, 9, 333. 10.1186/1556-276X-9-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccio D. A.; Nugent J. L.; Schoenfisch M. H. Stöber Synthesis of Nitric Oxide-Releasing S-Nitrosothiol-Modified Silica Particles. Chem. Mater. 2011, 23, 1727–1735. 10.1021/cm102510q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Annich G. M.; Miskulin J.; Stankiewicz K.; Osterholzer K.; Merz S. I.; Bartlett R. H.; Meyerhoff M. E. Nitric Oxide-Releasing Fumed Silica Particles: Synthesis, Characterization, and Biomedical Application. J. Am. Chem. Soc. 2003, 125, 5015–5024. 10.1021/ja0291538. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Slomberg D. L.; Sun B.; Schoenfisch M. H. Shape- and Nitric Oxide Flux-Dependent Bactericidal Activity of Nitric Oxide-Releasing Silica Nanorods. Small 2013, 9, 2189–2198. 10.1002/smll.201201798. [DOI] [PubMed] [Google Scholar]
- Backlund C. J.; Worley B. V.; Sergesketter A. R.; Schoenfisch M. H. Kinetic-Dependent Killing of Oral Pathogens with Nitric Oxide. J. Dent. Res. 2015, 94, 1092. 10.1177/0022034515589314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh A.; Riccio D. A.; Sun B.; Carpenter A. W.; Nichols S. P.; Schoenfisch M. H. Fabrication of Nitric Oxide-Releasing Polyurethane Glucose Sensor Membranes. Biosens. Bioelectron. 2011, 28, 17–24. 10.1016/j.bios.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto R. J.; Privett B. J.; Schoenfisch M. H. In Vivo Analytical Performance of Nitric Oxide-Releasing Glucose Biosensors. Anal. Chem. 2014, 86, 7141–7149. 10.1021/ac5017425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck J. S.; Vartuli J. C.; Roth W. J.; Leonowicz M. E.; Kresge C. T.; Schmitt K. D.; Chu C. T. W.; Olson D. H.; Sheppard E. W. A New Family of Mesoporous Molecular Sieves Prepared with Liquid Crystal Templates. J. Am. Chem. Soc. 1992, 114, 10834–10843. 10.1021/ja00053a020. [DOI] [Google Scholar]
- Huo Q.; Margolese D. I.; Stucky G. D. Surfactant Control of Phases in the Synthesis of Mesoporous Silica-Based Materials. Chem. Mater. 1996, 8, 1147–1160. 10.1021/cm960137h. [DOI] [Google Scholar]
- Huh S.; Wiench J. W.; Yoo J.-C.; Pruski M.; Lin V. S. Y. Organic Functionalization and Morphology Control of Mesoporous Silicas Via a Co-Condensation Synthesis Method. Chem. Mater. 2003, 15, 4247–4256. 10.1021/cm0210041. [DOI] [Google Scholar]
- Burleigh M. C.; Markowitz M. A.; Spector M. S.; Gaber B. P. Direct Synthesis of Periodic Mesoporous Organosilicas: Functional Incorporation by Co-Condensation with Organosilanes. J. Phys. Chem. B 2001, 105, 9935–9942. 10.1021/jp011814k. [DOI] [Google Scholar]
- Trewyn B. G.; Slowing I. I.; Giri S.; Chen H.-T.; Lin V. S. Y. Synthesis and Functionalization of a Mesoporous Silica Nanoparticle Based on the Sol–Gel Process and Applications in Controlled Release. Acc. Chem. Res. 2007, 40, 846–853. 10.1021/ar600032u. [DOI] [PubMed] [Google Scholar]
- Chen J.; Li Q.; Xu R.; Xiao F. Distinguishing the Silanol Groups in the Mesoporous Molecular Sieve Mcm-41. Angew. Chem., Int. Ed. Engl. 1996, 34, 2694–2696. 10.1002/anie.199526941. [DOI] [Google Scholar]
- Gartmann N.; Brühwiler D. Controlling and Imaging the Functional-Group Distribution on Mesoporous Silica. Angew. Chem., Int. Ed. 2009, 48, 6354–6356. 10.1002/anie.200902436. [DOI] [PubMed] [Google Scholar]
- Sharma K. K.; Anan A.; Buckley R. P.; Ouellette W.; Asefa T. Toward Efficient Nanoporous Catalysts: Controlling Site-Isolation and Concentration of Grafted Catalytic Sites on Nanoporous Materials with Solvents and Colorimetric Elucidation of Their Site-Isolation. J. Am. Chem. Soc. 2008, 130, 218–228. 10.1021/ja074128t. [DOI] [PubMed] [Google Scholar]
