

Abstract
The lack of an effective detection method for lung circulating tumor cells (CTCs) presents a substantial challenge to elucidate the value of CTCs as a diagnostic or prognostic indicator in lung cancer, particularly in non-small cell lung cancer (NSCLC). In this study, we prepared a capture surface exploiting strong multivalent binding mediated by poly(amidoamine) (PAMAM) dendrimers to capture CTCs originating from lung cancers. Given that 85% of the tumor cells from NSCLC patients overexpress epidermal growth factor receptor (EGFR), anti-EGFR was chosen as a capture agent. Following in vitro confirmation using the murine lung cancer cell lines (ED-1 and ED1-SC), cyclin E-overexpressing (CEO) transgenic mice were employed as an in vivo lung tumor model to assess specificity and sensitivity of the capture surface. The numbers of CTCs in blood from the CEO transgenic mice were significantly higher than those from the healthy controls (on average 75.3 ± 14.9 vs. 4.4 ± 1.2 CTCs/100 μL of blood, p<0.005), indicating the high sensitivity and specificity of our surface. Furthermore, we found that the capture surface also offers a simple, effective method for monitoring treatment responses, as observed by the significant decrease in the CTC numbers from the CEO mice upon a treatment using a novel anti-miR-31 locked nucleic acid (LNA), compared to a vehicle treatment and a control-LNA treatment (p<0.05). This in vivo evaluation study confirms that our capture surface is highly efficient in detecting in vivo CTCs and thus has translational potential as a diagnostic and prognostic tool for lung cancer.
Late detection and frequent recurrence of metastatic lung cancer makes it the leading cause of cancer-related mortality in the United States.1,2 Approximately 85% of lung cancers can be histologically defined as non-small cell lung cancer (NSCLC) and the majority of patients with NSCLC are diagnosed with an inoperable and rarely curable disease. Following the discovery of abnormal chest radiographs or computed tomography (CT) scans, lung tissue biopsies or fludeoxyglucose-positron emission tomography (FDG-PET) scans are typically performed to determine respectively the pathology and the clinical stage.3 Complications with lung tissue biopsy may arise such as pneumothorax, bleeding, infection, air embolism, and, rarely, tumor spreading along the track of the needle.4 Instead, “liquid biopsy” via detection of circulating tumor cells (CTCs) from peripheral blood is a promising alternative to detect lung cancer and its metastasis since CTCs are known to correlate with clinical stage, metastasis, and recurrence of lung cancers.5-8
However, currently available CTC detection methods to capture CTCs from NSCLC patients are inefficient and thus need for improvement. Although the only FDA-approved CTC detection method, CellSearch®, relatively successfully captured CTCs from the small cell lung cancer (SCLC) patients, it exhibited much lower sensitivity for CTCs from NSCLC patients.6,7 Additionally, CellSearch® failed to detect CTCs in blood from many NSCLC cases including Stage IV patients.6,7,9 Considering that NSCLC patients represent approximately 85% of all lung cancer patients, an improved CTC detection method with clinically significant sensitivity toward CTCs from NSCLC patients is urgently required.
We developed a CTC capture system that takes advantage of strong multivalent binding and modularity offered by PAMAM dendrimers, in order to provide a platform technology for effective detection of various types of CTCs. We previously showed that a biomimetic approach that exploits the naturally occurring strong multivalent binding effect mediated by surface-modified generation 7 (G7) PAMAM dendrimers markedly improved efficiency of tumor cell capture.10 Moreover, following thorough screening of various cancer-specific capture agents, our platform could be functionalized with various tumor-cell-specific antibodies in pattern to detect CTCs from diverse cancer types with high sensitivity and specificity.11 Hence, it was hypothesized that a combination of the dendrimer-mediated multivalent binding effect and the NSCLC-specific capture agent would result in enhanced surface capture of the NSCLC cells.
Clinically relevant in vivo murine models of lung carcinogenesis can be used in place of human blood samples to validate the sensitivity and specificity of the lung cancer-adapted capture platform. Such murine transgenic mouse lines were generated to mimic aberrant cyclin E expression, a negative prognostic indicator in the NSCLC patients, and the cyclin E-overexpressing (CEO) mice have a higher incidence and rapid onset of lung carcinogenesis.12,13 The CEO mouse model mimics key features of clinical lung carcinogenesis allowing for better understanding of lung cancer biology and molecular therapeutics. For example, the CEO mice were used to develop and test an engineered antagonist (locked nucleic acid) against microRNA-31 (anti-miR-31) as a therapeutic agent.14 Additionally, the CEO mice were used to establish in vitro murine lung cancer cell lines: a wild-type cyclin E-driven lung cancer cell line (ED-1) and invasive ED1-SC harvested from tumor of FVB/N mice after subcutaneous injection of ED-1 cells.
In this study, we performed a series of experiments to assess our stated hypothesis. First, three capture agents for NSCLC were screened using ED-1 and ED1-SC to develop a multivalent capture platform that is optimized for effective detection of lung cancer cells (Figure 1a). The detection sensitivity and specificity of the chosen capture surfaces were then measured using blood samples drawn independently from both wild-type and transgenic CEO mice with lung tumors. Lastly, the NSCLC-specific capture surface was investigated in its applicability as a monitoring tool of therapeutic effects by counting CTCs in peripheral blood after anti-miR-31 treatment in CEO transgenic mice. This study provides evidence that validates the potential of our multivalent capture surface to be clinically translated into a diagnostic and prognostic tool to detect disseminated NSCLC CTCs.
Figure 1.
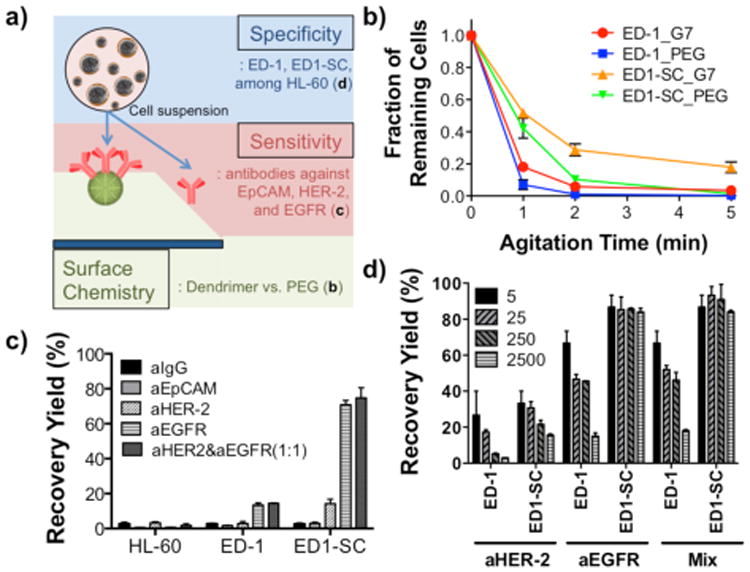
Selectivity and sensitivity screening of a G7 PAMAM dendrimer-immobilized platform with antibodies against surface markers using in vitro murine lung cancer cell lines, ED-1 and ED1-SC. a) A schematic illustration of this study investigating polymer chemistry, sensitivity, and specificity. b) Enhanced binding between anti-EGFR and cancer cells through dendrimer-mediated multivalent binding. c) Antibody screening for capturing of mouse lung cancer cells. All antibodies were functionalized on dendrimer-immobilized surfaces. Anti-EGFR antibody showed the highest binding with the both lung cancer cells, but not with HL-60, a leukocyte model. d) High recovery of the both cells from cell mixtures (in a range of 40-10,000 ED-1 or ED1-SC cells mixed with 1 × 107 HL-60 cells were used) using anti-EGFR. (n=3, All error bars: standard error (S.E.)).
Materials and Methods
Materials
Anti-human epithelial-cell-adhesion-molecule (EpCAM)/TROP1 antibody (anti-EpCAM) and anti-human epidermal growth factor receptor-2 (HER-2)/TROP1 antibody (anti-HER-2) were purchased from R&D systems (Minneapolis, MN). Anti-human epidermal growth factor receptor (EGFR) antibody (anti-EGFR, N-20) and unconjugated goat anti-mouse IgG antibody (H+L, anti-IgG) were acquired from Santa Cruz Biotech (Dallas, TX) and Pierce Biotechnology, Inc (Rockford, IL), respectively. All antibodies except anti-IgG used are polyclonal antibodies and cross-react with both mice and human proteins. Epoxy-functionalized glass surfaces (SuperEpoxy2®) were purchased from TeleChem International, Inc (Sunnyvale, CA). PAMAM dendrimers (G7), bovine serum albumin (BSA), Calcein AM, and all other chemicals were obtained from Sigma-Aldrich (St. Louis, MO).
Surface preparation
Antibodies against human epithelial markers, such as anti-EpCAM, anti-HER-2, and anti-EGFR, at a concentration of 5 μg/mL in PBS were immobilized on either PEG- or dendrimer-surfaces by overnight incubation. The PEG- and dendrimer-immobilized surfaces were prepared from an epoxy-functionalized glass slide under the optimized condition as we previously reported.10,11 Briefly, epoxy-functionalized slides were PEGylated by adding 0.5 μg/mL of a heterobifunctional PEG (NH2-(PEG)-COOH, 5 kDa, Nektar Therapeutics (Huntsville, AL)) in DDI water for 4 hrs. Some surfaces were then further functionalized with dendrimers by adding an excess amount of partially carboxylated G7 PAMAM dendrimers (50 μM in PBS buffer (pH 9.0)) for 6 hrs after EDC/NHS activation (an 1:1 mixture of EDC/NHS in DDI water for 1 hr). Note that G7 PAMAM dendrimers were used to accommodate multiple antibodies per dendrimer, considering their sizes (molecular weights of typical antibodies and G7 PAMAM dendrimers are both approximately 100 kDa). To prevent non-specific binding, the antibody-immobilized surfaces were treated with 1 (w/v)% bovine serum albumin in PBS or 1 μg/mL methoxy PEG-NH2 (Nektar Therapeutics) in DDI water for 4 hrs. The volume of all reagents used in this preparation was fixed at 300 μL. All incubation processes were carried out at room temperature with constant, gentle shaking, and the surfaces were washed between steps with DDI water and PBS to remove residual chemicals from the surfaces. The functionalized surfaces were immediately used for the subsequent experiments.
Cell culture and Binding assays
ED-1 and ED1-SC cells were grown in RPMI media supplemented with 10% (v/v) FBS and 1% (v/v) penicillin/streptomycin in a humidified incubator at 37°C and 5% CO2. HL-60 cells purchased from ATCC (anassas, VA) were cultured in Iscove's Modified Dulbecco's Medium (IMDM) supplemented with 10% (v/v) FBS and 1% (v/v) penicillin/streptomycin. Prior to all experiments, the ED-1 and ED1-SC cells were transferred into tissue culture dishes at concentrations of 1 × 106 cells/mL two days before experiments. To fluorescently label the cancer cells for the binding assays, the ED-1 and ED1-SC cells were stained while on the flask with 4 μM of calcein AM in Ca2+-included PBS for 30 min before all binding experiments were conducted. The final cell suspensions, at a concentration of 8 × 105 cells/ml, were placed on ice throughout the subsequent cell experiments.