- Bruhwiler D. Postsynthetic Functionalization of Mesoporous Silica. Nanoscale 2010, 2, 887–892. 10.1039/c0nr00039f. [DOI] [PubMed] [Google Scholar]
- Lang N.; Tuel A. A Fast and Efficient Ion-Exchange Procedure to Remove Surfactant Molecules from Mcm-41 Materials. Chem. Mater. 2004, 16, 1961–1966. 10.1021/cm030633n. [DOI] [Google Scholar]
- Kecht J.; Bein T. Oxidative Removal of Template Molecules and Organic Functionalities in Mesoporous Silica Nanoparticles by Peroxide Treatment. Microporous Mesoporous Mater. 2008, 116, 123–130. 10.1016/j.micromeso.2008.03.027. [DOI] [Google Scholar]
- Webb P. A.; Orr C.. Analytical Methods in Fine Particle Technology. Micromeritics Instrument Corporation: Norcross, GA, 1997. [Google Scholar]
- Sing K. S. W.; Everett D. H.; Haul R. A. W.; Moscou L.; Pierotti R. A.; Rouquerol J.; Siemieniewska T. Reporting Physisorption Data for Gas/Solid Systems with Special Reference to the Determination of Surface Area and Porosity. Pure Appl. Chem. 1985, 57, 603–613. 10.1351/pac198557040603. [DOI] [Google Scholar]
- Nooney R. I.; Thirunavukkarasu D.; Chen Y.; Josephs R.; Ostafin A. E. Synthesis of Nanoscale Mesoporous Silica Spheres with Controlled Particle Size. Chem. Mater. 2002, 14, 4721–4728. 10.1021/cm0204371. [DOI] [Google Scholar]
- Lee C.-H.; Lo L.-W.; Mou C.-Y.; Yang C.-S. Synthesis and Characterization of Positive-Charge Functionalized Mesoporous Silica Nanoparticles for Oral Drug Delivery of an Anti-Inflammatory Drug. Adv. Funct. Mater. 2008, 18, 3283–3292. 10.1002/adfm.200800521. [DOI] [Google Scholar]
- Pauly T. R.; Pinnavaia T. J. Pore Size Modification of Mesoporous Hms Molecular Sieve Silicas with Wormhole Framework Structures. Chem. Mater. 2001, 13, 987–993. 10.1021/cm000762t. [DOI] [Google Scholar]
- Kruk M.; Jaroniec M. Gas Adsorption Characterization of Ordered Organic–Inorganic Nanocomposite Materials. Chem. Mater. 2001, 13, 3169–3183. 10.1021/cm0101069. [DOI] [Google Scholar]
- Albert K.; Bayer E. Characterization of Bonded Phases by Solid-State Nmr Spectroscopy. J. Chromatogr. A 1991, 544, 345–370. 10.1016/S0021-9673(01)83995-9. [DOI] [Google Scholar]
- Kobler J.; Möller K.; Bein T. Colloidal Suspensions of Functionalized Mesoporous Silica Nanoparticles. ACS Nano 2008, 2, 791–799. 10.1021/nn700008s. [DOI] [PubMed] [Google Scholar]
- Smarsly B.; Groenewolt M.; Antonietti M.. Saxs Analysis of Mesoporous Model Materials: A Validation of Data Evaluation Techniques to Characterize Pore Size, Shape, Surface Area, and Curvature of the Interface. In Scattering Methods and the Properties of Polymer Materials; Springer: Berlin Heidelberg, 2005; Vol. 130, pp 105–113. [Google Scholar]
- Kresge C. T.; Leonowicz M. E.; Roth W. J.; Vartuli J. C.; Beck J. S. Ordered Mesoporous Molecular Sieves Synthesized by a Liquid-Crystal Template Mechanism. Nature 1992, 359, 710–712. 10.1038/359710a0. [DOI] [Google Scholar]
- Huo Q.; Margolese D. I.; Ciesla U.; Demuth D. G.; Feng P.; Gier T. E.; Sieger P.; Firouzi A.; Chmelka B. F. Organization of Organic Molecules with Inorganic Molecular Species into Nanocomposite Biphase Arrays. Chem. Mater. 1994, 6, 1176–1191. 10.1021/cm00044a016. [DOI] [Google Scholar]
- Beck J. S.; Vartuli J. C.; Kennedy G. J.; Kresge C. T.; Roth W. J.; Schramm S. E. Molecular or Supramolecular Templating: Defining the Role of Surfactant Chemistry in the Formation of Microporous and Mesoporous Molecular Sieves. Chem. Mater. 1994, 6, 1816–1821. 10.1021/cm00046a040. [DOI] [Google Scholar]
- Davies K. M.; Wink D. A.; Saavedra J. E.; Keefer L. K. Chemistry of the Diazeniumdiolates. 2. Kinetics and Mechanism of Dissociation to Nitric Oxide in Aqueous Solution. J. Am. Chem. Soc. 2001, 123, 5473–5481. 10.1021/ja002899q. [DOI] [PubMed] [Google Scholar]