For the binding assay, each antibody-immobilized surface was incubated with cancer cell suspensions (total 2 × 105 cells per surface) for 10 min at 37°C. Unbound cells were removed from the surface by washing with PBS buffer. The bound cells on PEG- or dendrimer-immobilized surfaces were agitated using a shaker (600 rpm) for a total of 5 min, and the number of the remaining cells on the surfaces was recorded. To measure binding specificity, different numbers of Calcein AM-labeled cancer cells (20, 100, 1,000, and 10,000 cancer cells) were resuspended in 1 mL of PBS containing 2 × 107 cells/mL HL-60 cells. These mixed cell suspensions were added onto each antibody-immobilized surface and incubated at 37°C for 10 min. The unbound cells were removed from the surface by manual washing with PBS buffer. Based on the loading cell number, the recovery yield (%) was calculated.
Transgenic mice and their treatments
The described animal protocols were reviewed and approved by Dartmouth's Institutional Animal Care and Use Committee (IACUC). To compare the number of CTC in mouse blood from wild-type FVB mice and transgenic CEO mice that develop spontaneous lung tumors, six to seven mice of each arm were used. The following study was then performed to compare the number of CTC in mouse blood in correlation to lung pathology using anti-miR-31 LNA as a novel therapeutic strategy. Seven- month old, sex-matched CEO mice were each injected with either vehicle (PBS with 10% mouse serum (Invitrogen, Carlsbad, CA)), control LNA (3 mg/ml), or anti-miR-31 LNA (3 mg/ml) via tail veins once daily for 5 days before being euthanized using an IACUC-approved protocol at 24 hrs after the last treatment. Note that three mice treated with vehicle were 17 months old. Five to six mice of each treatment group were used. Blood for CTC detection was drawn via cardiac puncture and lung tissues were harvested, formalin-fixed and paraffin-embedded, processed and sectioned for histopathology and hematoxylin and eosin (H&E) staining. The pathologist was blinded to the treatment administered.
Buffy coat isolation from blood samples
The withdrawn mouse blood was kept at 4°C and the experiments were performed within 24 hrs after receiving the blood samples. In a round bottom-tube, a sequence of the following solutions was loaded without mixing: 1 mL of Ficoll-Paque Plus (Stemcell Technologies Inc., Vancouver, Canada) and up to 1 mL of the mouse blood diluted in 1 mL of PBS buffer (3 mL in total volume). The PBS buffer was supplemented with 2% FBS and EDTA. The round bottom-tubes were centrifuged at 20°C for 20 min at 1,500× g with brake function off. The upper plasma layers were removed without disturbing the plasma-Ficoll interphase. One milliliter of the mononuclear cell layer at the plasma-Ficoll interphase (buffy coat) was transferred into a new round bottom-tube without disturbing the erythrocyte/granulocyte pellets in the bottom layer. The buffy coat including the cancer cells was washed twice with FBS/EDTA-included PBS buffer for 10 min at 2,000 rpm with the brake function on. The recovered cells were resuspended in 500 μL of RPMI culture media.
CTC capture and identification
The antibody-immobilized surfaces were incubated with the blood samples at 37°C for 1 hr. Unbound cells were removed from the surfaces by washing with PBS buffer. After fixing the captured cells with 4% paraformaldehyde for 15 min and washing with PBS, the fixed cells were permeabilized with 1% TritonX in PBS for 15 min and treated with 5% BSA for 1 hr. CTCs were identified using a multi-step process. First, the cells were immunostained overnight with the first primary antibodies: rabbit anti-wide spectrum cytokeratin (1:50). Second, the first secondary immunofluorescent-tagged antibodies, goat anti-rabbit Alexa Fluor 594 (red, 1:500), were incubated for 1 hr for signal amplification. Then the cells were immunostained with the second primary antibodies, mouse IgG1 anti-CD45 (1:1,000), for 1 hr. Finally, the second secondary immunofluorescent-tagged antibodies, goat anti-mouse IgG1 Alexa Fluor 488 (green, 1:500), were incubated for 1 hr. Antibodies were diluted using 5% BSA-PBS buffer, and the surfaces were washed with PBS buffer three times. After staining the nuclei with DAPI for 2 min, the surfaces were sealed with mounting media to obtain images under a fluorescence microscope (Olympus IX70 inverted microscope, Olympus America, Inc., Center Valley, PA). Images were recorded using a 10× objective and a CCD camera (QImaging Retiga 1300B, Olympus America, Inc.). The number of cells on the surfaces was counted, based on the images taken in independent observations/measurements using ImageJ (NIH).
Results and Discussions
Enhanced binding stability through dendrimer-mediated multivalent binding
For our dendrimer-functionalized capture platform to be used for detection of NSCLC-derived CTCs, the surface had to be customized via immobilization of lung cancer-specific CTC surface markers selected from literature and binding affinity screenings of antibodies using in vitro lung cancer cell lines (Figure 1). For NSCLC cells, binding counterparts for several surface markers of the tumor cells (e.g. epidermal growth factor receptor (EGFR), human epidermal growth factor receptor-2 (HER-2), and epithelial cell adhesion molecule (EpCAM)) would be reasonable for use as capture agents. The expression of EGFR, a member of the ErbB family of transmembrane tyrosine kinase receptors, is detected in up to 85% of NSCLC.15 Abundant expression of EGFR in NSCLC tissue relative to adjacent normal lung tissues is reported to associate with a poor survival outcome for lung cancer patients and an increased recurrence after tissue resection.15,16 HER-2 has been found to be overexpressed on NSCLC cell lines such as NCI-H1650 and NCI-H1466,17 and EpCAM is commonly targeted for CTC detection including those originated from lung cancer.6,7
Binding enhancement through dendrimer-mediated multivalent binding effect was first observed using ED-1 and ED1-SC via a static agitation assay. Given that the significant enhancement of cancer cell binding was shown with anti-EpCAM and anti-HER-2 in our previous publications,10,11 the newly employed capture agent, anti-EGFR, was tested to confirm whether the multivalent binding effect is applicable to the use with this antibody. Compared to the linear poly(ethylene glycol) (PEG)-coated surface with anti-EGFR, the G7 PAMAM dendrimer-immobilized surface with anti-EGFR showed modestly improved recovery yields (percentage of the number captured on the surface out of the initial number of cells added) for both lung cancer cell lines, ED-1 (4.6% increase) and ED1-SC (9.8% increase), as shown in Supporting Information, Figure S1. However, one of the most unique characteristics of multivalent binding is its substantially decreased dissociation kinetics, rather than association kinetics.10,18 By measuring fractions of remaining cells (Figure 1b) on the surfaces after agitation, the dissociation rate constants (kd, Supporting Information, Table S1) were calculated, which can be used to determine relative binding strength between the cells and the surface.10 Our data showed that the binding between the cancer cells and the anti-EGFR-dendrimer-immobilized surface was much more stable than that on the anti-EGFR-PEG-coated surface, as demonstrated by the increased fraction of the remaining cells and decreased dissociation rate constant (∼1.5 fold decrease) over static agitation. It is notable that the more invasive ED1-SC cells generally showed the greater degree of enhanced binding stability on the anti-EGFR-dendrimer-immobilized surfaces than ED-1 cells. Although the enhancement in binding kinetics as a result of incorporation of dendrimers was less significant, compared to our prior report,10 the improved dissociation kinetics support that the use of dendrimers as a linker for antibody immobilization provides enhanced detection.
In addition to the multivalent binding effect, the enhanced capture efficiency of the dendrimer surface observed in this study could be due to the high local density of the antibodies immobilized on the surface enabled by the nano-scale topography created by the dendrimers.10 We previously showed that the highly localized, many functional groups of the dendrimers significantly increased the local density of the antibodies, improving the overall capture efficiency.10,11
Next, the recovery yields of ED-1 and ED1-SC were compared among the capture surfaces functionalized with different proteins, as shown in Figure 1c. Four antibodies, anti-IgG, anti-EpCAM, anti-HER-2, and anti-EGFR, were separately immobilized onto individual dendrimer-functionalized surfaces. Among the four surfaces, the anti-IgG-immobilized surface was used as a negative control. The EpCAM–/HER-2–/EGFR– HL-60 cells exhibited little-to-no binding to any of the antibody-immobilized surfaces, confirming that all the bindings observed in this experiment was a result of specific interactions. Both ED-1 and ED1-SC showed low binding on the anti-IgG- immobilized surface (2.7 ± 0.3% for ED-1 and 2.6 ± 0.6% for ED1-SC of recovery yields). The anti-EpCAM-immobilized surface also induced a similarly low degree of capture of ED-1 (1.6 ± 0.2%) and ED1-SC cells (2.9 ± 0.7%). These surface-marker-screening results demonstrated the limitation of EpCAM-based lung CTC detection, explaining in part why the anti-EpCAM-coated immunomagnetic beads (CellSearch®) exhibited low detection efficiency in clinical trials for NSCLC patients.5-7,19 The anti-HER-2-immobilized surface allowed for higher binding of ED1-SC (14.2 ± 2.6%), yet the binding of ED-1 (2.9 ± 1.2%) on the same surface was comparable to that on the anti-IgG- or anti-EpCAM-coated surfaces. Interestingly, anti-EGFR showed the highest recovery yields of both cells among the selected antibodies tested. ED1-SC cells displayed higher binding to the anti-EGFR-immobilized surface (70.8 ± 2.4% recovery yield) than ED-1 cells (13.5 ± 1.2% recovery yield). In addition, a mild synergistic enhancement was observed with an antibody mixture of anti-HER-2 and anti-EGFR (1:1, (v/v)) where marginal increases in recovery yield by 0.9 and 4.0% were observed for ED-1 and ED1-SC, respectively.
The binding specificity of the surfaces was then tested using ED-1 and ED1-SC cells suspended with the leukocyte model, HL-60 cells. Different numbers of the cancer cells ranging between 40-10,000 cells were spiked into 1 mL of PBS solution containing ten million HL-60 cells to simulate the physiological density of CTCs among leukocytes.19 The recovery yields of ED1-SC cells on all anti-body-immobilized surfaces were generally higher than those of ED-1 cells (Figure 1d). Under the three conditions using the two antibodies (anti-HER-2, anti-EGFR, and a 1:1 mixture of anti-HER-2 and anti-EGFR), the recovery yield of ED1-SC cells was as high as 93.3%. The recovery yield on the anti-HER-2-coated surfaces was the lowest at less than 33.3% regardless of the number of cancer cells added into the cell mixtures. Only marginal improvement (less than 6% increase) was observed using the mixture of anti-EpCAM and anti-EGFR (1:1, (v/v)). The highest binding of the tumorigenic ED1-SC cells on anti-EGFR and only modest antibody cocktail effect led us to use the anti-EGFR-immobilized dendrimer surfaces for subsequent experiments.