- Coneski P. N.; Schoenfisch M. H. Nitric Oxide Release Part Iii. Measurement and Reporting. Chem. Soc. Rev. 2012, 41, 3753–3758. 10.1039/c2cs15271a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson J.; Rosenholm J.; Areva S.; Lindén M. Influences of Material Characteristics on Ibuprofen Drug Loading and Release Profiles from Ordered Micro- and Mesoporous Silica Matrices. Chem. Mater. 2004, 16, 4160–4167. 10.1021/cm0401490. [DOI] [Google Scholar]
- Lai C.-Y.; Trewyn B. G.; Jeftinija D. M.; Jeftinija K.; Xu S.; Jeftinija S.; Lin V. S. Y. A Mesoporous Silica Nanosphere-Based Carrier System with Chemically Removable Cds Nanoparticle Caps for Stimuli-Responsive Controlled Release of Neurotransmitters and Drug Molecules. J. Am. Chem. Soc. 2003, 125, 4451–4459. 10.1021/ja028650l. [DOI] [PubMed] [Google Scholar]
- Winkler H.; Birkner A.; Hagen V.; Wolf I.; Schmechel R.; Seggern H. v.; Fischer R. A. Quantum-Confined Gallium Nitride in Mcm-41. Adv. Mater. 1999, 11, 1444–1448. 10.1002/(SICI)1521-4095(199912)11:17<1444::AID-ADMA1444>3.3.CO;2-8. [DOI] [Google Scholar]
- Cheng K.; Landry C. C. Diffusion-Based Deprotection in Mesoporous Materials: A Strategy for Differential Functionalization of Porous Silica Particles. J. Am. Chem. Soc. 2007, 129, 9674–9685. 10.1021/ja070598b. [DOI] [PubMed] [Google Scholar]
- Kecht J.; Schlossbauer A.; Bein T. Selective Functionalization of the Outer and Inner Surfaces in Mesoporous Silica Nanoparticles. Chem. Mater. 2008, 20, 7207–7214. 10.1021/cm801484r. [DOI] [Google Scholar]
- de Juan F.; Ruiz-Hitzky E. Selective Functionalization of Mesoporous Silica. Adv. Mater. 2000, 12, 430–432. 10.1002/(SICI)1521-4095(200003)12:6<430::AID-ADMA430>3.0.CO;2-3. [DOI] [Google Scholar]
- Dai S.; Shin Y.; Ju Y.; Burleigh M. C.; Lin J. S.; Barnes C. E.; Xue Z. A New Methodology to Functionalize Surfaces of Ordered Mesoporous Materials Based on Ion Exchange Reactions. Adv. Mater. 1999, 11, 1226–1230. 10.1002/(SICI)1521-4095(199910)11:14<1226::AID-ADMA1226>3.3.CO;2-H. [DOI] [Google Scholar]
- Bourlinos A. B.; Karakostas T.; Petridis D. Side Chain” Modification of Mcm-41 Silica through the Exchange of the Surfactant Template with Charged Functionalized Organosiloxanes: An Efficient Route to Valuable Reconstructed Mcm-41 Derivatives. J. Phys. Chem. B 2003, 107, 920–925. 10.1021/jp0205113. [DOI] [Google Scholar]
- Dance I. G. The Structural Chemistry of Metal Thiolate Complexes. Polyhedron 1986, 5, 1037–1104. 10.1016/S0277-5387(00)84307-7. [DOI] [Google Scholar]
- Son D.; Wolosiuk A.; Braun P. V. Double Direct Templated Hollow Zns Microspheres Formed on Chemically Modified Silica Colloids. Chem. Mater. 2009, 21, 628–634. 10.1021/cm8017852. [DOI] [Google Scholar]
- Riccio D. A.; Schoenfisch M. H. Nitric Oxide Release Part I. Macromolecular Scaffolds. Chem. Soc. Rev. 2012, 41, 3731–3741. 10.1039/c2cs15272j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheatley P. S.; Butler A. R.; Crane M. S.; Fox S.; Xiao B.; Rossi A. G.; Megson I. L.; Morris R. E. No-Releasing Zeolites and Their Antithrombotic Properties. J. Am. Chem. Soc. 2006, 128, 502–9. 10.1021/ja0503579. [DOI] [PubMed] [Google Scholar]
- Stasko N. A.; Fischer T. H.; Schoenfisch M. H. S-Nitrosothiol-Modified Dendrimers as Nitric Oxide Delivery Vehicles. Biomacromolecules 2008, 9, 834–841. 10.1021/bm7011746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossi L.; Montevecchi P. C. A Kinetic Study of S-Nitrosothiol Decomposition. Chem. - Eur. J. 2002, 8, 380–387. 10.1002/1521-3765(20020118)8:2<380::AID-CHEM380>3.3.CO;2-G. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.