CTC Capture from blood of the CEO transgenic mice
CTC capture directly from blood of wild-type FVB/N and CEO transgenic mice with lung tumors was performed using the adapted platform with anti-EGFR (Figure 2a). The numbers of the captured CTCs per 100 μL of blood from both healthy and transgenic mice with lung tumors are plotted in Figure 2b. Significantly more CTCs were captured from the transgenic mice with macroscopic lung cancer (75.3 ± 14.9 CTCs per 100 μL, n=7) than the wild-type controls (4.4 ± 1.2 CTCs per 100 μL, n=6, p=0.0011), demonstrating the applicability of our technology to detect in vivo CTCs for the first time. Given that the blood from mice was collected via cardiac puncture, the detected CTCs from the wild-type mice (4.4 ± 1.2 CTCs) could be due to the contamination with normal epithelial cells during the blood harvest.20 Among the captured cells, CTCs were identified via a series of immunostaining (Figure 2c), as previously reported.21,22 The cytokeratin-and DAPI-positive cells were identified as CTCs (red-fluorescent cells in Figure 2d) whereas the CD45-positive, nucleated cells on the surface were identified as leukocytes (green-fluorescent cells in Figure 2d). In addition, detailed information regarding the histological analysis and the CTC numbers are summarized in Supporting Information, Table S2. The histology results corresponded to the number of captured CTCs from blood, i.e., the more tumorigenic properties such as multiple lung and metastatic tumors, the more CTCs in peripheral blood (Supporting Information, Table S2). Additional fluorescence images of the captured cells on the anti-EGFR-G7 surfaces are shown in Supporting Information Figure S1, which indicates that significantly more CTCs were captured from the blood of the CEO mice than that of the healthy control.
Figure 2.
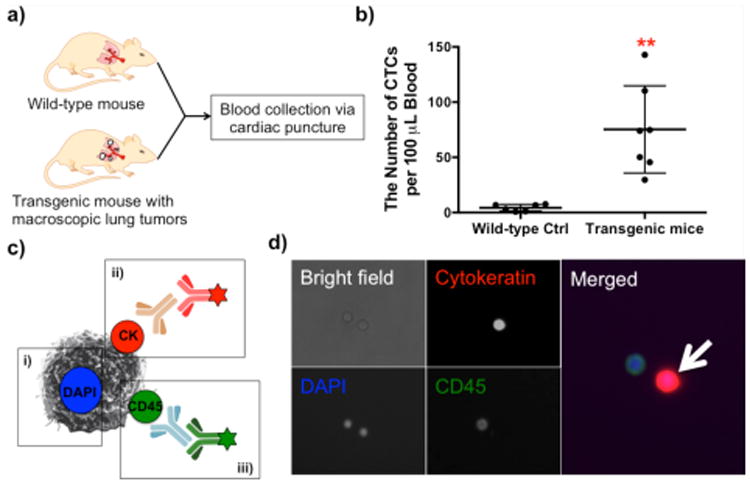
CTC detection from healthy and transgenic mice using an anti-EGFR-G7 dendrimer-immobilized platform. a) An illustration of the experiment independently using wild-type FVB/N mice and CEO transgenic mice. b) Comparison of the captured cell numbers per 100 μL blood between two groups on the anti-EGFR-dendrimer surfaces. A significantly higher number of CTCs (75.3 ± 14.9 CTCs per 100 μL, n=7) was captured from the transgenic mice than that (4.4 ± 1.2 CTCs per 100 μL, n=6, p=0.0011) from wild-type controls. (Mean ± S.E., **p<0.005) c) An immunostaining scheme of the captured cells and d) its representative images: i) DAPI staining, ii) cytokeratin staining, and iii) CD45 staining. The cell with a white arrow was identified as a CTC.
The results obtained using the CEO transgenic mouse model present advantages for in vivo validation for our CTC device to be eventually used for human NSCLC detection. First, the CEO transgenic mouse model has similar features to human lung cancer. Other carcinogen-induced or genetically engineered models of lung cancer rarely exhibit metastasis and frequently develop pulmonary adenomas rather than invasive tumors.23,24 The CEO transgenic mice mimic many features of the human lung carcinogenesis including premalignancy and multiple sites of lung cancers, providing a good model mimicking the human disease.12 Secondly, the correlation between the number of CTCs and NSCLC metastasis in multiple sites of dissemination can be confirmed by histological analysis of the CEO transgenic mice. Lung cancer metastasis is correlated to the number of CTCs in peripheral blood, which is supported by our observation of the substantially higher number of CTCs in the CEO mice than in the wild-type mice (Figure 2b). In fact, the CEO transgenic mice with a higher number of CTCs in peripheral blood showed correlation with more macroscopic tumors, as confirmed by the histology analysis (Supporting Information, Table S2). Thirdly, the validation of a CTC detection system using the CEO mice can overcome limitations of in vitro validation that lacks the heterogeneity of the surface marker expression on CTCs and the high number and variety of hematological cells in blood.
Therapeutic effect monitored by changes in CTC numbers in the blood of transgenic mice
To evaluate the ability of our CTC detection platform for therapeutic effect monitoring, anti-miR-31 locked nucleic acid (LNA) was chosen as a representative therapeutic approach for the treatment of the transgenic mice. miR-31 is overexpressed by NLSLC cells and targets tumor suppressors, such as large tumor suppressor 2 (Lats2) and PP2A regulatory subunit B alpha isoform (Ppp2r2a), consistent with miR-31 acting as an oncomir.14 Targeting miR-31 by anti-miR oligonucleotides is reported to decrease cellular proliferation in mouse and human lung cancer, increase the expression of the tumor suppressors, and subsequently suppress lung cancer proliferation in a dose-dependent manner.14 Therefore, the anti-miR-31 LNA has potential as a therapeutic agent in NLSLC cancer. In an effort to validate our CTC detection platform as a tool for monitoring in vivo responses to therapeutic treatments, changes in CTC numbers were measured in blood from the CEO transgenic mice and correlated to the therapeutic effect of anti-miR-31 LNA (Figure 3a).
Figure 3.
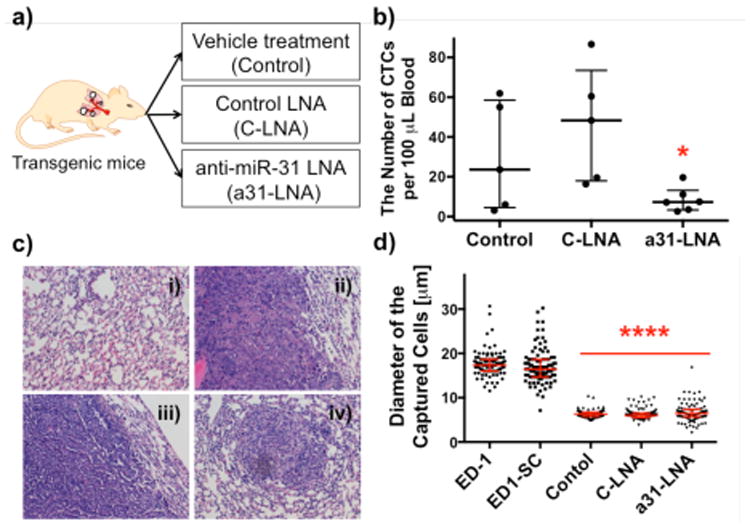
Comparison of the number and size of captured cells among three experimental groups on anti-EGFR-G7 dendrimer-immobilized surfaces. a) A schematic illustration of the three experimental groups of CEO transgenic mice to monitor therapeutic effects of anti-miR-31 LNA (a31-LNA). b) CTC numbers detected from the three groups. The group treated with a31-LNA (8.6 ± 2.5 CTCs, n=6, p= 0.0484) showed a significant decrease in CTC number compared to the other two groups. The CTC number in the group treated with C-LNA (46.3 ± 13.1 CTCs, n=5) was not statistically significantly different from that in the control CEO mice with vehicle treatment (29.96 ± 12.2 CTCs, n=5). (mean ± S.E., *p<0.05) c) Representative histology images of lung tissues of the CEO mice: i) healthy lung and ii) lung tumors formed in the vehicle-treated control; iii) lung tumors formed in the control LNA (C-LNA) group; and iv) shrunk lung tumor and close-to-the-normal lung tissue morphology found in the a31-LNA group. All images were taken at 20× magnification. d) Size distribution of the captured cells on the anti-EGFR-dendrimer surfaces. The cells from in vitro cell lines (17.7 ± 2.8 um for ED-1 and 17.1 ± 3.9 μm for ED1-SC) were larger than those from in vivo animal blood (6.4 ± 0.9 μm for control, 6.3 ± 1.2 μm for C-LNA, and 6.9 ± 2.1 μm for a31-LNA). (mean ± S.D., ****p<0.0001).
To monitor the therapeutic effect of anti-miR-31 LNA, we counted the number of captured CTCs per 100 μL blood of the transgenic mice after anti-miR-31LNA, control LNA, or vehicle treatment, as shown in Figure 3b. The average number of CTCs in the anti-miR-31 LNA-treated group was measured to be 8.6 ± 2.5, which was substantially lower than other groups (vehicle- and control LNA-treated groups). The control LNA-treated mice showed 46.3 ± 13.1 CTCs that were higher than the vehicle-treated group (29.9 ± 12.2 CTCs), although the difference was not statistically significant. Generally, our CTC capture surface detected the lowest number of CTCs in blood as well as the lowest occurrence of lung cancers from the anti-miR-31 LNA-treated mice. The histology results along with the number of CTCs are summarized in Supporting Information, Table S3. Note that not all mice developed lung cancer, which was typically observed12; however, once the tumor was developed, the mice treated with either vehicle or control LNA exhibited more advanced tumors than the a31-LNA-treated mice (Figure 3c). Although we did not observe that the anti-miR-31 LNA treatment reduced tumors in the CEO mice during the study period, the anti-miR-31 LNA-treated mice exhibited less lung tumors than the control LNA group, which was correlated to the lower number of CTCs counted from the anti-miR-31 LNA group.
Our results indicate that the unique combination of the CEO transgenic mouse model and the highly sensitive CTC device can provide a promising method for preclinical drug screening to develop tools to identify, validate, and optimize new therapeutic and preventive regimens for lung cancers. We have observed that the high number of CTCs in blood of the CEO transgenic mice indicates an adverse prognosis after therapy (Figure 3b). The results shown in this paper prove the sensitivity and specificity of our dendrimer-based system with anti-EGFR in capturing CTCs from blood as well as in examining the therapeutic effects of drug candidates.
Size difference between in vivo CTCs and in vitro cancer cells
It is notable that the sizes of the captured tumor cells from in vitro cell lines and in vivo CEO transgenic mice were significantly different, as shown in Figure 3d. The captured ED-1 and ED1-SC cells were measured to be 17.7 ± 2.8 μm and 17.1 ± 3.9 μm in diameter, respectively, which were significantly larger than the in vivo CTCs (6.4 ± 0.9 μm for the vehicle-treated control and 6.3 ± 1.2 μm for the control LNA group). Furthermore, CTCs from the anti-miR-31 LNA treatment group (6.9 ± 2.1 μm) had a variation in cell size than other groups tested in this study. The average diameters of the captured CTCs between in vitro cell lines and transgenic mice were significantly different (p<0.0001).
Most of the existing size-based filtration systems are based on an assumption that epithelial tumor cells would be larger than normal hematological cells including leukocytes. However, the significantly smaller size of the CTCs from mouse blood than the in vitro cell lines shown in Figure 3d suggests that the size-based separations may present limitations, especially in clinical situations.25-27 This potential issue is further supported by a study where, in human cancer patients, the CTC size was measured in a broad range of 4-30 μm in diameter.8 It is also reported that, due to the portion of CTCs that is smaller than leukocytes, a size-based filtration system showed the limited capture efficiency.8 It is therefore obvious that our approach using the antibody-functionalized dendrimer surface is of high potential in providing highly sensitive and specific detection of CTCs that may not be achieved by some of the size-based filtration systems.29,30
Conclusions
In conclusion, a versatile, dendrimer-based CTC detection platform was adapted for lung cancer CTCs by immobilization of anti-EGFR that is specific to NSCLC cells including ED-1 and ED1-SC, and used to measure murine CTCs from peripheral blood of CEO transgenic mice. The correlation between the number of CTCs and lung cancer development in the transgenic mice was determined, showing that CTCs detected using our platform can serve as an indicator for lung cancer development. We also validated this CTC detection platform as a way to monitor the therapeutic effect of anti-miR-31 using the transgenic mouse model, which demonstrates that CTC monitoring has potential for translational studies in lung cancer biology and therapy. All in all, our study presents a new CTC methodology for NSCLC, offering a promising way to monitor cancer progress and responsiveness to therapeutic intervention.
Supplementary Material
Acknowledgments
This work was supported by National Cancer Institute (NCI), National Institutes of Health (NIH) under grant # R01-CA182528 (SH) and National Science Foundation (NSF) under grant # DMR-1409161 (SH). This work was also supported in part by NIH and NCI grants R01-CA062275 (ED and SJF) and R01-CA087546 (ED and SJF), and R01-CA190722 (ED and SJF) as well as by a Samuel Waxman Cancer Research Foundation Award (ED), a UT-STARS award (ED) and by a grant from Uniting Against Lung Cancer with Mary Jo's Fund to Fight Cancer (SJF).
Footnotes
Supporting Information: Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
Dissociation rate constants of the surfaces, histology results and measured CTC numbers from blood of healthy and transgenic mice as well as anti-miR-31 treated mice, and representative fluorescence images of the captured CTCs
Conflict of Interest: S.H. is a co-founder of Capio Biosciences, Inc. and has a significant equity share of the company.
Author Contributions: J.H.M., M.R., E.D., S.J.F., and S.H. planned and designed the experiments. J.H.M. conducted the experiments to develop CTC capture platforms and to capture CTCs from blood samples. M.R., and S.J.F. conducted the experiments for animal studies. T.M. and V.A.M conducted H&E staining for mouse tissue samples. J.H.M., K.A.T., and S.H. analyzed the data and wrote the manuscript. S.H. supervised the overall project.
References
- 1.Jemal A, Thun MJ, Ries LAG, Howe HL, Weir HK, Center MM, Ward E, Wu XC, Eheman C, Anderson R, Ajani UA, Kohler B, Edwards BK. J Natl Cancer I. 2008;100:1672–1694. doi: 10.1093/jnci/djn389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Edwards BK, Noone AM, Mariotto AB, Simard EP, Boscoe FP, Henley SJ, Jemal A, Cho H, Anderson RN, Kohler BA, Eheman CR, Ward EM. Cancer-Am Cancer Soc. 2014;120:1290–1314. doi: 10.1002/cncr.28509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pfister DG, Johnson DH, Azzoli CG, Sause W, Smith TJ, Baker S, Jr, Olak J, Stover D, Strawn JR, Turrisi AT, Somerfield MR. J Clin Oncol. 2004;22:330–353. doi: 10.1200/JCO.2004.09.053. [DOI] [PubMed] [Google Scholar]
- 4.Robertson EG, Baxter G. Clin Radiol. 2011;66:1007–1014. doi: 10.1016/j.crad.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 5.Tanaka F, Yoneda K, Kondo N, Hashimoto M, Takuwa T, Matsumoto S, Okumura Y, Rahman S, Tsubota N, Tsujimura T, Kuribayashi K, Fukuoka K, Nakano T, Hasegawa S. Clin Cancer Res. 2009;15:6980–6986. doi: 10.1158/1078-0432.CCR-09-1095. [DOI] [PubMed] [Google Scholar]
- 6.Hou JM, Greystoke A, Lancashire L, Cummings J, Ward T, Board R, Amir E, Hughes S, Krebs M, Hughes A, Ranson M, Lorigan P, Dive C, Blackhall FH. Am J Pathol. 2009;175:808–816. doi: 10.2353/ajpath.2009.090078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krebs MG, Sloane R, Priest L, Lancashire L, Hou JM, Greystoke A, Ward TH, Ferraldeschi R, Hughes A, Clack G, Ranson M, Dive C, Blackhall FH. J Clin Oncol. 2011;29:1556–1563. doi: 10.1200/JCO.2010.28.7045. [DOI] [PubMed] [Google Scholar]
- 8.Allison M. Nat Biotechnol. 2010;28:999–1002. doi: 10.1038/nbt1010-999. [DOI] [PubMed] [Google Scholar]
- 9.O'Flaherty JD, Gray S, Richard D, Fennell D, O'Leary JJ, Blackhall FH, O'Byrne KJ. Lung Cancer. 2012;76:19–25. doi: 10.1016/j.lungcan.2011.10.018. [DOI] [PubMed] [Google Scholar]
- 10.Myung JH, Gajjar KA, Saric J, Eddington DT, Hong S. Angew Chem Int Edit. 2011;50:11769–11772. doi: 10.1002/anie.201105508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Myung JH, Gajjar KA, Chen J, Molokie RE, Hong S. Anal Chem. 2014;86:6088–6094. doi: 10.1021/ac501243a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma Y, Fiering S, Black C, Liu X, Yuan Z, Memoli VA, Robbins DJ, Bentley HA, Tsongalis GJ, Demidenko E, Freemantle SJ, Dmitrovsky E. Proc Natl Acad Sci USA. 2007;104:4089–4094. doi: 10.1073/pnas.0606537104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freemantle SJ, Dmitrovsky E. Cancer Prev Res. 2010;3:1513–1518. doi: 10.1158/1940-6207.CAPR-10-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu X, Sempere LF, Ouyang H, Memoli VA, Andrew AS, Luo Y, Demidenko E, Korc M, Shi W, Preis M, Dragnev KH, Li H, Direnzo J, Bak M, Freemantle SJ, Kauppinen S, Dmitrovsky E. J Clin Invest. 2010;120:1298–1309. doi: 10.1172/JCI39566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rusch V, Baselga J, Cordon-Cardo C, Orazem J, Zaman M, Hoda S, McIntosh J, Kurie J, Dmitrovsky E. Cancer Res. 1993;53:2379–2385. [PubMed] [Google Scholar]
- 16.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 17.Wroblewski JM, Bixby DL, Borowski C, Yannelli JR. Lung Cancer. 2001;33:181–194. doi: 10.1016/s0169-5002(01)00210-0. [DOI] [PubMed] [Google Scholar]
- 18.Hong S, Leroueil PR, Majoros IJ, Orr BG, Baker JR, Jr, Banaszak Holl MM. Chem Biol. 2007;14:107–115. doi: 10.1016/j.chembiol.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 19.Allard WJ, Matera J, Miller MC, Repollet M, Connelly MC, Rao C, Tibbe AGJ, Uhr JW, Terstappen LWMM. Clin Cancer Res. 2004;10:6897–6904. doi: 10.1158/1078-0432.CCR-04-0378. [DOI] [PubMed] [Google Scholar]
- 20.Zhang L, Ridgway LD, Wetzel MD, Ngo J, Yin W, Kumar D, Goodman JC, Groves MD, Marchetti D. Sci Transl Med. 2013;5 doi: 10.1126/scitranslmed.3005109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L, Smith MR, Kwak EL, Digumarthy S, Muzikansky A, Ryan P, Balis UJ, Tompkins RG, Haber DA, Toner M. Nature. 2007;450:1235–U1210. doi: 10.1038/nature06385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu M, Ting DT, Stott SL, Wittner BS, Ozsolak F, Paul S, Ciciliano JC, Smas ME, Winokur D, Gilman AJ, Ulman MJ, Xega K, Contino G, Alagesan B, Brannigan BW, Milos PM, Ryan DP, Sequist LV, Bardeesy N, Ramaswamy S, Toner M, Maheswaran S, Haber DA. Nature. 2012;490 doi: 10.1038/nature11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meuwissen R, Berns A. Genes Dev. 2005;19:643–664. doi: 10.1101/gad.1284505. [DOI] [PubMed] [Google Scholar]
- 24.Frese KK, Tuveson DA. Nat Rev Cancer. 2007;7:645–658. doi: 10.1038/nrc2192. [DOI] [PubMed] [Google Scholar]
- 25.Krebs M, Sloane R, Priest L, Lancashire L, Hou J, Clack G, Hughes A, Dive C, Blackhall F. J Thorac Oncol. 2010;50:S57–S57. doi: 10.1097/JTO.0b013e31823c5c16. [DOI] [PubMed] [Google Scholar]
- 26.Riethdorf S, Fritsche H, Muller V, Rau T, Schindibeck C, Rack B, Janni W, Coith C, Beck K, Janicke F, Jackson S, Gornet T, Cristofanilli M, Pantel K. Clin Cancer Res. 2007;13:920–928. doi: 10.1158/1078-0432.CCR-06-1695. [DOI] [PubMed] [Google Scholar]
- 27.Alix-Panabieres C, Pantel K. Nat Rev Cancer. 2014;14:623–631. doi: 10.1038/nrc3820. [DOI] [PubMed] [Google Scholar]
- 28.Alix-Panabieres C, Pantel K. Clin Chem. 2013;59:110–118. doi: 10.1373/clinchem.2012.194258. [DOI] [PubMed] [Google Scholar]
- 29.Myung JH, Gajjar KA, Han YE, Hong S. Polym Chem. 2012;3:2336–2341. [Google Scholar]
- 30.Lin MX, Hyun KA, Moon HS, Sim TS, Lee JG, Park JC, Lee SS, Jung HI. Biosens Bioelectron. 2013;40:63–67. doi: 10.1016/j.bios.2012.06.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.